

Abstract
In vitro genotoxicity testing plays an important role in chemical risk assessment. The human B-lymphoblastoid cell line TK6 is widely used as a standard cell line for regulatory safety evaluations. Like many other mammalian cell lines, TK6 cells have limited metabolic capacity; therefore, usually require a source of exogenous metabolic activation for use in genotoxicity testing. Previously, we developed a set of TK6-derived cell lines that individually express one of fourteen cytochrome P450s (CYPs). In the present study, we surveyed a panel of major Phase II drug-metabolizing enzymes to characterize their baseline expression in TK6 cells. These results may serve as a reference enzymatic profile of this commonly used cell line.
Keywords: Human TK6 cells, genotoxicity testing, biotransformation, Phase II drug-metabolizing enzymes
Introduction
In vitro genotoxicity testing has been successfully used to predict genotoxicity and plays an important role in chemical risk assessment. The human B-lymphoblastoid TK6 cell line was established in 1978 from the parental WI-L2 cells, which are diploid lymphoblast cells derived from a 5-year-old male with hereditary spherocytosis. The relevance of TK6 cells to genetic toxicology stems from their heterozygosity at the thymidine kinase (TK) locus on human chromosome 17 and the presence of the hypoxanthine phosphoribosyl transferase (HPRT) gene on the X chromosome. Due to these specific features, the TK6 cell line was originally used in the TK mutation assay to detect point mutations, deletions, and recombination, and is also well suited for the HPRT gene mutation assay. Both TK and HPRT gene mutation assays have been recommended by international authorities such as the Organization for Economic Co-operation and Development (OECD) and described in the OECD testing guidelines (TG) 476 and 490.1,2 Currently, the human TK6 cell line is widely used as a standard cell line for regulatory safety assessments to conduct TK and HPRT gene mutation assays, chromosome aberration tests (e.g., OECD TG473), micronucleus assays (e.g., OECD TG487), and comet assays.3 In fact, a recent international survey indicated that the human TK6 cells and mouse lymphoma L5178Y cells are the most used cell lines for in vitro genotoxicity testing.4 In addition, TK6 cells not only demonstrate negligible genetic variability to the human reference genome, but also harbor a homozygous wild type p53 gene, making them more physiologically relevant than other cell models used in genotoxicity testing.5 TK6 cells have the potential for high-throughput genotoxicity screening since they readily expand in standard RPMI 1640 cell culture media in suspension.6
However, one drawback of TK6 cells is the lack of metabolic competency, notably low levels of Phase I drug-metabolizing enzymes (CYPs), which can impact the results of genotoxicity testing, particularly for detection of drugs/chemicals that must go through biotransformation to a DNA reactive metabolite.7,8 To overcome the deficiencies of low biotransformation capacity, we developed a panel of TK6-derived cell lines that individually express one of each of fourteen human CYPs (CYP1A1, 1A2, 1B1, 2A6, 2B6, 2C8, 2C18, 2C9, 2C19, 2D6, 2E1, 3A4, 3A5, and 3A7) and demonstrated that this cell system is able to identify DNA damage and micronucleus formation induced by the bioactivation of prototypical genotoxicants and pyrrolizidine alkaloids, without addition of rat liver S9 fraction.7,9
On the other hand, the important role of Phase II enzymes in metabolism cannot be overlooked. For example, it is well-documented that sulfotransferases (SULT) and N-acetyltransferase (NAT) are required for the bioactivation of heterocyclic amines.10 In our recent study, we surprisingly found that TK6 cells can biotransform luteolin, a flavonoid, to a less toxic metabolite, diosmetin, in a time-dependent manner without addition of any exogenous enzyme systems.11 This O-methylation reaction is likely catalyzed by a Phase II enzyme – catechol O-methyltransferase, suggesting that TK6 cells may not be completely “incompetent” in exogenous drug/chemical metabolism. However, no study has systematically investigated the Phase II enzymatic profile of TK6 cells. Therefore, to fill this knowledge gap, the current short communication surveyed the gene and protein expression profiles of a panel of major Phase II drug-metabolizing enzymes in TK6 cells. Primary human hepatocytes (PHHs), which are considered the in vitro gold-standard for liver drug metabolism enzyme expression and activity,12 and the human hepatoma-derived cell line HepG2, which lacks drug-metabolizing activities,13 were included for comparison to the TK6 cell line. This study characterized the baseline expression of Phase II drug metabolizing enzymes in TK6 cells and can serve as a reference for the Phase II enzyme expression profile of this commonly used cell line.
Materials and methods
Cell culture
The human lymphoblastoid TK6 cells were purchased from ATCC (Manassas, VA) and cultured in RPMI 1640 medium supplemented with 0.5% L-glutamine (Gibco, Gaithersburg, MD), 10% fetal bovine serum (FBS), 100 U/mL penicillin (Gibco), and 100 μg/mL streptomycin (Gibco). Cells were routinely maintained at a density of 2 × 105 to 1.5 × 106 cells/mL.
The HepG2 cell line was purchased from ATCC (Manassas, VA) and were cultured in Williams’ Medium E complete media (Sigma-Aldrich, St. Louis, MO) containing 10% FBS and antibiotics (100 units/mL penicillin, 100 μg/mL streptomycin, and 0.25 μg/mL amphotericin B). Unless otherwise specified, cells were maintained below 80% confluency and passaged as needed.
Cryopreserved PHHs pooled from 10 donors were purchased from In Vitro ADMET Laboratories (Columbia, MD). Cell culture dishes were pre-coated with PureColV® (Advanced BioMatrix, Carlsbad, CA) for PHHs. Cells were thawed and maintained in Universal Primary Cell Plating Medium (UPCM™) provided by the supplier. Cells were seeded in 60 mm-tissue culture dishes as previously described before RNA or protein isolation.14
Real-time PCR array
RNA was extracted using the RNeasy Mini kit (Qiagen, Valencia, CA). The quantity and purity of RNA were measured with a NanoDrop 8000 spectrophotometer (Thermo Fisher Scientific, Wilmington, DE). cDNA was synthesized from 2 μg RNA using the High-Capacity cDNA Reverse Transcription kit (Applied Biosystems, Foster City, CA) according to the manufacturer’s instructions. The Drug Metabolism Phase II Enzymes RT2 Profiler PCR Array (Qiagen, Valencia, CA) was used to determine mRNA levels. The qPCR reactions were performed in a 25-μL volume using SsoAdvanced Universal SYBR Green Supermix (Bio-Rad Laboratories, Hercules, CA) on a ViiA 7 Real-Time PCR System (Applied Biosystems). The geometric mean of a panel of five reference genes (ACTB, B2M, GAPDH, HPRT1, and RPLP0) were used for normalization. The Ct values for all tested genes are shown in Supplementary Table 1 and a gene was considered non-detectable when a Ct value > 35. Expression value implied the relative mRNA expression abundance of a gene, arbitrarily assuming the averaged expression level of the five reference genes being 10,000 copies. The Expression value of each gene was defined using the equation: E = 2−(Ct of test gene−averaged Ct of reference genes) × 10,000. The data were expressed as the mean and standard deviation (SD) values of three independent samples for each cell type. To examine whether the gene expression levels are significantly different from each cell line, two-way ANOVA followed by Dunnett’s multiple comparison test was performed using GraphPad Prism version 6.07 (GraphPad Software, La Jolla, CA).
Western blot
Two to five million cells were used for protein extraction. Whole-cell lysates were prepared using RIPA buffer containing Halt Protease Inhibitor Cocktail (Thermo Fisher Scientific). For the detection of Uridine 5′-diphospho-glucuronosyltransferase (UGTs) in TK6 and HepG2 cells, microsomes were isolated following the method described previously.15 Briefly, 5 × 107 cells were homogenized in lysis buffer by TissueLyser II (Qiagen). After centrifuging at 10,000 × g to remove cell debris, the supernatant was further centrifuged at 100,000 × g for 45 min to separate microsome. The protein concentrations of the whole cell lysate or microsome samples were determined using a Pierce BCA Protein Assay kit (Thermo Fisher Scientific). Standard Western blots protocols, SDS-PAGE, transfer, and antibody staining were performed.16 Primary antibodies were selected against COMT1 (ab129504, Abcam, Cambridge, MA), GSTP1 (3369S, Cell Signaling Technology, Danvers, MA), MGST1 (MA5–34942, Thermo Fisher Scientific), NQO1 (62262S, Cell Signaling Technology), SULT1A1 (ab155012, Abcam), UGT1A1 (ab170858), UGT2B7 (ab126269), and GAPDH (5174S, Cell Signaling Technology) and diluted in blocking reagents at 1:1000. GAPDH was used as the internal control. The protein signals were visualized with a FluorChem E System and quantified with AlphaView software (ProteinSimple, San Jose, CA).
Results and discussion
The gene expression of 84 Phase II drug-metabolizing enzymes in TK6 cells, HepG2 cells, and PHHs were profiled, and the summarized results are shown in Table 1 and Supplementary Table 2. The relative mRNA expression value of each gene was calculated by assuming the averaged expression level of the five reference genes (ACTB, B2M, GAPDH, HPRT1, and RPLP0) as being 10,000 copies. Representative data for Western blot analysis of several key Phase II drug-metabolizing enzymes expression in TK6 cells, HepG2 cells, and PHHs is shown in Figure 1.
Table 1.
Gene expression valuea of 84 Phase II drug-metabolizing enzymes in TK6 cells, HepG2 cells, and primary human hepatocytes (PHHs).
| Gene | TK6 | HepG2 | PHHs | Gene | TK6 | HepG2 | PHHs |
|---|---|---|---|---|---|---|---|
| AANAT | 1.1 | 1.3 | 1.1 | MGAT1 | 213.9 | 615.9b | 170.2 |
| ACSL1 | 161.3 | 14.5b | 1195.3b | MGAT2 | 56.5 | 45.9 | 132.1 |
| ACSL3 | 317.5 | 1844.7b | 1290.0b | MGST1 | 742.8 | 1602.8 b | 12881.9 b |
| ACSL4 | 155.3 | 1781.6b | 419.6b | MGST2 | 6.4 | 737.9b | 153.4b |
| ACSM1 | 0.9 | 1.4 | 2.7 | MGST3 | 75.3 | 201.2b | 309.1b |
| ACSM2B | 0.7 | 1.9 | 78.4 | NAA20 | 43.7 | 120.2 | 65.7 |
| ACSM3 | 38.1 | 32.1 | 119.7 | NAT1 | 5.8 | 3.0 | 4.9 |
| AGXT | 0.3 | 136.2b | 1025.9b | NAT2 | 29.2 | 25.3 | 319.4b |
| AS3MT | 0.6 | 62.8 | 11.0 | NNMT | 0.3 | 0.2 | 11.7 |
| ASMT | 0.4 | 0.6 | 0.8 | NQO1 | 344.2 | 9128.6 b | 133.0 b |
| BAAT | 0.3 | 3.3 | 1742.6b | NQO2 | 16.1 | 258.2b | 418.4b |
| CCBL1 | 14.8 | 66.4 | 10.8 | PNMT | 0.5 | 0.7 | 0.4 |
| CES1 | 0.4 | 57.4 | 7330.7b | POMGNT1 | 36.2 | 129.7 | 98.8 |
| CES2 | 64.2 | 362.7b | 594.0b | PTGES | 1.3 | 0.6 | 2.4 |
| CES3 | 10.6 | 49.3 | 26.3 | SAT1 | 133.5 | 1960.6b | 12427.8b |
| CES5A | 10.1 | 0.3 | 5.8 | SULT1A1 | 0.5 | 568.6 b | 66.1 |
| CHST7 | N.D. | N.D. | N.D. | SULT1A2 | 1.9 | 882.3b | 137.7b |
| COMT | 256.1 | 1148.9 b | 631.0 b | SULT1B1 | 0.4 | 1.1 | 40.0 |
| DDOST | 434.8 | 3524.1b | 454.2 | SULT1C2 | 0.6 | 68.6 | 8.8 |
| EPHX1 | 4.9 | 534.1b | 3830.4b | SULT1C3 | 0.5 | 0.5 | 0.8 |
| EPHX2 | 0.3 | 185.6b | 195.6b | SULT1C4 | 0.6 | 31.6 | 2.8 |
| GALNT1 | 301.7 | 392.9 | 464.5b | SULT1E1 | 0.3 | 3.2 | 18.9 |
| GALNT4 | 2.6 | 67.5 | 120.1b | SULT2A1 | 0.4 | 503.6b | 1290.3b |
| GAMT | 126.7 | 533.4b | 385.3b | SULT2B1 | 0.7 | 2.6 | 0.3 |
| GCNT1 | 0.7 | 6.3 | 7.3 | SULT4A1 | 0.5 | 0.4 | 1.4 |
| GLYAT | 0.3 | 0.2 | 317.0b | SULT6B1 | 1.4 | 1.0 | 0.7 |
| GNMT | 4.6 | 25.9 | 377.5b | TPMT | 3.9 | 36.1 | 60.7 |
| GSTA1 | 0.6 | 10.4 | 1732.4b | TST | 40.3 | 275.7b | 357.9b |
| GSTA3 | 0.5 | 0.7 | 18.2 | UGCG | 113.3 | 104.2 | 476.6b |
| GSTA4 | 1.1 | 47.1 | 12.0 | UGT1A1 | 0.8 | 13.9 | 2364.1 b |
| GSTA5 | 1.0 | 0.8 | 0.8 | UGT1A4 | 0.5 | 0.7 | 846.2b |
| GSTK1 | 197.3 | 337.9b | 600.9b | UGT1A9 | 1.1 | 1.4 | 496.9b |
| GSTM2 | 0.8 | 12.6 | 3.3 | UGT2A1 | 0.6 | 0.4 | 0.4 |
| GSTM3 | 0.2 | 39.7 | 31.0 | UGT2A3 | 2.3 | 794.0b | 203.0b |
| GSTM4 | 17.1 | 78.8 | 66.8 | UGT2B10 | 0.5 | 301.2b | 910.9b |
| GSTM5 | 0.2 | 0.1 | 0.3 | UGT2B17 | 41.0 | 5.7 | 490.7b |
| GSTO1 | 254.4 | 2034.3b | 2100.3b | UGT2B28 | 8.0 | 327.4b | 1163.4b |
| GSTO2 | 0.6 | 22.3 | 20.6 | UGT2B4 | 0.9 | 2.3 | 5442.2b |
| GSTP1 | 1481.4 | 4.1 b | 14.1 b | UGT2B7 | 14.5 | 1014.5 b | 4313.3 b |
| GSTT1 | 4.3 | 19.8 | 9.2 | UGT3A1 | 1.2 | 1.1 | 105.9 |
| HNMT | 0.6 | 5.1 | 4.8 | UGT8 | 0.7 | 0.4 | 0.5 |
| INMT | 0.5 | 0.3 | 0.3 | XDH | 0.6 | 0.5 | 170.5b |
N.D.: not detectable.
Genes in bold font were selected for Western blot analysis.
The data were expressed as the mean values of three independent samples for each cell line.
Indicates the expression level is statistically different compared to TK6 cells.
Figure 1.
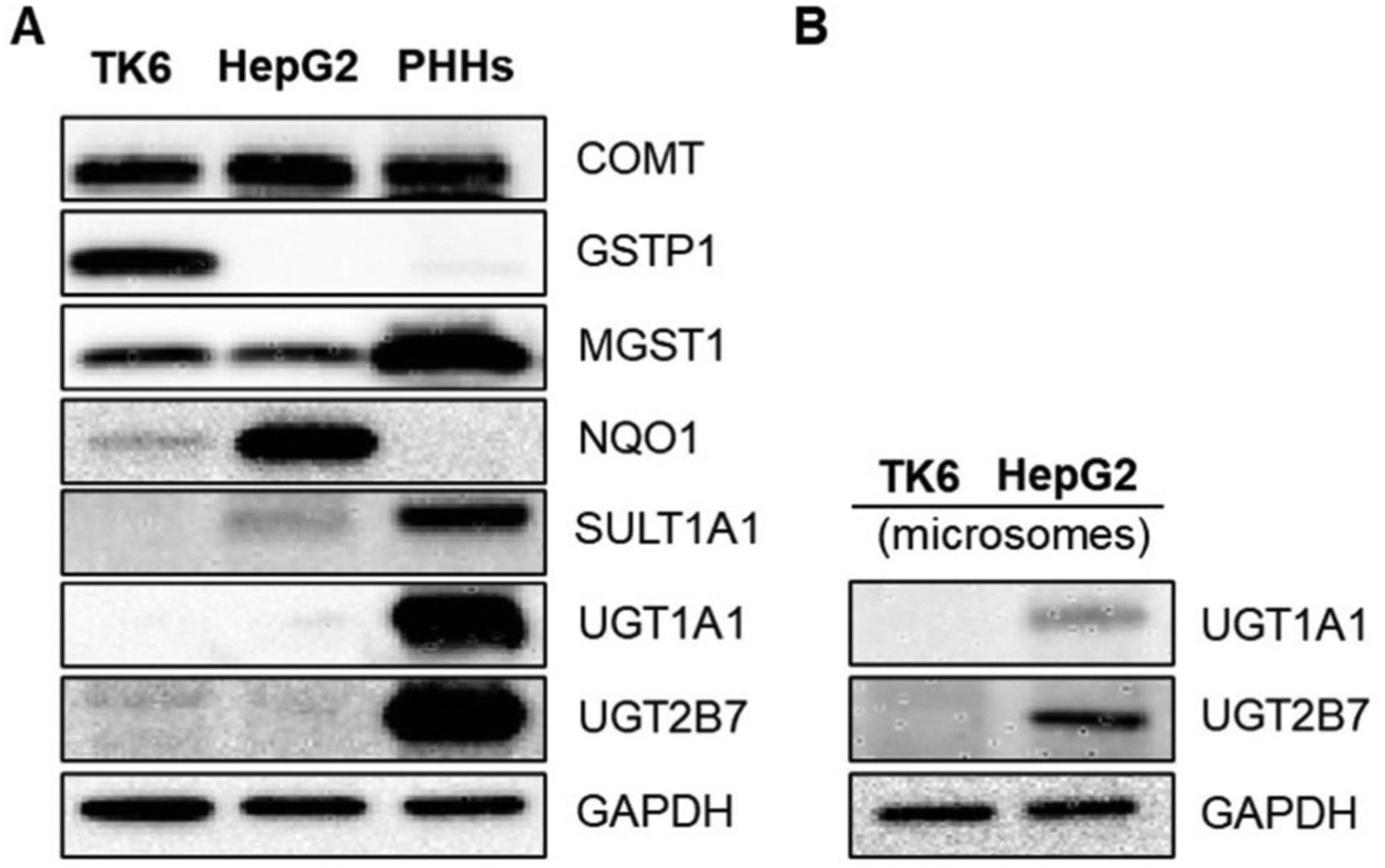
The protein levels of several key Phase II drug-metabolizing enzymes in TK6 cells, HepG2 cells, and PHHs. Representative Western blot images are shown from one experiment and similar results were obtained from three independent experiments. A total of 20-μg protein samples from whole-cell lysates (A) or microsomes (B) were loaded for each lane.
Sulfotransferases (SULTs)
SULTs are major Phase II enzymes that catalyze the sulfate conjugation of many drugs and xenobiotics. Generally, sulfonation is recognized as a detoxification process. For instance, SULT1A1 is involved in the detoxification of genotoxicants such as acrolein.17 However, it is well-documented that some SULT isoforms, particularly SULT1A1 and 1A2, play critical roles in bioactivating hydroxymethyl polyaromatic hydrocarbons, heteroaromatic amines, and 2-acetylaminofluorene, leading to the formation of DNA-damaging sulfuric acid esters that can cause mutagenicity or carcinogenicity.18,19 As shown in Table 1, TK6 cells have negligible expression of all 11 SULTs investigated. HepG2 cells and PHHs, on the other hand, had appreciable basal expression levels of SULT1A1, 1A2, and 2A1. Western blot assay also demonstrated that no SULT1A1 protein was found in TK6 cells, while the presence of SULT1A1 protein in HepG2 cells was confirmed (Figure 1A). As expected, PHHs express high level of SULT1A1 protein. These results suggest uninduced TK6 cells are devoid of SULTs.
Uridine 5’-diphospho-glucuronosyltransferase (UGTs)
Glucuronidation catalyzed by UGTs is another major Phase II drug metabolic reaction. UGTs are mainly involved in the detoxification of genotoxicants by conjugating potential DNA reactive intermediates. For example, multiple studies have shown that impaired expression of UGTs, particularly UGT1A isoforms, increased the genotoxicity of benzo(a)pyrene.20,21 The proposed mechanism is that the formation of glucuronide conjugate of benzo(a)pyrene-diol reduces the production of benzo(a)pyrene diol epoxide, the ultimate mutagenic carcinogen that covalently binds to DNA and forms DNA adducts. UGT1A9 also seemed to reduce the mutagenicity of N-hydroxy-2-acetylaminofluorene in the Ames test, although the exact mechanism remains unknown.22 In contrast, one study showed that UGTs bioactivated several carboxylic acid compounds, forming acyl glucuronides that cause DNA damage (measured with the comet assay) in mouse hepatocytes.23 Therefore, whether UGTs reduce or increase the genotoxicity of a compound depends on the structure of chemicals. In the current study, we found TK6 cells had negligible gene expression levels of selected UGT1A isoenzymes (Table 1). UGT2B17 is expressed in TK6 cells whose function is associated with the metabolism of steroid hormones, bilirubin, and some flavonoids.24 Recently, a study suggested that UGT2B17 is the main lymphoid glucuronosyltransferase in human lymphoid cells and its expression may lead to the resistance to anti-cancer drugs in leukemic cells.25 In liver cells, as expected, PHHs had high expression levels of major hepatic UGTs including UGT1A1, 1A4, 1A9, 2B4, 2B7, 2B10, 2B17, and 2B28 (Table 1). Western blotting also showed abundant protein expression of UGT1A1 and 2B7 in PHHs (Figure 1A). As is well documented in the literature, HepG2 cells had a distinct UGT expression profile when compared to PHHs. More specifically, the mRNA levels of UGT1A1, 1A4, 1A9, 2B4, and 2B17 were much lower in HepG2 cells, confirming that without genetic modification this cell line may not be a suitable model for metabolism studies.13 Previous studies showed that HepG2 may have detectable level of UGT1A1 in microsomes.15 Therefore, we performed Western blot using microsomal fractions from TK6 and HepG2 cells. The results showed that UGT1A1 was not expressed in TK6 cells and lowly expressed in HepG2 cells (Figure 1B).
Glutathione S-transferases (GSTs)
Several classes of GSTs including alpha (GSTA), kappa (GSTK), mu (GSTM), omega (GSTO), pi (GSTP), and theta (GSTT), as well as membrane-bound microsomal GSTs (MGST) levels were investigated in TK6 cells. We found that TK6 cells had low or negligible gene expression of GSTA, GSTM, and GSTT. Interestingly, TK6 cells had a very high mRNA level of GSTP1, and the expression level was significantly higher than that of HepG2 cells or PHHs (Table 1). Western blot analysis also showed high protein expression of GSTP1 in TK6 cells, whereas no GSTP1 protein was detected in HepG2 cells and PHHs (Figure 1A). Consistent with our results, a previous study showed that human lymphoid lines (e.g., T-lymphoblast cell lines) had relatively higher GSTP1 gene expression and enzymatic activity levels.26 This finding may have an important implication in genotoxicity testing using TK6 cells. A role for GSTP has long been suggested in chemical carcinogenesis. GSTP catalyzes the detoxification of electrophilic diol epoxides produced by the bioactivation of polyaromatic hydrocarbons (PAHs) such as benzo(a)pyrene.27 Upon benzo(a)pyrene exposure, Gstp knockout mice had more than 8-fold increased numbers of lung adenomas when compared to wild-type mice. Considering that TK6 cells exhibit a high basal level of GSTP expression, they may be less sensitive to the genotoxicity of PAHs when compared to other cells with low GSTP expression. In addition, MGST1 is also highly expressed in TK6 cells. Western blot confirmed the expression of MGST1 protein in both TK6 and HepG2 cells, although the levels were lower compared to PHHs (Figure 1A). MGST1 is mainly localized to the endoplasmic reticulum and in the outer membrane of mitochondria; and is considered an antioxidant enzyme. Studies have shown that MGST1 decreased the toxicity of DNA alkylating cytostatic agents, such as cisplatin and melphalan, as well as silica nanoparticles, by reducing intracellular oxidative stress.28,29 TK6 cells also expressed detectable levels of GSTO1 and GSTK1; however, the role of these two GST enzymes in genotoxicity remains unclear.
Catechol-O-methyltransferase (COMT)
In a previous study, we demonstrated that TK6 cells without any exogenous enzyme system were able to methylate luteolin into diosmetin. Diosmetin showed lower genotoxicity and cytotoxicity compared to its parental form luteolin.11 After a 24-h exposure, more than 90% of luteolin in the medium was converted to diosmetin measured by liquid chromatography with tandem mass spectrometry (LC-MS/MS). This observation made us question whether some traditionally recognized “metabolically incompetent” cell lines may possess exogenous drug/chemical metabolizing enzymes. As shown in Table 1 and Figure 1A, TK6 cells had a basal level expression of COMT at both the mRNA and protein levels, further confirming our postulation in the previous study that COMT present in TK6 cells accounted for the biotransformation of luteolin. HepG2 cells and PHHs also showed high expression of COMT in both mRNA and protein levels, which is consistent with previous findings.13,30 The implication of COMT in genotoxicity assessment is mainly associated with its O-methylation reaction in flavonoids. As seen in the case of quercetin, one of the most well-studied flavonoids, COMT is likely to explain the in vitro and in vivo discrepancy in its carcinogenic effects. Quercetin is highly mutagenic in vitro, but not in animals. COMT was proposed to rapidly methylate quercetin and provides sufficient inactivation in vivo.31 Since such a detoxification process is missing or less efficient in the Ames test, quercetin constantly produces positive results with or without the rat liver S9 fraction. Mammalian cell models such as TK6 cells and HepG2 cells constitutively express COMT, and thus may serve as complementary in vitro tools to the Ames test for studying the genotoxicity of flavonoids.
NAD(P)H quinone dehydrogenases (NQOs)
The NQO gene family consists of two members, NQO1 and NQO2. Both of them encode cytosolic flavoenzymes that catalyze the reduction of quinone to hydroquinone, which is considered an antioxidation and detoxification process.32 For example, NQO1 deficiency leads to increased genotoxicity (as measured by the micronucleus assay) of benzene in mice.33 However, such a reduction is not always protective in cells. It is well documented that NQO1 catalyzes the bioreductive activation of mitomycin C, generating leucomitomycin C that causes DNA interstrand crosslinking.34 We found the mRNA of NQO1 was highly expressed in TK6 cells – the expression value ranked the 3rd highest among the 84 Phase II enzymes investigated (Table 1). NQO1 protein was also identified in TK6 cells, although the band intensity was weak due to an extremely high level of NQO1 expressed in HepG2 cells (Figure 1A). Notably, the gene expression level of NQO1 in HepG2 cells was significantly higher than that in TK6 cells and PHHs, respectively (Table 1). PHHs had no detectable protein level of NQO1, and the trend was consistent with the gene expression level (Figure 1A). Since many genotoxicants, especially dietary supplements, exert their DNA-damaging effects via oxidative stress, HepG2 cells may be less sensitive in detecting the genotoxicity of these agents due to a relatively high level of NQO1.
Spermidine/spermine N1-acetyltransferase 1 (SAT1)
SAT1 is the rate-limiting enzyme for polyamine catabolism. The level of polyamines often increases in tumor cells, stimulating their growth and proliferation. Recent studies have demonstrated that a reduced level of polyamines may increase the susceptibility of cells in response to many double-strand break (DSB)-inducing agents, including ionizing radiation, ultraviolet, and etoposide.35 The underlying mechanism has been associated with the role of polyamine in stimulating homologous recombination mediated DSB repair by enhancing the DNA strand exchange activity of RAD51. SAT1 governs the transportation of polyamines in cells. The over-expression of SAT1 leads to excessive export of polyamines, which may further impair homologous recombination and sensitize cells to genotoxic stresses. Although TK6 cells expressed SAT1 mRNA, the level was significantly lower than that in HepG2 cells or PHHs (Table 1). The impact of this difference on sensitivity of cells to various DNA DSB-inducing agents warrants further investigation.
Other phase II enzymes
When arbitrarily using an expression value >100 as the cutoff (over 1% of the reference genes), another seven genes, including ACSL1, ACSL3, and ACSL4, DDOST, GAMT, MGAT1, and UGCG were considered to have detectable expression in TK6 cells with appreciable levels. These genes mainly encode proteins involved in the Phase II metabolism of carbohydrates and fatty acids. Their association with genotoxicity remains to be elucidated.
Conclusion
Although TK6 cells are largely devoid of major Phase I drug-metabolizing enzymes and Phase II SULTs and UGTs, they express several key genotoxicity-relevant Phase II enzymes, such as COMT, GSTP1, MGST1, and NQO1. Since the TK6 cell line has served as, and will continue to serve as, one of the most important cell models for genotoxicity screening, the current study provides a reference for the TK6 Phase II enzyme expression profile. These results provide a better understanding of this cell line and assist with the interpretation of genotoxicity testing results. Considering some Phase II enzyme activities in TK6 cells are restricted, it is necessary to be careful when evaluating the compounds that require biotransformation to DNA reactive metabolites. Based on the goal of future studies, the measurement of selected Phase II enzyme activities in TK6 cells and comparison of these activities with those in human peripheral lymphocytes or primary human hepatocytes are warranted.
Supplementary Material
Acknowledgments
This manuscript reflects the views of the authors and does not necessarily reflect those of the U.S. FDA. We thank Drs. Dayton M. Petibone and Baitang Ning for their critical review of this manuscript.
Funding
XL and YL were supported by an appointment to the Postgraduate Research Program at the National Center for Toxicological Research (NCTR), and KGN was supported by an appointment to the Summer Student Research Program; both programs are administered by the Oak Ridge Institute for Science Education through an interagency agreement between the U.S. Department of Energy and the U.S. Food and Drug Administration (FDA).
References
- [1].OECD. In vitro mammalian cell gene mutation test, OECD guideline for testing of chemicals, No. 476. 1997. https://www.oecd-ilibrary.org/environment/test-no-476-in-vitro-mammalian-cell-gene-mutation-test_9789264071322-en. (Accessed 19 February 2022). [Google Scholar]
- [2].OECD. In vitro mammalian cell gene mutation tests using the thymidine kinase gene, OECD guideline for testing of chemicals, No. 490. 2015. http://www.oecd-ili-brary.org/environment/test-no-490-in-vitro-mammalian-cell-gene-mutation-tests-using-the-thymidine-kinase-gene_9789264242241-en. (Accessed 19 February 2022). [Google Scholar]
- [3].Muruzabal D, Collins A, Azqueta A. The enzyme-modified comet assay: Past, present and future. Food Chem Toxicol. 2021;147:111865. doi: 10.1016/j.fct.2020.111865. [DOI] [PubMed] [Google Scholar]
- [4].Lorge E, Moore MM, Clements J, et al. Standardized cell sources and recommendations for good cell culture practices in genotoxicity testing. Mutat Res Genet Toxicol Environ Mutagen. 2016;809:1–15. doi: 10.1016/j.mrgentox.2016.08.001. [DOI] [PubMed] [Google Scholar]
- [5].Whitwell J, Smith R, Jenner K, et al. Relationships between p53 status, apoptosis and induction of micronuclei in different human and mouse cell lines in vitro: implications for improving existing assays. Mutat Res Genet Toxicol Environ Mutagen. 2015;789–790:7–27. doi: 10.1016/j.mrgentox.2015.05.011. [DOI] [PubMed] [Google Scholar]
- [6].Hsieh JH, Smith-Roe SL, Huang R, et al. Identifying compounds with genotoxicity potential using Tox21 high-throughput screening assays. Chem Res Toxicol. 2019; 32(7):1384–1401. doi: 10.1021/acs.chemrestox.9b00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Li X, Chen S, Guo X, et al. Development and application of TK6-derived cells expressing human cytochrome p450s for genotoxicity testing. Toxicol Sci. 2020; 175(2):251–265. doi: 10.1093/toxsci/kfaa035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Shah UK, Seager AL, Fowler P, et al. A comparison of the genotoxicity of benzo[a]pyrene in four cell lines with differing metabolic capacity. Mutat Res Genet Toxicol Environ Mutagen. 2016;808:8–19. doi: 10.1016/j.mrgentox.2016.06.009. [DOI] [PubMed] [Google Scholar]
- [9].Li X, He X, Chen S, et al. Evaluation of pyrrolizidine alkaloid-induced genotoxicity using metabolically competent TK6 cell lines. Food Chem Toxicol. 2020; 145:111662. doi: 10.1016/j.fct.2020.111662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Chevereau M, Glatt H, Zalko D, Cravedi JP, Audebert M. Role of human sulfotransferase 1A1 and N-acetyltransferase 2 in the metabolic activation of 16 heterocyclic amines and related heterocyclics to genotoxicants in recombinant V79 cells. Arch Toxicol. 2017;91(9):3175–3184. doi: 10.1007/s00204-017-1935-8. [DOI] [PubMed] [Google Scholar]
- [11].Li X, He X, Chen S, et al. The genotoxicity potential of luteolin is enhanced by CYP1A1 and CYP1A2 in human lymphoblastoid TK6 cells. Toxicol Lett. 2021;344: 58–68. doi: 10.1016/j.toxlet.2021.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Guo X, Seo JE, Li X, Mei N. Genetic toxicity assessment using liver cell models: past, present, and future. J Toxicol Environ Health B Crit Rev. 2020;23(1):27–50. doi: 10.1080/10937404.2019.1692744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Guo L, Dial S, Shi L, et al. Similarities and differences in the expression of drug-metabolizing enzymes between human hepatic cell lines and primary human hepatocytes. Drug Metab Dispos. 2011;39(3):528–538. doi: 10.1124/dmd.110.035873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Chen S, Wu Q, Li X, et al. Characterization of cytochrome P450s (CYP)-overexpressing HepG2 cells for assessing drug and chemical-induced liver toxicity. J Environ Sci Health C Toxicol Carcinog. 2021;39(1):68–86. doi: 10.1080/26896583.2021.1880242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Yueh MF, Bonzo JA, Tukey RH. The role of Ah receptor in induction of human UDP-glucuronosyltransferase 1A1. Methods Enzymol. 2005;400:75–91. doi: 10.1016/S0076-6879(05)00005-4. [DOI] [PubMed] [Google Scholar]
- [16].Mahmood T, Yang PC. Western blot: technique, theory, and trouble shooting. N Am J Med Sci. 2012;4(9):429–434. doi: 10.4103/1947-2714.100998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Oda Y. Development and progress for three decades in UMU test systems. Genes Environ. 2016;38:24. doi: 10.1186/s41021-016-0054-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Oda Y, Zhang Y, Buchinger S, Reifferscheid G, Yang M. Roles of human sulfotransferases in genotoxicity of carcinogens using genetically engineered UMU test strains. Environ Mol Mutagen. 2012;53(2):152–164. doi: 10.1002/em.20696. [DOI] [PubMed] [Google Scholar]
- [19].Bendadani C, Meinl W, Monien B, et al. Determination of sulfotransferase forms involved in the metabolic activation of the genotoxicant 1-hydroxymethylpyrene using bacterially expressed enzymes and genetically modified mouse models. Chem Res Toxicol. 2014;27(6):1060–1069. doi: 10.1021/tx500129g. [DOI] [PubMed] [Google Scholar]
- [20].Kalthoff S, Landerer S, Reich J, Strassburg CP. Protective effects of coffee against oxidative stress induced by the tobacco carcinogen benzo[α]pyrene. Free Radic Biol Med. 2017;108:66–76. doi: 10.1016/j.freeradbiomed.2017.03.006. [DOI] [PubMed] [Google Scholar]
- [21].Hu Z, Wells PG. Human interindividual variation in lymphocyte UDP-glucuronosyltransferases as a determinant of in vitro benzo[a]pyrene covalent binding and cytotoxicity. Toxicol Sci. 2004;78(1):32–40. doi: 10.1093/toxsci/kfh010. [DOI] [PubMed] [Google Scholar]
- [22].Yueh MF, Nguyen N, Famourzadeh M, et al. The contribution of UDP-glucuronosyltransferase 1A9 on CYP1A2-mediated genotoxicity by aromatic and heterocyclic amines. Carcinogenesis. 2001;22(6):943–950. doi: 10.1093/carcin/22.6.943. [DOI] [PubMed] [Google Scholar]
- [23].Sallustio BC, DeGraaf YC, Weekley JS, Burcham PC. Bioactivation of carboxylic acid compounds by UDP-glucuronosyltransferases to DNA-damaging intermediates: role of glycoxidation and oxidative stress in genotoxicity. Chem Res Toxicol. 2006;19(5):683–691. doi: 10.1021/tx060022k. [DOI] [PubMed] [Google Scholar]
- [24].Meech R, Hu DG, McKinnon RA, et al. The UDP-glycosyltransferase (UGT) superfamily: new members, new functions, and novel paradigms. Physiol Rev. 2019;99(2): 1153–1222. doi: 10.1152/physrev.00058.2017. [DOI] [PubMed] [Google Scholar]
- [25].Allain EP, Rouleau M, Vanura K, et al. UGT2B17 modifies drug response in chronic lymphocytic leukaemia. Br J Cancer. 2020;123(2):240–251. doi: 10.1038/s41416-020-0887-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wang L, Groves MJ, Hepburn MD, Bowen DT. Glutathione S-transferase enzyme expression in hematopoietic cell lines implies a differential protective role for T1 and A1 isoenzymes in erythroid and for M1 in lymphoid lineages. Haematologica. 2000;85(6):573–579. [PubMed] [Google Scholar]
- [27].Ritchie KJ, Henderson CJ, Wang XJ, et al. Glutathione transferase pi plays a critical role in the development of lung carcinogenesis following exposure to tobacco-related carcinogens and urethane. Cancer Res. 2007;67(19):9248–9257. doi: 10.1158/0008-5472.CAN-07-1764. [DOI] [PubMed] [Google Scholar]
- [28].Johansson K, Åhlen K, Rinaldi R, Sahlander K, Siritantikorn A, Morgenstern R. Microsomal glutathione transferase 1 in anticancer drug resistance. Carcinogenesis. 2007;28(2):465–470. doi: 10.1093/carcin/bgl148. [DOI] [PubMed] [Google Scholar]
- [29].Shi J, Karlsson HL, Johansson K, et al. Microsomal glutathione transferase 1 protects against toxicity induced by silica nanoparticles but not by zinc oxide nanoparticles. ACS Nano. 2012;6(3):1925–1938. doi: 10.1021/nn2021056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Zhu BT, Wang P, Nagai M, Wen Y, Bai HW. Inhibition of human catechol-O-methyltransferase (COMT)-mediated O-methylation of catechol estrogens by major polyphenolic components present in coffee. J Steroid Biochem Mol Biol. 2009;113(1–2):65–74. doi: 10.1016/j.jsbmb.2008.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Zhu BT, Ezell EL, Liehr JG. Catechol-O-methyltransferase-catalyzed rapid O-methylation of mutagenic flavonoids: metabolic inactivation as a possible reason for their lack of carcinogenicity in vivo. J Biol Chem. 1994;269(1):292–299. doi: 10.1016/S0021-9258(17)42348-9. [DOI] [PubMed] [Google Scholar]
- [32].Vasiliou V, Ross D, Nebert DW. Update of the NAD(P)H:quinone oxidoreductase (NQO) gene family. Hum Genomics. 2006;2(5):329–335. doi: 10.1186/1479-7364-2-5-329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Bauer AK, Faiola B, Abernethy DJ, et al. Genetic susceptibility to benzene-induced toxicity: role of NADPH: quinone oxidoreductase-1. Cancer Res. 2003;63(5):929–935. [PubMed] [Google Scholar]
- [34].Siegel D, Yan C, Ross D. NAD(P)H:quinone oxidoreductase 1 (NQO1) in the sensitivity and resistance to antitumor quinones. Biochem Pharmacol. 2012;83(8): 1033–1040. doi: 10.1016/j.bcp.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lee C-Y, Su G-C, Huang W-Y, et al. Promotion of homology-directed DNA repair by polyamines. Nat Commun. 2019;10(1):65. doi: 10.1038/s41467-018-08011-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.