

Abstract
Mutations in the human kinesin family member 5A (KIF5A) gene were recently identified as a genetic cause of amyotrophic lateral sclerosis (ALS). Several KIF5A ALS variants cause exon 27 skipping and are predicted to produce motor proteins with an altered C‐terminal tail (referred to as ΔExon27). However, the underlying pathogenic mechanism is still unknown. Here, we confirm the expression of KIF5A mutant proteins in patient iPSC‐derived motor neurons. We perform a comprehensive analysis of ΔExon27 at the single‐molecule, cellular, and organism levels. Our results show that ΔExon27 is prone to form cytoplasmic aggregates and is neurotoxic. The mutation relieves motor autoinhibition and increases motor self‐association, leading to drastically enhanced processivity on microtubules. Finally, ectopic expression of ΔExon27 in Drosophila melanogaster causes wing defects, motor impairment, paralysis, and premature death. Our results suggest gain‐of‐function as an underlying disease mechanism in KIF5A‐associated ALS.
Keywords: aggregation, ALS, autoinhibition, KIF5A, microtubules
Subject Categories: Membranes & Trafficking, Molecular Biology of Disease, Neuroscience
The ALS‐associated KIF5A ΔExon27 mutation drives the disease via a gain‐of‐function mechanism caused by constitutive activation and increased motor association and aggregation of mutant KIF5A.
Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease characterized by loss of upper and lower motor neurons (MNs) leading to paralysis and death within 3–5 years after diagnosis (Brenner & Weishaupt, 2019). Most of the ALS patients are classified as sporadic ALS (sALS), while about 10% of patients show a clear family history (fALS). Although varying in disease onset and duration, fALS and sALS patients cannot be differentiated by their clinical features. The mechanisms of disease pathogenesis leading to the exclusive demise of MNs remain unclear and there is no effective therapy. Over 30 genes have been linked to ALS, and several of these genes, such as PFN1 (profilin 1) and TUBA4A (tubulin, alpha 4A) are involved in cytoskeletal function and intracellular transport (Brenner & Weishaupt, 2019). Motor neurons are highly polarized cells with neurites that can exceed 1 m in length. Various materials, such as mRNA, proteins, lipids, membrane‐bound vesicles, and organelles commute between cell bodies and synaptic terminals as they are transported along microtubules by two major families of motor proteins. Kinesin family (KIF) proteins mediate anterograde transport away from the cell body to neurite terminals whereas cytoplasmic dynein‐1 drives the retrograde transport in the opposite direction. In 2018, three KIF5A splice site mutations (OMIM# 602821) were identified among 426 ALS families by the Weishaupt group (Brenner et al, 2018). Additional ALS patients with KIF5A mutations were subsequently reported in American and Asian cohorts (Nicolas et al, 2018; Faruq et al, 2019; Zhang et al, 2019; Saez‐Atienzar et al, 2020; Nakamura et al, 2021; Naruse et al, 2021).
Human KIF5A, together with KIF5B and KIF5C, belongs to the kinesin‐1 family and is one of the founding members of the kinesin superfamily with a total of 45 members (Hirokawa et al, 2009). The KIF5A motor complex is a heterotetramer consisting of two kinesin heavy chains (KHCs) and two kinesin light chains (KLCs). It uses the energy derived from ATP binding and hydrolysis to transport a variety of cargos processively (the ability to take hundreds of steps along microtubules before dissociation) to the plus‐ends of microtubules. Studies in animal models support that KIF5A has essential functions in the development and functioning of the nervous system (Tanaka et al, 1998; Kanai et al, 2000; Xia et al, 2003). The KIF5A KHC contains an N‐terminal catalytic motor domain, an α‐helical stalk region, and a C‐terminal tail region (Fig 1A). KIF5A was previously identified as the causative gene for hereditary spastic paraplegia type10 (SPG10) and Charcot–Marie–Tooth type 2 (CMT2), with mutations mostly located in the N‐terminal motor domain (Fichera et al, 2004; Blair et al, 2006; Crimella et al, 2012). Moreover, de novo heterozygous frameshift rare variants in the C‐terminal domain of KIF5A were reported to be associated with neonatal intractable myoclonus (NEIMY), a severe infantile‐onset neurologic disorder characterized by myoclonic seizures, hypotonia, dysphagia, and early development arrest (Duis et al, 2016; Rydzanicz et al, 2017). Interestingly, the ALS‐associated variants reported in KIF5A are mainly clustered at the 5′ and 3′ splice junctions of exon 27 (Brenner et al, 2018; Nicolas et al, 2018). These variants cause a complete skipping of exon 27, yielding a protein with the normal C‐terminal 34 amino acids replaced with a “neopeptide” of 39 aberrant amino acids (referred to as ΔExon27 hereafter; Fig 1A). Alternatively, some KIF5A‐ALS mutations are predicted to cause a frameshift in exon 27 without exon skipping, also leading to mutant motor proteins with altered C‐terminal tails including the 39 aberrant amino acids, which had not been experimentally proven thus far (Appendix Fig S1).
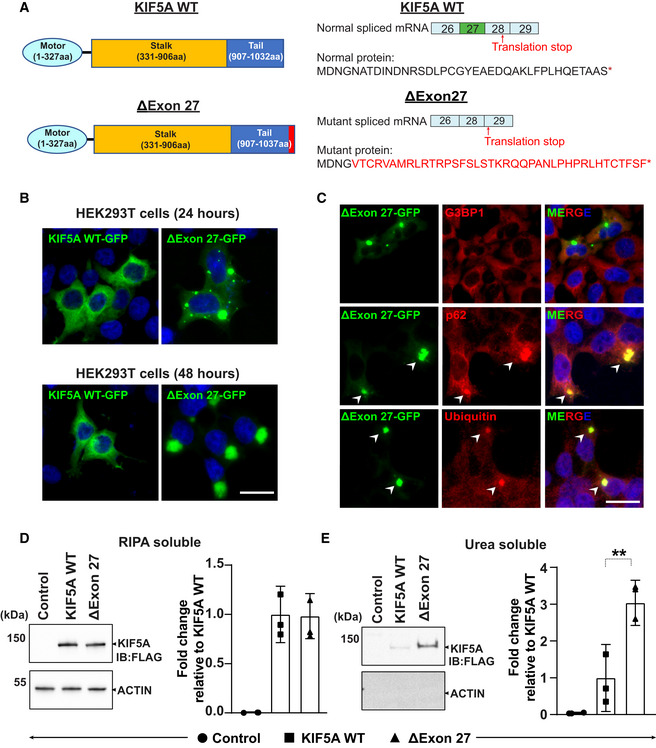
Figure 1. ALS‐associated KIF5A ΔExon27 is prone to form cytoplasmic aggregates.
-
ASchematic illustration of KIF5A WT and ΔExon27 showing the motor, stalk, and cargo domains (left). Several ALS‐associated KIF5A variants disrupt the C‐terminal tail by skipping exon 27 during mRNA splicing and produce a neopeptide of aberrant 39 amino acids (red) replacing the normal tail of 34 amino acids (right).
-
BHEK293T cells were transfected with KIF5A WT‐GFP or ΔExon27‐GFP for 24 and 48 h. Nuclei were stained with DAPI, three biological replicates, n = 200 cells per experiment. Scale bar: 20 μm.
-
CCo‐staining of G3BP1, p62, and Ubiquitin with KIF5A ∆Exon27 granules, three biological replicates, n = 250 cells per experiment. Scale bar: 20 μm.
-
D, EWestern blot analysis of RIPA‐soluble fraction and RIPA‐insoluble, urea‐soluble fraction of proteins prepared from HEK293T cells expressing empty plasmid (control), FLAG‐KIF5A WT, FLAG‐ΔExon27. Insoluble/soluble KIF5A fractions were detected with anti‐FLAG and actin was used as a loading control, three biological replicates. Statistical analysis was performed using student's t‐test (**P < 0.01). Data represent the mean ± SD.
Currently, the molecular mechanism of ΔExon27 underlying ALS pathogenesis is unclear. All known KIF5A ALS variants are autosomal dominant, and it is hypothesized that defective KIF5A alleles may lead to dysfunctional kinesin and impaired cargo transport. Alternatively, the altered C‐terminal tail in the ΔExon27 mutant might confer detrimental effects through a toxic gain‐of‐function mechanism. The latter is supported by the tight genotype/phenotype correlation with the accumulation of all KIF5A‐ALS mutations in a narrow hot spot. Moreover, these mutations are predicted to result in the same aberrant peptide at the C‐terminus.
In this study, we show the expression of mutant KIF5A proteins in iPSC‐derived motor neurons from KIF5A ALS patients using a custom antibody generated against the neopeptide of 39 amino acids. We performed a comprehensive analysis of the ΔExon27 mutation by genetic, biochemical, and single‐molecule methods. We find that ΔExon27 is prone to aggregate formation and is neurotoxic. Transgenic Drosophila expressing ΔExon27 display wing deficits, motor impairment, paralysis, and premature death. Mechanistically, ΔExon27 is constitutively active and exhibits an increased motor self‐association and a drastically enhanced processivity on microtubules. Together, these results suggest that the ΔExon27 protein is expressed in KIF5A ALS patients and leads to neuronal toxicity via gain‐of‐function caused by constitutive activation and increased motor association and aggregation.
Results
KIF5A ΔExon27 is aggregation prone
We first expressed human wild‐type KIF5A (WT) and the ALS mutant ΔExon27 with C‐terminal GFP tags in HEK293T cells. Twenty‐four hours after transfection, WT is mainly diffuse or filamentous in the cytoplasm (Fig 1B), consistent with previous reports (Rahman et al, 1999; Kamata et al, 2017; Yoo et al, 2019). Interestingly, a major population of cells (∼90%) expressing ΔExon27 show cytoplasmic granules of various sizes (Fig 1B). At 48 h, ∼4% WT expressing cells also show a few granules, though significantly less abundant than those expressing ΔExon27 which usually have one single large inclusion in the cytoplasm (Fig 1B). Similar results were also observed when these constructs were expressed in mouse neuroblastoma cells (N2a; Fig EV1A). To exclude the possibility that this phenotype is due to the additional C‐terminal GFP tag, we generated N‐terminally FLAG‐tagged KIF5A constructs. Like the C‐terminally GFP‐tagged ones, the N‐terminally FLAG‐tagged ΔExon27 also forms robust cytoplasmic granules (Fig EV1B). In contrast, the missense mutation (R280C) in motor region, which is the most common genetic cause for SPG10, is diffuse or filamentous in the cytoplasm, as with the FLAG‐tagged WT (Fig EV1B).
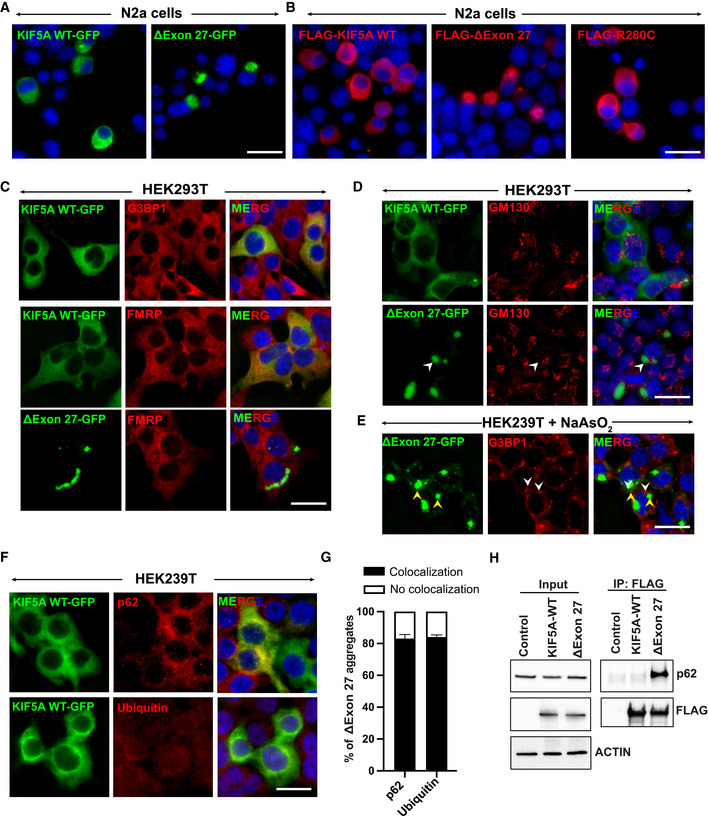
Figure EV1. KIF5A ΔExon27 is prone to form cytoplasmic aggregates.
-
A, BExpression of human KIF5A WT and disease‐associated mutants with C‐terminal GFP (A) or N‐terminal FLAG (B) tags in N2a cells. Scale bar: 20 μm.
-
CColocalization of G3BP1 and FMRP, markers of stress granules, with KIF5A WT or ∆Exon27 expressed in HEK293T cells. Scale bar: 20 μm.
-
DGM130 (a Golgi marker) staining in HEK293T cells expressing either KIF5A WT or ∆Exon27. Scale bar: 20 μm.
-
EG3BP1 staining in HEK239T cells expressing ∆Exon27 and treated with sodium arsenite (200 μM) for 30 min. White and yellow arrowheads highlight stress granules and ∆Exon27 aggregates respectively. Scale bar: 20 μm.
-
Fp62 and ubiquitin staining in KIF5A WT expressing HEK293T cells. Scale bar: 20 μm.
-
GPercent of cells with p62 and ubiquitin colocalized with cytoplasmic ∆Exon27 granules. Bars indicate mean ± SD.
-
HImmunoprecipitation of KIF5A using antibodies against the N‐terminal FLAG tag also pulled down endogenous p62 only in cells expressing Δexon27, but not WT KIF5A. Three biological replicates, n = 200 cells per experiment.
To identify the properties of these granules, we first determined whether ΔExon27 can induce stress granule formation and colocalize with them. We performed immunostaining against Ras GTPase‐activating protein‐binding protein 1 (G3BP1) and Fragile X mental retardation protein (FMRP), classical markers of stress granules that are implicated in ALS pathogenesis (Li et al, 2013). Few stress granules are observed in cells expressing either WT (Fig EV1C) or ΔExon27 and merely ∼2% ΔExon27 granules co‐stain with G3BP1 or FMRP (Figs 1C and EV1C), nor do they localize in cellular organelles such as Golgi (Fig EV1D). We further induced stress granules with sodium arsenite (NaAsO2) and did not see ΔExon27 granules colocalize with G3BP1‐positive stress granules (Fig EV1E). Protein aggregations are characteristic features of many neurodegenerative diseases (Ross & Poirier, 2004). We found that ∼83% of ΔExon27 granules are positive for p62 and ubiquitin (Figs 1C, and EV1F and G), common components of cytoplasmic inclusions found in many protein aggregation diseases (Ciechanover & Kwon, 2015). Immunoprecipitation of KIF5A using an antibody against the N‐terminal FLAG tag also pulls down endogenous p62 only in cells expressing ΔExon27, but not WT (Fig EV1H). We further determined the solubility of WT and ΔExon27 in detergents of different strength. In the RIPA‐soluble fraction, the expression level of ΔExon27 is comparable to that of WT, suggesting that the propensity of ΔExon27 to form cytoplasmic granules is not due to its higher expression (Fig 1D). In the RIPA‐insoluble and urea‐soluble fraction, we detected significantly higher signal of ΔExon27 compared with WT (Fig 1E). Because cytoplasmic inclusions of TAR DNA‐binding protein 43 (TDP‐43) were observed in > 95% of ALS patients (Neumann et al, 2006), we performed immunofluorescence staining to detect TDP‐43. In cells expressing either WT or ΔExon27, TDP‐43 remains within the nucleus (Fig EV2A). To circumvent possible transient transfection effects, we expressed WT or ΔExon27 via lentivirus‐mediated transduction. ΔExon27 level is reduced by > 100 folds and is comparable to the endogenous KIF5A (Fig EV2B). We still detect a robust accumulation of p62‐positive ΔExon27 aggregates in the cytoplasm (Fig EV2C). These results support that KIF5A ΔExon27 with the altered C‐terminal tail is aggregation prone.
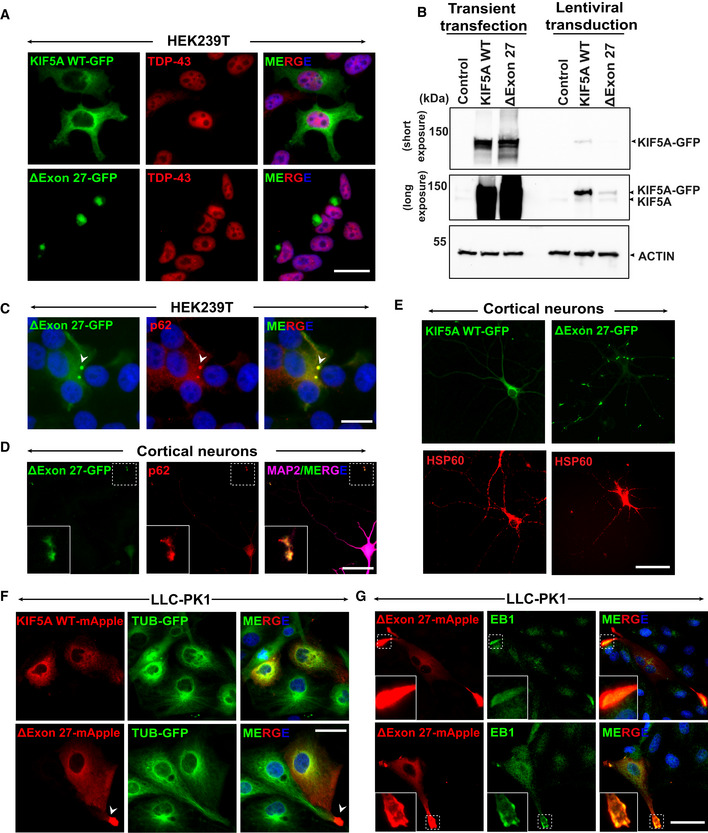
Figure EV2. KIF5A ΔExon27 accumulates at the plus‐ends of microtubules.
-
AStaining of TDP‐43 in HEK293T cells expressing either KIF5A WT or ∆Exon27 showed nuclear TDP‐43. Three biological replicates, n = 200 cells per experiment. Scale bar: 20 μm.
-
BWestern blot detecting KIF5A expression with transient transfection and lentiviral transduction. Two biological replicates.
-
C∆Exon27 expressed in HEK293T cells via lentiviral transduction forms small aggregates that colocalize with p62 (white arrowhead). Two biological replicates. Scale bar: 20 μm.
-
DColocalization of p62 with KIF5A ∆Exon27 granules. Scale bar: 20 μm.
-
EMitochondria distribution in primary cortical neurons expressing KIF5A WT or ∆Exon27. Scale bar: 20 μm.
-
FExpression of KIF5A WT‐mApple and ∆exon27‐mApple in LLC‐PK1 cells stably expressing tubulin‐GFP. KIF5A ∆exon27 shows aggregates at proximal tubule regions as highlighted by the white arrowhead. Scale bar: 20 μm.
-
G∆exon27 expressed in LLC‐PK1 cells shows aggregates that colocalize with microtubule plus‐end‐tracking protein EB1 at the proximal extrusions. Three biological replicates, n = 30 cells per experiment. Scale bar: 20 μm.
ΔExon27 interacts with WT KIF5A and causes neuronal toxicity
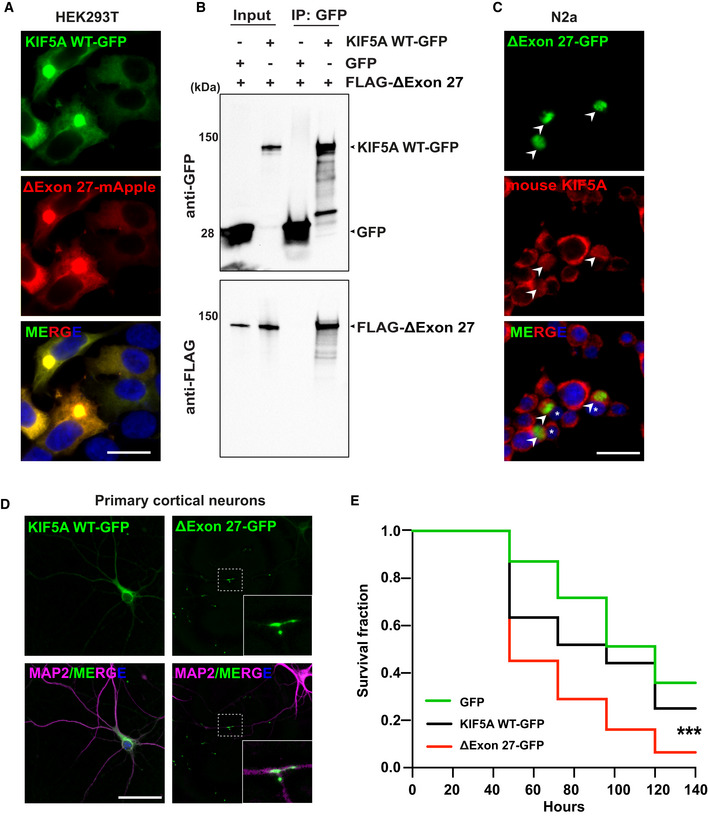
KIF5A has been proposed to function as homodimers or heterodimers, with the dimerization domain located in the stalk region (Kanai et al, 2000). Since ALS‐associated KIF5A variants are autosomal dominant, pathogenicity may be caused by mutant‐WT KIF5A complexes. To mimic the patient scenario and determine whether ΔExon27 interacts with WT KIF5A, we co‐expressed GFP‐tagged WT and mApple‐tagged ΔExon27 in HEK293T cells. WT and ΔExon27 colocalize near‐perfectly, forming robust cytoplasmic aggregates 48 h after transfection (Fig 2A). In addition, immunoprecipitation of WT KIF5A pulls down ΔExon27 when the two constructs are co‐expressed (Fig 2B). We also expressed ΔExon27‐GFP in N2a cells and stained for the endogenous KIF5A WT using an antibody generated with a peptide corresponding to the C‐terminus of mouse KIF5A WT (amino acids 1,007–1,027), which does not recognize ΔExon27. While the endogenous KIF5A WT is uniformly distributed in the cytoplasm of nontransfected cells, it colocalizes with ΔExon27 aggregates in the transfected cells (Fig 2C). These results suggest that KIF5A ΔExon27 interacts with WT and sequester them into aggregates.
Figure 2. ΔExon27 interacts with WT KIF5A to form aggregates and causes neuronal toxicity.
-
AExpression of KIF5A WT and ΔExon27 tagged with GFP and mApple, respectively, in HEK293T cells. Images were taken 48 h after transfection, two biological replicates. Scale bar: 20 μm.
-
BPull‐down analysis showing the association of ΔExon27 with WT KIF5A, two biological replicates.
-
CStaining of mouse endogenous KIF5A in N2a cells transfected with ΔExon27‐GFP using an antibody generated with a peptide corresponding to the C‐terminus of KIF5A WT (amino acids 1007–1027), which does not recognize ΔExon27. ΔExon27 granules are highlighted with arrowheads. The nuclei of cells expressing ΔExon27 are indicated with asterisk, two biological replicates. Scale bar: 20 μm.
-
DMouse primary neurons transfected with KIF5A WT‐GFP and ΔExon27‐GFP were probed using anti‐GFP antibody 24 h after transfection. Neurons were identified by microtubule‐associated protein 2 (MAP2) and nuclei were stained with DAPI, three biological replicates, n = 200 cells per experiment. Scale bar: 20 μm.
-
EFive‐day‐old mouse cortical neurons were transfected with mApple together with either GFP (n = 150), KIF5A WT‐GFP (n = 160), or ΔExon27‐GFP (n = 135), three biological replicates. Images were taken every 24 h after transfection. Survival of neurons was analyzed by Kaplan–Meier survival analysis (***P < 0.0001, log‐rank test).
We further analyzed the expression pattern of WT and ΔExon27 in primary mouse cortical neurons. Twenty‐four hours after transfection, ΔExon27 accumulates along the neurites and at the terminal ends in almost all transfected neurons (Fig 2D). In rare populations, we also found ΔExon27 granules in the soma of neurons. In contrast, WT KIF5A exhibits a diffuse pattern in the cytoplasm and along neurites (Fig 2D). Immunostaining showed that almost all of the ΔExon27 inclusions robustly colocalize with p62, further suggesting they are likely caused by motor protein aggregations (Fig EV2D). We next checked whether the distribution of mitochondria, a well‐known cargo of KIF5A, is altered by ΔExon27. The ΔExon27 granules do not colocalize with mitochondria, nor do we observe any obvious difference in gross mitochondrial distribution in neurons expressing either WT or ΔExon27 (Fig EV2E). Given the robust accumulation of ΔExon27 along the neurites and at the terminal ends, we hypothesized that ΔExon27 preferentially accumulates at the plus‐ends of microtubules. To test this, we obtained a highly polarized porcine kidney cell line (LLC‐PK1) stably expressing tubulin‐GFP (Rusan et al, 2001). Indeed, ΔExon27 granules are observed mainly at the peripheral extrusions (Fig EV2F) and colocalize with microtubule plus‐end‐tracking protein EB1 (Fig EV2G).
Protein inclusions formed by several disease‐related proteins have been shown to trigger cellular toxicity (Soto & Pritzkow, 2018). To test whether aggregation‐prone ΔExon27 causes neuronal toxicity, we co‐transfected GFP, KIF5A WT‐GFP, or ΔExon27‐GFP together with mApple in mouse primary cortical neurons at DIV (days in vitro) 5 and used automated longitudinal microscopy to track the survival of hundreds of neurons indicated by mApple fluorescence over 6 days. The rate of cell death is significantly higher in neurons expressing ΔExon27‐GFP than those expressing WT‐GFP or GFP alone (Fig 2E), indicating that ΔExon27 is neurotoxic.
KIF5A ΔExon27 displays relieved autoinhibition, increased motor self‐association, and enhanced processivity on microtubules
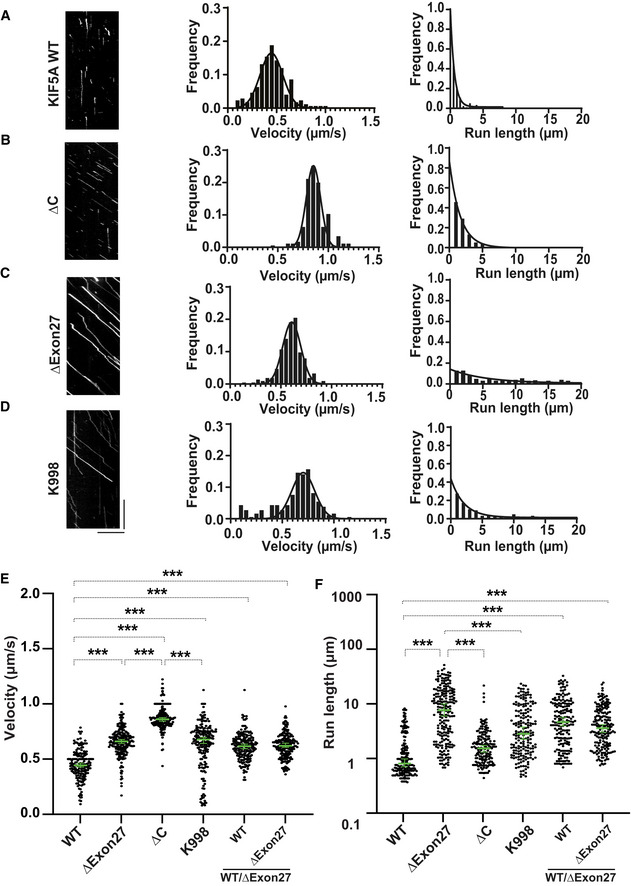
The accumulation of ΔExon27 granules at the plus‐ends of microtubules suggests that the localization of granules observed in distal neurites may be driven by aberrant motility and/or association of the mutant protein along microtubules. Kinesin‐1 motors transport cargoes by converting chemical energy from ATP to mechanical energy to drive processive movement on microtubules. When not transporting cargos, kinesin is autoinhibited to prevent ATP squandering. Autoinhibition is achieved by the C‐terminal tail domain binding to the motor domain to keep it in a folded autoinhibited state (Dietrich et al, 2008; Kaan et al, 2011). Given that ΔExon27 possesses an altered C‐terminal tail, we hypothesized that the autoinhibition of KIF5A might be compromised. To test this, we performed single‐molecule total internal reflection fluorescence (smTIRF) microscopy assays to assess the mobility of three fluorophore‐tagged KIF5A proteins: WT, ∆C (amino acids 1–906, Fig EV3A, a KIF5A construct in which the C‐terminal cargo domain was removed rendering the motor incapable of autoinhibition (Seiler et al, 2000; Cai et al, 2007)), and ∆Exon27. On microtubule filaments bound to the cover glass of the slide chamber, KIF5A WT motors mostly show nonmotile interactions with the microtubule (Fig 3A), suggesting the majority of motors are primarily in the autoinhibited state. The few WT motors that show motility display an average velocity of 0.45 ± 0.11 μm/s and processivity of 0.6 [0.57, 0.71] μm (median with 95% CI). In both HEK293T and N2a cells, ∆C also shows diffused/filamentous cytoplasmic expression like WT (Fig EV3B). As expected, and in contrast to WT, ∆C exhibits enhanced motility and significantly increased velocity (0.85 ± 0.07 μm/s) and processivity (1.63 [1.47, 1.81] μm) compared with WT (Fig 3B and Table 1). Similar to ∆C, ∆Exon27 motors also exhibit enhanced motility events along microtubules in the smTIRF assay, supporting the hypothesis that autoinhibition is relieved in ∆Exon27. Intriguingly, while ∆Exon27 displays increased velocity (0.66 ± 0.10 μm/s) compared with WT, its velocity is slower than ∆C and it exhibits drastically increased processivity (5.47 [3.84, 7.80] μm) relative to ∆C (P < 0.0001, Fig 3C and Table 1). Analysis of the initiation of motor movement indicates that ∆Exon27 does not have a preference over the landing location on microtubules (Fig EV3C).
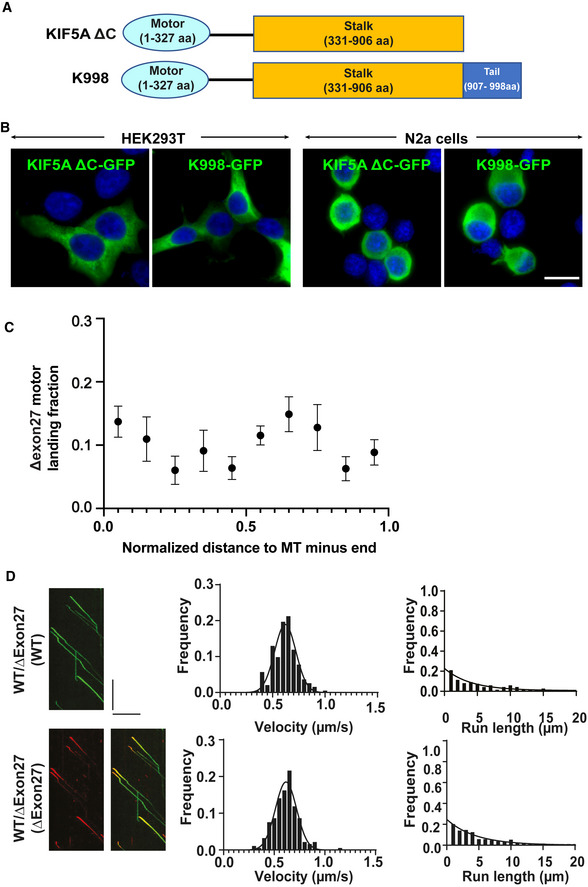
Figure EV3. Motile properties of KIF5A ΔExon27 and characterization of truncation mutants K998 and ∆C.
-
ASchematic illustrations of KIF5A truncation mutants, K998 and ∆C.
-
BBoth KIF5A K998 and ∆C truncated proteins diffuse in the cytoplasm when expressed in HEK293T and N2a cells. Two biological replicates, n = 100 cells per experiment. Scale bar: 20 μm.
-
CMotor landing rate of KIF5A ∆Exon27 along the microtube was accessed. ∆Exon27 does not have a preference over the landing location on microtubules.
-
DMotor velocities (middle) and run‐lengths (right) were determined based on kymographs (example on the left) for KIF5A WT and ∆Exon27 complexes. HEK293T cells were transfected with both KIF5A WT‐GFP and ∆Exon27‐mApple. The movements of WT and ∆Exon27 were assessed by tracking either GFP or mApple, respectively. The right kymograph in D is the overlay of the GFP and mApple of moving WT‐GFP/∆Exon27‐mApple complexes. The x‐axis scale for the processivity graphs was limited to 20 μm to permit a direct comparison of the run‐lengths of the different constructs. Diagonal lines in the kymograph represent KIF5A molecules moving over time. The velocity data were fit with Gaussian distribution and the processivity data were fit with an exponential decay function. The depicted scale bars for all kymographs shown in this figure is 5 μm (horizontal line) and 10 s (vertical line). The measured values for the velocities and run‐lengths are listed in Table 1.
Figure 3. KIF5A ∆Exon27 relieves motor autoinhibition and displays increased velocity and processivity.
-
AKIF5A WT‐GFP. From kymographs (example on the left), single‐motor velocities (middle) and run‐lengths (right) were determined.
-
BAs in (A), but for ∆C‐GFP.
-
CAs in (A), but for ∆Exon27‐GFP.
-
DAs in (A), but for K998‐GFP. The x‐axis scale for the processivity graphs was limited to 20 μm to permit a direct comparison of the run‐lengths of different constructs. Diagonal lines in the kymograph represent KIF5A molecules moving over time. The depicted scale bars shown for all kymographs in this figure are 5 μm (horizontal line) and 10 s (vertical line). The velocity data were fit with Gaussian distribution and the processivity data were fit with an exponential decay function.
-
E, FStatistical analysis of the velocity (E) and processivity (F) of various KIF5A proteins. The green bars represent the median with 95% CI. The statistical comparison of velocity was performed using unpaired parametric t‐test (***P < 0.001). The run‐length is in log scale. The statistical evaluation of processivity was performed using one‐way ANOVA (***P < 0.001). The measured values for the velocities and run‐lengths are listed in Table 1. WT: n = 176; ∆Exon27: n = 215; ∆C: n = 175; K998: n = 185; WT/∆Exon27 (WT): n = 185; WT/∆Exon27 (∆Exon27): n = 193. Three biological replicates. Data represent the mean ± SD.
Table 1.
Measured velocities and run‐lengths of all KIF5A constructs.
| Construct | Velocity (mean ± SD) (μm/s) | Processivity (lifetime [95% CI]) (μm) |
|---|---|---|
| KIF5A WT‐GFP | 0.45 ± 0.11 | 0.60 [0.51, 0.71] |
| ∆Exon27‐GFP | 0.66 ± 0.10 | 5.47 [3.84, 7.80] |
| ∆C‐GFP | 0.85 ± 0.07 | 1.63 [1.47, 1.81] |
| K998‐GFP | 0.70 ± 0.11 | 2.07 [1.72, 2.50] |
| KIF5A WT‐GFP/∆Exon27‐mCherry (KIF5A WT‐GFP a ) | 0.62 ± 0.10 | 3.95 [2.81, 5.53] |
| KIF5A WT‐GFP/∆Exon27‐mCherry (∆Exon27‐mCherry a ) | 0.62 ± 0.10 | 4.49 [3.64, 5.63] |
The velocity data were fit with Gaussian distribution, and the run‐length data were fit with an exponential decay function.
Denotes the fluorophore with which KIF5A movement was tracked.
WT and ∆Exon27 share the same sequence up to amino acid 998 but diverge in the C‐terminal tail region. To determine whether the neopeptide of ∆Exon27 is required for the drastically increased processivity, we made a construct that contains the same sequence for WT and ∆Exon27 (amino acids 1–998, referred to as K998, Fig EV3A). When expressed in HEK293T and N2a cells, K998 has diffused cytoplasmic localization as WT and ∆C (Fig EV3B). In the smTIRF assay, K998 displays a similar velocity (0.70 ± 0.11 μm/s) and processivity (2.07 [1.72, 2.50] μm) as those observed for ∆C. Importantly, the processivity of K998 is significantly lower than that of ∆Exon27 (Fig 3D and Table 1).
We next characterized the motility of WT–∆Exon27 complexes. WT and ∆Exon27, being fluorescently tagged by GFP and mApple, respectively, were co‐expressed in HEK293T cell. In the presence of ∆Exon27, WT displays motile properties that resemble ∆Exon27. Analysis using either WT‐GFP or ∆Exon27‐mApple to assess motility yields similar results for both velocity (0.62 ± 0.10 and 0.62 ± 0.10 μm/s, respectively) and processivity (3.95 [2.81, 5.53] and 4.49 [3.64, 5.63] μm, respectively; Figs 3E and F, and EV3D). Overlay of the kymograph of WT‐GFP and ∆Exon27‐mApple demonstrates that most of the moving spots consist of both WT and ∆Exon27 (Fig EV3D), consistent with our earlier observation that ∆Exon27 and WT form complexes with each other (Fig 2A–C).
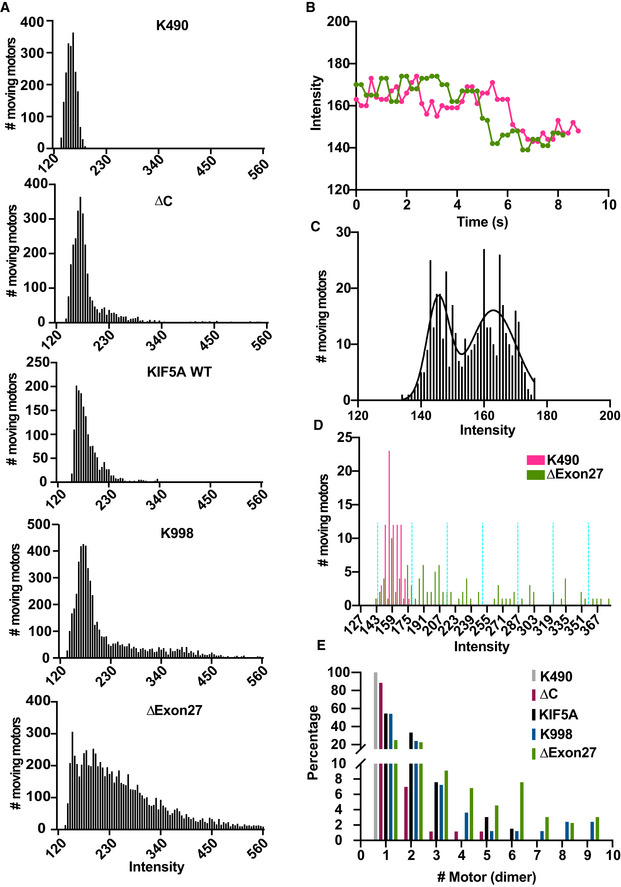
Since ∆Exon27 is aggregation prone, we hypothesized that the increased processivity by ∆Exon27 may result from increased motor association. Using a truncated KIF5A (K490, amino acids 1–490) as a standard for fluorescence intensity of dimeric motors (Tomishige et al, 2006), we observed that the fluorescence intensities of many moving ∆Exon27 motors are much stronger than those of WT and ∆C (Fig EV4A), indicating that ∆Exon27 promotes self‐association (or aggregation) among KIF5A motors and moves along the microtubules as multimotor complexes. The recruitment of additional motors would increase the number of motor domains near the microtubules thereby decreasing the chance of complete dissociation of the motor complex from the microtubules during a run. Interestingly, both WT and K998 show motor molecules with higher fluorescence intensities compared with dimeric K490 (Fig EV4B–E), suggesting that KIF5A has an intrinsic tendency to oligomerize, which agrees with a recent study (Chiba et al, 2022). Nonetheless, these results suggest that the neopeptide confers a gain‐of‐function via the relief of KIF5A autoinhibition and promotion of motor self‐association, which in turn causes KIF5A ∆Exon27 motor complexes to move super‐processively along microtubules.
Figure EV4. Enhanced motor self‐association in KIF5A ΔExon27.
-
AHistograms of the measured fluorescence intensities of the various moving kinesin motors. The fluorescence intensity in every frame of each moving spot was measured, accumulated, and plotted in a histogram. Numerous studies have shown that truncated kinesin‐1 motors are dimeric and active (Friedman & Vale, 1999; Tomishige et al, 2006; Lam et al, 2021), and kinesin‐1 family is highly conserved in the motor domain. Hence, a truncated KIF5A motor (K490, amino acid 1–490) was generated and used as the standard for fluorescence intensity of dimeric motors. Moving KIF5A ∆Exon27 molecules exhibit a much larger fraction of motors with high fluorescence intensities. The majority of moving KIF5A ∆Exon27 spots consist of multiple motors. K490: repeated two times, n = 189; other constructs are described in Fig. 4. Number of frames that have been analyzed: K490, n = 1,953; ∆C, n = 2,964; WT, n = 1,634; K998, n = 5,462; ∆Exon27, n = 8,345.
-
BTwo example traces of K490‐EGFP showing photobleaching events. K490‐EGFP was used as the standard dimer molecule to determine the fluorescent intensity of EGFP.
-
CHistogram of measured intensities of moving K490‐EGFP molecules that showed a single photobleaching step. The intensity distribution was fit with two Gaussian functions, resulting in mean values of 145.5 ± 3.6 (mean ± SD) and 163.0 ± 7.3. With an average background fluorescence signal of 127.6 ± 0.7, one obtains an average fluorescent intensity is 17.7 for EGFP (in a.u.).
-
D, EDetermination of the percentage of the number of motors for each motor complex. KIF5A K490 and KIF5A ∆Exon27 are depicted here as examples (D). The same method was applied to the other motors. Each moving spot was assigned to a single intensity value that corresponded to the average intensity measured over the first 20 frames of the moving spot so the intensity would be less likely to be averaged down due to photobleaching. The measured values were then accumulated and plotted in a histogram. Based on the mean intensity of EGFP, the measured intensity values were sorted into intervals of 35.4 (depicted by the turquoise dashed lines), with the first interval centered at 163 (163 ± 17.7). The percentage of each bin was calculated and then assigned to the calculated number of dimers (E). The percentages of a single dimer for all KIF5A constructs are: K490, 100%; ∆C, 88%; K998, 54%; WT, 54%; ∆Exon27, 25%.
Ectopic expression of KIF5A ΔExon27 in Drosophila leads to abnormal wings, motor deficits, paralysis, and premature death
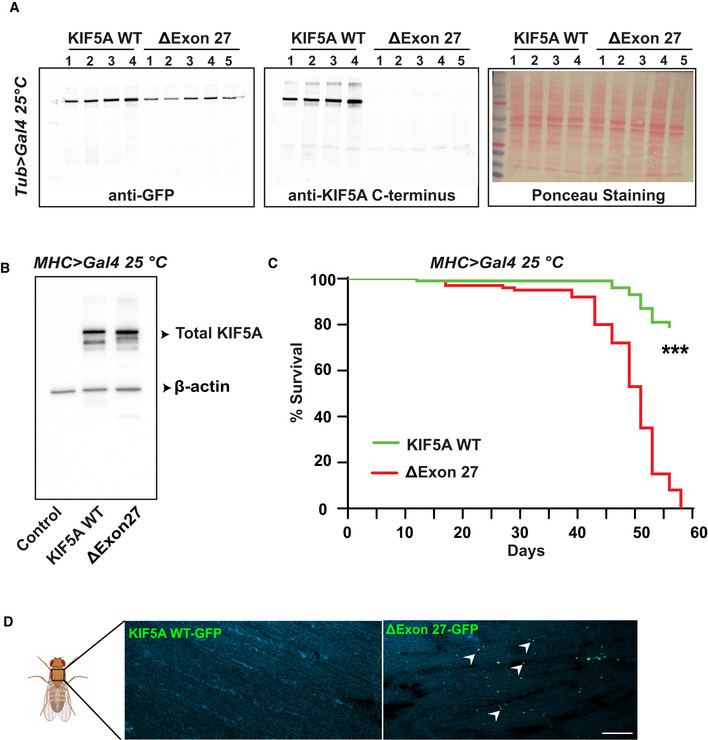
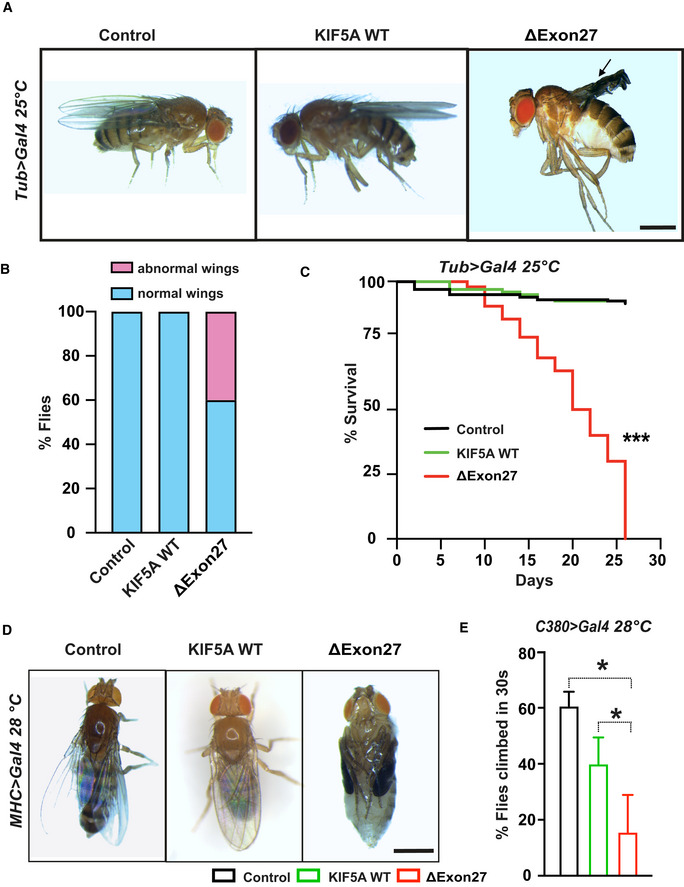
To further investigate the role of KIF5A ALS mutants in vivo, we generated transgenic Drosophila expressing either WT or ΔExon27 with a C‐terminal GFP tag. To avoid any influence of the genomic environment on transgene expressions, a single copy of UAS > hKIF5A transgenes was targeted to the well‐established 68A4 landing site on chromosome III by PhiC31 integrase‐mediated insertion. These transgenes are not expressed at baseline but will be transcribed when Gal4 is introduced by genetic crossing. The protein levels of WT and ΔExon27 are comparable when driven by the ubiquitously expressed tubulin‐Gal4 (Fig EV5A). Constitutively ubiquitous expression of ΔExon27 in Drosophila causes immature‐looking adult escapers with unexpanded wings in ∼40% of flies and an early lethality (Fig 4A–C). In contrast, neither the tubulin‐Gal4 itself nor KIF5A WT causes any defects, confirming that the phenotype is exclusively due to ΔExon27 expression.
Figure EV5. Muscle‐specific expression of ∆Exon27 in Drosophila leads to early lethality.
-
AWestern blot detecting the level of human KIF5A proteins in Drosophila ubiquitously expressing WT or ∆Exon27 driven by tubulin‐Gal4. Genotypes were: KIF5A WT (UAS‐KIF5A WT‐GFP/ tubulin‐Gal4); ∆Exon27 (UAS‐∆Exon27‐GFP/ tubulin‐Gal4). Samples from 4 (WT) and 5 (∆Exon27) different induvial lines were assessed with similar results, two independent experiments.
-
BWestern blot detecting the expression of human KIF5A proteins in Drosophila expressing WT or ∆Exon27 in muscle tissues driven by MHC‐Gal4. Genotypes were: Control (MHC‐Gal4/+); KIF5A WT (UAS‐KIF5A WT‐GFP/MHC‐Gal4); ∆Exon27 (UAS‐∆Exon27‐GFP/MHC‐Gal4), two independent experiments.
-
CLifespan of male flies expressing KIF5A WT or ∆Exon27 in muscles driven by MHC‐Gal4 at 25°C (n = 50 flies per group, two independent experiment ***P < 0.001, log‐rank test).
-
DAggregates accumulate in thorax muscles at 7 days post‐eclosion in flies expressing ΔExon27, but not in those expressing KIF5A WT, n = 10 flies per group. Scale bar: 10 μm.
Figure 4. KIF5A ∆Exon27 is toxic in Drosophila melanogaster .
-
AUbiquitous overexpression of ΔExon27 leads to wing defects (black arrows). Scale bar: 0.5 mm.
-
BPercent of flies with normal and abnormal wings (n = 50 flies per group, two independent experiments).
-
CLife spans were analyzed for flies expressing KIF5A WT‐GFP and ∆Exon27‐GFP, and control flies expressing tubulin‐Gal4 only. Expression of ∆Exon27‐GFP by the tubulin driver causes a substantial decrease in viability (***P < 0.001, log‐rank test), n = 50 flies per group, two independent experiments.
-
DExpression of ∆Exon27‐GFP in Drosophila muscles leads to complete paralysis. The pupal case of the fly expressing ∆Exon27‐GFP has been removed for this picture. The folded wings and legs are characteristic of the pupal state in flies expressing ∆Exon27. Scale bar: 0.5 mm.
-
ENegative geotaxis assay showing reduced motor function in 25‐day‐old flies expressing ∆Exon27‐GFP driven by a motor neuron‐specific driver (C380‐Gal4; n = 50 flies each group, two independent experiments). Bars indicate mean ± SD. Statistical analysis was performed using one‐way ANOVA (*P < 0.05).
Since ALS is a neuromuscular degenerative disease, we confined the expression of transgenes in fly muscles using MHC‐Gal4 driver. Contrary to KIF5A WT or MHC‐Gal4, all the ΔExon27‐expressing flies fully develop to the pupal stage as pharate adults but are unable to hatch from their pupal cases when raised at 28°C. After dissecting the ΔExon27‐expressing flies from their pupal case, we found that they have no obvious defects other than what could be expected from being paralyzed with folded wings and legs, wet appearance, and no movement (Fig 4D). When raised at 25°C, ΔExon27‐expressing flies can develop to adults and have similar expression levels as WT (Fig EV5B). However, they have dramatically reduced life span (Fig EV5C). Accompany this, apparent aggregates accumulate in thorax muscles at 7 days post‐eclosion in flies expressing ΔExon27, but not in those expressing KIF5A WT (Fig EV5D). We further expressed KIF5A transgenes specifically in motor neurons using the commonly used Drosophila motor neuron driver (C380‐Gal4) and assessed their motor function with negative geotaxis assays. Flies expressing ΔExon27 show impaired climbing activity compared with those expressing KIF5A WT or control flies expressing C380‐Gal4 (Fig 4E). Of note, we did not observe intraneuronal ΔExon27 aggregation in the ventral nerve cord at 7 days post‐eclosion. Overall, our results highlight an imperative role of KIF5A ΔExon27 in mediating toxicity.
KIF5A ALS patient iPSC‐derived motor neurons (iMNs) express mutant proteins and have increased KIF5A inclusion in neurites
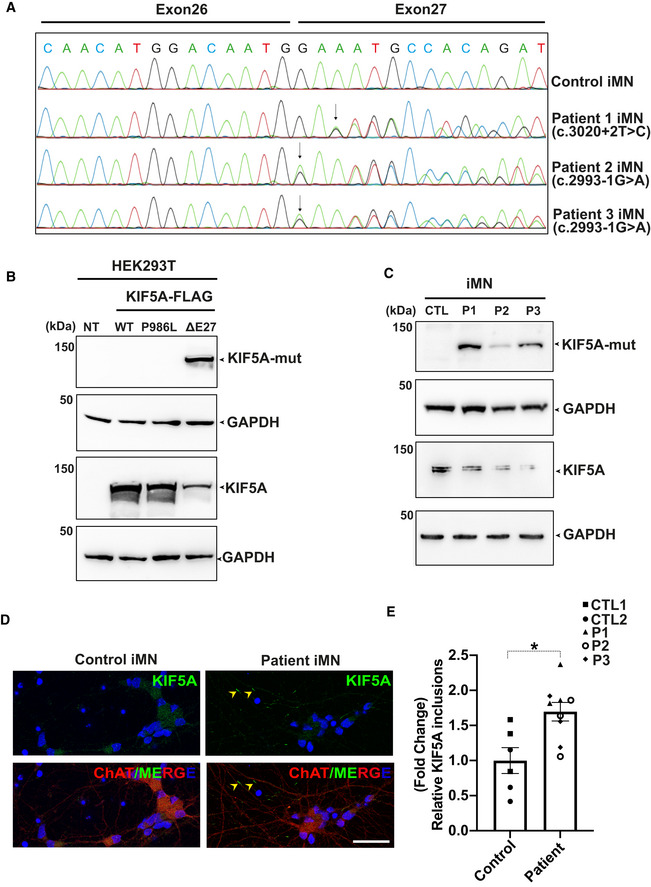
Next, we investigated the impact of KIF5A ALS mutations in patient iMNs on mRNA and protein expressions and on KIF5A protein distribution. To this end, we obtained primary blood mononuclear cells (PBMCs) from one pre‐manifest carrier of a heterozygous ΔExon27 mutation (c.3020+2T>C) and two ALS affected members of a family with a heterozygous mutation (c.2993‐1G>A). The effect of the latter mutation on splicing has not been shown before. PBMCs were reprogrammed to iPSCs and differentiated into motor neurons until DIV20. PCR amplification from cDNA of iMNs and subsequent Sanger sequencing of the PCR product not only proved exon 27 skipping in the c.3020+2T>C mutant line, but also showed the splice acceptor site shift by one nucleotide and otherwise preservation of exon 27 at the mRNA level in the two c.2993‐1A>G mutant lines (Fig 5A). Nevertheless, despite preservation of exon 27 in the c.2993‐1A>G mutant lines, the respective predicted protein product still comprises the same C‐terminal aberrant 39 amino acid end as the c.3020+2T>C mutation (Appendix Fig S1).
Figure 5. Expression of endogenous mutant KIF5A proteins and increased KIF5A inclusions in three patient‐derived iMN cell lines.
-
ASanger sequencing chromatograms of cDNA from control and patient‐derived iMNs (P1–P3) showing the effect of mutations on KIF5A transcripts. While the c.3020+2T>C mutation causes exon 27 skipping, the c.2993‐1G>A mutation results in an aberrant transcript with a splice acceptor shifted only by a single nucleotide, leading to a frameshift but containing exon 27. The respective protein product predicted for both mutations comprise the same C‐terminal aberrant 39 amino acid end.
-
BWestern blot analysis for validation of the custom mutant‐specific antibody using transfected HEK293T cells. While an antibody against the total KIF5A (lower panel) recognized both wild‐type and mutant proteins, the custom antibody targeting the aberrant “neopeptide” detected ΔExon27 specifically (upper panel). NT: No transfection.
-
CMutant KIF5A proteins are endogenously expressed in iMNs, detected by using the custom mutant‐specific antibody directed against the C‐terminal aberrant 39 amino acid sequence. CTL, Control; P1, Patient 1; P2, Patient 2; P3, Patient 3, two independent experiments.
-
DRepresentative images of KIF5A distribution (arrowheads) in control and patient iMNs. Scale bar: 20 μm.
-
EAbundance of KIF5A inclusions after normalizing to total neuron numbers. Graph represents data from n = 146 control 1 (CTL1), n = 134 control 2 (CTL2), n = 154 patient 1 (P1), n = 162 patient 2 (P2) and n = 133 patient 3 (P3) iMNs from three individual experiments. Bars indicate mean ± SD. Statistical analysis was performed using Mann–Whitney Wilcoxon Test (*P < 0.05).
To determine whether these altered RNA transcripts are translated to proteins, a polyclonal custom antibody against the aberrant 39 amino acids predicted at the C‐terminus of ALS mutant KIF5A proteins was generated. This antibody specifically recognized ΔExon27 ALS‐associated KIF5A mutant proteins with the aberrant 39 amino acid sequence, but not the WT KIF5A protein or KIF5A with the single amino acid polymorphism p.P986L (Fig 5B). Using this antibody, we were able to detect endogenous expression of the aberrant 39 amino acid sequence predicted for both ALS‐linked mutations in motor neurons derived from KIF5A ALS patients and the pre‐manifest mutation carrier, respectively (Fig 5C). Immunostaining using an antibody that detects both WT and mutant KIF5A proteins revealed KIF5A expression in two control and three patient iMNs. However, the presence of KIF5A with the C‐terminal aberrant neopeptide results in a significantly increased KIF5A inclusions in neurites of patient iMNs compared with controls (Fig 5D and E). Thus, we were able to experimentally show for the first time the existence of the ALS‐associated KIF5A mutant proteins with a C‐terminal neopeptide as well as increased KIF5A inclusions in patient iPSC‐derived motor neurons at DIV20.
Discussion
Two different types of KIF5A variants in ALS patients were initially highlighted: a single nucleotide variant (SNV) caused by a missense mutation (rs113247976 [c.2957C > T]) that changes amino acid 986 from proline to leucine, and variants predicted to affect splicing of exon 27 or lead to a frameshift in exon 27 (Brenner et al, 2018; Nicolas et al, 2018). The identified SNV most likely represents a risk factor with low effect size that may play a role in oligogenic modes of ALS causation. Indeed, 11 of 29 patients carrying this SNV in the first study also carry genetic variants in other known ALS genes. Most of them are also of reduced penetrance/lower effect size, so synergy of several SNV/weaker mutations might be causative in these patients (Brenner et al, 2018). In contrast, variants predicted to affect exon 27, although far less common than rs113247976, are highly penetrant and were subsequently identified in multiple patient cohorts around the world (Faruq et al, 2019; Zhang et al, 2019; Nakamura et al, 2021; Naruse et al, 2021). These variants were termed as “loss‐of‐function” mutations in the original studies since they replace the normal C‐terminal 34 amino acids with a new tail containing aberrant 39 amino acids at the protein level. However, the underlying disease mechanism is currently unknown. The C‐terminal “neopeptide” is predicted to result from either exon 27 skipping (ΔExon27) or non‐exon27‐skipping (frameshifting) ALS mutations. Using a custom antibody generated against the neopeptide, we provided evidence of its endogenous expression at the protein level for both types of KIF5A‐ALS mutations in patient iPSC‐derived motor neurons. Mechanistically, our results suggest that KIF5A ΔExon27 causes toxicity mainly through gain‐of‐function. ΔExon27 is particularly prone to form cytoplasmic aggregates compared with WT or KIF5A with the deletion of C‐terminal tails. In addition, ΔExon27 is relieved from autoinhibition and displays drastically increased processivity through enhanced motor self‐association caused by the altered C‐terminal tail. As a result, un‐inhibited motors bind to microtubules and move processively toward the microtubule plus‐ends and are therefore effectively removed from the pool of cytoplasmic motors needed for plus‐end‐directed cargo transport. It has been shown that homozygous Kif5a knockout in mice is embryonic lethal while heterozygous Kif5a +/− animals are completely normal, indicating that having 50% of normal KIF5A levels is sufficient for its normal physiological functions (Xia et al, 2003). Moreover, deleterious mutations in other regions of KIF5A besides the narrow ALS‐linked C‐terminal mutational hot spot have never been observed in ALS patients. These results further suggest that KIF5A ΔExon27 causes ALS through a toxic gain‐of‐function mechanism rather than loss‐of‐function/expression.
In recent years, researchers have discovered that proteins bearing intrinsically disordered and low‐complexity domains coalesce into liquid droplets, a phenotype called liquid–liquid phase separation (LLPS). LLPS was initially recognized to play an important role in the formation and function of membrane‐less organelles, such as Cajal bodies, stress granules and nucleolus, to name a few (Pakravan et al, 2021; Wang et al, 2021). It has also been shown to regulate local enrichment of molecules to activate cell signaling and acceleration of biochemical reactions (Su et al, 2016; Sheu‐Gruttadauria & MacRae, 2018). By using seven different algorithms to predict proteins with intrinsic disordered domains, Seeger & Rice (2013) showed that intrinsically disordered residues are present throughout the motor, stalk, and tail domains of the kinesin family proteins. Strikingly, ∼71.8% of kinesin tail domain residues are intrinsically disordered. When expressed in mammalian cells, we found that KIF5A WT diffuses in the cytoplasm whereas ΔExon27 forms small granules at an earlier time point, which eventually increase in size likely by the fusion of multiple inclusions (Fig 1B). It is interesting to note that similar but significantly fewer granules were also observed for KIF5A WT with time and continuous protein accumulation. Indeed, we observed KIF5A WT motor molecules with higher fluorescence intensities than expected dimers in the smTIRF assay (Fig EV4), consistent with a recent study suggesting that KIF5A WT has an intrinsic property to form oligomers (Chiba et al, 2022). Whether KIF5A WT forms local condensates through LLPS and how this impacts its role in transporting cargos warrants further study. Importantly, dysfunctional LLPS of DNA‐ and RNA‐binding proteins such as TDP‐43 and FUS have been implicated in ALS pathogenesis, leading to pathological protein aggregates in patients (Pakravan et al, 2021). We showed that ΔExon27 granules are positive for p62 and ubiquitin and are only soluble in stringent detergent such as urea (Fig 1C–E). These data support that ΔExon27 is prone to form cytoplasmic aggregates that recruit p62‐ and ubiquitin‐dependent proteolytic pathways.
Mutations of KIF5A have been previously linked to SPG10 and CMT2, two neurological diseases with neuronal axonopathy (Crimella et al, 2012). While all the ALS‐related KIF5A variants are within the C‐terminal tail, most SPG10/CMT2 mutations are located at the N‐terminal motor region with a few in the stalk domain. The unique positions of KIF5A mutations that are associated with either SPG10/CMT2 or ALS provide an unprecedented opportunity to delineate pathogenic mechanisms underlying ALS. The kinesin‐1 family of motor proteins are the best‐understood members with respect to their mechanisms of motility at the single‐molecular level (Gennerich & Vale, 2009). The fundamental regulatory mechanism for kinesin‐1 is the transition from a “folded” (inhibited) to an “open” (activated) state (Fig 6). In the folded state, the tail/cargo‐binding domain binds to the motor domain (or “head”) and inhibits its activity by blocking the release of ADP (autoinhibition; Hackney et al, 2009; Seeger & Rice, 2010). When the motor is activated presumably by cofactors or regulators, one of the two heads releases ADP upon microtubule binding, which starts a productive processive run (Gennerich & Vale, 2009; Hancock, 2016). Using single‐molecule assays, we found that KIF5A ΔExon27 exhibits an increased motility due to a release of autoinhibition and a striking five‐fold increase in processivity compared with KIF5A WT as a result of oligomerization or aggregation (Fig 3 and Table 1). The processivity of ΔExon27 is also much higher than ∆C, a motor construct that is incapable of assuming an autoinhibited state due to the removal of the C‐terminal domain. This contrasts with the previous studies on the motile properties of SPG10 mutants, which exhibited reduced microtubule affinities and reduced velocities in microtubule‐gliding assays (Ebbing et al, 2008). In addition, KIF5A WT and ΔExon27 interact to form oligomeric motors (Fig 2A–C) and display increased velocity and processivity like ΔExon27, suggesting that when complexed, the mutant KIF5A confers its gain‐of‐function property to the WT protein. The consequence of this drastically increased processivity of KIF5A ΔExon27 to neuronal health is currently unknown. In primary cortical neurons, p62‐positive ΔExon27 granules are highly accumulated along the neurites and at the neurite terminals (Figs 2D and EV2D). Consistent with this, ∆Exon27 granules are mainly observed at the peripheral extrusions of highly polarized LLC‐PK1 cells and colocalize with microtubule plus‐end‐tracking protein EB1 (Fig EV2F and G). In patient iPSC‐derived motor neurons, we also detected increased KIF5A inclusions along the neurites consistently from multiple cell lines and multiple differentiations (Fig 5D and E). We hypothesize that these inclusions could be signatures of constitutively active endogenous KIF5A, which is in accordance with the studies shown by Hirokawa group (Shima et al, 2018). We further performed single‐molecule assays for endogenous KIF5A from patient‐derived motor neurons. However, this assay relies on antibodies that recognize unlabeled endogenous KIF5A. Due to the low abundance of endogenous KIF5A and the modest affinity of commercially available KIF5A antibodies, we failed to detect enough moving KIF5A molecules from iMNs that would have permitted us to perform a statistically meaningful analysis. Nevertheless, these studies suggest that enhanced processivity may result in an aberrant distribution of the motor along the neurites, with an increase in the local concentration of ∆Exon27 near the microtubule plus‐ends, which in turn could increase motor self‐association and aggregation. In addition, kinesin proteins need to be transported back to the cell body by dynein/dynactin‐based retrograde transport after delivering cargos to distal ends. It is thus possible that an imbalance between the movement of antegrade and retrograde motor proteins will eventually disrupt intracellular transport. Importantly, we demonstrated that K998 with the truncated tail behaves more like ∆C than ΔExon27. These results support the idea that the neopeptide in ΔExon27 is required to confer gain of toxicity through the relief of autoinhibition and through self‐association/aggregation of the mutant motors. In line with this notion, several single‐nucleotide deletion variants of KIF5A have also been identified in ALS patients (Nicolas et al, 2018; He et al, 2020) which are predicted to result in a C‐terminal tail containing 39 amino acids identical to that of the exon 27‐skipping variants (Appendix Fig S1).
Figure 6. Proposed mechanisms of neuronal toxicity caused by KIF5A ∆Exon27 gain‐of‐function.
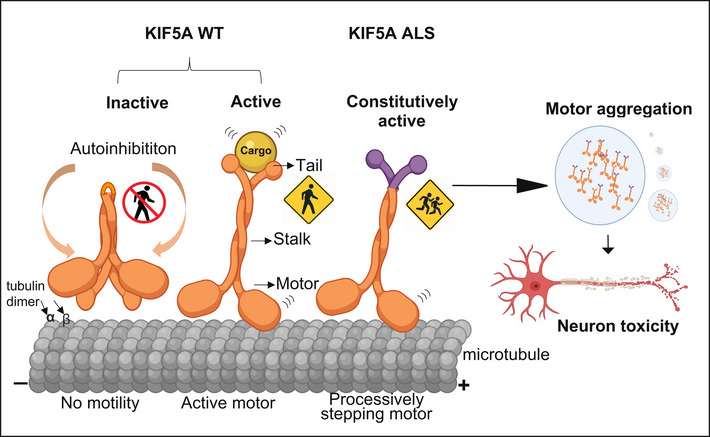
In the absence of tethered cargos, KIF5A WT is autoinhibited with the C‐terminal tail binding to the N‐terminal motor domain. When cargo binds to the C‐terminal tail, the motor associates with microtubules and starts a processive run toward the plus‐ends. ALS‐associated KIF5A ∆Exon27 with the aberrant C‐terminal tail relieves from the “autoinhibited” state even without cargo and self‐associates to form multiple motors, leading to a drastically increased run‐length on microtubules and accumulation at the plus‐ends. ∆Exon27 also forms complexes with WT KIF5A and enhances motor self‐association and aggregation. The image was created at BioRender.com.
The identification of ALS‐associated KIF5A mutations adds KIF5A to a growing list of known cytoskeletal‐related genes implicated in ALS. We characterized ΔExon27‐mediated toxicity in a transgenic Drosophila model. Drosophila is a versatile model system that has been widely used in the ALS field to study the molecular mechanisms of key biological functions given their genetic and overall experimental tractability (Liguori et al, 2021). We showed that ubiquitous expression of ΔExon27 in Drosophila causes wing abnormalities and shortened lifespan (Fig 4A–C). Tissue‐specific expression revealed higher susceptibility to ΔExon27 expression in muscles and led to paralysis (Fig 4D). Motor neuron‐specific expression of ΔExon27 also impairs fly climbing activities in negative geotaxis assays (Fig 4E). The toxicity of ΔExon27 is further supported by observations that mouse cortical neurons expressing mutant KIF5A have reduced survival (Fig 2E). Additional genetic studies for disease modifiers of toxicity using our Drosophila KIF5A ALS models will likely yield new mechanistic insights into disease pathogenesis and identify therapeutic strategies for this detrimental disease.
While our manuscript was under review, two independent groups further supported that ΔExon27 causes neuronal toxicity through gain‐of‐function in different cellular and animal systems (Baron et al, 2022; Nakano et al, 2022).
Materials and Methods
DNA constructs
Human KIF5A cDNA was a gift from Dr. Gary Bassell (Emory University) and was used as a template to generate the following GFP (EGFP), mApple, and FLAG‐tagged constructs: KIF5A‐GFP, KIF5A‐mApple, FLAG‐KIF5A [full‐length wild‐type (WT), Δexon27, Δcargo (amino acids 1–906, ΔC), K998 (amino acids 1–998), R280C (the most common SPG10 mutation)]. A synthetic oligo gene block with a sequence corresponding to the Δexon27 tail was ordered from Integrated DNA Technologies. Constructs were generated using standard PCR and using Gibson cloning (NEB). Healthy (control) and KIF5A patient cells were used to obtain KIF5A cDNA for cloning the pcDNA3‐KIF5A‐FLAG [full‐length KIF5A, SNP (P986L, Δexon27). Constructs used for in vitro studies were cloned into the pGW1‐EGFP backbone (a gift from Dr. Sami Bermada, University of Michigan) by PCR‐based cloning (Invitrogen). For lentivirus production, KIF5A‐GFP (WT, Δexon27) was subcloned into the pFUGW backbone (provided by Emory Viral Vector Core) and lentiviruses were produced with third‐generation packaging plasmids (Vector ID: (pLV201205‐1002epb)‐C, VectorBuilder). For longitudinal fluorescence microscopy pGW1‐mApple was used. The human KIF5A WT and Δexon27 were subcloned to the pUAST‐attB backbone, a gift from Dr. Ken Moberg (Emory University), for the generation of transgenic Drosophila. All plasmids were verified by Sanger sequencing (Genewiz, USA or Eurofins, Germany).
Drosophila stocks and genetics
Fly crosses were maintained at 25°C in a humidified chamber with 12‐h light–dark cycles. Fly food was prepared with a standard recipe (water; cornmeal; yeast; agar; molasses; propionic acid). Parental stocks were maintained at room temperature. Transgenic flies UAS > hKIF5A WT‐GFP and UAS > hKIF5A Δexon27‐GFP were generated by inserting the respective transgenes into the attP2 site of y 1 w 67c23 ; P[CaryP]attP2 (BI #8622) by phiC31 integration (BestGene Inc, USA). The tubulin‐gal4 and MHC‐Gal4 line were generously provided by Dr. Ken Moberg (Emory University) and Dr. Udai Pandey (University of Pittsburgh), respectively. The motor neuron driver line C380‐Gal4 was purchased from Bloomington Drosophila Stock Center (BI # 80580). Flies were imaged using a Leica MC170 HD digital camera mounted on a Nikon SMZ800N stereo microscope.
Drosophila survival assay
For the survival assay, parental flies were raised with standard food and under 12‐h day/night cycles at 25°C unless otherwise stated. The parents were allowed to mate and lay eggs for 3 days before being discarded. The offspring from these parents were collected over a period of 24 h and sorted by sex. Up to 12 male flies were kept in individual vials containing standard food. For each genotype, multiple replicate vials were set up so that the total sample size was 75 for each genotype. Flies were transferred onto fresh food every 2–3 days. The number of deaths was recorded each day. All survival assays were performed at 25°C.
Negative geotaxis assay
Fly climbing activity was assessed using negative geotaxis assay as previously described (Nichols et al, 2012). Briefly, 15 flies were transferred, without anesthetization, into each plastic vial and placed in the apparatus. The vials were tapped down against the bench and the climbing was recorded on video for 1 min. The percentage of flies climbing to the top of vials in 30 s was determined manually in a blinded manner.
Cell culture and transfection
Human embryonic kidney (HEK293T), and mouse neuroblastoma (N2a) cell lines from ATCC. The porcine kidney cell line (LLC‐PK1) was a gift from Dr. Melissa Gardner (University of Minnesota). Cells were cultured in high glucose DMEM (Invitrogen) supplemented with 10% fetal bovine serum (Corning), 4 mM Glutamax (Invitrogen), penicillin (100 U/ml), streptomycin (100 μg/ml) and nonessential amino acids (1%). Cells were grown at 37°C in a humidified atmosphere with 5% CO2. Cells were transiently transfected using polyethylenimine (1 mg/ml) or Lipofectamine 2000 (Invitrogen). Experiments were performed either 24 or 48 h after transfection.
Primary cortical neuronal culture and transfection
Primary cortical neurons were prepared from C57BL/6J mouse embryos (Charles River) of either sex on embryonic day 17. Cerebral cortices were dissected and enzymatically dissociated using trypsin w/ EDTA (Thermo Fisher Scientific; 10 min), mechanically dissociated in Minimum Essential Media (MEM; Fisher) supplemented with 0.6% glucose (Sigma) and 10% Fetal Bovine Serum (FBS; Hyclone). Neurons were plated on coverslips (Matsunami Inc., 22 mm) or MatTek dishes coated with poly‐l‐lysine (Sigma). A total of 50,000 neurons were plated as a “spot” on the center of the coverslip to create a small, high‐density network. Neurons were cultured in standard growth medium [glial conditioned neurobasal plus medium (Fisher) supplemented with Glutamax (GIBCO) and B27 plus (Invitrogen)], and half of the media was exchanged 2–3 times a week until the experiment endpoints. No antibiotics or antimycotics were used. Cultures were maintained in an incubator regulated at 37°C, 5% CO2 and 95% relative humidity as described (Valdez‐Sinon et al, 2020). Cells were transiently transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions.
Longitudinal fluorescence microscopy
Mouse primary cortical neurons were transfected with mApple and various KIF5A constructs and imaged by fluorescence microscopy at 24 h intervals for 7 days as described (Weskamp et al, 2019). Custom scripts were used to automatically generate regions of interest corresponding to each cell and determine time of death based on rounding of the soma, retraction of neurites, or loss of fluorescence. The time of death for individual neurons was used to calculate the risk of death in each population relative to a reference group. Images were acquired using Keyence BZ‐X810 microscope with a 10× objective and analyzed by Image J. The images were stitched and stacked, and cell death was scored using the criteria mentioned above.
Immunofluorescence
Cells were fixed in 4% paraformaldehyde (Electron Microscopy Sciences) for 20 min, washed three times for 5 min with phosphate buffer saline (1× PBS, Corning) and treated with 0.1% Triton X 100 (Sigma) in PBS. Cells were blocked for 30 min in a blocking solution consisting of 4% bovine serum albumin (Sigma) in PBS. Cells were incubated overnight in primary antibodies: rabbit anti‐G3BP1 (Proteintech), mouse anti‐FMRP (BioLegend), rabbit anti‐GM130 (Abclonal), rabbit anti‐p62 (Proteintech), mouse anti‐p62 (Novus Biologicals), mouse anti‐p62 (abcam), rabbit anti‐TDP‐43 (Proteintech), rabbit anti‐synapsin I (Sigma), guinea pig anti‐MAP2 (Synaptic Systems), goat anti‐ChAT (Sigma‐Aldrich), mouse anti‐SOX2 (DHSB), mouse anti‐OCT3/4 (Santa cruz) diluted in blocking solution (1:500). The next day, cells were washed three times for 5 min in PBS and incubated in secondary antibodies in blocking solution for 1 h at room temperature. After washing three times for 5 min, coverslips with the cells were mounted using Prolong Gold Antifade mounting media (Invitrogen). Images were acquired with Keyence BZ‐X810 microscope with a 60× oil objective. For image analysis, around 100–150 transfected cells were counted for each genotype in each experiment. Quantification of colocalization of Δexon27 aggregates with G3BP1, p62, and ubiquitin were performed manually in a blinded manner using ImageJ analysis. In Drosophila, adult fly thoraxes were dissected in 1× PBS, fixed in 4% PFA for 1 h and subsequently mounted in Prolong Gold Antifade mounting media (Invitrogen) and imaged with Nikon A1 confocal microscope with z‐stacks measuring 0.2 μm steps (around 10 total steps) with a 60× oil magnification lens. For the analysis of KIF5A‐positive inclusions in iPSC‐derived MNs, we acquired images with a 40× oil immersion objective with z‐stacks measuring 0.5 μm steps (around 8 total steps) using a Leica SP8 confocal upright microscope. The images were analyzed In ImageJ.
Western blotting and immunoprecipitation
Whole cell extracts were isolated using RIPA Lysis Buffer pH 7.4 (Bio‐world, USA) supplemented with Halt™ protease and phosphatase inhibitor cocktail (ThermoFisher Scientific), followed by DNA shearing. After centrifuge, the pellet was washed three times and dissolved in 8 M urea buffer (10 mM Tris, pH 8.0; 8 M urea). The concentration of the isolated proteins was determined using BCA Protein Assay Reagent (Pierce, USA). Twenty microgram of protein was resolved in 4–20% precast polyacrylamide gel (Bio‐Rad, USA). For immunoprecipitation, HEK293T cells were collected and lysed using Pierce™ IP Lysis Buffer (ThermoFisher Scientific) supplemented with Halt™ protease and phosphatase inhibitor cocktail (ThermoFisher Scientific), and 500 μg of protein lysate was precleared with 25 μl of Chromotek GFP‐Trap® or Pierce™ Anti‐FLAG magnetic agarose beads. Following the manufacture's instructions, protein lysate was immunoprecipitated for 2 h at 4°C with the following antibodies: mouse anti‐GFP (1:1,000; Takara Bio), mouse anti‐FLAG (1:1,000; Sigma). The input 10% was analyzed with Western blot. Proteins were transferred to nitrocellulose membranes (0.2 μm, Bio‐Rad) and incubated with primary antibodies: mouse anti‐GFP (1:2,000; Clontech), mouse anti‐FLAG (1:1,000; Sigma), rabbit anti‐KIF5A N‐terminus (1:1,000; GeneTex), rabbit anti‐KIF5A C‐terminus (1:1,000; Abcam), rabbit anti‐KIF5A custom neopeptide antibody (1:2,000; ThermoFisher Scientific), rabbit anti‐β actin (1:2,000; GeneTex), and rabbit anti‐GAPDH (1:2,000; Cell Signalling Technology), overnight at 4°C followed by HRP‐conjugated secondary antibodies (aBclonal) or IRDye secondary antibodies (Li‐cor) at room temperature for 1 h. Super Signal West Pico (Pierce, USA) was used for the detection of peroxidase activity. Molecular masses were determined by comparison to protein standards (Thermo Scientific). Band intensities were measured using ImageJ and normalized to tubulin or actin.
Generation, expression, and purification of K490‐EGFP
K490‐EGFP gene fragment was generated by PCR with 25 bp overlap with a modified NEB SNAP‐tag backbone in which the SNAP‐tag was replaced by a C‐term 6His tag. The plasmid was generated by Gibson assembly. The insertion was confirmed by enzymatic digestion and agarose gel electrophoresis. Protein expression and purification were performed as previously described (Budaitis et al, 2021). Briefly, the plasmid was transformed into BL21‐CodonPlus (DE3)‐RIPL competent cells (Agilent, #230280). A single colony was inoculated in 1 ml terrific broth (TB) medium with 50 μg/ml chloramphenicol and 25 μg/ml carbenicillin. After being shaken at 37°C overnight, the 1 ml culture was inoculated into 400 ml TB medium and was shaken at 37°C for 5 h. The cell culture was then cooled down on ice to < 18°C, and IPTG was added to final 0.1 mM concentration. The protein expression was induced at 18°C overnight with vigorous shaking. Afterwards, the cell was harvested by centrifugation at 3,000 g for 10 min. The supernatant was discarded, and the pellet was resuspended in 5 mL B‐PER™ complete bacterial protein extraction reagent (Thermo Scientific, #89821) supplemented with 2 mM DTT, 0.2 mM ATP, 4 mM MgCl2, 2 mM EGTA, and 2 mM PMSF. The resuspended solution was flash frozen and stored at −80°C before purification.
To purify K490‐EGFP, the cell solution was thawed at 37°C, followed by nutation at room temperature for 20 min. The cells were further lysed via douncing for 10 strokes on ice, and then cleared by centrifuging at 270,000 g for 10 min at 4°C using a Beckman Coulter tabletop centrifuge. At the same time, 0.5 ml Roche Ni‐NTA resin (MilliporeSigma, #5893682001) was washed with 2 × 1 ml of wash buffer [50 mM HEPES, 300 mM KCl, 2 mM MgCl2, 1 mM EGTA, 10% glycerol, 1 mM DTT, 0.1 mM ATP, 1 mM PMSF, 0.1% Pluronic F‐127 (w/v), pH 7.2]. The cleared lysate was carefully added to the Ni‐NTA resin and allowed to flow through. The resin was then washed with 5 × 2 ml wash buffer. The protein was eluted with elution buffer [50 mM HEPES (pH 7.2), 150 mM KCl, 2 mM MgCl2, 1 mM EGTA, 250 mM imidazole (pH 8.0), 1 mM DTT, 0.1 mM ATP, 1 mM PMSF, 0.1% Pluronic F‐127 (w/v), 10% glycerol].
Microtubule polymerization
Microtubule polymerization was performed as described (Rao et al, 2018). Briefly, 2 μl of 1 mg/ml Cy5‐labeled tubulin (prepared as described in Nicholas et al, 2014), 2 μl of 1 mg/ml biotinylated tubulin (Cytoskeleton Inc., #T333P), and 2 μl of 10 mg/ml unlabeled tubulin (Cytoskeleton Inc., #T240) was mixed on ice. 0.6 μl of 10 mM GTP was added to the tubulin solution, and the mixture was incubated at 37°C for 15 min. Then 0.72 μl of 0.1 mM taxol in DMSO was added to the mixture, which was further incubated at 37°C for 15 min. Tubulin that was not incorporated into the microtubules was removed by centrifugation through a glycerol cushion [BRB80 (80 mM PIPES, 2 mM MgCl2, 1 mM EGTA) with 60% (v/v) glycerol, 10 μM taxol, and 1 mM DTT, pH 6.8]. The pellet was re‐suspended in resuspension buffer [BRB80 with 10% (v/v) glycerol, 10 μM taxol, and 1 mM DTT, pH 6.8] to final concentration of 1 mg/ml microtubules. The microtubule solution was stored at room temperature in the dark for several days.
Single‐molecule total internal reflection fluorescence (smTIRF) microscopy assay
HEK293T cells were grown in a 100‐mm tissue culture dish (VWR). After 48 h post‐transfection, cells were washed with sterile PBS and detached from the dish using a cell scraper. Cells were resuspended in ice cold RIPA lysis buffer supplemented with Halt™ protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific). Lysates were centrifuged at 17,000 g for 5 min at 4°C. The supernatant was aliquoted, flash frozen in liquid nitrogen, and stored at –80°C. smTIRF assay was carried out similarly as described before (Rao et al, 2018). Briefly, a coverslip (No. 1.5, Zeiss, #474030–9,000‐000) was cleaned with ethanol, and a flow chamber was assembled using the cleaned coverslip, a glass slide, and two stripes of cut parafilm as described before (Nicholas et al, 2014). 0.5 mg/ml of BSA‐biotin was flown into the chamber, and the slide was incubated in a humidity box for 5 min. Afterwards, the chamber was washed with 3 × 20 μl of blocking buffer (BB) [BRB80 with 1% Pluronic F‐127 (w/v), 10 μM taxol, 2 mg/ml BSA, 1 mg/ml α‐casein, pH 6.8], and incubated for 10 min in the humidity box. After the surface was passivated, 0.25 mg/ml of streptavidin was flown into the chamber and incubated for 5 min. The chamber was then washed with 3 × 20 μl BB; 0.5 μl of 0.2 mg/ml microtubules was diluted in 19.5 μl BB and subsequently flown through the chamber. The chamber was washed with 2 × 20 μl BB and 20 μl motility buffer (MB) [60 mM HEPES, 50 mM KAc, mM MgCl2, 1 mM EGTA, 0.5% Pluronic F‐127 (w/v), 10% glycerol (v/v), 10 μM taxol, 1 mM DTT, 2 mg/ml BSA, 1 mg/ml α‐casein, pH 7.2]. The final assay solution was prepared by mixing 3 μl of 50 mg/ml BSA, 1 μl of 100 mM ATP, 2 μl of 25× protease inhibitor cocktail (Roche, #4693159001), 1 μl of 50 mM biotin, 1 μl of appropriated diluted HEK293T cell lysate or K490, and 42 μl of MB. The final dilution of the lysate was 200× to 1,000× depending on the construct. The final dilution of K490 was 10,000× (200 pM). 2 × 20 μl of the assay solution was flown through the chamber, and the chamber was sealed by vacuum grease. The images were acquired by 200 ms/frame. The data were analyzed using a home‐built MATLAB program.
Generation and characterization of iPSC lines from control and KIF5A patients
Skin fibroblasts were obtained from two healthy donors (a 44‐year‐old healthy female and a 25‐year‐old healthy female). Cells were derived with given informed consent within the collaborative research center project SFB636 B7 (ID number B7_028#4 and B7_068#3 respectively). Peripheral Blood Mononuclear Cells (PBMCs) were obtained from one pre‐manifest mutation carrier and two patients harboring a heterozygous mutation in the KIF5A gene. The (pre‐symptomatic) mutation carrier (named Patient 1 or P1) is a 28‐year‐old female who carries a c.3020 + 2 T > C nucleotide mutation. One disease‐affected individual (Patient 2 or P2) is a 63‐year‐old male who carries a heterozygous c.2993‐1G > A mutation. He is the cousin of the second ALS patient (Patient 3 or P3), a 38‐year‐old male who also carries a heterozygous c.2993‐1G > A mutation. All three individuals provided blood samples for iPSC models with informed consent. The respective mutations/families are described (Brenner et al, 2018). Fibroblasts and PBMCs were reprogrammed by nonintegrative delivery of OCT4, SOX2, KLF4, and c‐MYC using Sendai virus (SeV) vectors (CytoTune‐iPS 2.1 Sendai Reprogramming Kit, Thermo Fisher (Ban et al, 2011); Ethics Committee II of Heidelberg University approval no. 2018‐617 N‐MA for hiPSC generation). Pluripotent stem cell identity was confirmed by immunofluorescence staining of the pluripotency markers Oct4 and Sox2 as described (Appendix Fig S2) (Marsoner et al, 2016). iPSCs were cultured as colonies in Essential 8 (E8) DMEM/F12 with Hepes (Thermo Fisher Scientific), sodium selenite (14 ng/ml; Sigma‐Aldrich), l‐ascorbic acid phosphate (64 μg/ml; Sigma‐Aldrich), insulin (20 μg/ml; Sigma‐Aldrich), transferrin (11 μg/ml; Sigma‐Aldrich), FGF2 (100 ng/ml; Cell Guidance Systems), and transforming growth factor–β (2 ng/ml; Cell Guidance Systems) medium on Geltrex‐coated (Thermo Fisher Scientific) cell culture plates with daily medium change. Cells were passaged using EDTA (Thermo Fisher Scientific) and 5 μM Y‐27632 (Cell Guidance Systems) was added to the medium for 24 h. All human iPS cell lines were regularly checked and confirmed negative for mycoplasma.
Generation of iPSC‐derived motor neurons (iMN)
Motor neuron differentiation was based on the protocol of Du et al (2015) with slight modifications. Briefly, at 50% confluency, hiPSCs were switched to a chemically defined basal media comprised of Neurobasal medium and DMEM/F12 medium without HEPES (Life Technologies) in a 1:1 ratio, 1% Penicillin–Streptomycin (Thermo Fisher Scientific), 1% GlutaMAX (Thermo Fisher Scientific), 0.5 × B27‐Supplement (Thermo Fisher Scientific), and 0.5 × N2 Supplement (Thermo Fisher Scientific, 17502048) and 0.1 mM ascorbic acid (Sigma‐Aldrich) basal media supplemented with 3 μM CHIR99021 (Cell Guidance), 2 μM DMH1 (Selleck Chemicals), and 2 μM SB431542 (Cell Guidance Systems). Media were changed daily, and media volume was increased according to cell density. After 6 days, cells were passaged using Accutase (Thermo Fisher Scientific) and plated at a 1:6 ratio onto Geltrex‐coated 6‐well plates in basal media supplemented with 1 μM CHIR99021, 2 μM DMH1, 2 μM SB431542, 0.5 μM purmorphamine (Cell Guidance Systems), and 0.1 μM retinoic acid (Sigma‐Aldrich). Media was changed daily for 6 days, and media volume was increased according to cell density. Following this 12‐day induction, cells differentiated in motor neuron precursors (MNPs). At day 0, MNPs were passaged using Accutase and plated in a ration 1:3 onto Geltrex‐coated 6‐well plates in basal media supplemented with 0.1 μM purmorphamine and 0.5 μM retinoic acid. Media were changed daily, and media volume was increased according to cell density. At day 6, cells were passaged using Accutase and seeded in the final format onto PEI (Sigma‐Aldrich)/Laminin (Sigma‐Aldrich)‐coated plates in basal media supplemented with 0.1 μM purmorphamine, 0.5 μM retinoic acid, 5 μM DAPT (Cell Guidance Systems), 1 μM LM22A (Sigma‐Aldrich), 1 μM LM22B (Tocris), 10 ng/ml GDNF (Cell Guidance Systems), and 10 ng/ml IGF‐1 (Cell Guidance Systems). A half‐medium change was performed twice a week. MNPs were cryopreserved in basal media supplemented with 10% DMSO (Carl Roth) and 10 μM Y‐27632. For expansion, MNPs were passaged with Accutase at a 1:6 ratio and cultured for 6 days in basal media supplemented with 1 μM CHIR99021, 2 μM DMH1, 2 μM SB431542, 0.5 μM purmorphamine (Cell Guidance Systems), 1 μM retinoic acid (Sigma‐Aldrich), and 0.5 mM valproic acid (Thermo Fisher Scientific). Throughout the protocol, 10 μM Y‐27632 was added to the medium for 24 h following each replating. Media containing retinoic acid were kept away from direct light. The identity of mature motor neurons was confirmed from choline acetyltransferase (ChAT) immunostaining.
RNA extraction
iMNs were harvested in RNA‐Solv Reagent (Omega), and RNA was isolated according to the manufacturer's protocol. Briefly, chloroform was added in the lysates, and the mixture was incubated for 15 min at room temperature. Samples were centrifuged at 12,000 g for 5 min leading to separation of the different phases; isopropanol was added to the watery phase. RNA was precipitated overnight at −20°C and spun down at 12,000 g for 15 min. After two washing steps with 75% ethanol, RNA pellets were air‐dried and resuspended in diethyl pyrocarbonate‐treated water. Contaminations with genomic DNA were removed by treatment with deoxyribonuclease I (Sigma‐Aldrich). An iScript complementary DNA (cDNA) synthesis kit (Bio‐Rad) was used according to the manufacturer's protocol to generate cDNA. All clones were verified by Sanger sequencing (Eurofins, Germany).
Single‐nucleotide polymorphism karyotyping
Genomic DNA was prepared using the DNeasy Blood & Tissue Kit according to manufacturer's instructions (Qiagen). Whole‐genome single‐nucleotide polymorphism (SNP) genotyping was performed at the University of Heidelberg, Mannheim. Genomic DNA at a concentration of 50 ng/μl was used for whole‐genome amplification. Afterward, the amplified DNA was fragmented and hybridized to sequence‐specific oligomers bound to beads on an Illumina HumanOmniExpress BeadChip 12 V1.0 chip (Illumina, San Diego, CA, USA). Data were analyzed using Illumina GenomeStudio V2011.1 with the cnvPartition 3.2.0 plugin. Normal karyotypes were verified in controls previously (Jabali et al, 2022) and KIF5A iPSC lines in this study (Appendix Figs S3–S5).
Statistical analysis
Statistical analyses and graphs were prepared in GraphPad Prism (version 9). Data are expressed as mean ± SD or mean ± SEM as shown in figure legends. Student's t‐test or one‐way ANOVA was used for statistical analysis unless specified in figure legends. P‐value less than 0.05 was considered significant (*P < 0.05, **P < 0.01, ***P < 0.001).
Author contributions
Jie Jiang: Conceptualization; formal analysis; supervision; funding acquisition; investigation. Devesh C Pant: Conceptualization; resources; formal analysis; investigation; methodology. Janani Parameswaran: Conceptualization; formal analysis; funding acquisition; investigation; methodology. Lu Rao: Formal analysis; methodology. Liang Shi: Resources. Ganesh Chilukuri: Formal analysis. Jonathan D Glass: Resources. Gary J Bassell: Resources. Arne Gennerich: Formal analysis; supervision; methodology. Isabel Loss: Investigation; methodology. Rosanna Parlato: Investigation; methodology. Philipp Koch: Resources; supervision. Rüstem Yilmaz: Funding acquisition; investigation; methodology. Jochen H Weishaupt: Resources; supervision; investigation.
In addition to the CRediT author contributions listed above, the contributions in detail are:
DCP, JP, and JJ performed and analyzed all in vivo and cell‐based experiments. LR performed and analyzed the single‐molecule motility assays and edited the manuscript. IL characterized the iPSC with oversight from PK. RY and JHW designed and validated the custom neopeptide antibody. JHW provided access to the patient samples and edited the manuscript. IL and RP performed confocal imaging. AG designed and interpreted single‐molecule data and edited the manuscript. GC helped in data analysis and Drosophila experiments. LS, GJB, and JDG provided biological reagents. DCP, JP, and JJ wrote the manuscript.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
We are grateful to the patients and their families for participating in this study. We are grateful for the support and the private fundraising activity “Sternenlicht” of Thorsten Voß. The authors would like to thank Drs. David Brenner, Zachary McEachin, members of Jie Jiang, Gary Bassell, Jochen Weishaupt, Philipp Koch, Kenneth Moberg, and Victor Faundez labs for many helpful discussions. We also thank Laura Fox‐Goharioon (Emory Integrated Cellular Imaging Core) as well as Gina Tillmann, Lukas Eckrich, and Melita Simic for their technical support. We acknowledge the support of the Core Facility Live Cell Imaging Mannheim (LIMa) at the Medical Faculty Mannheim in using the TCS SP8 confocal microscope (Leica Microsystems). DCP and JP are supported by the Milton Safenowitz Postdoctoral Fellowship from the ALS association (Grants# 22‐PDF‐605 to DCP and 21‐PDF‐585 to JP). RY is supported by German Research Foundation (Deutsche Forschungsgemeinschaft, DFG) Walter Benjamin Programme (492655220). LR and AG are supported by National Institutes of Health (NIH) grants R01GM098469 and R01NS114636. LS and GJB are supported by the NIH R01 (R01NS114253 to GJB). The work is partially supported by the NIH R01 grant R01AG068247 to JJ.
EMBO reports (2022) 23: e54234
Data availability
This study includes no data deposited in external repositories.
References
- Ban H, Nishishita N, Fusaki N, Tabata T, Saeki K, Shikamura M, Takada N, Inoue M, Hasegawa M, Kawamata S et al (2011) Efficient generation of transgene‐free human induced pluripotent stem cells (iPSCs) by temperature‐sensitive Sendai virus vectors. Proc Natl Acad Sci USA 108: 14234–14239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron DM, Fenton AR, Saez‐Atienzar S, Giampetruzzi A, Sreeram A, Shankaracharya, Keagle PJ, Doocy VR, Smith NJ, Danielson EW et al (2022) ALS‐associated KIF5A mutations abolish autoinhibition resulting in a toxic gain of function. Cell Rep 39: 110598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair MA, Ma S, Hedera P (2006) Mutation in KIF5A can also cause adult‐onset hereditary spastic paraplegia. Neurogenetics 7: 47–50 [DOI] [PubMed] [Google Scholar]
- Brenner D, Weishaupt JH (2019) Update on amyotrophic lateral sclerosis genetics. Curr Opin Neurol 32: 735–739 [DOI] [PubMed] [Google Scholar]
- Brenner D, Yilmaz R, Muller K, Grehl T, Petri S, Meyer T, Grosskreutz J, Weydt P, Ruf W, Neuwirth C et al (2018) Hot‐spot KIF5A mutations cause familial ALS. Brain 141: 688–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budaitis BG, Jariwala S, Rao L, Yue Y, Sept D, Verhey KJ, Gennerich A (2021) Pathogenic mutations in the kinesin‐3 motor KIF1A diminish force generation and movement through allosteric mechanisms. J Cell Biol 220: e202004227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai D, Hoppe AD, Swanson JA, Verhey KJ (2007) Kinesin‐1 structural organization and conformational changes revealed by FRET stoichiometry in live cells. J Cell Biol 176: 51–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba K, Ori‐McKenney KM, Niwa S, McKenney RJ (2022) Synergistic autoinhibition and activation mechanisms control kinesin‐1 motor activity. Cell Rep 39: 110900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciechanover A, Kwon YT (2015) Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Exp Mol Med 47: e147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crimella C, Baschirotto C, Arnoldi A, Tonelli A, Tenderini E, Airoldi G, Martinuzzi A, Trabacca A, Losito L, Scarlato M et al (2012) Mutations in the motor and stalk domains of KIF5A in spastic paraplegia type 10 and in axonal Charcot‐Marie‐tooth type 2. Clin Genet 82: 157–164 [DOI] [PubMed] [Google Scholar]
- Dietrich KA, Sindelar CV, Brewer PD, Downing KH, Cremo CR, Rice SE (2008) The kinesin‐1 motor protein is regulated by a direct interaction of its head and tail. Proc Natl Acad Sci USA 105: 8938–8943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du ZW, Chen H, Liu H, Lu J, Qian K, Huang CL, Zhong X, Fan F, Zhang SC (2015) Generation and expansion of highly pure motor neuron progenitors from human pluripotent stem cells. Nat Commun 6: 6626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duis J, Dean S, Applegate C, Harper A, Xiao R, He W, Dollar JD, Sun LR, Waberski MB, Crawford TO et al (2016) KIF5A mutations cause an infantile onset phenotype including severe myoclonus with evidence of mitochondrial dysfunction. Ann Neurol 80: 633–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebbing B, Mann K, Starosta A, Jaud J, Schols L, Schule R, Woehlke G (2008) Effect of spastic paraplegia mutations in KIF5A kinesin on transport activity. Hum Mol Genet 17: 1245–1252 [DOI] [PubMed] [Google Scholar]
- Faruq M, Kumar D, Wadhwa S, Shamim U, Mathur A, Parveen S, Garg A, Srivastava AK (2019) Intrafamilial variable spastic paraplegia/ataxia/ALS phenotype linked to a novel KIF5A mutation. Clin Genet 96: 271–273 [DOI] [PubMed] [Google Scholar]
- Fichera M, Lo Giudice M, Falco M, Sturnio M, Amata S, Calabrese O, Bigoni S, Calzolari E, Neri M (2004) Evidence of kinesin heavy chain (KIF5A) involvement in pure hereditary spastic paraplegia. Neurology 63: 1108–1110 [DOI] [PubMed] [Google Scholar]
- Friedman DS, Vale RD (1999) Single‐molecule analysis of kinesin motility reveals regulation by the cargo‐binding tail domain. Nat Cell Biol 1: 293–297 [DOI] [PubMed] [Google Scholar]
- Gennerich A, Vale RD (2009) Walking the walk: How kinesin and dynein coordinate their steps. Curr Opin Cell Biol 21: 59–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackney DD, Baek N, Snyder AC (2009) Half‐site inhibition of dimeric kinesin head domains by monomeric tail domains. Biochemistry 48: 3448–3456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock WO (2016) The Kinesin‐1 Chemomechanical cycle: Stepping toward a consensus. Biophys J 110: 1216–1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Liu X, Tang L, Zhao C, He J, Fan D (2020) Whole‐exome sequencing identified novel KIF5A mutations in Chinese patients with amyotrophic lateral sclerosis and Charcot‐Marie‐tooth type 2. J Neurol Neurosurg Psychiatry 91: 326–328 [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Noda Y, Tanaka Y, Niwa S (2009) Kinesin superfamily motor proteins and intracellular transport. Nat Rev Mol Cell Biol 10: 682–696 [DOI] [PubMed] [Google Scholar]
- Jabali A, Hoffrichter A, Uzquiano A, Marsoner F, Wilkens R, Siekmann M, Bohl B, Rossetti AC, Horschitz S, Koch P et al (2022) Human cerebral organoids reveal progenitor pathology in EML1‐linked cortical malformation. EMBO Rep 23: e54027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaan HY, Hackney DD, Kozielski F (2011) The structure of the kinesin‐1 motor‐tail complex reveals the mechanism of autoinhibition. Science 333: 883–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamata H, Tsukasaki Y, Sakai T, Ikebe R, Wang J, Jeffers A, Boren J, Owens S, Suzuki T, Higashihara M et al (2017) KIF5A transports collagen vesicles of myofibroblasts during pleural fibrosis. Sci Rep 7: 4556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai Y, Okada Y, Tanaka Y, Harada A, Terada S, Hirokawa N (2000) KIF5C, a novel neuronal kinesin enriched in motor neurons. J Neurosci 20: 6374–6384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam AJ, Rao L, Anazawa Y, Okada K, Chiba K, Dacy M, Niwa S, Gennerich A, Nowakowski DW, McKenney RJ (2021) A highly conserved 310 helix within the kinesin motor domain is critical for kinesin function and human health. Sci Adv 7: eabf1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YR, King OD, Shorter J, Gitler AD (2013) Stress granules as crucibles of ALS pathogenesis. J Cell Biol 201: 361–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liguori F, Amadio S, Volonté C (2021) Fly for ALS: Drosophila modeling on the route to amyotrophic lateral sclerosis modifiers. Cell Mol Life Sci 78: 6143–6160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsoner F, Marcatili M, Karnavas T, Bottai D, D'Agostino A, Scarone S, Conti L (2016) Generation and characterization of an induced pluripotent stem cell (iPSC) line from a patient with clozapine‐resistant schizophrenia. Stem Cell Res 17: 661–664 [DOI] [PubMed] [Google Scholar]
- Nakamura R, Tohnai G, Atsuta N, Nakatochi M, Hayashi N, Watanabe H, Yokoi D, Watanabe H, Katsuno M, Izumi Y et al (2021) Genetic and functional analysis of KIF5A variants in Japanese patients with sporadic amyotrophic lateral sclerosis. Neurobiol Aging 97: 147 [DOI] [PubMed] [Google Scholar]
- Nakano J, Chiba K, Niwa S (2022) An ALS‐associated KIF5A mutant forms oligomers and aggregates and induces neuronal toxicity. Genes Cells 27: 421–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naruse H, Ishiura H, Mitsui J, Takahashi Y, Matsukawa T, Sakuishi K, Nakamagoe K, Miyake Z, Tamaoka A, Goto J et al (2021) Splice‐site mutations in KIF5A in the Japanese case series of amyotrophic lateral sclerosis. Neurogenetics 22: 11–17 [DOI] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM et al (2006) Ubiquitinated TDP‐43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314: 130–133 [DOI] [PubMed] [Google Scholar]
- Nicholas MP, Rao L, Gennerich A (2014) Covalent immobilization of microtubules on glass surfaces for molecular motor force measurements and other single‐molecule assays. Methods Mol Biol 1136: 137–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols CD, Becnel J, Pandey UB (2012) Methods to assay Drosophila behavior. J Vis Exp 7: 3795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas A, Kenna KP, Renton AE, Ticozzi N, Faghri F, Chia R, Dominov JA, Kenna BJ, Nalls MA, Keagle P et al (2018) Genome‐wide analyses identify KIF5A as a novel ALS gene. Neuron 97: e1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pakravan D, Orlando G, Bercier V, Van Den Bosch L (2021) Role and therapeutic potential of liquid‐liquid phase separation in amyotrophic lateral sclerosis. J Mol Cell Biol 13: 15–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman A, Kamal A, Roberts EA, Goldstein LS (1999) Defective kinesin heavy chain behavior in mouse kinesin light chain mutants. J Cell Biol 146: 1277–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao L, Hulsemann M, Gennerich A (2018) Combining structure‐function and single‐molecule studies on cytoplasmic dynein. Methods Mol Biol 1665: 53–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross CA, Poirier MA (2004) Protein aggregation and neurodegenerative disease. Nat Med 10: S10–S17 [DOI] [PubMed] [Google Scholar]
- Rusan NM, Fagerstrom CJ, Yvon AM, Wadsworth P (2001) Cell cycle‐dependent changes in microtubule dynamics in living cells expressing green fluorescent protein‐alpha tubulin. Mol Biol Cell 12: 971–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rydzanicz M, Jagla M, Kosinska J, Tomasik T, Sobczak A, Pollak A, Herman‐Sucharska I, Walczak A, Kwinta P, Ploski R (2017) KIF5A de novo mutation associated with myoclonic seizures and neonatal onset progressive leukoencephalopathy. Clin Genet 91: 769–773 [DOI] [PubMed] [Google Scholar]
- Saez‐Atienzar S, Dalgard CL, Ding J, Chiò A, Alba C, Hupalo DN, Wilkerson MD, Bowser R, Pioro EP, Bedlack R et al (2020) Identification of a pathogenic intronic KIF5A mutation in an ALS‐FTD kindred. Neurology 95: 1015–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeger MA, Rice SE (2010) Microtubule‐associated protein‐like binding of the kinesin‐1 tail to microtubules. J Biol Chem 285: 8155–8162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeger MA, Rice SE (2013) Intrinsic disorder in the kinesin superfamily. Biophys Rev 5: 233–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiler S, Kirchner J, Horn C, Kallipolitou A, Woehlke G, Schliwa M (2000) Cargo binding and regulatory sites in the tail of fungal conventional kinesin. Nat Cell Biol 2: 333–338 [DOI] [PubMed] [Google Scholar]
- Sheu‐Gruttadauria J, MacRae IJ (2018) Phase transitions in the assembly and function of human miRISC. Cell 173: e916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shima T, Morikawa M, Kaneshiro J, Kambara T, Kamimura S, Yagi T, Iwamoto H, Uemura S, Shigematsu H, Shirouzu M et al (2018) Kinesin‐binding‐triggered conformation switching of microtubules contributes to polarized transport. J Cell Biol 217: 4164–4183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto C, Pritzkow S (2018) Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat Neurosci 21: 1332–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su X, Ditlev JA, Hui E, Xing W, Banjade S, Okrut J, King DS, Taunton J, Rosen MK, Vale RD (2016) Phase separation of signaling molecules promotes T cell receptor signal transduction. Science 352: 595–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Kanai Y, Okada Y, Nonaka S, Takeda S, Harada A, Hirokawa N (1998) Targeted disruption of mouse conventional kinesin heavy chain, kif5B, results in abnormal perinuclear clustering of mitochondria. Cell 93: 1147–1158 [DOI] [PubMed] [Google Scholar]
- Tomishige M, Stuurman N, Vale RD (2006) Single‐molecule observations of neck linker conformational changes in the kinesin motor protein. Nat Struct Mol Biol 13: 887–894 [DOI] [PubMed] [Google Scholar]
- Valdez‐Sinon AN, Lai A, Shi L, Lancaster CL, Gokhale A, Faundez V, Bassell GJ (2020) Cdh1‐APC regulates protein synthesis and stress granules in neurons through an FMRP‐dependent mechanism. iScience 23: 101132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Zhang L, Dai T, Qin Z, Lu H, Zhang L, Zhou F (2021) Liquid‐liquid phase separation in human health and diseases. Signal Transduct Target Ther 6: 290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weskamp K, Safren N, Miguez R, Barmada S (2019) Monitoring neuronal survival via longitudinal fluorescence microscopy. J Vis Exp 19: 3791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia CH, Roberts EA, Her LS, Liu X, Williams DS, Cleveland DW, Goldstein LS (2003) Abnormal neurofilament transport caused by targeted disruption of neuronal kinesin heavy chain KIF5A. J Cell Biol 161: 55–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo KS, Lee K, Oh JY, Lee H, Park H, Park YS, Kim HK (2019) Postsynaptic density protein 95 (PSD‐95) is transported by KIF5 to dendritic regions. Mol Brain 12: 97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Liu Q, Shen D, Tai H, Liu S, Wang Z, Shi J, Fu H, Wu S, Ding Q et al (2019) Mutation analysis of KIF5A in Chinese amyotrophic lateral sclerosis patients. Neurobiol Aging 73: 229 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File
Data Availability Statement
This study includes no data deposited in external repositories.