

Abstract
Mixed lineage kinase domain‐like protein (MLKL) is the terminal effector of necroptosis, a form of regulated necrosis. Optimal activation of necroptosis, which eliminates infected cells, is critical for antiviral host defense. MicroRNAs (miRNAs) regulate the expression of genes involved in various biological and pathological processes. However, the roles of miRNAs in necroptosis‐associated host defense remain largely unknown. We screened a library of miRNAs and identified miR‐324‐5p as the most effective suppressor of necroptosis. MiR‐324‐5p downregulates human MLKL expression by specifically targeting the 3′UTR in a seed region‐independent manner. In response to interferons (IFNs), miR‐324‐5p is downregulated via the JAK/STAT signaling pathway, which removes the posttranscriptional suppression of MLKL mRNA and facilitates the activation of necroptosis. In influenza A virus (IAV)‐infected human primary macrophages, IFNs are induced, leading to the downregulation of miR‐324‐5p. MiR‐324‐5p overexpression attenuates IAV‐associated necroptosis and enhances viral replication, whereas deletion of miR‐324‐5p potentiates necroptosis and suppresses viral replication. Hence, miR‐324‐5p negatively regulates necroptosis by manipulating MLKL expression, and its downregulation by IFNs orchestrates optimal activation of necroptosis in host antiviral defense.
Keywords: antiviral defense, interferon, miR‐324‐5p, MLKL, necroptosis
Subject Categories: Autophagy & Cell Death; Microbiology, Virology & Host Pathogen Interaction; RNA Biology
Expression of MLKL, the terminal effector of necroptosis, is reduced by miR‐324‐5p. Interferon signaling decreases miR‐324‐5p levels via the JAK/STAT1 pathway, thereby stabilizing MLKL and potentiating necroptosis in influenza virus‐infected cells to limit viral replication.
Introduction
Cell death of pathogen‐infected cells plays a critical role in the defense of multicellular organisms against infection by eliminating pathogens (Guo et al, 2015a; Man et al, 2017; Nailwal & Chan, 2019; He & Han, 2020). Apoptosis is a form of programmed cell death controlled by a group of cysteine proteases called caspases (Kerr et al, 1972). Inhibition of caspase activity in some cells can switch cell fate from TNF‐induced apoptosis to necroptosis, a form of regulated necrosis (Degterev et al, 2005; He & Wang, 2018; Mifflin et al, 2020). Necroptosis is regulated by receptor‐interacting kinase 1 (RIP1 or RIPK1), RIPK3 (or RIP3), and pseudokinase mixed lineage kinase domain‐like protein (MLKL) (Degterev et al, 2005; He & Wang, 2018; Mifflin et al, 2020). Previous studies have shown that necroptosis mediates the premature death of infected cells leading to the restriction of viral replication; it is involved in the host defense against viruses including the vaccinia virus (Cho et al, 2009), murine cytomegalovirus (Upton et al, 2010), human herpes simplex viruses (Wang et al, 2014; Huang et al, 2015; Guo et al, 2015b), and influenza A viruses (IAVs) (Kuriakose et al, 2016; Nogusa et al, 2016; Thapa et al, 2016). Thus, necroptosis plays a crucial role in host defense by eliminating pathogen‐infected cells.
Necroptosis can be induced by the activation of tumor necrosis factor (TNF) family death receptors (Laster et al, 1988; Holler et al, 2000), interferon (IFN) receptors (Robinson et al, 2012), and Toll‐like receptors (TLRs) (He et al, 2011; Kaiser et al, 2013) and by pathogen infection (Nailwal & Chan, 2019) and endogenous retroviruses (Jiao et al, 2020; Wang et al, 2020). In TNF‐induced necroptosis, RIPK3 is activated by forming a protein complex (necrosome) with RIPK1 via their RIP homotypic interaction motif (RHIM) domains (Cho et al, 2009; He et al, 2009; Zhang et al, 2009). In IFN‐induced necroptosis, RIPK3 can be activated by the RHIM‐containing protein ZBP1 without RIPK1 (Kaiser et al, 2008; Ingram et al, 2019; Upton et al, 2019). The activated RIPK3 recruits and phosphorylates the substrate MLKL, resulting in MLKL oligomerization and translocation to the plasma membrane where it mediates necroptosis (Sun et al, 2012; He & Wang, 2018). As MLKL is a key effector molecule in the execution of necroptosis, its full activation is important for necroptosis‐mediated protective host defense.
Interferons are crucial for host defense against a broad range of pathogens (Karki et al, 2021; Shannon et al, 2021). The type I (IFN‐α/β) and type II (IFN‐γ) IFNs are secreted from pathogen‐infected cells and enhance immune responses by activating the Janus kinase (JAK)‐signal transducer and activator of transcription (STAT) signaling pathway, which triggers the expression of various interferon‐stimulated genes (ISGs) including pro‐inflammatory cytokines and chemokines and anti‐microbial and antigen‐presenting molecules (Barrat et al, 2019; Xu et al, 2019; Yang & Li, 2020; Wang et al, 2021). Previous studies have shown that MLKL and ZBP1 can be transcriptionally induced by IFN stimulation, leading to increased sensitivity of cells to necroptosis (Ingram et al, 2019; Knuth et al, 2019; Sarhan et al, 2019; Chen et al, 2019a). Therefore, IFN signaling can coordinate the death of pathogen‐infected cells by regulating the expression of pro‐necroptosis ISGs. In addition to the induction of ISGs, IFNs repress the transcription of numerous genes, termed IFN‐repressed genes (IRepGs) (Trilling et al, 2013; Megger et al, 2017). In contrast to ISGs, the functions of IRepGs remain poorly understood, and their role in necroptosis has not been documented.
MicroRNAs (miRNAs) are a class of endogenous small noncoding RNAs of 19–22 nucleotides that negatively regulate target genes by binding to complementary sequences of the 3′ untranslated region (3′UTR) of target mRNAs, thereby leading to mRNA degradation or translation suppression (Lewis et al, 2005; Agarwal et al, 2015). In general, nucleotides 2–8 at the 5′ end of the miRNA (called the seed region) are considered to be critical for canonical target mRNA recognition and binding (Bartel, 2018). MiRNAs are involved in cellular processes including cell proliferation, cell differentiation, and cell death (Roy et al, 2017; Farina et al, 2020; Zhang et al, 2020). Studies have shown that some miRNAs are involved in the modulation of necroptosis. For example, miR‐874 targets caspase‐8 to promote necroptosis (Wang et al, 2013). MiR‐21 deficiency attenuates necroptosis in mouse models of acute pancreatitis injury and liver injury (Ma et al, 2015; Afonso et al, 2018). MiR‐155 targets RIPK1 to inhibit necroptosis in human cardiomyocyte progenitor cells and osteosarcoma (Liu et al, 2011). MiR‐325‐3p suppresses cardiomyocyte necroptosis by targeting RIPK3 (Zhang et al, 2019). However, the roles of miRNAs in regulating necroptosis remain largely unknown. In particular, it is unclear whether miRNAs mediating necroptosis specifically regulate MLKL expression.
In the present study, we demonstrate that miR‐324‐5p negatively regulates necroptosis by directly downregulating the expression of human MLKL. We also identified miR‐324‐5p as being repressed by IFN through the JAK/STAT1 signaling pathway. Expression of miR‐324‐5p was downregulated in IAV‐infected human peripheral blood mononuclear cell (PBMC)‐derived macrophages. Overexpression of miR‐324‐5p significantly inhibited necroptosis of infected cells and promoted IAV replication, whereas deletion of miR‐324‐5p enhanced necroptosis and thus contributed to the control of viral infection. These results indicate that miR‐324‐5p is a cellular suppressor of MLKL‐mediated necroptosis and highlight the role of IFN‐mediated repression of miR‐324‐5p in host defense against infection.
Results
Identification of miR‐324‐5p as a suppressor of necroptosis
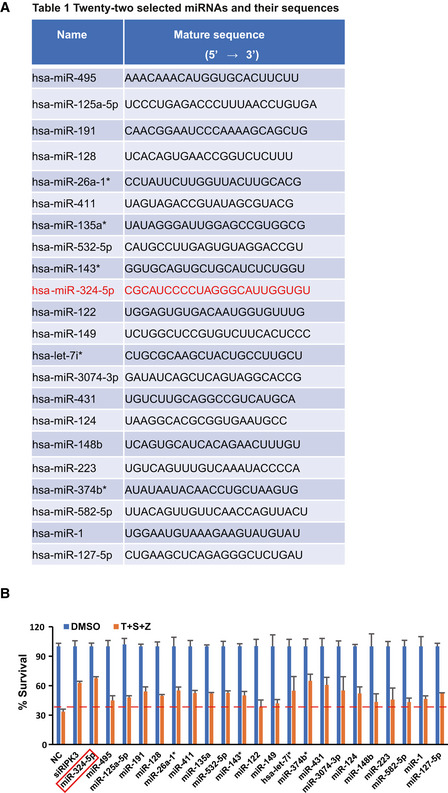
To identify miRNAs involved in the necroptosis pathway, we screened a library of ~1,000 miRNAs for candidate miRNAs regulating TNF‐induced necroptosis, a classical necroptotic pathway induced by stimulation of TNF‐α, Smac mimetic, and the pan‐caspase inhibitor z‐VAD (He et al, 2009). We performed this screen using human colon cancer HT‐29 cells, which were transfected with individual miRNAs and subsequently treated with the control (DMSO) or TNF‐α, Smac mimetic, and z‐VAD, then analyzed for cell viability (Fig 1A). A small interfering RNA (siRNA) oligo targeting RIPK3 was used as the positive control (Fig 1A). We found that 22 miRNAs could block TNF‐induced necroptosis. Cells transfected with these miRNAs showed a significantly higher survival rate compared with control miRNA‐transfected cells (Figs 1B and EV1A). Among these miRNAs, miR‐324‐5p has the greatest inhibitory effect on necroptosis (Figs 1C and EV1B). MiR‐324‐5p and miR‐324‐3p are transcribed from the same hairpin RNA structure. Unlike miR‐324‐5p, miR‐324‐3p had no effect on TNF‐induced necroptosis (Fig 1C), indicating that miR‐324‐5p and miR‐324‐3p have diverse effects on necroptosis. We further confirmed that miR‐324‐5p significantly inhibited necroptosis in multiple cell lines, including gastric carcinoma MKN45 cells and human colon cancer 174T cells (Fig 1D and E).
Figure 1. Identification of miR‐324‐5p as a suppressor of necroptosis.

-
ASchematic description of a screening for miRNAs blocking necroptosis. Human miRNA mimics, nontarget miRNA (the negative control, NC), and the positive control RIPK3 siRNA oligos (siRIPK3) were transferred into HT‐29. After 48 h, cells were treated with 40 ng/ml TNF‐α (T), 100 nM Smac mimetic (S), and 20 μM z‐VAD (Z) for 24 h, and then cell viability was determined by measuring ATP levels. Identical concentrations were used in later experiments unless otherwise stated. T + S + Z: TNF‐α, Smac mimetic and z‐VAD.
-
BGraphical representation of the screen results with each miRNA identified from the library (842 miRNAs). Arrowheads point to siRIPK3, miR‐324‐5p, and the negative control (NC).
-
CThe RNA sequences of miR‐324‐5p and miR‐324‐3p were aligned by DNAman. HT‐29 cells were transfected with NC, siRIPK3, miR‐324‐5p, or miR‐324‐3p. After 48 h, cells were treated with T + S + Z for 24 h. Cell viability was determined by measuring ATP levels.
-
DHuman gastric carcinoma MKN45 cells were transfected with NC, siRIPK1, or miR‐324‐5p. After 48 h, cells were treated with T + S + Z for 24 h. Cell viability was determined by measuring ATP levels.
-
EHuman colon cancer 174T cells were transfected with NC or miR‐324‐5p. After 48 h, cells were treated with T + S + Z for 24 h. Cell viability was determined by measuring ATP levels.
-
FHuman glioblastoma T98G cells were transfected with NC, siRIPK1, or miR‐324‐5p. After 48 h, cells were treated with T + S for 24 h. Cell viability was determined by measuring ATP levels.
-
GHuman monocytic leukemia U937 cells were transfected with NC, siRIPK3, or miR‐324‐5p. At 48 h, cells were treated with 100 ng/ml LPS (L) and 20 μM z‐VAD (Z) for an additional 24 h. Cell viability was determined by measuring ATP levels.
Data information: In (C–G), data are represented as the means ± SD of three biological replicates. Statistical analyses were performed using unpaired Student's t‐test. All experiments were performed at least three times, and representative data are shown.
Figure EV1. Screening for miRNAs regulating necroptosis.
-
ATwenty‐two miRNAs blocking necroptosis and their sequences.
-
BHT‐29 cells were transfected with NC, siRIPK3, or the indicated miRNAs at a final concentration of 50 nM. After 48 h, cells were treated with T + S + Z for an additional 24 h. Cell viability was determined by measuring ATP levels. Data are represented as the means ± SD of three biological replicates. Statistical analyses were performed using unpaired Student's t‐test.
Since TNF‐induced necroptosis and apoptosis share some common components such as TNFR and RIPK1, we examined the effect of miR‐324‐5p on TNF‐induced apoptosis, which is known to be induced by treatment with TNF‐α plus Smac mimetic (Wang et al, 2008). We observed that miR‐324‐5p had no obvious effect on TNF‐induced apoptosis in human glioblastoma T98G cells (Fig 1F), suggesting that miR‐324‐5p interferes with TNF‐induced necroptosis, but not with TNF‐induced apoptosis. TLR activation has been shown to activate necroptosis as well (He et al, 2011). We further evaluated the effect of miR‐324‐5p on TLR4‐mediated necroptosis induced by lipopolysaccharide (LPS), a specific ligand of TLR4, in the presence of z‐VAD in human monocytic leukemia U937 cells. Similar to TNF‐induced necroptosis, TLR4‐mediated necroptosis of U937 cells was blocked by miR‐324‐5p (Fig 1G). Collectively, these results demonstrate that miR‐324‐5p acts as a suppressor of necroptosis.
MiR‐324‐5p negatively regulates MLKL expression
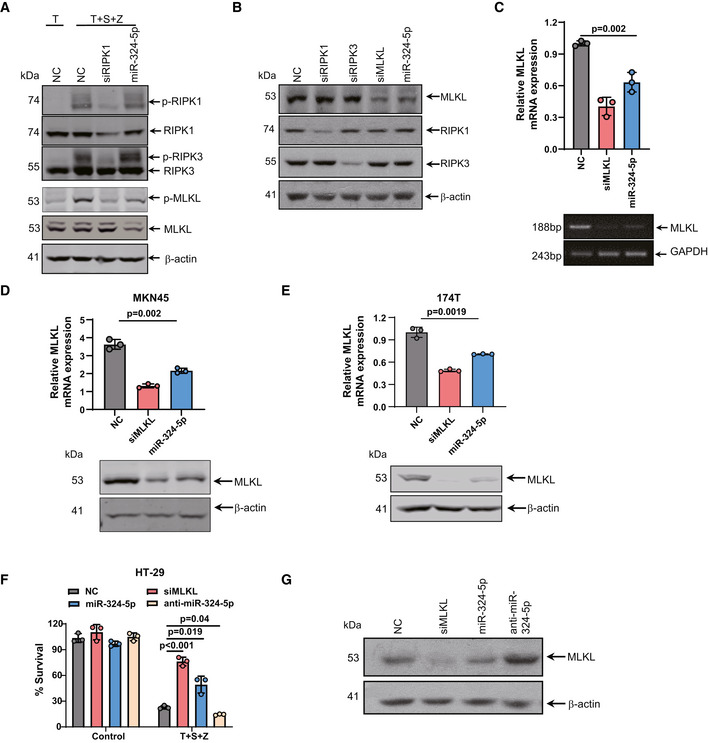
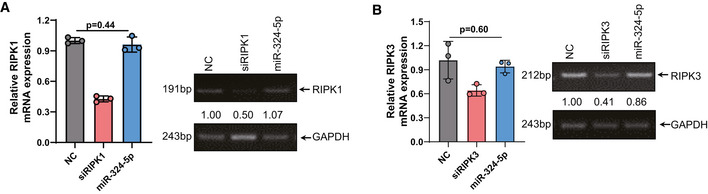
To investigate how miR‐324‐5p regulates necroptosis, we examined the effect of miR‐324‐5p on the activation of RIPK1, RIPK3, and MLKL during necroptosis by measuring the levels of phosphorylated RIPK1, RIPK3, and MLKL. Upon application of necroptotic stimuli, phosphorylation of RIPK1, RIPK3, and MLKL was detected in HT‐29 cells transfected with the control miRNA (Fig 2A). We observed that phosphorylation of MLKL was reduced in HT‐29 cells transfected with miR‐324‐5p, while phosphorylation of RIPK1 and RIPK3 was not affected under the same conditions (Fig 2A). We also detected a decrease of MLKL protein level in the cells transfected with miR‐324‐5p, while the expression levels of RIPK1 and RIPK3 were not affected in these cells (Fig 2B). Moreover, transfection of miR‐324‐5p led to a significant reduction in Mlkl mRNA expression (Fig 2C) but had no effect on either Ripk1 or Ripk3 mRNA levels (Fig EV2A and B). Like HT‐29 cells, MKN45 and 174T cells showed a decreased level of MLKL after transfection of miR‐324‐5p (Fig 2D and E), suggesting that miR‐324‐5p is capable of selectively downregulating MLKL expression. Furthermore, transfection of cells with anti‐miR‐324‐5p, an antisense inhibitor of endogenous miR‐324‐5p, induced the expression of MLKL and enhanced the sensitivity of cells to TNF‐induced necroptosis (Fig 2F and G). These results demonstrate that miR‐324‐5p is a negative regulator of MLKL expression during necroptosis.
Figure 2. MiR‐324‐5p negatively regulates MLKL expression.
-
AHT‐29 cells were transfected with NC, siRIPK1 or miR‐324‐5p. After 48 h, cells were treated with TNF‐α (T) or T + S + Z for an additional 6 h. Western blotting analysis of p‐RIPK1, RIPK1, p‐RIPK3, RIPK3, p‐MLKL, MLKL, and β‐actin.
-
BWestern blotting analysis of RIPK1, RIPK3, MLKL, and β‐actin levels in HT‐29 cells that were transfected with NC, siRIPK1, siRIPK3, MLKL siRNA oliogs (siMLKL), and miR‐324‐5p.
-
CHT‐29 cells were harvested 48 h after transfection with NC, siMLKL, and miR‐324‐5p. MLKL expression was analyzed by qPCR (upper) or RT‐PCR (lower).
-
D, EMKN45 (D) and 174T (E) cells were harvested 48 h after transfection with NC, siMLKL, and miR‐324‐5p. qPCR analysis for the expression of MLKL (upper) and western blotting analysis of MLKL and β‐actin (lower).
-
FHT‐29 cells were transfected with NC, siMLKL, miR‐324‐5p, or anti‐miR‐324‐5p. After 48 h, cells were treated with DMSO or T + S + Z for an additional 24 h. Cell survival was determined by measuring ATP levels.
-
GWestern blotting analysis of MLKL and β‐actin in HT‐29 cells that were harvested 48 h after transfection with NC, siMLKL, miR‐324‐5p, or an anti‐miR‐324‐5p.
Data information: Data are represented as the means ± SD of three biological replicates. Statistical analyses were performed using unpaired Student's t‐test. All experiments were performed at least three times, and representative data are shown.
Source data are available online for this figure.
Figure EV2. MiR‐324‐5p does not regulate the expression of RIPK1 or RIPK3.
-
AHT‐29 cells were transfected with NC, siRIPK1, or miR‐324‐5p mimics for 48h. qPCR analysis for the expression of RIPK1 (left) and RT‐PCR analysis of RIPK1 and GAPDH (right). Quantification of RIPK1 normalized to GAPDH (under the band of RIPK1).
-
BHT‐29 cells were transfected with NC, siRIPK3, or miR‐324‐5p mimics for 48h. qPCR analysis for the expression of RIPK3 (left) and RT‐PCR analysis of RIPK3 and GAPDH (right). Quantification of RIPK3 normalized to GAPDH (under the band of RIPK3).
Data information: Data are represented as the means ± SD of three biological replicates. Statistical analyses were performed using unpaired Student's t‐test. All experiments were performed at least three times, and representative data are shown.
MiRNA‐324‐5p targets the 3′UTR of MLKL mRNA in a seed region‐independent manner
To investigate whether miR‐324‐5p directly binds to the 3′UTR of MLKL mRNA, we constructed a recombinant plasmid (pmirGLO‐MLKL‐3′UTR reporter) containing the 3′UTR of MLKL fused to a firefly luciferase reporter gene (Fig 3A). HEK293T cells were transfected with pmirGLO‐MLKL‐3′UTR reporter combined with the control miRNA or miR‐324‐5p. The transfection efficiency was normalized by co‐transfection with a Renilla luciferase reporter. As shown in Fig 3B, miR‐324‐5p transfection significantly decreased the luciferase activity of MLKL‐3′UTR compared with control miRNA transfection, suggesting that miR‐324‐5p can bind to the 3′UTR of MLKL mRNA. We observed that miR‐324‐5p did not bind to the 3′UTR of Ripk1 or Ripk3 mRNA (Fig EV3A and B).
Figure 3. MiRNA‐324‐5p targets the 3′UTR of MLKL mRNA in a seed region‐independent manner.
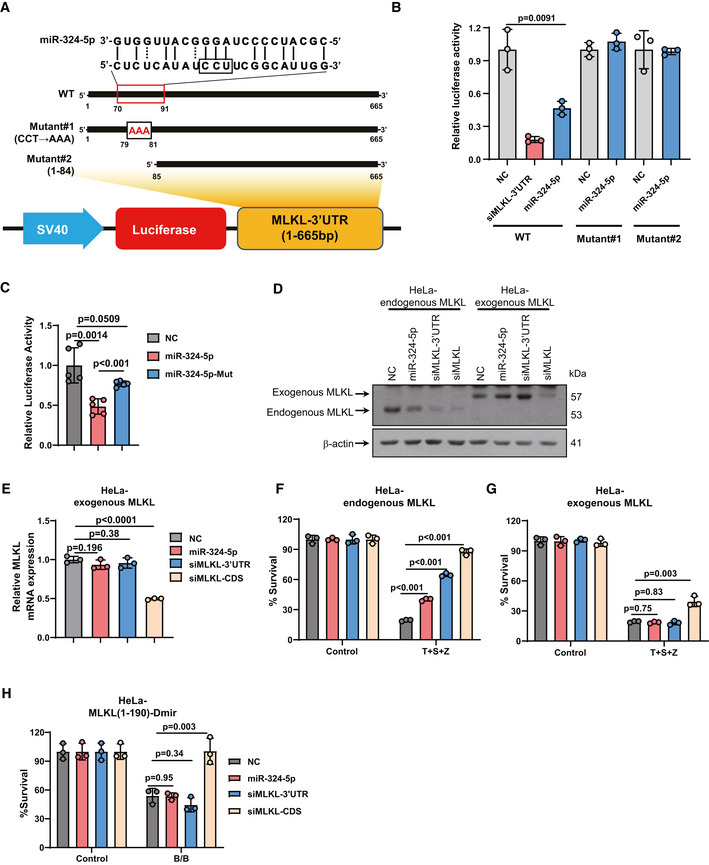
-
ASchematic representation of the miR‐324‐5p target binding site in the 3′UTR of MLKL. Two mutated forms (mutant#1 and mutant#2) of MLKL‐3′UTR were generated.
-
BHEK293T cells were transfected with NC, MLKL siRNA oligos targeting the 3′UTR of MLKL (siMLKL‐3′UTR), or miR‐324‐5p, together with pmirGLO‐MLKL‐3′UTR or the mutated form of MLKL‐3′UTR shown in A. Relative luciferase activity analysis of MLKL‐3′UTR. Data are presented as firefly luciferase activity/renilla luciferase activity ± SD.
-
CHEK293T cells were transfected with NC, miR‐324‐5p, or the mutated form of miR‐324‐5p together with pmirGLO‐MLKL‐3′UTR. Relative luciferase activity analysis of MLKL‐3′UTR. Data are presented as firefly luciferase activity/renilla luciferase activity ± SD (n = 5, biological replicates).
-
DWestern blotting analysis of MLKL and β‐actin in HeLa‐endogenous MLKL cells (HeLa cells stably expressing human RIPK3) and HeLa‐exogenous MLKL cells (Mlkl −/− HeLa cells stably expressing human RIPK3 and the coding sequence (CDS) of MLKL) that were harvested 48 h after transfection with NC, miR‐324‐5p, or MLKL siRNA oligos targeting the CDS region of MLKL (siMLKL‐CDS).
-
EqPCR analysis for the expression of MLKL in HeLa‐exogenous MLKL cells that were harvested 48 h after transfection with NC, miR‐324‐5p, siMLKL‐3′UTR, or siMLKL‐CDS.
-
F, GHeLa‐endogenous MLKL cells (F) and HeLa‐exogenous MLKL (G) were transfected with NC, miR‐324‐5p, siMLKL‐3′UTR, or siMLKL‐CDS. After 48 h, cells were treated with T + S + Z for 24 h. Cell survival was determined by measuring ATP levels.
-
HHeLa cells expressing MLKL(1–190 aa) fused to DmrB (HeLa‐MLKL(1–190)‐Dmir cells) were transfected with NC, miR‐324‐5p, siMLKL‐3′UTR, or siMLKL‐CDS. After 48 h, cells were treated with DMSO or the dimerization agent AP20187 (60 nM) for 24 h. Cell survival was determined by measuring ATP levels.
Data information: The number of surviving cells was normalized to the number of surviving control cells, which were treated with DMSO. In (B), and (E–H), data are represented as the means ± SD of three biological replicates. Statistical analyses were performed using unpaired Student's t‐test. All experiments were performed at least three times, and representative data are shown.
Source data are available online for this figure.
Figure EV3. MiR‐324‐5p cannot bind to the 3′UTR of RIPK1 or RIPK3 mRNA.
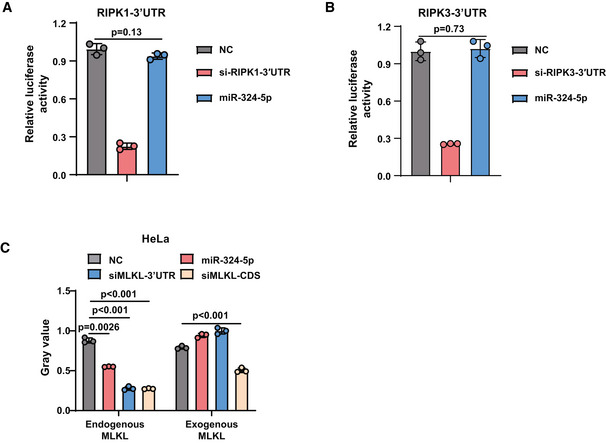
-
A, BRelative luciferase activity analysis in HEK‐293T cells after co‐transfection with indicated oligos and pmirGLO‐RIPK1‐3′UTR (A) or pmirGLO‐RIPK3‐3′UTR (B) for 48 h.
-
CHeLa‐endogenous MLKL cells and HeLa‐exogenous MLKL were transfected with NC, miR‐324‐5p, siMLKL‐3′UTR, or siMLKL‐CDS. Western blotting analysis of MLKL and β‐actin. Quantification of MLKL normalized to β‐actin levels.
Data information: Data are represented as the means ± SD of three biological replicates. Statistical analyses were performed using unpaired Student's t‐test. All experiments were performed at least three times, and representative data are shown.
We next predicted the potential miR‐324‐5p binding site in the 3′UTR of MLKL using the RNAhybrid program, which showed that the 79–81 bp (CCU) region of MLKL‐3′UTR could be the putative binding site for miR‐324‐5p. To validate the predicted miR‐324‐5p binding site in MLKL‐3′UTR, we generated two luciferase fusion constructs containing mutant forms of MLKL‐3′UTR: (i) mutant#1 in which CCU in the predicted binding site was replaced by AAA; (ii) mutant#2 in which the predicted binding site was removed by deleting the 1–84 bp region of MLKL‐3′UTR. Notably, disruption or deletion of the predicted miR‐324‐5p binding site in MLKL‐3′UTR completely abolished the inhibitory effect of miR‐324‐5p on MLKL‐3′UTR (Fig 3A and B), indicating that the 79–81 bp (CCU) region in the 3′UTR of MLKL is crucial for its binding to miR‐324‐5p at nucleotides 11–13. Moreover, we synthesized a mutant form of miR‐324‐5p in which AGG in the predicted binding site was replaced by UCC. This mutant form of miR‐324‐5p failed to inhibit the luciferase activity of human MLKL‐3′UTR, supporting that AGG at nucleotides 11–13 of miR‐324‐5p is required for its regulation of human MLKL (Fig 3C). Taken together, these results suggest that miR‐324‐5p targets MLKL in a seed region‐independent manner.
We next evaluated whether MLKL is the functional target of miR‐324‐5p responsible for its inhibitory effect on necroptosis. To this end, we generated two HeLa cell lines: (i) HeLa cells stably expressing human RIPK3 (HeLa‐endogenous MLKL), and (ii) Mlkl −/− HeLa cells stably expressing human RIPK3 and the coding sequence (CDS) of MLKL (HeLa‐exogenous MLKL). As shown in Fig 3D and E, and EV3C, the level of endogenous MLKL in HeLa‐endogenous MLKL cells was reduced by transfection of miR‐324‐5p or an siRNA oligo targeting either the MLKL CDS or 3′UTR. In HeLa‐exogenous MLKL cells, the level of exogenous MLKL was knocked down by transfection of an siRNA oligo targeting the CDS of MLKL (siMLKL‐CDS), but it was not affected by transfection of miR‐324‐5p or an siRNA oligo targeting the 3′UTR of MLKL (siMLKL‐3′UTR). Notably, miR‐324‐5p and siMLKL‐3′UTR significantly blocked TNF‐induced necroptosis in HeLa‐endogenous MLKL cells (Fig 3F), but they failed to affect TNF‐induced necroptosis in HeLa‐exogenous MLKL cells (Fig 3G). As expected, siMLKL‐CDS inhibited TNF‐induced necroptosis in both HeLa‐endogenous MLKL cells and HeLa‐exogenous MLKL cells (Fig 3F and G). It has been shown that HeLa cells expressing MLKL (1–190 aa) fused to DmrB undergo MLKL polymerization‐induced necroptosis in the presence of the dimerization agent AP20187. Consistent with this, neither miR‐324‐5p nor siMLKL‐3′UTR blocked necroptosis induced by MLKL polymerization in HeLa cells expressing MLKL (1–190 aa), while this necroptotic phenotype was greatly inhibited by siMLKL‐CDS (Fig 3H). Taken together, these results demonstrate that miR‐324‐5p‐mediated suppression of necroptosis completely depends on its regulation of MLKL.
MiR‐324‐5p‐mediated regulation of MLKL is species specific
Having shown that the 79–81 bp of CCU (CCT) in the 3′UTR region of MLKL is critical for binding to miR‐324‐5p, we next examined whether these nucleotides are conserved among mammalian species. As shown in Fig 4A, these nucleotides are conserved in the 3′UTR of MLKL genes from monkey (Cercocebus atys), cow (Bos taurus), horse (Equus caballus), and sheep (Ovis aries). However, this CCT region is not conserved in the 3′UTR of MLKL genes from pig (Sus scrofa) and rodent species including mouse (Mus musculus) and rat (Rattus norvegicus). Furthermore, we found that the CCT region of MLKL‐3′UTR is highly conserved among primates including chimpanzee (Pan troglodytes) and green monkey (Chlorocebus sabaeus) (Fig 4B).
Figure 4. MiR‐324‐5p‐mediated regulation of MLKL is species specific.
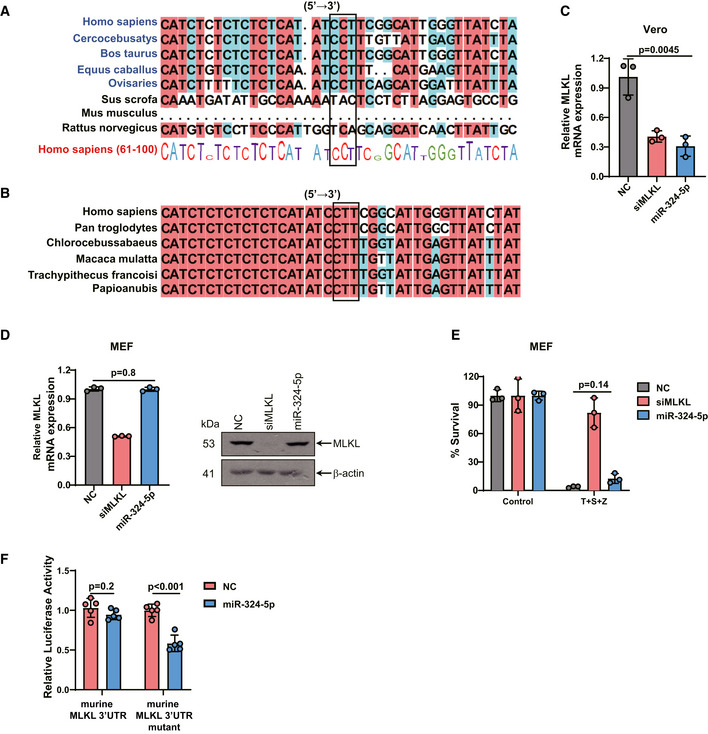
-
A, BEvolutionary conservation of the CCT(CCU) region in MLKL‐3′UTR. The DNA sequences of MLKL flanking the CCT(CCU) region from various species were aligned by DNAman.
-
CAfrican green monkey kidney epithelial (Vero) cells were transfected with NC, siMLKL, or miR‐324‐5p for 48 h. qPCR analysis for the expression of MLKL.
-
DMEFs were transfected with NC, siMLKL, or miR‐324‐5p for 48 h. qPCR analysis for the expression of MLKL and western blotting analysis of MLKL and β‐actin.
-
EMEFs were transfected with NC, siMLKL, or miR‐324‐5p. After 48 h, cells were treated with DMSO or T + S + Z for 24 h. Cell viability was determined by measuring ATP levels.
-
FHEK293T cells were transfected with NC or miR‐324‐5p, together with pmirGLO‐mouse MLKL‐3′UTR or the mutated form of mouse MLKL‐3′UTR. Relative luciferase activity analysis of MLKL‐3′UTR. Data are presented as firefly luciferase activity/renilla luciferase activity ± SD (n = 5, biological replicates).
Data information: In (C–E), data are represented as the means ± SD of three biological replicates. Statistical analyses were performed using unpaired Student's t‐test. All experiments were performed at least three times, and representative data are shown. β‐Actin serves as an internal control (D).
Source data are available online for this figure.
We next used African green monkey kidney epithelial (Vero) cells to examine the effect of miR‐324‐5p on MLKL expression. As expected, transfection of miR‐324‐5p resulted in the downregulation of MLKL in Vero cells (Fig 4C). In contrast, transfection of miR‐324‐5p did not affect the expression of MLKL in mouse embryonic fibroblasts (MEFs), in which MLKL does not have the miR‐324‐5p binding region (Fig 4D). Consistent with this finding, miR‐324‐5p could not block TNF‐α induced necroptosis in MEFs (Fig 4E). Further, we generated a mutant form of murine MLKL‐3′UTR by introducing the miR‐324‐5p binding region of human MLKL‐3′UTR into the start site of mouse MLKL‐3′UTR. We observed that miR‐324‐5p transfection significantly reduced the luciferase activity of this mutant form of mouse MLKL‐3′UTR, suggesting that introduction of the miR‐324‐5p binding region is sufficient to potentiate the regulation of mouse MLKL by miR324‐5p (Fig 4F). These results suggest that the CCU region is responsible for species‐dependent recognition of MLKL by miR‐324‐5p. Thus, it is likely that manipulation of MLKL‐mediated necroptosis by miR‐324‐5p has evolved in higher mammals.
The IFN‐JAK‐STAT1 signaling pathway negatively regulates miR‐324‐5p
Previous studies have shown that IFN treatment upregulates MLKL (Thapa et al, 2013; Stutz et al, 2018; Knuth et al, 2019; Chen et al, 2019a). Consistent with this, we observed induced expression of MLKL at both the mRNA and protein levels in response to IFN‐α, IFN‐β, and IFN‐γ in U937 cells, while MLKL expression was not affected by treatment with LPS, or poly(I:C) (Fig 5A and B). Interestingly, we found that IFN‐α, IFN‐β, and IFN‐γ could significantly downregulate the level of miR‐324‐5p (Fig 5C). Moreover, overexpression of miR‐324‐5p reduced both IFN‐β‐ and IFN‐γ‐induced expression of MLKL (Figs 5D and E, and EV4A and B).
Figure 5. The IFN‐JAK‐STAT1 signaling pathway negatively regulates miR‐324‐5p.
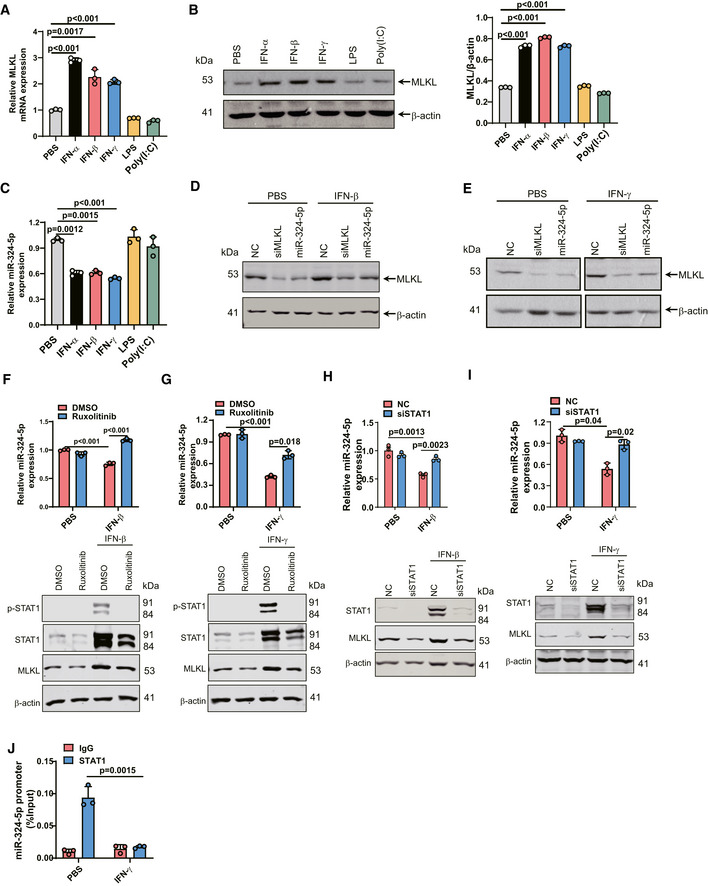
-
AU937 cells were treated with 100 ng/ml IFN‐α, 100 ng/ml IFN‐β, 100 ng/ml IFN‐γ, 100 ng/ml LPS, or 25 μg/ml poly(I:C) for 24 h. Identical concentrations were used in later experiments unless otherwise stated. qPCR analysis for the expression of MLKL.
-
BU937 cells were treated with IFN‐α, IFN‐β, IFN‐γ, LPS, or poly(I:C) for 24 h. and western blotting analysis of MLKL and β‐actin (left). Quantification of MLKL normalized to β‐actin levels (right).
-
CU937 cells were treated with PBS, IFN‐α, IFN‐β, IFN‐γ, LPS, or poly(I:C) for 24 h. qPCR analysis for the expression of miR‐324‐5p.
-
D, EU937 cells were transfected with NC, siMLKL, or miR‐324‐5p. After 48 h, cells were treated with IFN‐β (D) or IFN‐γ (E) for 24 h. Western blotting analysis of MLKL and β‐actin.
-
F, GU937 cells were incubated with 300 nM ruxolitinib for 2 h prior to IFN‐β (F) or IFN‐γ (G) treatment. After 24 h, qPCR analysis for the expression of miR‐324‐5p. After 48 h, western blotting analysis of p‐STAT1, STAT1, MLKL, and β‐actin.
-
H, IU937 cells were transfected with NC, or STAT1 siRNA oliogs (siSTAT1). After 48 h, cells were treated with IFN‐β (Η) or IFN‐γ (Ι). qPCR analysis for the expression of miR‐324‐5p. Western blotting analysis of STAT1, MLKL and β‐actin.
-
JHT‐29 cells were treated with IFN‐γ for 24 h. The cell lysate was collected for ChIP analysis. STAT1 binding to miR‐324‐5p promoter DNA region was determined by Chip‐qPCR. The amount of precipitated DNA was calculated as percent input.
Data information: Data are represented as the means ± SD of three biological replicates. Statistical analyses were performed using unpaired Student's t‐test. All experiments were performed at least three times, and representative data are shown.
Source data are available online for this figure.
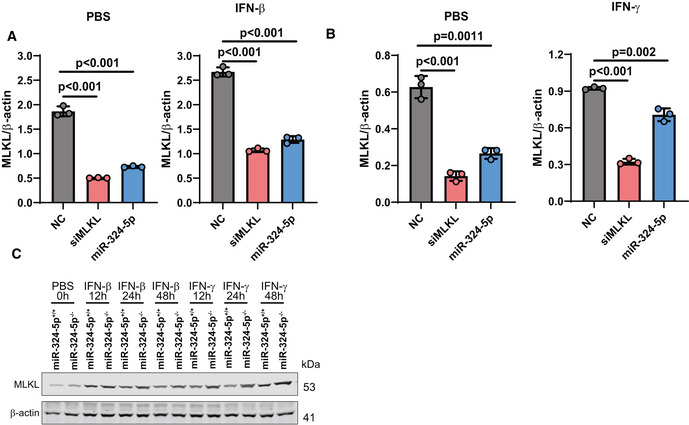
Figure EV4. IFNs regulates the expression levels of miR‐324‐5p and MLKL.
-
A, BU937 cells were electroporated with NC or miR‐324‐5p. After 48 h, cells were exposed to IFN‐β(Α) or IFN‐γ(Β) for 24 h. Western blotting analysis for the expression of MLKL and β‐actin. Quantification of MLKL normalized to β‐actin levels.
-
CMiR‐324‐5p+/+ HT‐29, and miR‐324‐5p−/− HT‐29 cells were treated with IFN‐β or IFN‐γ for the indicated time. Western blotting analysis of MLKL and β‐actin.
Data information: Data are represented as the means ± SD of three biological replicates. Statistical analyses were performed using unpaired Student's t‐test. All experiments were performed at least three times, and representative data are shown.
Source data are available online for this figure.
Interferons are known to signal through the JAK/STAT pathway (Krause et al, 2006). To further investigate the role of the JAK/STAT pathway in miR‐324‐5p transcription, we pretreated U937 cells with the JAK1/JAK2 inhibitor ruxolitinib prior to treatment with IFN‐β or IFN‐γ. The addition of ruxolitinib blocked both IFN‐β‐ and IFN‐γ‐induced downregulation of miR‐324‐5p as well as both IFN‐β‐ and IFN‐γ‐induced upregulation of MLKL (Fig 5F and G). STAT1 is an important mediator of type I and type II IFN signaling. We performed knockdown of STAT1 in U937 cells prior to treatment with IFN‐γ or IFN‐β. STAT1 levels were significantly reduced by transfection of STAT1 siRNA oligos (Fig 5H and I). Knocking down STAT1 abolished IFNs‐induced downregulation of miR‐324‐5p as well as IFNs‐induced upregulation of MLKL (Fig 5H and I). Moreover, a chromatin immunoprecipitation (ChIP) assay showed reduced interaction between STAT1 and the promoter of miR‐324‐5p in response to IFN‐γ stimulation (Fig 5J), indicating that the IFN‐JAK‐STAT1 signaling pathway negatively regulates miR‐324‐5p expression.
IFN‐mediated repression of miR‐324‐5p enhances necroptosis
Since miR‐324‐5p is an IFN‐repressed gene, we examined the effect of miR‐324‐5p on necroptosis upon stimulation with IFN‐γ. IFN‐γ induced cell death in U937 cells in the presence of z‐VAD, and cell death was inhibited by knockdown of MLKL (Fig 6A), indicating that IFN‐γ/z‐VAD activates MLKL‐mediated necroptosis in U937 cells. Transfection of U937 cells with miR‐324‐5p significantly blocked IFN‐γ‐induced necroptosis (Fig 6A). We next stimulated cultured human PBMC‐derived macrophages with IFN‐γ. IFN‐γ significantly reduced miR‐324‐5p expression and upregulated MLKL expression in human primary monocytes (Fig 6B). MiR‐324‐5p overexpression significantly inhibited necroptosis in both PBMC‐derived macrophages and HT‐29 cells in the presence of IFN‐γ (Fig 6C and D). Next, we generated miR‐324‐5p knockout HT‐29 cell lines using the CRISPR/Cas9 system to assess the role of cellular miR‐324‐5p in necroptosis. Two clones showed obvious deletion of endogenous miR‐324‐5p (Fig 6E). Deletion of miR‐324‐5p in HT‐29 cells increased the sensitivity of cells to TNF‐induced necroptosis (Fig 6F). HT‐29 miR‐324‐5p−/− cells also showed increased sensitivity to the treatment of IFN‐β or IFN‐γ in the presence of z‐VAD compared to HT‐29 miR‐324‐5p+/+ cells (Fig 6G and H). Deletion of miR‐324‐5p led to increased expression of MLKL upon stimulation of IFN‐β or IFN‐γ (Fig EV4C). Taken together, these results indicate that IFN‐induced repression of miR‐324‐5p promotes the activation of necroptosis.
Figure 6. IFN‐mediated repression of miR‐324‐5p enhances necroptosis.

-
AU937 cells were transfected with NC, siMLKL‐3′UTR, or miR‐324‐5p. After 48 h, cells were exposed to 100 ng/ml IFN‐γ plus 20 μM z‐VAD for 24 h. Identical concentrations were used in later experiments unless otherwise stated. Cell viability was determined by measuring ATP levels.
-
BqPCR analysis for the expression of MLKL (left) and miR‐324‐5p (middle) in PBMC‐derived macrophages that were treated with PBS or IFN‐γ for 24 h. The corresponding western blotting analysis of MLKL and β‐actin (right).
-
C, DPBMC‐derived macrophages (C) and HT‐29 cells (D) were transfected with NC, siMLKL, or miR‐324‐5p. After 48 h, cells were exposed to S + Z or IFN‐γ + S + Z for 24 h. Cell viability was determined by measuring ATP levels (left). The corresponding western blotting analysis of MLKL and β‐actin (right).
-
EqPCR analysis for the expression of miR‐324‐5p in miR‐324‐5p knockout HT‐29 cells (miR‐324‐5p−/− cells). Altered miR‐324‐5p DNA sequences were shown in miR‐324‐5p−/− clone 19# and 36#.
-
FMiR‐324‐5p+/+ HT‐29, miR‐324‐5p−/− −19#, and miR‐324‐5p−/− −36# cells were treated with T + S + Z for the indicated times. Cell viability was determined by measuring ATP levels.
-
G, HMiR‐324‐5p+/+ HT‐29, and miR‐324‐5p−/− HT‐29 cells were treated with IFN‐β (G) or IFN‐γ (H) plus 20 μM z‐VAD for 24 h. Cell viability was determined by measuring ATP levels.
Data information: Data are represented as the means ± SD of three biological replicates. Statistical analyses were performed using unpaired Student's t‐test. All experiments were performed at least three times, and representative data are shown.
Source data are available online for this figure.
MiR‐324‐5p inhibition sensitizes IAV‐infected cells to necroptosis and reduces viral replication
Necroptosis has an important role in host defense against IAV infection (Kuriakose et al, 2016; Nogusa et al, 2016; Thapa et al, 2016). We therefore examined the effect of miR‐324‐5p in IAV (H1N1 strain PR8)‐infected human PBMC‐derived macrophages. IFN‐β and IFN‐γ were induced in human macrophages infected with IAV (Fig 7A). We observed that miR‐324‐5p expression was decreased post‐IAV infection, while MLKL was upregulated in IAV‐infected cells (Fig 7B). It has been shown that IAV‐induced IFN‐β plays an important role in host defense against IAV (Koerner et al, 2007). IFNs secreted by infected cells trigger signal transduction through the JAK/STAT cascade (Schoggins et al, 2014). We found that upregulation of MLKL and downregulation of miR‐324‐5p in IAV‐infected macrophages were largely reversed by treatment with ruxolitinib (Fig 7B). IAV infection enhances necroptosis induced by the treatment of Smac mimetic plus z‐VAD in macrophages, and the phenomenon of necroptosis was significantly inhibited by miR‐324‐5p overexpression (Fig 7C). Overexpression of miR‐324‐5p significantly promoted IAV replication (Fig 7D). It is known that IAV can induce apoptosis as well as necroptosis (Nogusa et al, 2016). Transfection of either miR‐324‐5p or siMLKL oligos did not affect the level of cleavage of caspase‐3 (Fig EV5A). It has been shown that HT‐29 cells are susceptible to IAV subtypes such as H9N2 and H1N1(Qu et al, 2012). We observed that IFN‐β was also upregulated in HT‐29 cells infected with H1N1 strain PR8 (Fig 7E). IAV infection led to increased levels of MLKL and phosphorylated MLKL (Fig EV5B). Overexpression of miR‐324‐5p inhibited necroptosis induced by Smac mimetic plus z‐VAD or in combination with IAV infection in HT‐29 cells (Fig 7F). Knockout of miR‐324‐5p in HT‐29 cells significantly increased sensitivity of cells to IAV infection (Fig 7G). Importantly, deficiency of miR‐324‐5p resulted in a significant reduction in viral replication in IAV‐infected cells with or without z‐VAD treatment (Fig 7H). These results suggest that IFN‐mediated reduction of miR‐324‐5p promotes the necroptosis of IAV‐infected cells and limits viral replication.
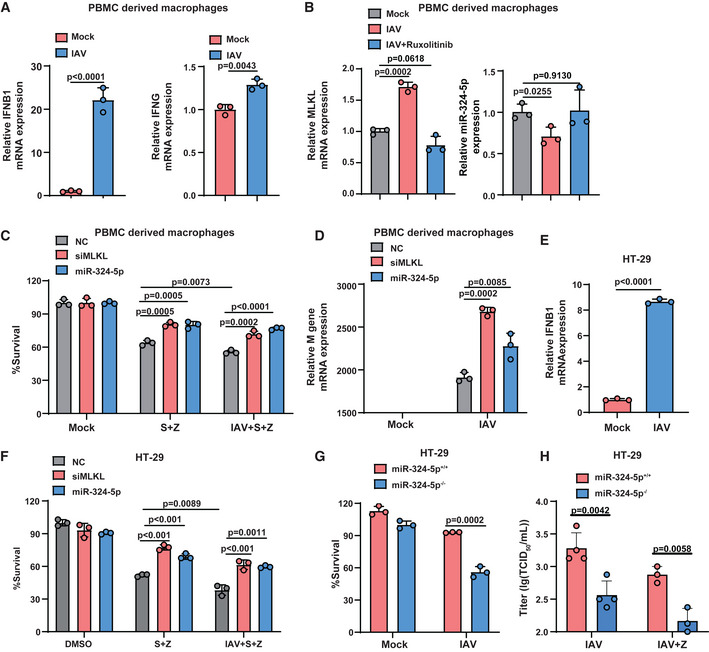
Figure 7. MiR‐324‐5p inhibition sensitizes IAV‐infected cells to necroptosis and reduces viral replication.
-
AqPCR analysis for the expression of IFN‐β (left) and IFN‐γ (right) in PBMC‐derived macrophages that were infected with IAV (H1N1 strain PR8) at an multiplicity of infection (MOI) of 0.2 for 24 h.
-
BqPCR analysis for the expression of MLKL (left) and miR‐324‐5p (right) in PBMC‐derived macrophages that were treated with the 300 nM ruxolitinib for 2 h prior to the infection with IAV (MOI = 0.2) for 24 h.
-
CPBMC‐derived macrophages were transfected with NC, siMLKL, or miR‐324‐5p. After 48 h, cells were treated with S + Z in the presence or absence of IAV (MOI = 0.2) for 24 h. Cell viability was determined by measuring ATP levels.
-
DqPCR analysis for the expression of M gene of IAV in PBMC‐derived macrophages that were transfected with NC, siMLKL or miR‐324‐5p for 48 h, followed by the infection with IAV (MOI = 0.2) for 24 h.
-
EHT‐29 cells were infected with IAV (MOI = 0.2) for 24 h. qPCR analysis for the expression of IFN‐β.
-
FHT‐29 cells were transfected with NC, siMLKL, or miR‐324‐5p. After 48 h, cells were treated with DMSO, S + Z in the presence or absence of IAV (MOI = 0.2) for an additional 24 h. Cell survival was determined by measuring ATP levels.
-
GMiR‐324‐5p+/+ and miR‐324‐5p−/− HT‐29 cells were infected with IAV (MOI = 0.2) for 24 h. Cell viability was determined by measuring ATP levels.
-
HMiR‐324‐5p+/+ and miR‐324‐5p−/− HT‐29 cells were infected with IAV (MOI = 0.2) with or without z‐VAD for 48 h (n = 4, biological replicates). IAV TCID50 analysis for the virus titer in the cell culture supernatant.
Data information: In (A–G), data are represented as the means ± SD of three biological replicates. Statistical analyses were performed using unpaired Student's t‐test. All experiments were performed at least three times, and representative data are shown.
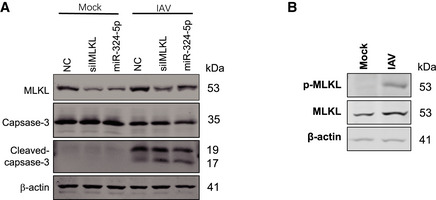
Figure EV5. MiR‐324‐5p inhibition sensitizes IAV‐infected cells to necroptosis and reduces viral replication.
-
APBMC cells were transfected with NC, siMLKL, or miR‐324‐5p. After 48h, cells were infected with mock or IAV (MOI = 0.2) for 24 h. Western blotting analysis of MLKL, caspase 3, cleaved caspase 3 and β‐actin.
-
BHT‐29 cells were infected with mock or IAV (MOI = 0.5) for 24 h. Western blotting analysis of MLKL, p‐MLKL and β‐actin.
Source data are available online for this figure.
Discussion
Eliminating pathogen‐infected cells by necroptosis is a critical mechanism of host defense against pathogen infection (Wang et al, 2014). MiRNAs play important roles in controlling the response to pathogen infection by regulating the expression of target genes (Othumpangat et al, 2021; Zhang et al, 2021). Our study revealed that miR‐324‐5p functions as a suppressor of necroptosis by directly inhibiting MLKL expression and that induction of IFNs by pathogens such as IAV downregulates the expression of miR‐324‐5p through the JAK/STAT1 signaling pathway (Fig 8). Downregulation of miR‐324‐5p by IFNs promotes optimal activation of MLKL‐mediated necroptosis, eliminating IAV‐infected cells (Fig 8).
Figure 8. Schematic of the IFN‐miR‐324‐5p‐MLKL axis protecting the host against IAV infection.
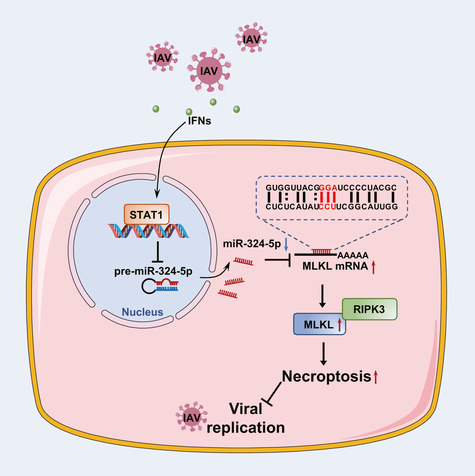
Upon IAV infection, IFNs are induced in host cells and activate the JAK/STAT1 pathway, resulting in a decrease in miR‐324‐5p expression. MiR‐324‐5p negatively regulates human MLKL by directly targeting the 79–81 bp (CCU) region in the 3′UTR. The reduction of miR‐324‐5p relieves its suppression of MLKL mRNA and thus promotes necroptosis of IAV‐infected cells, leading to restriction of viral replication.
MiR‐324‐5p has been reported to regulate cell proliferation and apoptosis under diverse conditions by targeting different mRNAs (Wang et al, 2015; Lin et al, 2018; Chen et al, 2019b; Wan et al, 2020; Zheng et al, 2021). For example, miR‐324‐5p inhibits mitochondrial fusion and apoptosis in cardiomyocytes and endothelial progenitor cells by downregulating Mtfr1 (Wang et al, 2015; Chen et al, 2019b; Huang et al, 2020). MiR‐324‐5p promotes cell proliferation and suppresses apoptosis in pancreatic cancer cells by targeting KLF3 (Wan et al, 2020). Conversely, miR‐324‐5p promotes apoptosis in gastric cancer cells by modulating the expression of TSPAN8 (Lin et al, 2018). Recently, miR‐324‐5p has been shown to boost apoptosis and promote the development of glioblastomas by regulating Bcl2 (Li et al, 2021). However, the role of miR‐324‐5p in necroptosis has remained unknown. We performed a cell‐based screen for miRNAs regulating necroptosis and identified miR‐324‐5p as a suppressor of necroptosis in multiple human cell lines. Overexpression of miR‐324‐5p significantly blocked the phosphorylation of MLKL, a key step in necroptosis. Moreover, miR‐324‐5p directly targets the 3′UTR of human MLKL leading to downregulation of MLKL expression. Furthermore, we found that miR‐324‐5p inhibition of necroptosis depends on the 3′UTR of human MLKL, as evidenced by the fact that miR‐324‐5p does not affect necroptosis in Mlkl −/− HeLa cells expressing the CDS region of MLKL. Therefore, MLKL is the sole target of miR‐324‐5p in the inhibition of necroptosis. Notably, deletion of cellular miR‐324‐5p increased the sensitivity of cells to necroptotic stimuli. Thus, our findings demonstrate that miR‐324‐5p functions as a negative regulator of necroptosis by manipulating MLKL expression.
Our findings establish a unique regulatory mechanism for miR‐324‐5p modulation of MLKL. In general, the seed region (nucleotides 2–8 of the miRNA) is required for target mRNA recognition and binding (Bartel, 2018). Seed region‐dependent miRNA binding is known to promote the degradation and/or suppress the translation of canonical mRNA targets. As such, seed‐pairing potential is usually used for the prediction of mRNA targets. It is worth noting that noncanonical miRNA‐target recognition sites beyond the seed region have been identified (Agarwal et al, 2015). However, some of the identified noncanonical recognition sites do not repress the expression of bound targets (Agarwal et al, 2015). Interestingly, we found that the 79–81 bp (CCU) region of MLKL is essential for its binding to miR‐324‐5p. The 79–81 bp (CCU) region of MLKL pairs with nucleotides 11–13 (AGG) of miRNA‐324‐5p, which are located outside the seed region. The mutant miRNA‐324‐5p in which AGG was replaced by UCC failed to target the human MLKL‐3′UTR. Thus, our study reveals a seed‐region‐independent binding mode for miR‐324‐5p recognition of the target MLKL. Notably, we observed that the 79–81 bp (CCU) region of MLKL is conserved in higher mammals, including monkeys and horses. However, it is not conserved in rodent species, including mice and rats. MiR‐324‐5p negatively regulates MLKL expression in human and monkey cells, but does not affect MLKL expression and necroptosis in MEFs. Previous studies have shown that MLKL can be induced by IFNs in mouse cells (Thapa et al, 2013; Stutz et al, 2018; Knuth et al, 2019; Chen et al, 2019a). It has been reported that IFN‐γ could directly induce the binding of STAT1 to the mouse Mlkl promoter region in mouse cells (Gunther et al, 2016), suggesting that IFNs directly regulate Mlkl gene transcription via activation of STAT1 in mouse species. Of note, the mutant form of mouse MLKL‐3′UTR, which was generated by introduction of the miR‐324‐5p binding region of human MLKL‐3′UTR, could be negatively regulated by miR‐324‐5p. Therefore, our work demonstrates that the function of miR‐324‐5p in necroptosis has evolved in higher mammals, suggesting a species‐specific mechanism for the modulation of necroptosis.
Optimal activation of necroptosis is considered to be beneficial for killing infected host cells, leading to the restriction of pathogen proliferation. IFNs play pivotal roles in host defense in response to viral infection (Chen et al, 2019a). As miR‐324‐5p counteracts the necroptosis signaling pathway, we speculated that the IFN signaling pathway may regulate miR‐324‐5p. Indeed, we found that miR‐324‐5p expression was downregulated by stimulation of IFN‐α, IFN‐β, or IFN‐γ. The JAK/STAT signaling pathway is known to be activated by IFNs to regulate the expression of target genes (Darnell et al, 1994). Inhibition of JAK1 activity or suppression of STAT1 abolished IFN‐induced repression of miR‐324‐5p and IFN‐induced MLKL expression. Moreover, a ChIP assay revealed the IFN‐mediated dissociation between STAT1 and the promoter of miR‐324‐5p. Thus, our findings demonstrate that miR‐324‐5p is an IFN‐repressed gene that is controlled by the JAK/STAT1 signaling pathway. Our study indicates that the IFN‐mediated repression of miR‐324‐5p promotes the upregulation of MLKL and optimal activation of necroptosis.
Necroptosis has been demonstrated to be a mechanism for host defense against various viruses, including murine cytomegalovirus (Upton et al, 2010), human herpes simplex viruses (Wang et al, 2014; Huang et al, 2015; Guo et al, 2015b), and IAV (Kuriakose et al, 2016; Nogusa et al, 2016; Thapa et al, 2016). Our findings demonstrate the functional role of IFN‐regulated miR‐324‐5p in necroptosis‐associated host defense. We found that IAV (H1N1 strain PR8) infection led to the induction of IFN in both human PBMC‐derived macrophages and HT‐29 cells. Of note, upregulation of miR‐324‐5p attenuated necroptosis of infected cells and enhanced viral replication. Moreover, the deletion of miR‐324‐5p sensitized cells to IAV‐induced necroptosis and reduced viral replication. Therefore, our findings demonstrate that IFN‐mediated repression of miR‐324‐5p orchestrates the activation of necroptosis in host defense against viral infection. It has been reported that miR‐324‐5p suppresses IAV (H5N1) replication by targeting the viral PB1 and host CUED2 genes (Kumar et al, 2018). However, miR‐324‐5p does not bind to the PB1 gene of H1N1 strain PR8 (Kumar et al, 2018), suggesting that the mechanism of action of miR‐324‐5p in viral infection is context‐dependent. Further studies will be required to understand the precise role of the IFN‐miR‐324‐5p‐MLKL axis in the pathogenesis of infectious diseases.
Materials and Methods
Cell culture
Human colon cancer HT‐29 cells (ATCC Number: HTB‐38) were cultured in McCoy's 5A culture medium (Gibco). 174T (ATCC Number: CL‐188), HEK293T (ATCC Number: CRL‐3216), T98G (ATCC Number: CRL‐1690), Vero (ATCC Number: CCL‐81), and L929 (ATCC Number: CCL‐1) cells were obtained from ATCC and cultured in Dulbecco's Modified Eagle Medium (DMEM, Hyclone). MKN‐45 cells (1101HUM‐PUMC000229) were obtained from the Cell Resource Center of Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences &Peking Union Medical College, China. Human U937 myelomonocytic cells (ATCC Number: CRL‐1593.2) were grown in RPMI‐1640 medium (Hyclone). MEFs were generated as previously reported (He et al, 2009). PBMCs were isolated from whole blood from five healthy adult donors. We isolated the PBMCs using Ficoll according to the manufacturer's instructions. Briefly, we diluted the blood with PBS containing 2% FBS and layered the diluted cells on Ficoll reagent in a tube. Then, the tube was centrifuged at 400 g for 30 min. The mononuclear cells at the interface would be harvested and cultured in RPMI‐1640 medium containing 10% FBS and 50 ng/ml MGSF. After 7 days, the macrophages were stimulated as indicated for further analysis. The use of PBMCs conforms to the guidelines of Zhejiang University (No. IRB‐2021‐028). HeLa‐MLKL knockout cells were a gift from Dr. Xiaodong Wang (National Institute of Biological Sciences, Beijing, China). The HeLa‐MLKL (1–190)‐Dmir cell line, which was kindly provided by Dr. Zhigao Wang (University of Texas Southwestern Medical Center at Dallas), was cultured in complete medium containing 10 μg/ml Blasticidin plus 1 μg/ml puromycin. All culture media were supplemented with 10% fetal bovine serum (Gibco), 1% penicillin/streptomycin (Invitrogen), and 2 mM L‐glutamine (Gibco). All cells were cultured in a 5% CO2 incubation chamber at 37°C.
Reagents, antibodies, and RNA oligos
The TNF‐α recombinant protein and Smac mimetic compound were synthesized as previously described (Wang et al, 2008). z‐VAD was purchased from Bachem. IFN‐α and IFN‐γ were bought from Novoprotein. IFN‐β was purchased from Peprotech. LPS and Poly(I:C) were purchased from Sigma and InvivoGen, respectively. Ruxolitinib was purchased from Selleck. The dimerization agent AP20187 was purchased from Sigma. The following antibodies were used for western blot analysis: RIPK1 (BD Biosciences, 610458, 1:8,000), RIPK3 (Prosci, 2283, 1:8,000), human MLKL (Huabio, ET1601, 1:1,000), mouse MLKL (Abgent, 142726, 1:2,000), p‐STAT1 (CST, 7649, 1:1,000), STAT1 (CST, 9175, 1:1,000), phospho‐human MLKL (Abcam, 187091, 1:1,000), Flag (Sigma, A8592, 1:10,000), ZBP1(SantaCruz, 67258, 1:2,000), caspase 3 (CST, 9662, 1:1,000), cleaved caspase 3 (CST, 9664, 1:1,000), and β‐actin (Sigma, A2066, 1:20,000). The following RNA sequences were used for in vitro transfection assay: MiRNA‐324‐5p (5′‐cgcauccccuagggcauuggugu‐3′), miRNA‐324‐3p (5′‐acugccccaggugcugcugg‐3′), negative control (5′‐uucuccgaacgugucacgutt‐3′), miRNA‐324‐5p inhibitor (5′‐acaccaaugcccuaggggaugcg‐3′), miRNA‐324‐5p‐mutant (5′‐cgcauccccuuccgcauuggugu‐3′), siRIPK3 (5′‐cccgacgaugucuucugucaa‐3′), siMLKL (5′‐gagauccaguucaacgaua‐3′), siRIPK1(5′‐ccacuagucugacugauga‐3′), siRIPK1‐3′‐UTR (5′‐gggcugauaacaguguugu‐3′), siRIPK3‐3′‐UTR (5′‐caggagucaauaaacauga‐3′), siMLKL‐3′‐UTR (5′‐gcuccucccuuccauaaau‐3′), siSTAT1(5′‐cagaaagagcuugacaguaaa‐3′), and control siRNA (5′‐aacguacgcggaauacuucga‐3′).
miRNA screening procedure
MicroRNA mimics are analogs of endogenous miRNAs that are synthesized by chemical synthesis and can enhance the function of endogenous miRNA. The miRNA mimics were synthesized by the Shanghai GenePharma Company. The control nontarget miRNA (negative control) and RIPK3 siRNA were used as the negative control and positive control, respectively. Human miRNAs (50 nM) were transferred into 96‐well plates containing Lipofectamine 2000 (0.2 μl per well). After 20 min of incubation, human colon cancer HT‐29 cells were added to the plates at a density of 3,000 cells per well. At 48 h post‐transfection, the cells were treated with 40 ng/ml TNF‐α, 100 nM Smac mimetic, and 20 μM z‐VAD for an additional 24 h and then cell viability was determined using the Cell Titer‐Glo Luminescent Cell Viability Assay Kit (Promega). Luminescence was calculated using SpectraMax i3x (Molecular Devices).
Generation of stable cell lines
All stable cell lines were generated via lentiviral infection. The plasmids (pCDH‐CMV‐MCS‐EF1‐copGFP) expressing MLKL, ΔR, VSVG, and Rev were co‐transfected into HEK293T using calcium phosphate. The medium was changed after 8 h, and cells were cultured for another 72 h. Conditioned medium was then collected daily for three more days. HeLa cells were infected with supernatant containing lentiviral particles expressing mCherry and RIPK3 (Genechem) to generate HeLa cells stably expressing RIPK3 (HeLa‐endogenous MLKL cells). HeLa‐MLKL knockout cells were infected with 1 ml of supernatant containing lentiviral particles expressing RIPK3 and MLKL (a DNA plasmid containing a HA and 3 × Flag tags at the C‐terminal of MLKL) to generate HeLa cells stably expressing RIPK3 and MLKL (HeLa‐exogenous MLKL cells). Three days postinfection, the infected cells were sorted by FACS according to GFP and mCherry fluorescence and then used as stable lines.
Electroporation
U937 cells (1 × 106) were centrifuged at 800 rpm for 3 min, resuspended in 100 μl of the desired electroporation buffer, and mixed with 4 μl of the oligo. The resuspended cells were transferred to cuvettes and immediately electroporated using the Nucleofector®. After electroporation, cells were incubated in a 6‐well plate with RPMI‐1640 medium supplemented containing 10% FBS. The day after electroporation, cells were centrifuged, and the medium was replaced with RPMI‐1640 medium containing 10% FBS.
RNA isolation and quantitative PCR
Total RNA was extracted from cell lines with TRNzol reagent (TIANGEN). For gene quantification, cDNA was synthesized from 1 μg of RNA using SuperScript III Reverse Transcriptase (Vazyme Biotech). For the detection of mature miR‐324‐5p, RNA was reverse‐transcribed using a specific reverse‐transcription primer (Applied Biosystems, CA). Quantitative PCR (qPCR) was performed using the Fast SYBR Green PCR Kit (Applied Biosystems, Carlsbad, CA, USA) and run on a real‐time PCR System (ABI 7500). qPCR was performed in triplicate for human RIPK1, human RIPK3, human MLKL, GAPDH, miRNA‐324‐5p, and U6. Gene and miRNA levels were normalized to the endogenous control, and the relative expression was calculated using the 2−ΔΔCT method. Differential miRNA expression was determined using a two‐sided Student's t‐test on a single miRNA basis. The following primers were used: for STAT1, 5′‐agggtcctctcatcgttac‐3′(forward) and 5′‐gaaagctgagcccatcgtg‐3′(reverse); for GAPDH, 5′‐gcaccaccaactgcttag‐3′(forward) and 5′‐ggccatgccagtgagctt‐3′(reverse); for MLKL, 5′‐ggccaggtcatccacaaac‐3′(forward) and 5′‐tatctccccattagcctcctc‐3′(reverse); for RIPK1, 5′‐ctcaccagccaacctcaag‐3′(forward) and 5′‐ggcatggtgggtgtatttc‐3′(reverse); for RIPK3, 5′‐cgggcgcaacataggaag‐3′(forward) and 5′‐tgcagcagccccgacaag‐3′(reverse); for IFN‐β, 5′‐ tgtggcaattgaatgggagg‐3′(forward) and 5′‐ cttccaggactgtcttcaga‐3′(reverse); for IFN‐γ, 5′‐gaagaattggaaagaggag‐3′(forward) and 5′‐gtattgctttgcgttggaca‐3′(reverse); for U6, 5′‐gcttcggcagcacatatactaa‐3′(forward) and 5′‐aacgcttcacgaatttgcgt‐3′(reverse); for miR‐324‐5p reverse transcription primer, 5′‐ctcaactggtgtcgtggagtcggcaattcagttgagacaccaat‐3′; for miR‐324‐5p qPCR primer, 5′‐acactccagctgggcgcatcccctagggc‐3′(forward) and 5′‐tggtgtcgtggagtcg‐3′(reverse); for the M gene of influenza A, 5′‐atgagccttctaaccgaggtcgaaacg‐3′(forward) and 5′‐tggacaaaacgtctacgctgcag‐3′(reverse).
Western blot analysis
For western blot analysis, cells were harvested and lysed in protein lysis buffer (20 mM Tris–HCl, 10% glycerol, 1% TritonX‐100, 150 mM NaCl, 1 mM Na3VO4, 25 mM β‐glycerol‐phosphate, 0.1 mM PMSF) supplemented with a protease inhibitor cocktail and a phosphatase inhibitor set (Roche). Cell lysates were fractionated on SDS‐PAGE gels and electrophoretically transferred to NC membranes. The NC membranes were blocked in 5% nonfat milk (PBS containing 0.1% Tween‐20) for 1 h at room temperature. Membranes were incubated with indicated primary antibody for 2 h at room temperature. After washing to remove any unbound primary antibody, membranes were incubated with goat anti‐rabbit (LI‐COR, 926–32211,1:10,000) or goat anti‐mouse (LI‐COR, 926–32210,1:10,000). β‐Actin was used as loading control. Blots were usually first stained for β‐actin, then the membrane was stripped and stained for the protein of interest. As the antibodies of STAT1 and β‐actin show no nonspecific bands at the location of target bands, membranes were incubated with anti‐STAT1 and anti‐β‐actin antibodies together, and both proteins were detected in the same blot. Proteins were visualized using Odyssey Imaging system (LI‐COR). Data were analyzed with ImageJ software. The band intensities of the analyzed proteins were normalized to that of β‐actin. Three independent experiments were performed, and representative results are shown.
Construction of the MLKL‐3′UTR luciferase reporter and activity analysis
According to the MLKL sequences and the predicted results from the Target Scan software, the luciferase reporter gene vector containing the human miR‐324‐5p binding site in the MLKL 3′UTR sequence was designed. The 3′UTR sequences of human and mouse MLKL were amplified by PCR and subcloned into the pmirGlo plasmid to construct the human pmirGlo‐MLKL‐3′UTR and murine pmirGlo‐MLKL‐3′UTR plasmid. The mutant form of murine MLKL‐3′UTR by introducing the miR‐324‐5p binding region “CTCTCATATCCTTCGGCATTG” of human MLKL‐3′UTR into the start site of mouse MLKL‐3′UTR. The 3′UTR sequences of RIPK1 and RIPK3 were subcloned into the pmirGlo plasmid to construct pmirGlo‐RIPK1‐3′UTR and pmirGlo‐RIPK3‐3′UTR plasmids, respectively. These constructs were checked by DNA sequencing. The following primers were used: for RIPK1‐3′UTR, 5′‐cgagctcccctggatgggctacggcagctgaagt‐3′(forward) and 5′‐ccgctcgagttgtggtataaatttttattttaaaaagtg‐3′(reverse); for RIPK3‐3′UTR, 5′‐cgagctcagcaccttccaagcttgcctccaag‐3′(forward) and 5′‐ccgctcgagtgactagcattccatcatgttta‐3′(reverse); for MLKL‐3′UTR, 5′‐cgagctctgtatcaaaatctaaaccaaggagtc‐3′(forward) and 5′‐ccgctcgagttagtgttaagacaacagtaattta‐3′(reverse); for MLKL‐3′UTR (mutant#2), 5′‐cgagctcgcattgggttatctatgggtg‐3′(forward); and for murine MLKL‐3′UTR, 5′‐cgagctcgaggacaaagtggacatttg‐3′(forward) and 5′‐ccgctcgagtttggtggtctatgcctttaa‐3′(reverse).
Logarithmically growing HEK293T cells were seeded in 96‐well plates, and the cell count for each well was 1 × 104. After 24 h, pmirGlo‐MLKL‐3′UTR‐wild type and pmirGlo‐MLKL‐3′UTR‐mutant were co‐transfected with miR‐324‐5p or nontarget miRNA using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). After 48–72 h, the cells were lysed, and the fluorescence intensity was determined using a dual‐luciferase reporter assay system (Promega). Luciferase activity was reported as firefly luciferase activity/renilla luciferase activity.
Site‐directed mutagenesis
Site‐directed mutagenesis using Pfu Turbo DNA Polymerase was used to modify the miR‐324‐5p binding sequence and to construct the mouse pmirGlo‐MLKL‐3′UTR mutant plasmid. The miR‐324‐5p binding region of human MLKL‐3′UTR (ctctcatatccttcggcattgg) was inserted before the start site of mouse MLKL‐3′UTR. DpnI was used to digest the nonmutated DNA template before transforming the mutated plasmids. The following primers were used for MLKL‐3′UTR (mutant#1), 5′‐gacatctctctctctcatataaatcggcattgggttatctatgg‐3′(forward) and 5′‐ccatagataacccaatgccgatttatatgagagagagagatgtc‐3′(reverse) and for murine MLKL‐3′UTR mutant, 5′‐ gttgtttaaacgagctcgctagcctctcatatccttcggcattgggaggacaaagtggacatttgtc −3′(forward) and 5′‐ gacaaatgtccactttgtcctcccaatgccgaaggatatgagaggctagcgagctcgtttaaacaac −3′(reverse).
ChIP assay
ChIP analysis of STAT1 binding to the miR‐324‐5p promoter was performed using an assay kit (R&D Systems, Minneapolis, MN, USA), following the manufacturer's instructions. In brief, after fixation with 1% formaldehyde, cell lysate was collected. The samples were then sonicated to shear chromatin and centrifuged. The supernatant was then collected for immunoprecipitation with an anti‐STAT antibody. After three times washes, the chromatin immunoprecipitate was subjected to PCR analysis using specific primers.
Virus and TCID50 assay
Influenza A virus (H1N1 strain PR8), which was a kind gift from Dr. Genhong Cheng (University of California, Los Angeles), was propagated in the allantoic cavities of 11‐day‐old specific pathogen‐free embryonated chicken eggs at 35°C. Freshly collected allantoic fluids were clarified by low‐speed centrifugation at 72 h postinoculation and then stored in small aliquots at −80°C. The virus titers were determined using a plaque‐forming assay in monolayers of Madin‐Darby canine kidney (MDCK) cells.
For the TCID50 assay, the IAV virus sample from infected HT‐29 cells was diluted in DMEM containing 1 μg/ml N‐tosyl‐L‐phenylalanine chloromethyl ketone‐trypsin (Sigma), 0.3% bovine albumin (Sigma), 1% penicillin/streptomycin, and 25 mM HEPES buffer (Gibco) across a 96‐well tissue culture plate with MDCK cells. After 48–72 h, TCID50 titer per 100 μl was determined using the Reed‐Muench method (Lei et al, 2021).
Generation of the miR‐324‐5p knockout cell line using CRISPR/Cas9
The chromosome region of mir‐324 (~12–30 bp) was deleted in HT‐29 cells using the CRISPR/Cas9 system. In HT‐29 cells, PX458 plasmid expressing sgRNA was transfected into cells using Lipofectamine 2000. The sequences of sgRNAs were 5′‐caccctagggcattggtgtaaagc‐3′ and 5′‐aaacgctttacaccaatgccctag‐3′. One day after transfection, cells were sorted by flow cytometry and reseeded in plates to allow individual clones to grow up. Clones were picked and verified by PCR genotyping and sequencing.
Statistical analysis
The GraphPad Prism software was used for all statistical analyses. An unpaired two‐tailed Student's t‐test was performed for two‐group comparisons. Statistical significance was set at *P < 0.05, **P < 0.01, or ***P < 0.001. Statistical significance was accepted for P‐values <0.05, and results are represented as means ± standard deviations (SD).
Author contributions
Xiaoyan Dou: Investigation; methodology. Xiaoliang Yu: Funding acquisition; investigation; methodology. Shujing Du: Investigation. Yu Han: Investigation. Liang Li: Investigation. Haoran Zhang: Investigation. Ying Yao: Investigation. Yayun Du: Investigation. Xinhui Wang: Investigation. Jingjing Li: Investigation. Tao Yang: Funding acquisition; investigation. Wei Zhang: Funding acquisition; investigation. Chengkui Yang: Investigation. Feng Ma: Supervision; funding acquisition. Sudan He: Supervision; funding acquisition.
In addition to the CRediT author contributions listed above, the contributions in detail are:
SH and FM designed this study and revised the manuscript; XD and XY carried out most of the experiments, analyzed the data, and drafted the manuscript; SD, YH, LL, HZ, and YY performed cell culture, qPCR, the CCID50 assay, and ChIP. YD, XW, and JL provided technical assistance; TY, WZ, and CY contributed to the discussion and supervised the project.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Source Data for Expanded View
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31830051, 31671436, 31900526, 31771533 and 3160010179), the National Key Research and Development Program of China (No.2018YFA0900803), the CAMS Innovation Fund for Medical Sciences (2021‐I2M‐1‐041, 2021‐I2M‐1‐047 and 2021‐I2M‐1‐061), Non‐profit Central Research Institute Fund of Chinese Academy of Medical Sciences (2021‐PT180‐001, 2019PT310028, 2017NL31004, 2017NL31002), a Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions, China Postdoctoral Science Foundation‐funded project (2019 M650563), Natural Science Foundation of Jiangsu Province Grant (BK20160314) and Fok Ying Tung Education Foundation for Young Teachers (151020). We appreciate the technical support from the RNA technology platform of the Suzhou Institute of Systems Medicine.
EMBO reports (2022) 23: e54438
Contributor Information
Feng Ma, Email: maf@ism.pumc.edu.cn.
Sudan He, Email: hesd@ism.pumc.edu.cn.
Data availability
No primary datasets have been generated and deposited.
References
- Afonso MB, Rodrigues PM, Simao AL, Gaspar MM, Carvalho T, Borralho P, Banales JM, Castro RE, Rodrigues CMP (2018) miRNA‐21 ablation protects against liver injury and necroptosis in cholestasis. Cell Death Differ 25: 857–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal V, Bell GW, Nam JW, Bartel DP (2015) Predicting effective microRNA target sites in mammalian mRNAs. elife 4: e05005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrat FJ, Crow MK, Ivashkiv LB (2019) Interferon target‐gene expression and epigenomic signatures in health and disease. Nat Immunol 20: 1574–1583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP (2018) Metazoan MicroRNAs. Cell 173: 20–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Kuroki S, Someda M, Yonehara S (2019a) Interferon‐gamma induces the cell surface exposure of phosphatidylserine by activating the protein MLKL in the absence of caspase‐8 activity. J Biol Chem 294: 11994–12006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P, Zhong J, Ye J, He Y, Liang Z, Cheng Y, Zheng J, Chen H, Chen C (2019b) miR‐324‐5p protects against oxidative stress‐induced endothelial progenitor cell injury by targeting Mtfr1. J Cell Physiol 234: 22082–22092 [DOI] [PubMed] [Google Scholar]
- Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK (2009) Phosphorylation‐driven assembly of the RIP1‐RIP3 complex regulates programmed necrosis and virus‐induced inflammation. Cell 137: 1112–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JE Jr, Kerr IM, Stark GR (1994) Jak‐STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 264: 1415–1421 [DOI] [PubMed] [Google Scholar]
- Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J (2005) Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol 1: 112–119 [DOI] [PubMed] [Google Scholar]
- Farina FM, Hall IF, Serio S, Zani S, Climent M, Salvarani N, Carullo P, Civilini E, Condorelli G, Elia L, et al (2020) miR‐128‐3p is a novel regulator of vascular smooth muscle cell phenotypic switch and vascular diseases. Circ Res 126: e120–e135 [DOI] [PubMed] [Google Scholar]
- Gunther C, He GW, Kremer AE, Murphy JM, Petrie EJ, Amann K, Vandenabeele P, Linkermann A, Poremba C, Schleicher U, et al (2016) The pseudokinase MLKL mediates programmed hepatocellular necrosis independently of RIPK3 during hepatitis. J Clin Invest 126: 4346–4360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Kaiser WJ, Mocarski ES (2015a) Manipulation of apoptosis and necroptosis signaling by herpesviruses. Med Microbiol Immunol 204: 439–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Omoto S, Harris PA, Finger JN, Bertin J, Gough PJ, Kaiser WJ, Mocarski ES (2015b) Herpes simplex virus suppresses necroptosis in human cells. Cell Host Microbe 17: 243–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X (2009) Receptor interacting protein kinase‐3 determines cellular necrotic response to TNF‐alpha. Cell 137: 1100–1111 [DOI] [PubMed] [Google Scholar]
- He S, Liang Y, Shao F, Wang X (2011) Toll‐like receptors activate programmed necrosis in macrophages through a receptor‐interacting kinase‐3‐mediated pathway. Proc Natl Acad Sci USA 108: 20054–20059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Wang X (2018) RIP kinases as modulators of inflammation and immunity. Nat Immunol 19: 912–922 [DOI] [PubMed] [Google Scholar]
- He S, Han J (2020) Manipulation of host cell death pathways by herpes simplex virus. Curr Top Microbiol Immunol 10.1007/82_2020_196 [DOI] [PubMed] [Google Scholar]
- Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, Bodmer JL, Schneider P, Seed B, Tschopp J (2000) Fas triggers an alternative, caspase‐8‐independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol 1: 489–495 [DOI] [PubMed] [Google Scholar]
- Huang Z, Wu SQ, Liang Y, Zhou X, Chen W, Li L, Wu J, Zhuang Q, Chen C, Li J, et al (2015) RIP1/RIP3 binding to HSV‐1 ICP6 initiates necroptosis to restrict virus propagation in mice. Cell Host Microbe 17: 229–242 [DOI] [PubMed] [Google Scholar]
- Huang L, Guo B, Liu S, Miao C, Li Y (2020) Inhibition of the LncRNA Gpr19 attenuates ischemia‐reperfusion injury after acute myocardial infarction by inhibiting apoptosis and oxidative stress via the miR‐324‐5p/Mtfr1 axis. IUBMB Life 72: 373–383 [DOI] [PubMed] [Google Scholar]
- Ingram JP, Thapa RJ, Fisher A, Tummers B, Zhang T, Yin C, Rodriguez DA, Guo H, Lane R, Williams R, et al (2019) ZBP1/DAI drives RIPK3‐mediated cell death induced by IFNs in the absence of RIPK1. J Immunol 203: 1348–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao H, Wachsmuth L, Kumari S, Schwarzer R, Lin J, Eren RO, Fisher A, Lane R, Young GR, Kassiotis G, et al (2020) Z‐nucleic‐acid sensing triggers ZBP1‐dependent necroptosis and inflammation. Nature 580: 391–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, Sehon CA, Marquis RW, Bertin J, Mocarski ES (2013) Toll‐like receptor 3‐mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem 288: 31268–31279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser WJ, Upton JW, Mocarski ES (2008) Receptor‐interacting protein homotypic interaction motif‐dependent control of NF‐kappa B activation via the DNA‐dependent activator of IFN regulatory factors. J Immunol 181: 6427–6434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karki R, Sharma BR, Tuladhar S, Williams EP, Zalduondo L, Samir P, Zheng M, Sundaram B, Banoth B, Malireddi RKS, et al (2021) Synergism of TNF‐alpha and IFN‐gamma triggers inflammatory cell death, tissue damage, and mortality in SARS‐CoV‐2 infection and cytokine shock syndromes. Cell 184: e117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr JF, Wyllie AH, Currie AR (1972) Apoptosis: a basic biological phenomenon with wide‐ranging implications in tissue kinetics. Br J Cancer 26: 239–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knuth AK, Rosler S, Schenk B, Kowald L, van Wijk SJL, Fulda S (2019) Interferons transcriptionally up‐regulate MLKL expression in cancer cells. Neoplasia 21: 74–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koerner I, Kochs G, Kalinke U, Weiss S, Staeheli P (2007) Protective role of beta interferon in host defense against influenza A virus. J Virol 81: 2025–2030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause CD, Lavnikova N, Xie J, Mei E, Mirochnitchenko OV, Jia Y, Hochstrasser RM, Pestka S (2006) Preassembly and ligand‐induced restructuring of the chains of the IFN‐gamma receptor complex: the roles of Jak kinases, Stat1 and the receptor chains. Cell Res 16: 55–69 [DOI] [PubMed] [Google Scholar]
- Kumar A, Kumar A, Ingle H, Kumar S, Mishra R, Verma MK, Biswas D, Kumar NS, Mishra A, Raut AA, et al (2018) MicroRNA hsa‐miR‐324‐5p suppresses H5N1 virus replication by targeting the viral PB1 and host CUEDC2. J Virol 92: e01057‐18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuriakose T, Man SM, Subbarao Malireddi RK, Karki R, Kesavardhana S, Place DE, Neale G, Vogel P, Kanneganti TD (2016) ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci Immunol 1: aag2045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laster SM, Wood JG, Gooding LR (1988) Tumor necrosis factor can induce both apoptic and necrotic forms of cell lysis. J Immunol 141: 2629–2634 [PubMed] [Google Scholar]
- Lei C, Yang J, Hu J, Sun X (2021) On the calculation of TCID50 for quantitation of virus infectivity. Virol Sin 36: 141–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis BP, Burge CB, Bartel DP (2005) Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120: 15–20 [DOI] [PubMed] [Google Scholar]
- Li D, Li L, Chen X, Yang W, Cao Y (2021) Circular RNA SERPINE2 promotes development of glioblastoma by regulating the miR‐361‐3p/miR‐324‐5p/BCL2 signaling pathway. Mol Ther Oncolytics 22: 483–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H, Zhou AJ, Zhang JY, Liu SF, Gu JX (2018) MiR‐324‐5p reduces viability and induces apoptosis in gastric cancer cells through modulating TSPAN8. J Pharm Pharmacol 70: 1513–1520 [DOI] [PubMed] [Google Scholar]
- Liu J, van Mil A, Vrijsen K, Zhao J, Gao L, Metz CH, Goumans MJ, Doevendans PA, Sluijter JP (2011) MicroRNA‐155 prevents necrotic cell death in human cardiomyocyte progenitor cells via targeting RIP1. J Cell Mol Med 15: 1474–1482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Conklin DJ, Li F, Dai Z, Hua X, Li Y, Xu‐Monette ZY, Young KH, Xiong W, Wysoczynski M, et al (2015) The oncogenic microRNA miR‐21 promotes regulated necrosis in mice. Nat Commun 6: 7151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man SM, Karki R, Kanneganti TD (2017) Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol Rev 277: 61–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megger DA, Philipp J, Le‐Trilling VTK, Sitek B, Trilling M (2017) Deciphering of the human interferon‐regulated proteome by mass spectrometry‐based quantitative analysis reveals extent and dynamics of protein induction and repression. Front Immunol 8: 1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mifflin L, Ofengeim D, Yuan J (2020) Receptor‐interacting protein kinase 1 (RIPK1) as a therapeutic target. Nat Rev Drug Discov 19: 553–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nailwal H, Chan FK (2019) Necroptosis in anti‐viral inflammation. Cell Death Differ 26: 4–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogusa S, Thapa RJ, Dillon CP, Liedmann S, Oguin TH 3rd, Ingram JP, Rodriguez DA, Kosoff R, Sharma S, Sturm O, et al (2016) RIPK3 activates parallel pathways of MLKL‐driven necroptosis and FADD‐mediated apoptosis to protect against influenza A virus. Cell Host Microbe 20: 13–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Othumpangat S, Beezhold DH, Umbright CM, Noti JD (2021) Influenza virus‐induced novel miRNAs regulate the STAT pathway. Viruses 13: 967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu B, Li X, Gao W, Sun W, Jin Y, Cardona CJ, Xing Z (2012) Human intestinal epithelial cells are susceptible to influenza virus subtype H9N2. Virus Res 163: 151–159 [DOI] [PubMed] [Google Scholar]
- Robinson N, McComb S, Mulligan R, Dudani R, Krishnan L, Sad S (2012) Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nat Immunol 13: 954–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S, Bantel H, Wandrer F, Schneider AT, Gautheron J, Vucur M, Tacke F, Trautwein C, Luedde T, Roderburg C (2017) miR‐1224 inhibits cell proliferation in acute liver failure by targeting the antiapoptotic gene Nfib. J Hepatol 67: 966–978 [DOI] [PubMed] [Google Scholar]
- Sarhan J, Liu BC, Muendlein HI, Weindel CG, Smirnova I, Tang AY, Ilyukha V, Sorokin M, Buzdin A, Fitzgerald KA, et al (2019) Constitutive interferon signaling maintains critical threshold of MLKL expression to license necroptosis. Cell Death Differ 26: 332–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoggins JW, MacDuff DA, Imanaka N, Gainey MD, Shrestha B, Eitson JL, Mar KB, Richardson RB, Ratushny AV, Litvak V, et al (2014) Pan‐viral specificity of IFN‐induced genes reveals new roles for cGAS in innate immunity. Nature 505: 691–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon JP, Vrba SM, Reynoso GV, Wynne‐Jones E, Kamenyeva O, Malo CS, Cherry CR, McManus DT, Hickman HD (2021) Group 1 innate lymphoid‐cell‐derived interferon‐gamma maintains anti‐viral vigilance in the mucosal epithelium. Immunity 54: e275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutz MD, Ojaimi S, Allison C, Preston S, Arandjelovic P, Hildebrand JM, Sandow JJ, Webb AI, Silke J, Alexander WS, et al (2018) Necroptotic signaling is primed in Mycobacterium tuberculosis‐infected macrophages, but its pathophysiological consequence in disease is restricted. Cell Death Differ 25: 951–965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X, et al (2012) Mixed lineage kinase domain‐like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148: 213–227 [DOI] [PubMed] [Google Scholar]
- Thapa RJ, Ingram JP, Ragan KB, Nogusa S, Boyd DF, Benitez AA, Sridharan H, Kosoff R, Shubina M, Landsteiner VJ, et al (2016) DAI senses influenza A virus genomic RNA and activates RIPK3‐dependent cell death. Cell Host Microbe 20: 674–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thapa RJ, Nogusa S, Chen P, Maki JL, Lerro A, Andrake M, Rall GF, Degterev A, Balachandran S (2013) Interferon‐induced RIP1/RIP3‐mediated necrosis requires PKR and is licensed by FADD and caspases. Proc Natl Acad Sci USA 110: E3109–E3118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trilling M, Bellora N, Rutkowski AJ, de Graaf M, Dickinson P, Robertson K, Prazeres da Costa O, Ghazal P, Friedel CC, Alba MM, et al (2013) Deciphering the modulation of gene expression by type I and II interferons combining 4sU‐tagging, translational arrest and in silico promoter analysis. Nucleic Acids Res 41: 8107–8125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton JW, Kaiser WJ, Mocarski ES (2010) Virus inhibition of RIP3‐dependent necrosis. Cell Host Microbe 7: 302–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton JW, Kaiser WJ, Mocarski ES (2019) DAI/ZBP1/DLM‐1 complexes with RIP3 to mediate virus‐induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 26: 564 [DOI] [PubMed] [Google Scholar]
- Wan Y, Luo H, Yang M, Tian X, Peng B, Zhan T, Chen X, Ding Y, He J, Cheng X, et al (2020) miR‐324‐5p contributes to cell proliferation and apoptosis in pancreatic cancer by targeting KLF3. Mol Ther Oncolytics 18: 432–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Liu F, Zhou LY, Ding SL, Long B, Liu CY, Sun T, Fan YY, Sun L, Li PF (2013) miR‐874 regulates myocardial necrosis by targeting caspase‐8. Cell Death Dis 4: e709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Zhang DL, Long B, An T, Zhang J, Zhou LY, Liu CY, Li PF (2015) NFAT4‐dependent miR‐324‐5p regulates mitochondrial morphology and cardiomyocyte cell death by targeting Mtfr1. Cell Death Dis 6: e2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Du F, Wang X (2008) TNF‐alpha induces two distinct caspase‐8 activation pathways. Cell 133: 693–703 [DOI] [PubMed] [Google Scholar]
- Wang R, Li H, Wu J, Cai ZY, Li B, Ni H, Qiu X, Chen H, Liu W, Yang ZH, et al (2020) Gut stem cell necroptosis by genome instability triggers bowel inflammation. Nature 580: 386–390 [DOI] [PubMed] [Google Scholar]
- Wang X, Li Y, Liu S, Yu X, Li L, Shi C, He W, Li J, Xu L, Hu Z, et al (2014) Direct activation of RIP3/MLKL‐dependent necrosis by herpes simplex virus 1 (HSV‐1) protein ICP6 triggers host antiviral defense. Proc Natl Acad Sci USA 111: 15438–15443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Song Q, Huang W, Lin Y, Wang X, Wang C, Willard B, Zhao C, Nan J, Holvey‐Bates E, et al (2021) A virus‐induced conformational switch of STAT1‐STAT2 dimers boosts antiviral defenses. Cell Res 31: 206–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Jiang Y, Xu X, Su X, Liu Y, Ma Y, Zhao Y, Shen Z, Huang B, Cao X (2019) Inducible degradation of lncRNA Sros1 promotes IFN‐gamma‐mediated activation of innate immune responses by stabilizing Stat1 mRNA. Nat Immunol 20: 1621–1630 [DOI] [PubMed] [Google Scholar]
- Yang E, Li MMH (2020) All About the RNA: Interferon‐Stimulated Genes That Interfere With Viral RNA Processes. Front Immunol 11: 605024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Zhu Z, Gao J, Yang L, Dang E, Fang H, Shao S, Zhang S, Xiao C, Yuan X, et al (2020) Plasma exosomal miR‐375‐3p regulates mitochondria‐dependent keratinocyte apoptosis by targeting XIAP in severe drug‐induced skin reactions. Sci Transl Med 12: eaaw6142 [DOI] [PubMed] [Google Scholar]
- Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, Dong MQ, Han J (2009) RIP3, an energy metabolism regulator that switches TNF‐induced cell death from apoptosis to necrosis. Science 325: 332–336 [DOI] [PubMed] [Google Scholar]
- Zhang DY, Wang BJ, Ma M, Yu K, Zhang Q, Zhang XW (2019) MicroRNA‐325‐3p protects the heart after myocardial infarction by inhibiting RIPK3 and programmed necrosis in mice. BMC Mol Biol 20: 17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Ma Y, Tian Y, Zhou Y, Tang Y, Hu S (2021) Downregulation of microRNA221 facilitates H1N1 influenza A virus replication through suppression of typeIFN response by targeting the SOCS1/NFkappaB pathway. Mol Med Rep 24: 497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Z, Li J, An J, Feng Y, Wang L (2021) High miR‐324‐5p expression predicts unfavorable prognosis of gastric cancer and facilitates tumor progression in tumor cells. Diagn Pathol 16: 5 [DOI] [PMC free article] [PubMed] [Google Scholar]