

Summary
Comparisons of genomes of different species are used to identify lineage-specific genes, those genes that appear unique to one species or clade. Lineage-specific genes are often thought to represent genetic novelty that underlies unique adaptations. Identification of these genes depends not only on genome sequences, but also on inferred gene annotations. Comparative analyses typically use available genomes that have been annotated using different methods, increasing the risk that orthologous DNA sequences may be erroneously annotated as a gene in one species but not another, appearing lineage-specific as a result. To evaluate the impact of such “annotation heterogeneity,” we identified four clades of species with sequenced genomes with more than one publicly available gene annotation, allowing us to compare the number of lineage-specific genes inferred when differing annotation methods are used to those resulting when annotation method is uniform across the clade. In these case studies, annotation heterogeneity increases the apparent number of lineage-specific genes by up to 15-fold, suggesting that annotation heterogeneity is a substantial source of potential artifact.
Introduction
Comparing the genome sequences of different organisms can yield inferences about the genetic basis of the biological differences between them. One such analysis aims to identify genes unique to a particular monophyletic group. Such genes, called “orphan genes” when restricted to one species and “lineage-specific” or “taxonomically-restricted” when restricted to a clade of several species, are interesting from the perspective of genetic and evolutionary novelty. For example, they have been previously hypothesized to underlie lineage-specific structural and functional innovations, and to be novel genes that have emerged from noncoding DNA 1–5.
Lineage-specific genes are typically identified by searching for homologs in outgroup species: genes for which homologs cannot be found are considered lineage-specific. Such analyses typically begin not with raw genome sequences, but with particular “annotations” of them: inferences about what genes they encode. Often, only genes included in these annotations are considered in the homology search 2,6,7.
Previous work has recognized two ways in which errors in genome annotations could produce spurious lineage-specific genes. A real gene could be annotated in the focal lineage, but its homologs incorrectly unannotated in outgroups 8–10. Conversely, a non-genic sequence could be incorrectly annotated as a gene in the lineage, but correctly omitted in outgroups 11. Such errors could occur even when all genomes in an analysis are consistently annotated by the same annotation methodology, but the potential for error is expected to increase if genomes are annotated by different methods, which use different criteria in determining which sequences are genic. We refer to this as “annotation heterogeneity.”
Annotation heterogeneity is common. Comparative analyses typically use existing annotations rather than producing their own, potentially uniform, ones. Available annotations have been generated by a wide variety of methods. Large consortia, such as bioinformatics institutes or model organism databases, all use different methods, including custom pipelines, (NCBI 12, Ensembl 13), hand curation (Flybase 14, Wormbase 15), and crowd-sourced annotation (VectorBase 16). Individual research groups also produce annotations, usually using heterogeneous selections of one or a combination of at least 30 available software tools 17 and custom parameters. Annotations from these varied sources can often be downloaded or accessed from large centralized databases (e.g. Refseq/Genbank, Uniprot), but are not homogenized when they are deposited into them: proteomes downloaded from such databases (including NCBI’s “non-redundant,” or “nr”, database, the default in a BLASTP web server search) or searches performed on them are therefore highly heterogeneous. Of 25 lineage-specific studies discussed in a prominent 2019 review [18], 18 (76%) depended on heterogeneous genome annotations, rather than on homogeneous re-annotations or on annotation-independent homology searches with six-frame translations of all ORFs (Supplemental Table 1). And of 33 studies published between 2019 and 2022, 21 (64%) depended on heterogeneous annotations, a rate not significantly different (p=0.58, Fisher’s exact test) than among the older studies (Supplemental Table 2).
Here we explore the effect of annotation heterogeneity on inferred numbers of lineage-specific genes. We identify four clades of species with available genome sequences for which multiple different annotations are publicly available. These enable us to conduct case studies in which we compare the number of lineage-specific genes when all species are annotated with the same method (“uniform annotations”) to when they are annotated with different methods (“heterogeneous annotation”). We find that annotation heterogeneity consistently and substantially increases the inferred number of lineage-specific genes. This effect is strongest when all species within the lineage are annotated with one method and all outgroup species with a different one. Our results suggest that annotation heterogeneity can produce many spurious lineage-specific genes, potentially a majority of those found in a study.
Results
Identification of clades of sequenced genomes with annotations from two methods
To directly compare lineage-specific genes found using uniform annotations and heterogeneous annotations, we manually searched the literature and bioinformatic databases for species groups in which all species were annotated with the same method, and, additionally, the same assembly of each species had been independently annotated with some other method. We used existing annotations from a variety of standard sources instead of generating our own to make results maximally representative of real studies. We identified four groups of five species: cichlids, primates, bats, and rodents. For cichlids and primates, all five species were annotated with the same two methods, whereas for bats and rodents, one method was applied to all five species, and the other available annotation was from three different methods, with each species being annotated by one of the three. Full details of these annotations, the underlying assemblies, and the methods used to annotate them can be found in Supplemental Table 3. Each of these four groups is less than approximately 60 My old.
Different annotations of the same genome have many proteins unique to each method
Spurious lineage-specific genes may result from annotation heterogeneity when different annotation methods differentially annotate homologous sequences. Spurious lineage-specific genes may also result from such erroneous differential annotation even when a single annotation method is used, as sequence differences between the species may alter a given method’s determination regarding genic status. To get a sense of how many spurious lineage-specific protein-coding genes annotation heterogeneity per se can produce, we compared two protein annotations of the same species to identify proteins appearing to be unique to one of the annotations. (This is a limiting case of the “phyletic annotation” described below.) Because the underlying genome sequences are identical, proteins can only appear to be “orphans,” unique to one of the annotations, as a result of annotation heterogeneity.
To mimic a typical analysis, for each species’ two annotations, we used BLASTP 18 for all proteins in one annotation to see if a significantly similar (E<0.001) homolog was present in the other annotation (Supplemental Table 4). On average, 1380 proteins in each annotation lacked a significantly similar sequence in the other. This represented an average of 3% of total proteins, with a range of between 0.6% and 9.7%. 19 of the 40 annotations, or nearly half, had over 1000 proteins without a significant homolog in the other annotation. In an extreme case of the cichlid Astatotilapia burtonii, one annotation (Broad Institute) found 4110 genes that had no significant similarities in the other (NCBI eukaryotic annotation pipeline), and 799 proteins in the NCBI annotation lacked significant similarities in the Broad annotation. These substantial differences between two annotations of one genome illustrate the potential for spurious lineage-specific genes in comparisons of different genomes.
Different patterns of annotation heterogeneity may differently affect the inferred number of lineage-specific genes
When different annotation methods are used for species within an analysis, different patterns in which those methods are arranged on the species topology are possible. These different patterns may differently affect the number of spurious lineage-specific genes produced by annotation heterogeneity. In particular, because a gene is called as “lineage-specific” if no significant homologs are found in any species outside the lineage, we expected that the number of spurious lineage-specific genes would be positively related to the overall degree of difference between the lineage and outgroup annotations.
We considered three such patterns. In the first, one annotation method is used for all ingroup species (in the lineage, the gray boxes in the figures), and a different method for all outgroup species (outside the lineage); we refer to this as “phyletic” annotation (Figure 1). In the second, one method is used for all ingroup species, but a mixture of methods is used for the outgroup species; we refer to this as “semi-phyletic” annotation (Figure 2). In the third, a mixture of methods is used for both the ingroup species and the outgroup species; we refer to this as “unpatterned” annotation (Figure 3).
Figure 1: Comparison of the number of lineage-specific genes found using uniform and heterogeneous (phyletic) annotations in a) cichlids and b) primates.
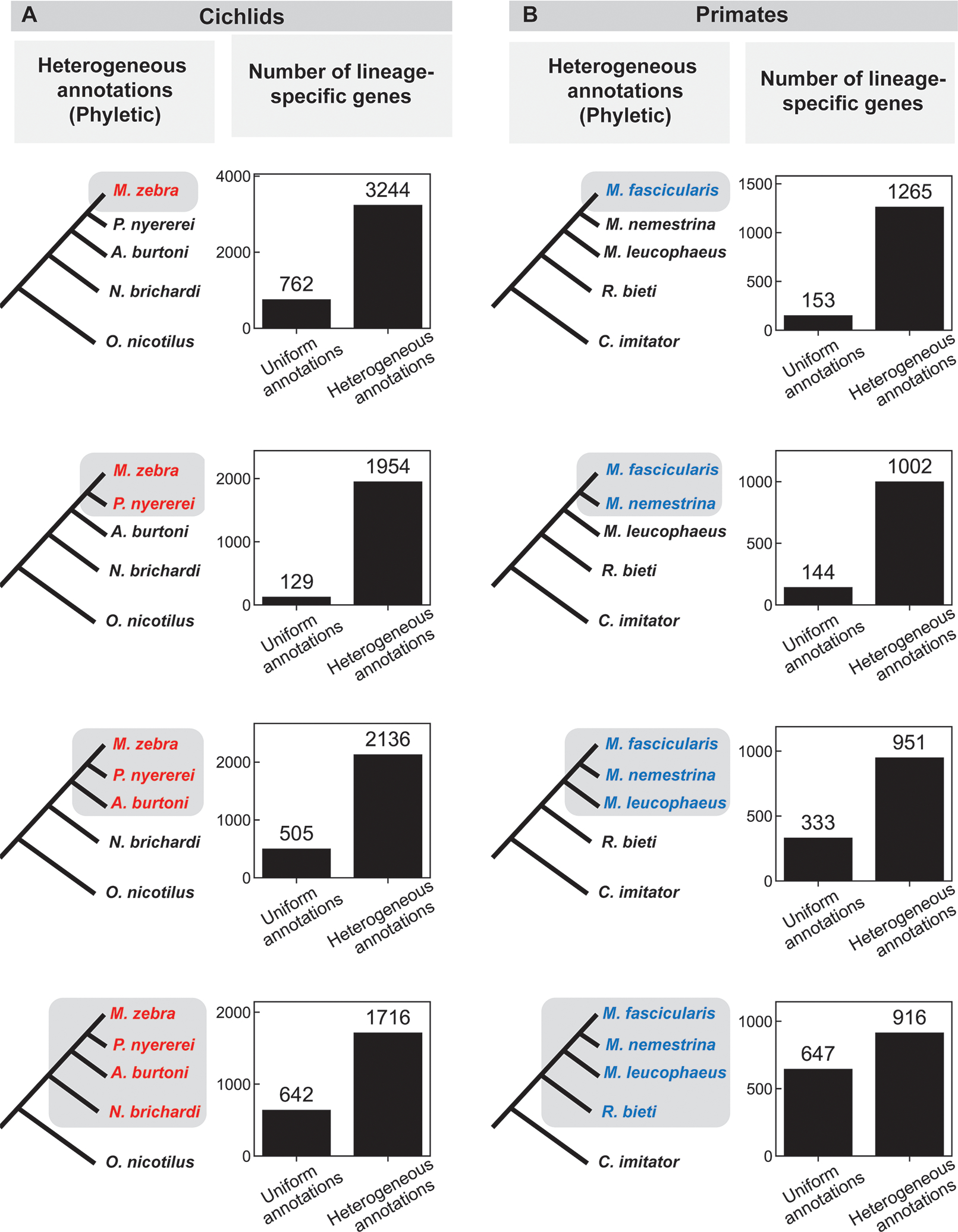
The species tree on the left indicates the lineage under consideration (grey shading); different text colors indicate different annotation sources in the heterogeneous annotation analysis (black, NCBI; red, research group at the Broad Institute; blue, Ensembl; see also Supplemental Table 3, 4). A depiction of the uniform annotation pattern, in which all annotations are from NCBI (black), is not shown. Bar graphs indicate the number of genes that appear specific to the lineage shaded on the species tree to the left using either uniform or heterogeneous annotations. See also Supplemental Table 5 for results of tBLASTx searches in this group.
Figure 2: Comparison of the number of lineage-specific genes found using uniform and heterogeneous (semi-phyletic) annotations in a) rodents and b) bats.
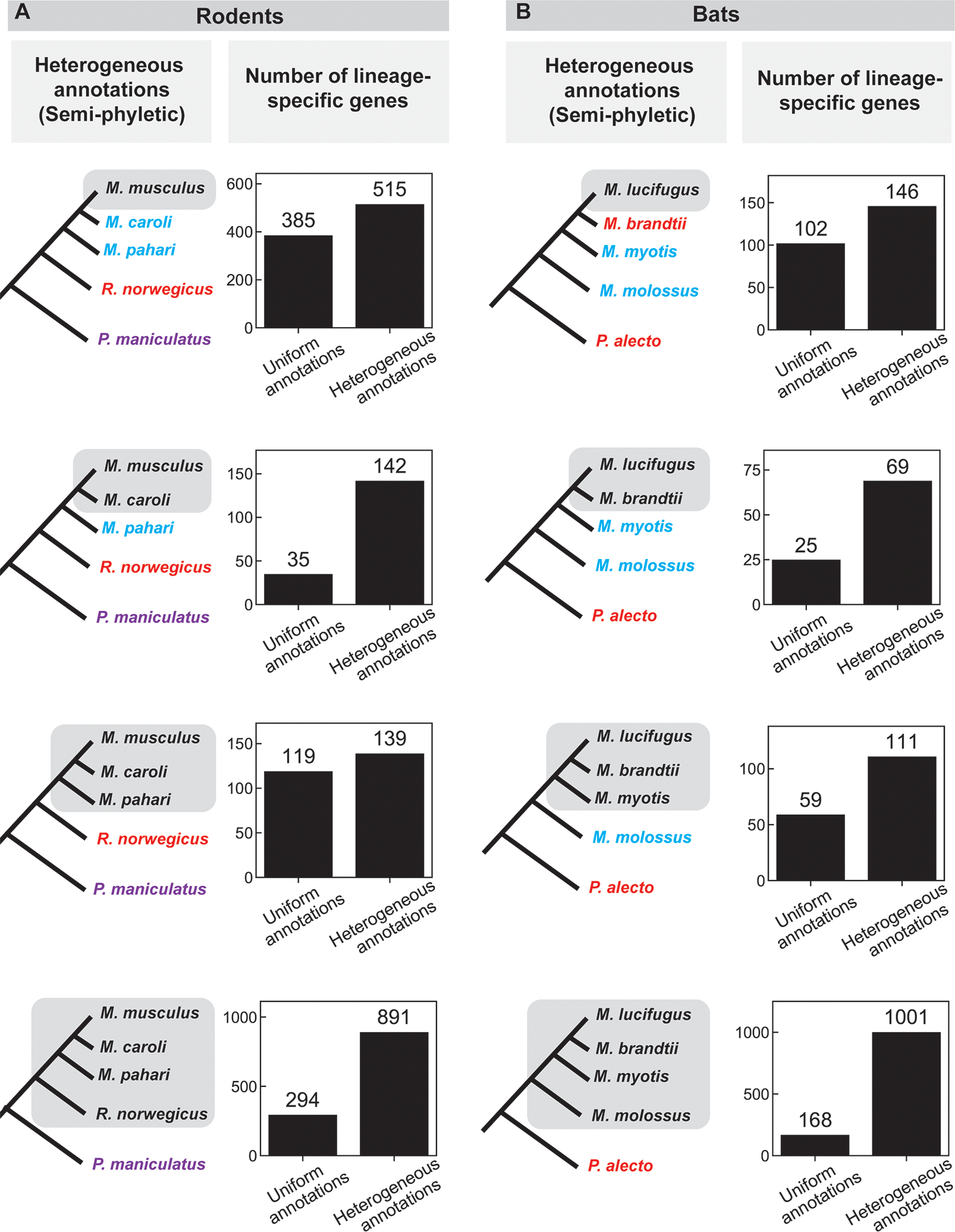
The species tree on the left indicates the lineage under consideration (grey shading); different text colors indicate different annotation sources in the heterogeneous annotation analysis (black, NCBI; blue, UCSC; red, Ensembl “mixed genebuild”; purple, Ensembl “full genebuild”; green, Bat1k; pink, Beijing Genomics Institute; see also Supplemental Table 3, 4). A depiction of the uniform annotation pattern, in which all annotations are from NCBI (black), is not shown. Bar graphs indicate the number of genes that appear specific to the lineage shaded on the species tree to the left using either uniform or heterogeneous annotations. See also Supplemental Table 5 for results of tBLASTx searches in this group.
Figure 3: Comparison of the number of lineage-specific genes found using uniform and heterogeneous (unpatterned) annotations in a) rodents and b) bats.

The species tree on the left indicates the lineage under consideration (grey shading); different text colors indicate different annotation sources in the heterogeneous annotation analysis (black, NCBI; blue, UCSC; red, Ensembl “mixed genebuild”; purple, Ensembl “full genebuild”; green, Bat1k; pink, Beijing Genomics Institute; see also Supplemental Table 3, 4). A depiction of the uniform annotation pattern, in which all annotations are from NCBI (black), is not shown. Bar graphs indicate the number of genes that appear specific to the lineage shaded on the species tree to the left using either uniform or heterogeneous annotations. See also Supplemental Table 5 for results of tBLASTx searches in this group.
The degree of difference between the annotations of ingroups and outgroups are largest for phyletic annotation, intermediate for semi-phyletic annotation, and smallest for unpatterned annotation. We expected the magnitude of the impact of annotation heterogeneity genes to scale accordingly. We used our four clades to create case studies for each pattern.
Annotating a lineage with one method and outgroups with a different method greatly increases the apparent number of lineage-specific genes
Phyletic annotation occurs in at least two scenarios. Studies that newly sequence a lineage often use their own method to annotate that lineage, and may then compare it to outgroup annotations from another single source (e.g. Ensembl). Additionally, studies using existing annotations may encounter a correlation between taxon and annotation method because genome sequencing groups (with their annotation teams) often select species taxonomically (e.g. studies of particular taxa, sequencing consortia/database initiatives for particular taxa).
We tested the impact of phyletic annotation on the apparent number of lineage-specific genes on two groups of species, where the same genome assembly for every species had been annotated by the same two methods: five cichlids, annotated both by the Broad Institute and NCBI; and five primates, annotated both by Ensembl and NCBI (Supplemental Table 3).
For each tree of five species, we exploited the ladder-like topology (Figure 1) of the tree to perform four analyses, comparing each of the four monophyletic groups including the focal species to the remaining outgroups. For each lineage that included the focal species, we conducted a typical analysis of lineage-specific genes by identifying genes in the focal species that have a significantly similar homolog in the deepest rooted member of the ingroup (and thus are “present” in that clade), but lack significant similarity to any protein in any outgroup species in a BLASTP search (Methods). We compared the number of lineage-specific genes found when all species (both ingroups and outgroups) were annotated with the same method to the number found when the annotations for all outgroup species were switched to the other method in a “phyletic” annotation pattern (Figure 1).
Heterogeneous annotation consistently caused a large increase of hundreds to thousands of apparent lineage-specific genes, typically about a 4-fold (ranging from 1.4-fold to 15-fold) difference relative to uniform annotation. In all but one of the eight cases in Figure 1, the increase is more than 2-fold, suggesting that the majority of lineage-specific genes inferred in heterogeneous annotations are artifacts of the heterogeneity.
Annotating a lineage with one method and outgroups with a mixture of other methods increases the apparent number of lineage-specific genes
“Semi-phyletic” annotation, where the ingroup is annotated with one method and outgroups with a mixture of methods, was the most common type of annotation heterogeneity in our review of the published literature (Supplemental Tables 1, 2). It often occurs in scenarios similar to phyletic annotation, but where outgroup annotations come from a mixture of sources (e.g. a combination of Ensembl and NCBI, or a large heterogeneous database like NCBI’s “non-redundant” sequenes).
We created case studies of semi-phyletic annotation using groups of species for which every species had been annotated both by the same method and by one of a mix of other methods: five rodents and five bats (Supplemental Table 3). We repeated the procedure described for phyletic annotation above to compare the number of lineage-specific genes in semi-phyletic annotations to those in uniform annotations (Figure 2).
Semi-phyletic annotation heterogeneity caused a smaller but still substantial increase in the number of apparent lineage-specific genes in all lineages in both groups (Figure 2). The magnitude of this effect ranged from 20 to 833 additional lineage-specific genes, corresponding to 1.2-fold to 6-fold increases.
Annotating species with a mixture of methods without taxonomic bias increases the apparent number of lineage-specific genes
Examples of what we call “unpatterned” annotation, where the annotation method varies within the ingroup as well as the outgroup, are also common. It can occur in scenarios similar to semi-phyletic annotation, but when studies use existing annotations for the desired species, which often come from a variety of sources.
We created case studies of unpatterned annotation using the same rodent and bat species we used for semi-phyletic annotation (Figure 2), with the difference that we always compared the uniform annotations to the full set of mixed annotations (Figure 3) to produce unpatterned annotation heterogeneity.
Unpatterned annotation heterogeneity usually caused an increase in apparent lineage-specific genes (Figure 3), though the effect was smaller than for phyletic or semi-phyletic annotations. Two cases showed equal numbers or slight decreases, and the other six cases showed increases of 1.1-fold to 5.7-fold; the largest increases were in the cases with a single outgroup species.
Sequence characteristics of genes affected by annotation heterogeneity
We wondered whether certain sequence characteristics of proteins may affect how likely they are to be heterogeneously annotated (included in one annotation of a genome assembly and omitted from another). We therefore classified all proteins in our four focal species as affected or unaffected by annotation heterogeneity. Affected proteins have significantly similar homologs only in one of the two annotations of at least one outgroup species; unaffected proteins either have homologs in both annotations or in neither.
We found that, in all species groups, proteins affected by annotation heterogeneity were shorter than those that were not (M. musculus, mean of 377 amino acids vs. 691 amino acids, t-test p-value=1.5*10−96; M. zebra, 193 vs 720, p=10−100; M. fascicularis, 136 vs 562, p=2.8*10−208; M. lucufugus, 299 vs. 608, p=2.1*10−154). We also found a significant association between annotation heterogeneity and protein disorder in all groups. However, the direction of this association was inconsistent: heterogeneously annotated proteins were more disordered in two taxa (M. musculus: mean IUpred disorder 0.38 vs. 0.33, p=3.2*10−41; M. lucufugus, 0.35 vs 0.32, p=10−5), and less disordered in two other taxa (M. zebra: 0.28 vs 0.35, p=6*10−154; M. fasciscularis, p=6*10−154; 0.30 vs 0.31, p=10−5).
We hypothesize that the consistent length effect is due to essentially all annotation methods being more likely to consider longer open reading frames as genes. Similarly, the inconsistent disorder effect may be due to different annotation programs weighting disorder, or some other characteristic with which it is correlated, differently in determining whether an open reading frame is a gene.
As expected, six-frame translation homology searches dramatically reduce the apparent number of lineage-specific genes
A homology search in which the query protein is compared directly to a six-frame translation of the target genome does not rely on an annotation of the target species, and so should reduce this source of spurious lineage-specific genes. Such translated searches have previously been shown to reduce the inferred number of lineage-specific genes 8,9. In agreement with these expectations, we find that, for all of the lineages described above (depicted in Figures 1–3), a search for the focal species’ proteins against six-frame translations of all comparator species genomes dramatically reduces the number of lineage-specific genes: to below the number inferred with uniform annotations, and often to less than one hundred (Supplemental Table 5).
Discussion
We used six case studies to ask if varying the annotation method across species in a comparative analysis (“annotation heterogeneity”), common in comparative analyses, alters the apparent number of lineage-specific genes. We found that switching from uniform to heterogeneous annotations consistently increased the number of genes that were classified as lineage-specific, with increases ranging from tens to thousands of genes, corresponding to increases of up to 15-fold. The largest increases were seen when one annotation method was used for all the ingroup species and another was used for all the outgroup species (“phyletic annotation”). The smallest increases were seen when a mixture of annotation methods were used in both ingroup and outgroup species. Even within types of annotation heterogeneity, however, we find substantial variation in effect size; this likely depends on the details of the particular annotation methods involved, making it difficult to estimate the impact of annotation heterogeneity within any specific study a priori. Our results suggest that the numbers of lineage-specific genes found in these studies may be inflated, especially in “phyletic annotation” cases. Annotation heterogeneity may also have consequences that we do not explore here, like producing spurious lineage-specific losses.
We find evidence that sequence characteristics of proteins correlate with their tendency to be heterogeneously annotated. Length and disorder are consistent correlates of heterogeneous annotation, but the direction of the association for disorder varies across our case studies. We speculate that which correlations arise in a particular analysis depends on the particulars of the annotation methods in use. The differences in our results for disorder are reminiscent of conflicting reports of the direction of correlation between apparent gene age and disorder found in previous studies 8,11,19–23. We speculate that annotation heterogeneity may contribute to these discrepancies, consistent with the previous finding that the direction of the correlation can be reversed when ORFs unlikely to be real genes are carefully excluded 11.
Our case studies consist of closely related groups of five species. We did not consider larger groups of more distantly related species because we were unable to find ones satisfying our requirement that each species have two available annotations of the same genome assembly, one of which is from the same method for all members of the group. The effects of annotation heterogeneity in larger and more distantly related cases could be less pronounced.
Recent work from us and others has shown that homology detection failure, in which homology searches fail to detect homologs that are actually present in outgroups, can also produce spurious lineage-specific genes independently of annotation errors 24,25. Previous studies have noted a surprisingly large number of “young” lineage-specific genes found in recently evolved clades 26, which, compared to older lineage-specific genes, are less readily explained by homology detection failure, which is minimized at short evolutionary distances. The results here are all for young (<60 My old) clades, showing that annotation heterogeneity can be a significant source of spurious lineage-specific genes in young clades.
In accordance with previous results, we show that annotation heterogeneity artifacts can be reduced by performing homology searches of six-frame translated genomic DNA sequence in search of unannotated homologs in target species. This approach has caveats. At short evolutionary distances, a sequence may be sufficiently similar for successful detection in such a search without having the same coding status as the query; for example, a truly de novo originated gene is expected to have significant nucleotide similarity to a homologous noncoding locus in close outgroup species. This approach also still relies on an accurate annotation of the focal species.
When annotation methods disagree, which is correct? Our results do not address this, only demonstrating a consequence of this disagreement. Even homogeneous annotations are imperfect. Of particular concern, methods in general rely on features (homology to known genes, length, expression level, codon optimization) that seem likely to be absent or weaker in newly evolved (de novo) genes, and so may fail to identify these genes. We consider annotation accuracy primarily accountable to experimental data. While we do not perform such analyses here, we propose that assessing transcription, translation, and function in all species in question is of ultimate importance in accurately identifying lineage-specific genes. In light of our results, we suggest more emphasis on these metrics. In the meantime, the true number of lineage-specific genes remains difficult to ascertain, but better understanding sources of spurious ones helps constrain it.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Caroline M. Weisman (cweisman@princeton.edu)
Materials availability
This study did not generate new unique reagents.
Data and code availability
All genome annotations on which these analyses were based are publicly available and are listed in the Key Resources Table and Supplemental Tables 3 and 4. All results summarized in Figures 1–3 are publicly available as of the date of publication at https://github.com/caraweisman/Annotation_homology.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Deposited Data | ||
| Cichlid genome annotations | Broad Institute | ftp://ftp.broadinstitute.org/pub/vgb/cichlids/Annotation/Protein_coding/Peptide_Files/ |
| Cichlid genome annotations | NCBI | GCF_000238955.1, GCF_000239375.1, GCF_000239415.1, GCF_000239395.1, GCF_000188235.2 |
| Primate genome annotations | Ensembl | Release 102, macaca_fascicularis; Release 103, macaca_nemestrina, mandrillus_leucophaeus, rhinopithecus_bieti, cebus_imitator |
| Primate genome annotations | NCBI | GCF_000364345.1, GCF_000956065.1, GCF_000951045.1, GCF_001698545.1, GCF_001604975.1 |
| Rodent genome annotations | Downloaded from Ensembl; generated by Ensemble and UCSC | Release 101, mus_musculus, mus_caroli, mus_pahari, rattus_norwegicus; Release 104, peromyscus_maniculatus |
| Rodent genome annotations | NCBI | GCF_000001635.26, GCF_900094665.1, GCF_900095145.1, GCF_000001895.5, GCF_000500345.1 |
| Bat genome annotations | Downloaded from Ensembl and NCBI; generated by Ensembl, BAT1K consortium, Beijing Genomics Institute | GCA_000412655.1, GCA_014108235.1, GCA_014108415.1, GCA_000325575.1, Ensembl release 103, myotis_lucifugus |
| Bat genome annotations | NCBI | GCF_000147115.1, GCF_000412655.1, GCF_014108235.1, GCF_014108415.1, GCF_000325575.1 |
| Software and Algorithms | ||
| Basic Local Alignment Search Tool (BLAST) | Altschul, Stephen F., et al. “Basic local alignment search tool.” Journal of molecular biology 21 5.3 (1990): 403–410. | Version 6.2.0 |
METHOD DETAILS
Identifying lineage-specific proteins
For each species group, we defined a protein as specific to a particular lineage if a search using BLASTP 18 version 6.2.0 had no similar protein at a significance threshold of E=0.001 in the annotation of any species that was an outgroup to that lineage. We did not require that a protein be present in all members of the lineage to be specific to that lineage: a protein was defined as specific to a lineage based on the most distant species in which it was detected. For example, if a protein in M. musculus was detected only in R. norvegicus, it was defined as specific to that lineage; if a gene in M. musculus was detected in M. caroli, M. pahari, and R. norvegicus, it was also defined as specific to that same lineage. If a protein was found in the earliest-branching member of the species group, it was considered “conserved” and so not counted as any kind of lineage-specific gene. This way of classifying lineage-specificity coheres with standard practice 6.
For the six-frame translated searches, we first generated a six-frame translation of the genome assembly of each species using the ‘esl-translate’ command in the hmmer easel package, and then used it as the target database in a BLASTP search, as described in the previous paragraph. Results from these searches are summarized in Supplemental Table 5.
Literature review of studies of lineage-specific genes
We considered each of the papers cited in Table 1 of a recent review paper on de novo genes27 and determined whether or not it used heterogeneously annotated genomes. A list of these papers, whether it uses heterogeneous annotations, and, if so, relevant details about the heterogeneous annotations are shown in Supplemental Table 1.
We performed a literature search for articles about lineage-specific genes published after 2019. We considered all articles returned by a Google Scholar search for publications between 2019 and present whose titles included the phrases “lineage-specific gene(s),” “orphan gene(s),” “taxonomically-restricted gene(s),” “de novo gene(s),” or “proto-gene(s).” We manually excluded results clearly on unrelated topics (e.g. “lineage-specific genes” in the context of development, where the “lineage” is a cell type and its progenitors). Some of these studies used lineage-specific genes identified in previous studies; in these cases, we considered whether the original study used heterogeneous annotations. A list of these papers, whether it uses heterogeneous annotations, and, if so, relevant details about the heterogeneous annotations are shown in Supplemental Table 2.
QUANTIFICATION AND STATISTICAL ANALYSIS
Frequency of annotation heterogeneity in published literature
To compare the rates of annotation heterogeneity among the two sets of papers on the subject of lineage-specific genes that we considered, one from pre-2020 and one from post-2020, we used a Fisher’s exact test, with N being the total number of papers in the two groups, as described in the text (Introduction) and as listed in Supplemental Tables 1 and 2. No data were excluded.
Sequence characteristics of genes affected by annotation heterogeneity
To assess the statistical association between genes’ tendency to be affected by annotation heterogeneity and gene length, GC content, and disorder, we used t-tests to compare the distribution of the lengths of genes affected and not affected by annotation heterogeneity. N was the total number of proteins in the annotation of the focal species, given in Supplemental Table 4, with the results for particular genes available on the Github as described above in Data and Code Availability. No data were excluded.
Supplementary Material
Table S1: List of papers from a 2019 review27 and whether they involve annotation heterogeneity. Related to STAR Methods: Literature review of studies of lineage-specific genes
Table S2: List of papers published in or later than 2019 and whether they involve annotation heterogeneity. Related to STAR Methods: Literature review of studies of lineage-specific genes
Table S3: Sources, brief descriptions, and links to protein annotations and genome assemblies used in this study. Related to Figures 1–3 and STAR Methods: Data and code availability.
Table S4: Brief description of annotation source, number of genes in the annotation, and the number and percentage of genes in each annotation with no significant homologs found by a BLASTP search in the other annotation for the given species are listed. Related to Figures 1–3 and STAR Methods: Data and code availability. Note that, where large differences in the number of proteins included in a pair of annotations occurs, this is often due in part to one annotation including a larger number of different isoforms of the same locus, all or many of which may have significant similarity to the same protein(s) in the other annotation.
Acknowledgements
This work was primarily funded by a Howard Hughes Medical Institute investigator award to SRE. SRE is also supported in part by NIH (R01-HG009116). AWM is supported by grants from the NIH (RO1-GM43987), and the NSF-Simons Center for the Mathematical and Statistical Analysis of Biology (NSF #1764269, Simons #594596). CMW is supported in part by a NSF-Simons Quantitative Biology PhD Student Fellowship. Computations were done on the Cannon cluster supported by the FAS Division of Science, Research Computing Group at Harvard University.
Footnotes
Declaration of interests
The authors declare no competing interests.
References
- 1.Khalturin K, Hemmrich G, Fraune S, Augustin R, and Bosch TC (2009). More than just orphans: are taxonomically-restricted genes important in evolution? Trends in Genetics 25, 404–413. [DOI] [PubMed] [Google Scholar]
- 2.Tautz D, and Domazet-Lošo T (2011). The evolutionary origin of orphan genes. Nature Reviews Genetics 12, 692. [DOI] [PubMed] [Google Scholar]
- 3.Wilson G, Bertrand N, Patel Y, Hughes J, Feil E, and Field D (2005). Orphans as taxonomically restricted and ecologically important genes. Microbiology 151, 2499–2501. [DOI] [PubMed] [Google Scholar]
- 4.McLysaght A, and Guerzoni D (2015). New genes from non-coding sequence: the role of de novo protein-coding genes in eukaryotic evolutionary innovation. Philosophical Transactions of the Royal Society B: Biological Sciences 370, 20140332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tautz D (2014). The discovery of de novo gene evolution. Perspectives in biology and medicine 57, 149–161. [DOI] [PubMed] [Google Scholar]
- 6.Domazet-Lošo T, Brajković J, and Tautz D (2007). A phylostratigraphy approach to uncover the genomic history of major adaptations in metazoan lineages. Trends in Genetics 23, 533–539. [DOI] [PubMed] [Google Scholar]
- 7.McLysaght A, and Hurst LD (2016). Open questions in the study of de novo genes: what, how and why. Nature Reviews Genetics 17, 567. [DOI] [PubMed] [Google Scholar]
- 8.Basile W, and Elofsson A (2017). The number of orphans in yeast and fly is drastically reduced by using combining searches in both proteomes and genomes. bioRxiv, 185983. [Google Scholar]
- 9.Casola C (2018). From de novo to “de nono”: the majority of novel protein-coding genes identified with phylostratigraphy are old genes or recent duplicates. Genome Biology and Evolution 10, 2906–2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zile K, Dessimoz C, Wurm Y, and Masel J (2020). Only a single taxonomically restricted gene family in the Drosophila melanogaster subgroup can be identified with high confidence. Genome Biology and Evolution. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilson BA, Foy SG, Neme R, and Masel J (2017). Young genes are highly disordered as predicted by the preadaptation hypothesis of de novo gene birth. Nature ecology & evolution 1, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thibaud-Nissen F, DiCuccio M, Hlavina W, Kimchi A, Kitts P, Murphy T, Pruitt K, and Souvorov A (2016). P8008 the NCBI eukaryotic genome annotation pipeline. Journal of Animal Science 94, 184–184. [Google Scholar]
- 13.Howe KL, Achuthan P, Allen J, Allen J, Alvarez-Jarreta J, Amode MR, Armean IM, Azov AG, Bennett R, and Bhai J (2021). Ensembl 2021. Nucleic acids research 49, D884–D891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Drysdale R, and Consortium F (2008). FlyBase. Drosophila, 45–59. [Google Scholar]
- 15.Howe K, Davis P, Paulini M, Tuli MA, Williams G, Yook K, Durbin R, Kersey P, and Sternberg PW (2012). WormBase: annotating many nematode genomes. In 1. (Taylor & Francis; ), pp. 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giraldo-Calderón GI, Emrich SJ, MacCallum RM, Maslen G, Dialynas E, Topalis P, Ho N, Gesing S, Consortium V, and Madey G (2015). VectorBase: an updated bioinformatics resource for invertebrate vectors and other organisms related with human diseases. Nucleic acids research 43, D707–D713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yandell M, and Ence D (2012). A beginner’s guide to eukaryotic genome annotation. Nature Reviews Genetics 13, 329–342. [DOI] [PubMed] [Google Scholar]
- 18.Altschul SF, Gish W, Miller W, Myers EW, and Lipman DJ (1990). Basic local alignment search tool. Journal of molecular biology 215, 403–410. [DOI] [PubMed] [Google Scholar]
- 19.James JE, Willis SM, Nelson PG, Weibel C, Kosinski LJ, and Masel J (2021). Universal and taxon-specific trends in protein sequences as a function of age. eLife 10, e57347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Foy SG, Wilson BA, Bertram J, Cordes MH, and Masel J (2019). A shift in aggregation avoidance strategy marks a long-term direction to protein evolution. Genetics 211, 1345–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Willis S, and Masel J (2018). Gene birth contributes to structural disorder encoded by overlapping genes. Genetics 210, 303–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carvunis A-R, Rolland T, Wapinski I, Calderwood MA, Yildirim MA, Simonis N, Charloteaux B, Hidalgo CA, Barbette J, and Santhanam B (2012). Proto-genes and de novo gene birth. Nature 487, 370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vakirlis N, Hebert AS, Opulente DA, Achaz G, Hittinger CT, Fischer G, Coon JJ, and Lafontaine I (2018). A molecular portrait of de novo genes in yeasts. Molecular Biology and Evolution 35, 631–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weisman CM, Murray AW, and Eddy SR (2020). Many, but not all, lineage-specific genes can be explained by homology detection failure. PLoS biology 18, e3000862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moyers BA, and Zhang J (2016). Evaluating phylostratigraphic evidence for widespread de novo gene birth in genome evolution. Molecular biology and evolution 33, 1245–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Neme R, and Tautz D (2013). Phylogenetic patterns of emergence of new genes support a model of frequent de novo evolution. BMC genomics 14, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Van Oss SB, and Carvunis A-R (2019). De novo gene birth. PLoS genetics 15, e1008160. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: List of papers from a 2019 review27 and whether they involve annotation heterogeneity. Related to STAR Methods: Literature review of studies of lineage-specific genes
Table S2: List of papers published in or later than 2019 and whether they involve annotation heterogeneity. Related to STAR Methods: Literature review of studies of lineage-specific genes
Table S3: Sources, brief descriptions, and links to protein annotations and genome assemblies used in this study. Related to Figures 1–3 and STAR Methods: Data and code availability.
Table S4: Brief description of annotation source, number of genes in the annotation, and the number and percentage of genes in each annotation with no significant homologs found by a BLASTP search in the other annotation for the given species are listed. Related to Figures 1–3 and STAR Methods: Data and code availability. Note that, where large differences in the number of proteins included in a pair of annotations occurs, this is often due in part to one annotation including a larger number of different isoforms of the same locus, all or many of which may have significant similarity to the same protein(s) in the other annotation.
Data Availability Statement
All genome annotations on which these analyses were based are publicly available and are listed in the Key Resources Table and Supplemental Tables 3 and 4. All results summarized in Figures 1–3 are publicly available as of the date of publication at https://github.com/caraweisman/Annotation_homology.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Deposited Data | ||
| Cichlid genome annotations | Broad Institute | ftp://ftp.broadinstitute.org/pub/vgb/cichlids/Annotation/Protein_coding/Peptide_Files/ |
| Cichlid genome annotations | NCBI | GCF_000238955.1, GCF_000239375.1, GCF_000239415.1, GCF_000239395.1, GCF_000188235.2 |
| Primate genome annotations | Ensembl | Release 102, macaca_fascicularis; Release 103, macaca_nemestrina, mandrillus_leucophaeus, rhinopithecus_bieti, cebus_imitator |
| Primate genome annotations | NCBI | GCF_000364345.1, GCF_000956065.1, GCF_000951045.1, GCF_001698545.1, GCF_001604975.1 |
| Rodent genome annotations | Downloaded from Ensembl; generated by Ensemble and UCSC | Release 101, mus_musculus, mus_caroli, mus_pahari, rattus_norwegicus; Release 104, peromyscus_maniculatus |
| Rodent genome annotations | NCBI | GCF_000001635.26, GCF_900094665.1, GCF_900095145.1, GCF_000001895.5, GCF_000500345.1 |
| Bat genome annotations | Downloaded from Ensembl and NCBI; generated by Ensembl, BAT1K consortium, Beijing Genomics Institute | GCA_000412655.1, GCA_014108235.1, GCA_014108415.1, GCA_000325575.1, Ensembl release 103, myotis_lucifugus |
| Bat genome annotations | NCBI | GCF_000147115.1, GCF_000412655.1, GCF_014108235.1, GCF_014108415.1, GCF_000325575.1 |
| Software and Algorithms | ||
| Basic Local Alignment Search Tool (BLAST) | Altschul, Stephen F., et al. “Basic local alignment search tool.” Journal of molecular biology 21 5.3 (1990): 403–410. | Version 6.2.0 |