

Abstract
The global crisis of untreatable microbial infections necessitates the design of new antibiotics. Drug repurposing is a promising strategy for expanding the antibiotic repertoire. In this study, we repurpose the clinically-approved anticancer agent cisplatin into a targeted antibiotic by conjugating its Pt(IV) prodrug to enterobactin (Ent), a triscatecholate siderophore employed by Enterobacteriaceae for iron (Fe) acquisition. The l-Ent-Pt(IV) conjugate (l-EP) exhibits antibacterial activity against Escherichia coli K12 and the uropathogenic isolate E. coli CFT073. Similar to cisplatin, l-EP causes filamentous morphology in E. coli and initiates lysis in lysogenic bacteria. Studies with E. coli mutants defective in Ent transport proteins show that Ent mediates the delivery of l-EP into the E. coli cytoplasm, where reduction of the Pt(IV) prodrug releases the cisplatin warhead, causing growth inhibition and filamentation of E. coli. Substitution of Ent with its enantiomer affords the d-Ent-Pt(IV) conjugate (d-EP), which displays enhanced antibacterial activity, presumably because d-Ent cannot be hydrolyzed by Ent esterases and thus iron cannot be released from this conjugate. E. coli treated with l/d-EP accumulate ≥10-fold more Pt as compared to cisplatin treatment. By contrast, human embryonic kidney cells (HEK293T) cells accumulate cisplatin but show negligible Pt uptake after treatment with either conjugate. Overall, this work demonstrates that attachment of a siderophore repurposes a Pt anticancer agent into a targeted antibiotic that is recognized and transported by siderophore uptake machinery, providing a design strategy for drug repurposing by siderophore modification and heavy-metal “trojan-horse” antibiotics.
Graphical Abstract
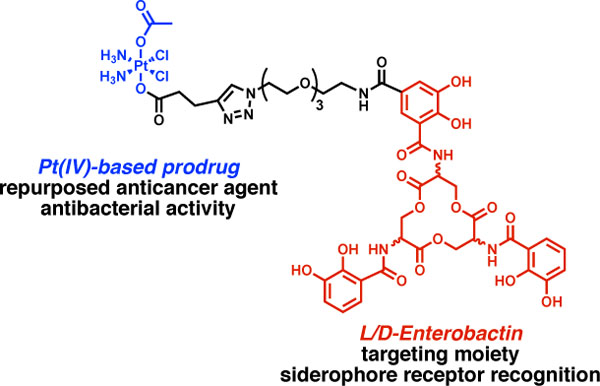
Introduction
Antibiotics are invaluable for treating bacterial infections; however, the extensive use of broad-spectrum antibiotics has resulted in widespread antibiotic resistance and negative effects on the human microbiota.1–3 The need for new classes of antibacterials is urgent. In addition to de novo antibiotic development, drug repurposing holds promise as a cost-effective strategy for identifying new clinical applications for approved drugs.4 Nevertheless, potential toxicity is one major concern in repurposing existing drugs for treating bacterial infections because antibiotics are typically dosed at levels considerably higher than non-antibacterial drugs.4 Accordingly, targeted delivery and controlled release strategies may address this challenge by enhancing therapeutic efficacy towards the target while minimizing off-target effects.5 Herein, we report the repurposing of cisplatin, a Pt anticancer agent, as an antibiotic using the strategy of siderophore-mediated delivery.
Siderophores are secondary metabolites produced and utilized by bacteria for iron (Fe) acquisition in the vertebrate host.6 Siderophore-mediated Fe acquisition is important for bacterial growth because Fe is an essential nutrient for almost all bacterial species and its availability is tightly regulated in the host environment.7–8 Moreover, during bacterial infection, Fe availability is further limited by the host to starve the pathogen in a host-defense strategy termed “nutritional immunity”.9–10 By scavenging Fe from the host environment, siderophores enable bacterial colonization and contribute to bacterial pathogenesis. To import Fe(III)-siderophore complexes, bacterial pathogens express siderophore uptake systems, and these transport systems can be hijacked for efficient and targeted antibiotic delivery.11 This strategy is exemplified by naturally occurring siderophore–antibiotic conjugates (SACs), which are deployed by some microbes to target their microbial competitors, such as sideromycins and class IIb microcins.12–16 These natural products inspire the “Trojan-horse” strategy by which a toxic agent can be disguised in a synthetic SAC and actively transported into bacteria expressing the cognate siderophore uptake machinery.17
Many examples of synthetic SACs have been reported over the past decades. The vast majority of these reports describe SACs where the antibacterial cargo is a clinically-approved antibiotic.18 For instance, we have designed and evaluated SACs based on the siderophore enterobactin (Ent, 1) that have β-lactam or fluoroquinolone cargos (Figure 1A,B).19–22 Ent 1 is a major siderophore produced by Enterobacteriaceae including Escherichia coli, Salmonella, and Klebsiella pneumoniae.23 Our studies of Ent-β-lactam and Ent-fluoroquinolone conjugates including Ent-Amp 2 and Ent-Cipro 3 (Figure 1B) demonstrated that the Ent transport machinery effectively recognizes and transports SACs, thereby delivering antibiotic cargos into the periplasm and cytoplasm of E. coli.19–22 Thus far, we have found that the antibacterial activity of these SACs is generally equal to or greater than that of the corresponding unmodified antibiotics. In particular, conjugation of Ent to β-lactam antibiotics accelerates the rate of bacterial killing relative to the unmodified β-lactams, presumably because Ent conjugation changes the uptake pathway of β-lactams from passive diffusion to active transport.19, 21–22 Moreover, the Ent modification narrows the antibacterial activity spectrum of these broad-spectrum antibiotics, which is one way to limit the spread of resistance across non-targeted bacteria and prevent secondary infections by preserving the host microbiota.1, 24
Figure 1.
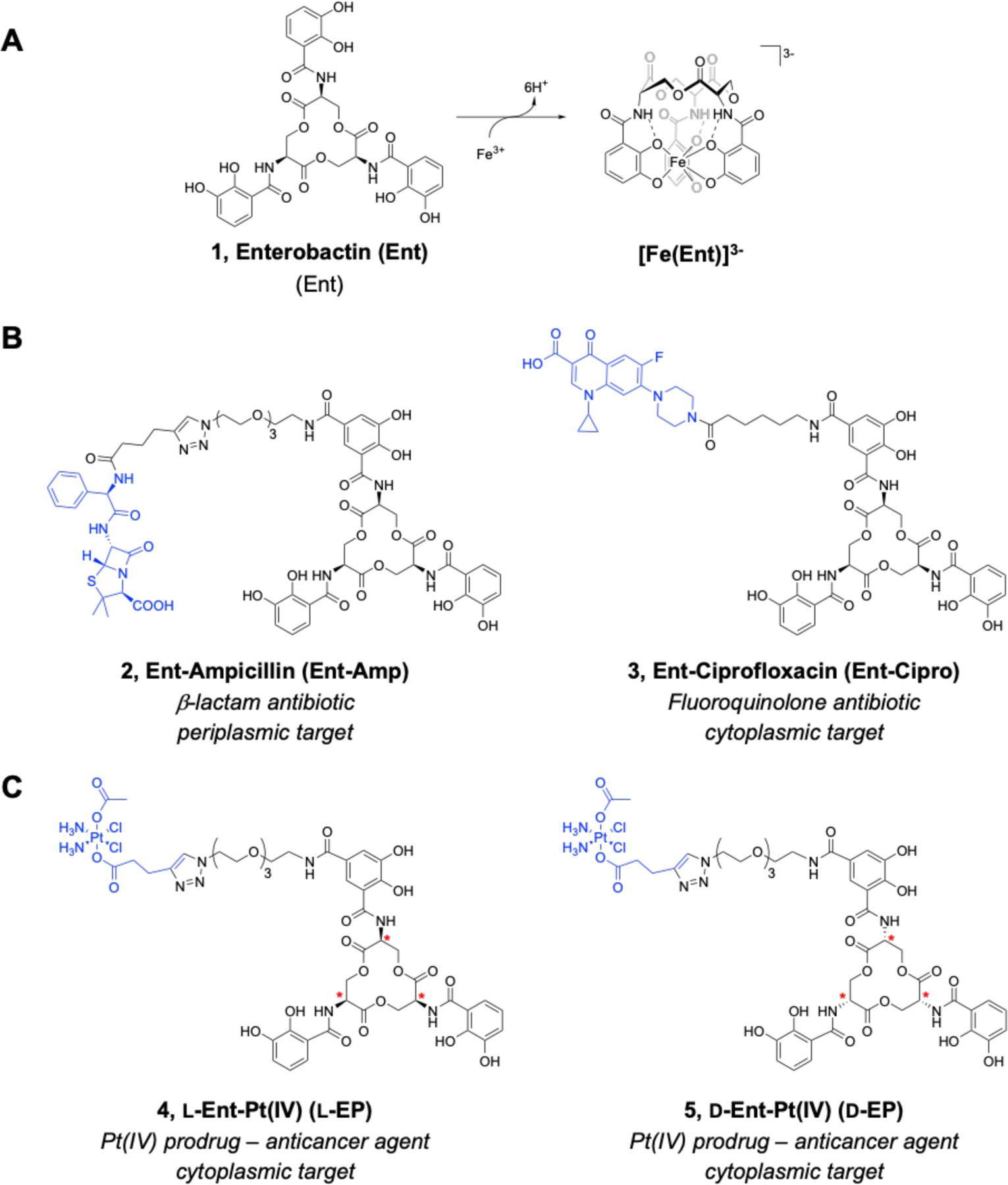
Overview of Ent and Ent-based SACs. (A) Chemical structure of l-Ent 1. Deprotonation and binding of Fe(III) by Ent to give ferric Ent [Fe(Ent)]3-. (B) Chemical structures of previously reported Ent-β-lactam 2 and Ent-fluoroquinolone 3 conjugates. (C) Chemical structures of the l- and d-Ent-Pt(IV) conjugates 4 and 5 described in this work. Stereocenters are designated with red asterisks. The antibiotic and Pt(IV) cargos are shown in blue.
During the course of these studies, we reasoned that SACs provide a framework for repurposing a broad range of toxic agents as antibiotics. Consequently, we looked beyond traditional antibiotics for new possibilities of expanding the cargo scope and hence the current antibiotic arsenal. Taking inspiration from medicinal inorganic chemistry, we focused on metallodrugs and selected the FDA-approved anticancer drug cisplatin as a case study. Cisplatin was discovered serendipitously by its ability to inhibit cell division and induce filamentous growth in E. coli.25 However, cisplatin exhibits low antibacterial activity and high toxicity towards human cells, which hinder its use as an antibiotic.26–27 To improve anticancer treatment, strategies for safer and more effective Pt-based anticancer agents are under exploration, including investigations of targeted Pt(IV) prodrugs.28 Guided by these studies, we reasoned that we could use Ent as a drug carrier to direct a Pt(IV) warhead to bacterial species expressing Ent receptors and thereby repurpose cisplatin into an antibiotic that targets Gram-negative bacteria via Ent transport machinery.
Herein, we report the design and synthesis two Ent-based Pt(IV) conjugates that combine targeted delivery by Ent and controlled release of Pt(IV) prodrugs (Figure 1C). We report that l-Ent-Pt(IV) (l-EP) 4 inhibits bacterial growth, causes filamentation in E. coli, and induces lysis in lysogenic bacteria. We demonstrate that l-EP is recognized and transported into the E. coli cytoplasm by the Ent uptake machinery FepABCDG. Moreover, we show that the enantiomer d-Ent-Pt(IV) (d-EP) 5 has improved potency against E. coli attributable to the d-Ent scaffold which is resistant to esterase-mediated cleavage, a key step in cytoplasmic Ent processing for Fe release. Both conjugates enhance Pt uptake in E. coli CFT073 cells and reduce Pt uptake in HEK293T cells. Overall, our results show that Ent conjugation repurposes a Pt anticancer agent as an antibacterial against E. coli, which provides a design strategy for a new family of heavy-metal “trojan-horse” antibiotics.
Results and Discussion
Design and synthesis of l-Ent-Pt(IV).
Guided by our prior research on Ent–antibiotic conjugates as well as work by others on Pt(IV) prodrugs,19–22, 27, 29–30 we designed l-EP 4, a conjugate that contains an Ent moiety for targeted delivery, a polyethylene glycol linker (PEG)3, and a cisplatin-derived Pt(IV) prodrug cargo (Figure 1C and Scheme 1). We note that the Pt(IV) prodrug strategy has been widely explored in Pt-based anticancer agents to mask the activity of Pt(II) species before entering target cells.27, 30 Unlike cisplatin and other square-planar Pt(II) species, Pt(IV) complexes are six-coordinate with octahedral geometries.27 The saturated coordination spheres of low-spin d6 Pt(IV) centers are substantially more kinetically inert than Pt(II), making Pt(IV) species more resistant to ligand substitution.27 Moreover, the two axial ligands are important for controlling the physiochemical properties of Pt(IV) complexes like reduction potential.30–35 In our design, we selected carboxylates as axial ligands because they have been shown to afford relatively good stability for Pt(IV) complexes in biological fluids and effective reductive activation upon cell entry.30, 33, 36–37 In the proposed working model (Figure 2), l-EP is actively imported into bacteria by the Ent transport system, including the outer membrane receptor FepA, the periplasmic binding protein FepB, and the inner membrane ATP-binding cassette transporter FepCDG. Upon entry into the reducing environment of the cytoplasm, the Pt(IV) prodrug is activated,38–40 leading to the release of the Pt(II) species and two axial ligands. The liberated active Pt(II) species targets bacterial DNA and causes cross-links, inhibiting cell division and ultimately resulting in cell death.41
Scheme 1.
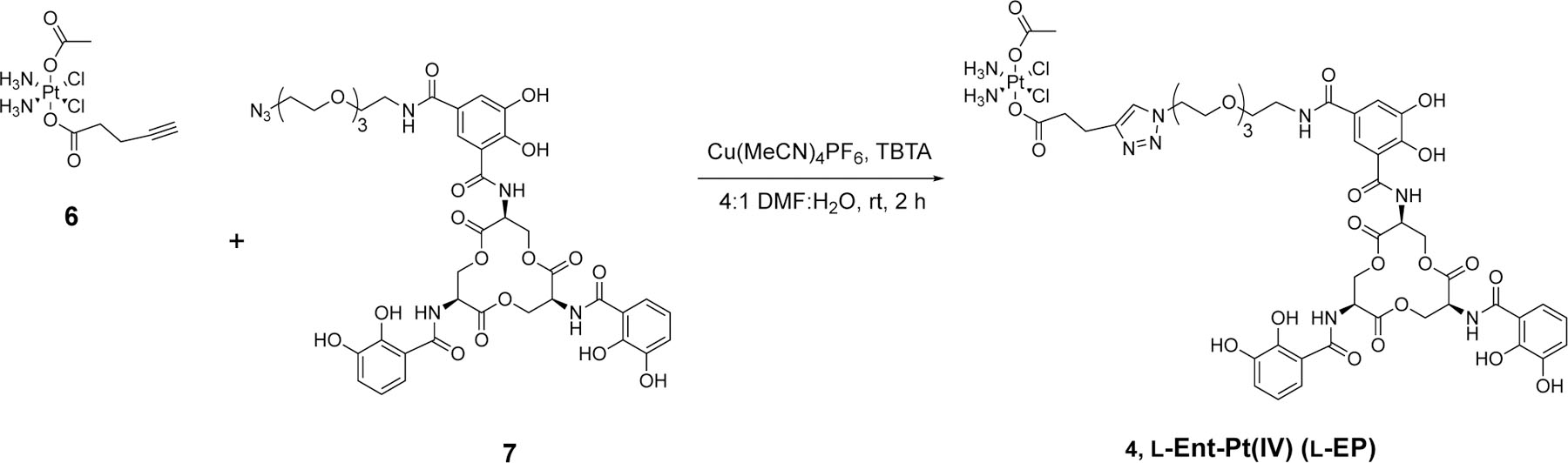
Synthesis of l-Ent-Pt(IV) via click chemistry.
Figure 2.
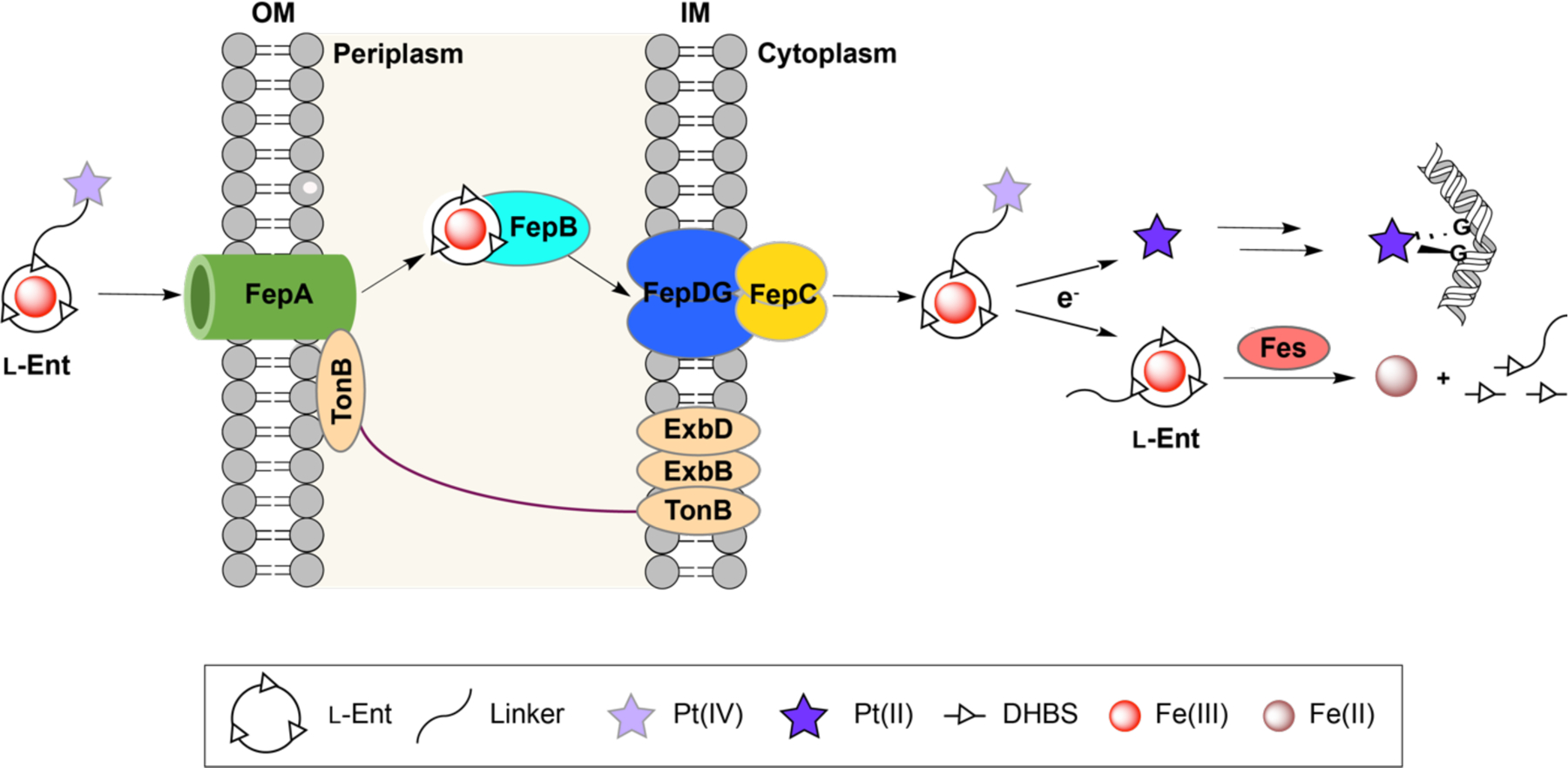
Cartoon depiction of working model for repurposing cisplatin as antibiotics that selectively target Gram-negative bacteria via Ent transport machinery. Ent transport and processing machinery is shown for E. coli K12. The TonB-ExbB-ExbD complex provides energy transduction for transport of ferric siderophores across the outer membrane. Fes is an esterase that hydrolyzes the trilactone ring of Fe-Ent. DHBS, 2,3-dihydroxybenzoyl serine; G, a guanine base. The intrastrand 1,2-d(GpG) crosslink shown is found as the major cisplatin-DNA adduct based on studies with salmon sperm DNA.30
We conjugated the Ent and Pt(IV) prodrug moieties by copper-catalyzed alkyne-azide cycloaddition (CuAAC) using alkyne-functionalized Pt(IV) 6 and l-Ent-N3 7 (Scheme 1).42 In our prior syntheses of Ent-β-lactam conjugates, we performed CuAAC by using the Cu(II) salt CuSO4 and sodium ascorbate (NaAsc) for in situ reduction, tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA) as the Cu chelator, and a mixture of DMSO and water as the solvent.19, 43 In the current work, we modified these conditions based on the properties of Pt(IV)-alkyne 6. We employed the Cu(I) salt Cu(MeCN)4PF6 as the catalyst to avoid potential Pt(IV) reduction caused by NaAsc,44 and we substituted DMF for DMSO in the solvent mixture to avoid potential ligand replacement of the chloro ligands of Pt(IV)-alkyne 6 by the nucleophilic sulfur atom of DMSO.45 We continued to use TBTA as a stabilizing ligand to protect Cu(I) from oxidation, disproportionation, and potential interactions with the two reactants.46 Using these conditions, the CuAAC reaction and subsequent purification by HPLC afforded l-EP 4 in high purity on a milligram scale for microbiology assays (Figure S1).
l-EP causes growth inhibition and induces filamentation in E. coli.
In prior investigations of Ent-antibiotic conjugates we employed a modified M9 growth medium which has low Fe content (600–700 nM by ICP-MS) and therefore induces expression of siderophore biosynthesis and transport machineries, including the enterobactin gene cluster that is responsible for Ent biosynthesis and transport.20, 29, 47 In this work, we modified this medium by decreasing its thiamine content for microbiology studies of conjugates 4 and 5 harboring the Pt(IV) prodrug (Figures S2–S4 and Supporting Discussion).
We first investigated the antibacterial activity of l-EP against two strains of E. coli, the laboratory strain K12 (BW25113) and the uropathogenic strain CFT073. Both strains biosynthesize Ent and express FepABCDG for the transport of Fe-Ent into the cytoplasm as well as the cytoplasmic esterase Fes that hydrolyzes the trilactone ring of Fe-Ent for Fe release (Figure 2).19, 43, 48 E. coli CFT073 also harbors the pathogen-associated iroA gene cluster, which is also expressed under conditions of Fe limitation and includes genes for the biosynthesis, transport, and hydrolysis of salmochelins, C-glucosylated derivatives of Ent.49–51 Consequently, CFT073 expresses the outer membrane receptor IroN, which binds and transports Fe-Ent and Fe-bound salmochelins,51 and the corresponding cytoplasmic esterase IroD that hydrolyzes these Fe-siderophore complexes.48 In addition, CFT073 expresses the outer membrane receptor IhA which also binds and transports Fe-Ent.52
Because the modified M9 medium contains less than 1 µM Fe, we treated E. coli with Fe(III)-bound l-EP, which was prepared by pre-incubating l-EP with 0.9 equiv of Fe(acac)3. We first evaluated E. coli growth in the absence and presence of l-EP by monitoring culture turbidity and observed modest growth inhibition for E. coli treated with the conjugate (Figures 3A, S5 and S6). Treatment of E. coli K12 with 7.5 or 15 µM l-EP resulted in a ~20% reduction in OD600 value compared to the untreated control, and the relative OD600 was further reduced by ~30% with 30 and 60 µM l-EP. l-EP showed a slightly larger growth inhibitory effect on E. coli CFT073, with the relative OD600 reduced by 37% with 60 µM l-EP (Figure 3A). In comparison, cisplatin was somewhat more potent against both E. coli strains (Figures 3A and S7). Treatment of K12 with 30 or 60 µM cisplatin reduced the relative OD600 by 41% or 68%, respectively. For E. coli CFT073, treatment with 30 or 60 µM cisplatin reduced the OD600 value by 51 or 58% compared to the untreated control, respectively.
Figure 3.
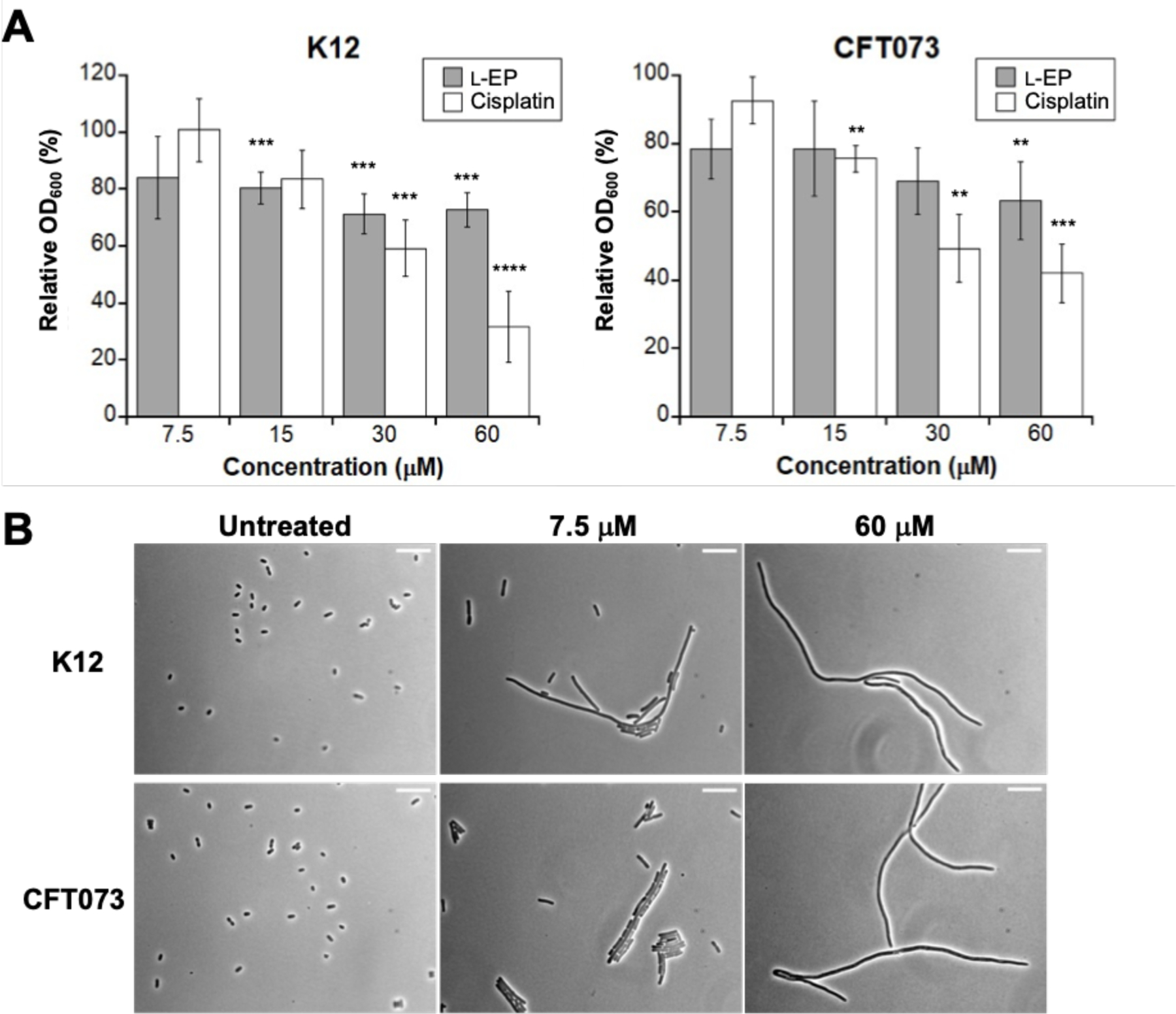
(A) Antibacterial activity of l-EP and cisplatin against E. coli K12 and CFT073. Relative OD600 refers to the OD600 value of the treated sample divided by that of the untreated control (mean ± standard deviation, n = 6 for K12 experiments, n = 7 for CFT073 and l-EP, n = 4 for CFT073 and cisplatin). Statistical differences compared to untreated controls were calculated using two-tailed student t test assuming unequal variances; ****P < 0.0001, ***P < 0.001, **P < 0.01; (B) Representative phase-contrast micrographs of E. coli K12 and CFT073 treated with l-EP. Scale bar: 10 µm. All assays were performed in modified M9 medium (11 h, 30 °C with shaking).
Filamentous morphology is a hallmark of E. coli treated with cisplatin and other Pt compounds.25, 41 Thus, we evaluated cellular morphology induced by l-EP using phase-contrast microscopy. Following treatment with l-EP, both K12 and CFT073 exhibited filamentous morphology similar to that observed for cisplatin-treated E. coli (Figures 3B and S8–S11). Moreover, filamentation occurred at the lowest l-EP concentration tested (7.5 µM, Figure 3B), and both the number of filaments and length of the filaments tended to increase with increasing l-EP concentration. Together, these initial experiments demonstrated that growth effects of l-EP on E. coli are similar to those observed for cisplatin, including growth inhibition and filamentation.
l-Ent-Pt(IV) induces lysis in lysogenic bacteria.
In early work, bacterial cell filamentation was a clue that cisplatin targets DNA because filamentation can also be a consequence of DNA damage caused by UV radiation, ionizing radiation and hydroxyurea.53–56 In prior studies, another clue for the interaction between cisplatin and DNA was the ability of cisplatin to initiate lysis of E. coli cells containing bacteriophage λ.41, 57–58 To compare l-EP to other reported Pt complexes and probe the origin its activity, we examined its ability to induce a lytic transformation in E. coli W3104, a lysogenic strain infected with bacteriophage λ. Under normal conditions, this bacteriophage grows in a lysogenic mode in which viral genes are silently duplicated along with bacterial genes.41 When E. coli W3104 sense DNA damage, signals generated by the SOS response initiate the lytic phase of bacteriophage λ and the resulting production of viral particles leads to E. coli cell lysis. When a suspension of E. coli W3104 treated by cisplatin or other Pt compounds was spotted on a lawn of non-lysogenic E. coli, viral particles released from lysed E. coli W3104 can lead to plaque formation, which served as an indicator of DNA damage triggered in E. coli W3104 by the Pt warheads.41, 57–58
Guided by these literature reports, we first treated aliquots of an exponentially growing E. coli W3104 culture with cisplatin (0–30 µM), diluted the resulting suspensions and spotted on a lawn of non-lysogenic E. coli CFT073 with a 10 µL drop of each diluted suspension.41 Consistent with prior work where plaques were observed for E. coli W3104 treated with cisplatin and some related Pt complexes at 1000-fold dilution,41 we observed plaque formation for E. coli W3104 treated with ≥15 µM cisplatin at 1000-fold dilution. We then incubated E. coli W3104 with 15 µM l-EP, 15 µM l-Ent, or 1% DMSO. We observed plaque formation for E. coli W3104 treated with 15 µM l-EP at 1000-fold dilution, but not for W3104 treated with l-Ent, 1% DMSO or the untreated control (Table 1, Figure S12). Moreover, no plaques formed where the lawn was spotted with solutions of the compounds only, indicating that the plaque formation was caused by viral particles released from lysed lysogenic bacteria rather than the compound alone. Collectively, the ability of l-EP to induce bacterial filamentation (Figures 3B) and initiate lysis in lysogenic bacteria (Table 1) is congruent with previously reported Pt complexes, indicating induction of the SOS response and suggesting that the antibacterial activity of this conjugate may be attributed to Pt-induced DNA damage.
Table 1.
Effect of l-EP on lysogenic E. colia
| Cisplatin | l-EP | Ent | 1% DMSO | Untreated | |
|---|---|---|---|---|---|
| E. coli W3104 suspension | + | + | − | − | − |
| Compound solution | − | − | − | − | − |
The development of plaques in a lawn of non-lysogenic E. coli CFT073 following spotting 1000-fold diluted suspensions of E. coli W3104 treated with 15 µM of each compound or solutions containing only the corresponding compound.
+, plaque formation; −, no plaque observed. Representative images of agar plates are shown in Figure S12.
The outer membrane receptor FepA is involved in the uptake of l-EP.
Next, we evaluated whether the outer membrane receptor FepA is important for l-EP activity against E. coli. We treated K12 and the fepA mutant (obtained from the Keio Collection)59 with 0–60 µM l-EP and then examined cellular morphology by microscopy. In contrast to the parent strain which showed filamentous morphology, most of the fepA mutant cells were normal-sized and rod-shaped following l-EP treatment (Figures 4A and S8), indicating that FepA is required for the uptake of l-EP by E. coli K12.
Figure 4.
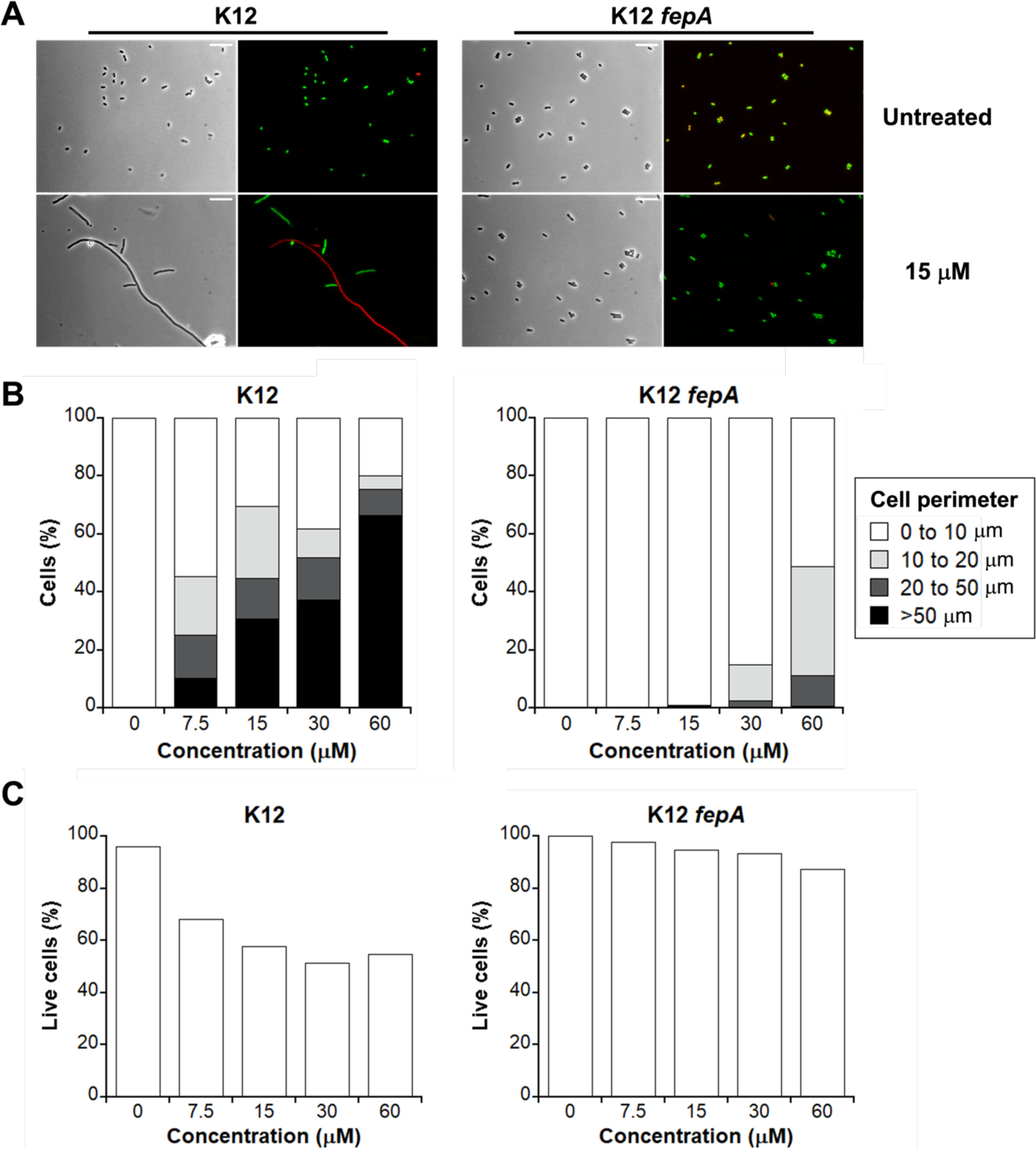
Activity of l-EP against E. coli K12 and the fepA mutant. (A) Representative phase-contrast and fluorescence micrographs of E. coli K12 and its outer membrane receptor mutant fepA treated with 15 µM l-EP in modified M9 medium for 11 h at 30 °C. Scale bar: 10 µm. (B) Quantification of the l-EP -induced bacterial morphologies in E. coli. (C) Viability of E. coli determined by LIVE/DEAD staining. For morphology and viability quantification, 500–1000 cells were manually counted for each condition from micrographs. For morphology quantification, cell size was measured and grouped by perimeter.
We also performed a quantitative analysis on the cellular morphology in K12 and the fepA mutant by measuring the perimeter of each bacterial cell and determining the relative percentage of filamented cells at each l-EP concentration evaluated (Figure 4B). Because the size of a normal E. coli cell is ~2 µm in length and ~0.6 µm in diameter,60 we grouped cells into four categories based on perimeter measurements: normal size (0–10 µm), elongation (10–20 µm), filamentation (20–50 µm), and extreme filamentation (>50 µm). With increasing concentration of l-EP, the percentage of normal-sized K12 cells decreased and more cells exhibited abnormal morphology, especially those showing extreme filamentation (10% at 7.5 µM and 66% at 60 µM) (Figure 4B left, Table S2). By contrast, more than 90% of the fepA mutant cells treated with ≤15 µM l-EP were normal-sized (Figure 4B right, Table S2). Though we observed some fepA cells with abnormal morphologies in the presence of 60 µM l-EP (49%), most of them were elongated (38%), and only 0.7% of the cells exhibited extreme filamentation. We reason that the abnormal morphologies observed for the fepA mutant treated with high l-EP concentrations occurred because of some decomposition of l-EP in the growth medium, resulting in the release of Pt(II) species that can enter bacterial cells by passive diffusion. We note that cisplatin caused comparable filamentation in both K12 and the fepA mutant (Figure S10).
We also examined the viability of K12 and the fepA mutant using the LIVE/DEAD viability assay. This assay distinguishes cells with intact and compromised outer membranes by using the fluorescent dyes SYTO 9 (green) and propidium iodide (red), respectively.21, 61 Quantification was performed by counting the number of green (live) and red (dead) cells, respectively (Figure 4C). This analysis indicated that K12 was much more susceptible to l-EP than the fepA mutant. For instance, ~50% of the K12 cells were stained red following treatment with ≥15 µM l-EP whereas 13% of the fepA cells were stained red following treatment with 60 µM l-EP (Figure 4C). Moreover, compared to the initial turbidity measurements (Figure 3A), examination of viability based on LIVE/DEAD staining and cell counting indicated more profound growth inhibition for K12. These microscopic results also corroborated that turbidity measurements are insufficient to describe growth effects caused by Pt agents because the cultures include live cells with different morphologies, cells with compromised membranes, cell debris, etc.
We extended these outer membrane receptor studies to CFT073, which presents a more complicated case because in addition to FepA, CFT073 expresses two other outer membrane receptors, IroN and IhA, that transport Fe-Ent.21, 51–52, 62 We observed that treatment of CFT073 with l-EP resulted in filamentation of both the parent strain and the fepA iroN double mutant (Figures S13). This result is consistent with our prior studies of Ent-β-lactam conjugate uptake by CFT073; we found that Ent-β-lactam conjugates retained some antibacterial activity against the fepA iroN double mutant.21–22 We note that deletion of ihA had negligible effect on cell killing by the Ent-β-lactams. One possible explanation is that l-EP or a hydrolysis product is recognized and transported by another as-yet undetermined outer membrane receptor expressed by CFT073. Further studies are warranted to investigation the origin of activity observed for the Ent-β-lactam and l-EP conjugates against the fepA iroN double mutant.
The inner membrane transporter FepCDG is involved in the uptake of l-EP.
Next, we examined whether FepCDG, the inner membrane ABC transporter that delivers Fe-Ent to the cytoplasm, is necessary for l-EP antibacterial activity. We focused these studies on the CFT073 fepC and fepDG mutants because the E. coli K12 fepC, fepD and fepG mutants obtained from the Keio Collection showed severe growth defects in the modified M9 medium. In contrast to the parent strain, which exhibited abnormal morphologies with a mixture of live and dead cells in the presence of l-EP (Figures 5A and S9), most of the fepC and fepDG mutant cells remained normal-sized and viable (Figures 5A, S9 and S14). For instance, following treatment with 30 µM l-EP, 61% of the CFT073 cells were normal-sized compared to 94% of the fepC cells (Figure 5B, Table S2). When 60 µM l-EP was employed, the percentage of normal-sized fepC mutant cells slightly decreased to 79%, presumably due to some decomposition of the conjugate (vide supra). LIVE/DEAD staining and cell counting indicated that following treatment with 60 µM l-EP, only 53% of the parent strain cells remained viable, whereas 97% of the fepC cells still remained viable (Figure 5C, Table S2). Morphology and cell viability analyses of the fepDG mutant provided similar trends (Figure S14, Table S2). Cisplatin, by contrast, induced comparable filamentation in all three strains (Figure S11), and Pt(IV)-alkyne 6 did not cause abnormal morphologies or cell death (Figure S15). Taken together, these results show that the inner membrane transporter FepCDG transports l-EP into the cytoplasm.
Figure 5.
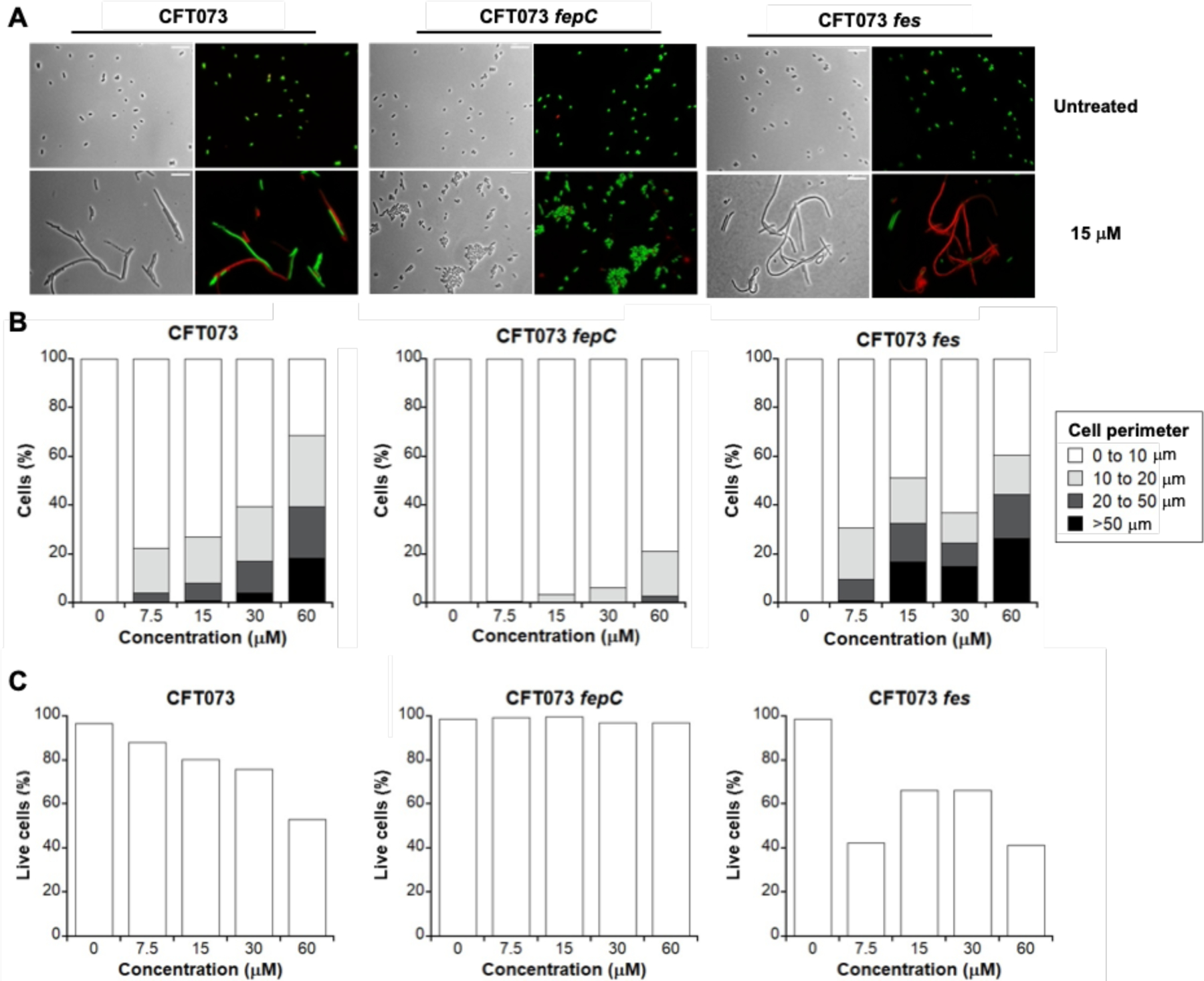
Activity of l-EP against E. coli CFT073 and the fepC and fes mutants. (A) Representative phase-contrast and fluorescence micrographs of E. coli CFT073, its inner membrane transporter mutant fepC, and its cytoplasmic esterase mutant fes treated with 15 µM l-EP in modified M9 medium for 11 h at 30 °C. Scale bar: 10 mm. (B) Quantification of the l-EP-induced bacterial morphologies in E. coli. (C) Viability of E. coli determined by LIVE/DEAD staining. For morphology and viability quantification, 500–1000 cells were manually counted for each condition from micrographs. For morphology quantification, cell size was measured and grouped by perimeter. Activity of l-EP against the inner membrane transporter mutant fepDG and the cytoplasmic esterase mutant iroD are shown in Figure S14.
Loss of the Ent esterase Fes enhances the susceptibility of E. coli to l-EP.
By evaluating the antibacterial activity and cellular morphology data we obtained for l-EP against E. coli, we demonstrated that Ent conjugation mediates the delivery of the Pt(IV) prodrug into bacterial cytoplasm via the Ent transport machinery. We next considered the fate of l-EP in the cytoplasm and questioned whether co-delivery of the toxic Pt(IV) payload with nutrient Fe bound by the siderophore compromised antibacterial activity, which would provide a potential explanation for why l-EP was less effective in reducing culture turbidity than cisplatin (Figure 3A). In addition to being an essential nutrient that contributes to overall fitness, Fe plays important roles in DNA metabolism, including DNA repair in E. coli. For instance, multiple DNA repair enzymes (helicases, nucleases, glycosylases, demethylases) use Fe as an indispensable cofactor.63 In considering the fate of l-EP in the cytoplasm, we reasoned that Fe(III)-bound l-EP is a substrate for Fes and other enterobactin esterases like IroD and that its hydrolysis would provide E. coli with Fe that can be used metabolically and increase bacterial fitness. Thus, we examined the consequences of fes deletion on the antibacterial activity of l-EP against CFT073. Turbidity measurements showed that the fes mutant is markedly more susceptible than the parent strain to l-EP treatment (Figures 6 and S6). In the fes mutant, treatment with 15 µM l-EP reduced culture turbidity by 56% and further reduction was seen following the treatment with 30 and 60 µM l-EP (70% and 77%, respectively). In contrast, only 37% reduction in turbidity occurred at 60 µM l-EP for the parent strain.
Figure 6.
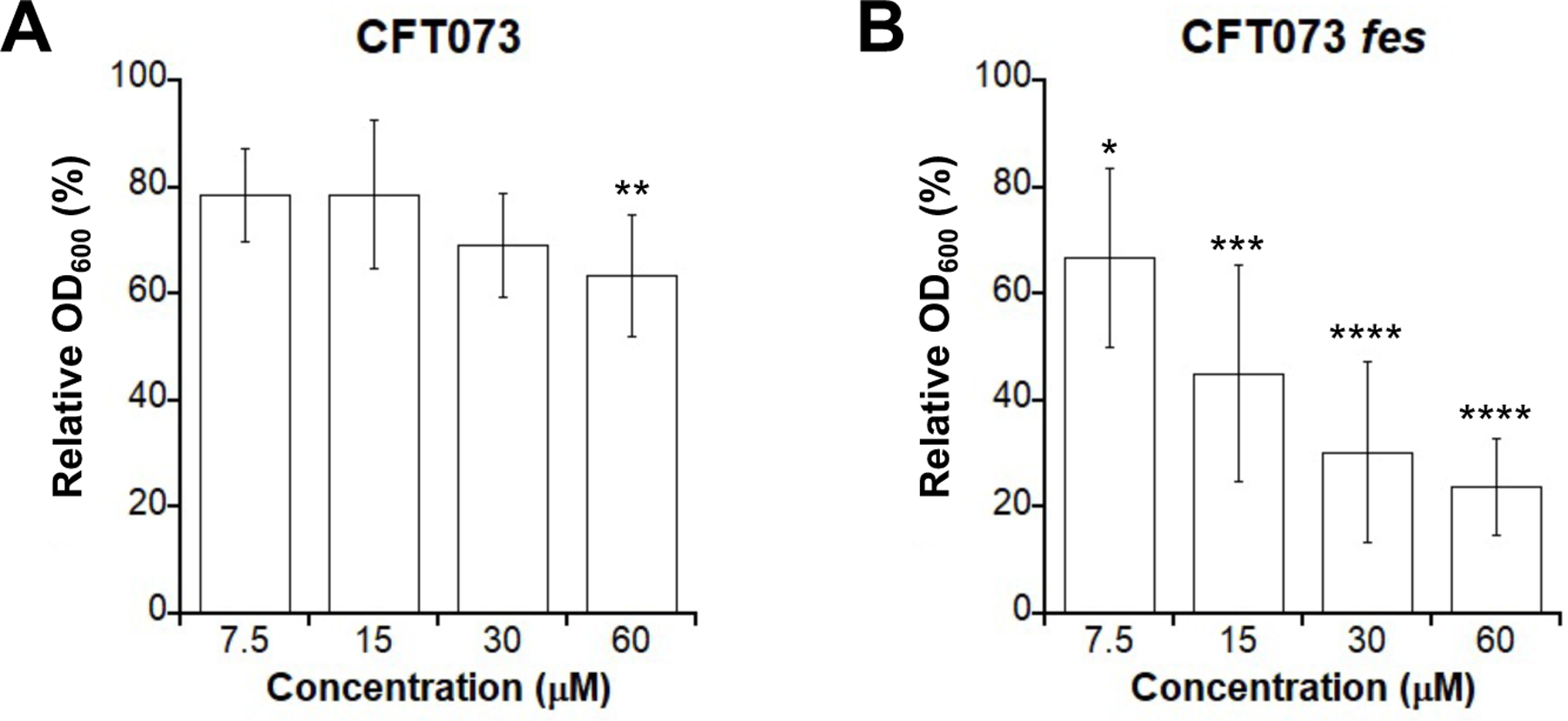
Antibacterial activity of l-EP against (A) E. coli CFT073 and (B) the fes mutant. Relative OD600 refers to the OD600 value of the treated sample divided by that of the untreated control; (mean ± standard deviation, n = 7 for CFT073, n = 8 for the fes mutant); statistical differences compared to untreated controls were calculated using two-tailed student t test assuming unequal variances; ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05. All assays were performed in modified M9 medium at 30 °C for 11 h.
Imaging of the fes mutant further supported its enhanced susceptibility to l-EP compared to the parent strain (Figures 5A, S16). Quantitative analysis of bacterial morphology demonstrated that the fes mutant exhibited more extremely long filaments in the presence of l-EP than the parent strain at all tested concentrations, especially at 15 and 30 µM (≥15% in the fes mutant compared to <5% in the parent strain, Figure 5B, Table S2). Moreover, LIVE/DEAD staining indicated that cell viability decreased by 10–15% for the fes mutant compared to the parent strain in the presence of ≥15 µM l-EP (Figure 5C, Table S2). These results suggested that Fes-catalyzed Ent hydrolysis with the release of Fe compromises the ability of the Pt cargo to inhibit cell division to some degree, likely by increasing the fitness of E. coli. We also tested iroD mutant in parallel, which also showed increased susceptibility to l-EP treatment compared to the parent strain (Figures S14 and S16).
Substitution of l-Ent with its enantiomer affords d-EP with enhanced antibacterial activity.
Prior studies demonstrated that the Fe(III)-bound d-enantiomer of Ent (d-Ent) is actively transported into the cytoplasm of E. coli at levels similar to that of the Fe(III)-bound l-form;64 however, d-Ent does not recover the growth of E. coli under Fe limitation because the cytoplasmic Fes does not accept d-Ent as a substrate and thus bound Fe(III) cannot be released.43, 64 As IroD shares sequence similarity with Fes,64–65 we speculated that IroD is also stereospecific and cannot hydrolyze d-Ent. Based on these studies and our observation that loss of fes or iroD enhanced the susceptibility of CFT073 to l-EP, we hypothesized that the enantiomer d-Ent-Pt(IV) (d-EP) 5 would display enhanced antibacterial activity compared to l-EP. We therefore synthesized d-EP from d-Ent-N3 and Pt(IV)-alkyne and evaluated its antibacterial activity.
In an initial antibacterial activity assay using a dilution series spanning 0–60 µM, we found that treatment of CFT073 with 7.5 µM d-EP resulted in a greater reduction in culture turbidity compared to treatment with 60 µM l-EP, indicating that d-EP is more potent than either l-EP or cisplatin (Figure 7A). We then examined the antibacterial activity of d-EP over a concentration range of 0–10 µM and found that the turbidity of CFT073 cultures decreased markedly in the presence of 1 µM d-EP (Figure 7B). This concentration of l-EP, cisplatin and Pt(IV)-alkyne had negligible effect on culture turbidity (Figure S17B). We also found that 10 µM d-EP almost fully reduced the OD600 value of E. coli CFT073 cultures to the baseline (OD600 = 0.05 compared to 0.2 for the untreated control, Figure S17B). In the case of K12, the culture turbidity was reduced by 72% in the presence of 0.1 µM d-EP and was fully reduced to the baseline when treated with 1 and 10 µM d-EP (Figure S17C). We also evaluated whether the outer membrane receptor FepA and the inner membrane transporter FepCDG are important for d-EP activity against E. coli (Figure S18). The results were consistent with our findings for l-EP. In particular, 10 µM d-EP had negligible effect on the growth of the K12 fepA mutant and the CFT073 inner membrane transporter mutants fepC and fepDG, indicating that the Ent transport machinery is required for d-EP to enter the E. coli cytoplasm and exert antibacterial activity.
Figure 7.
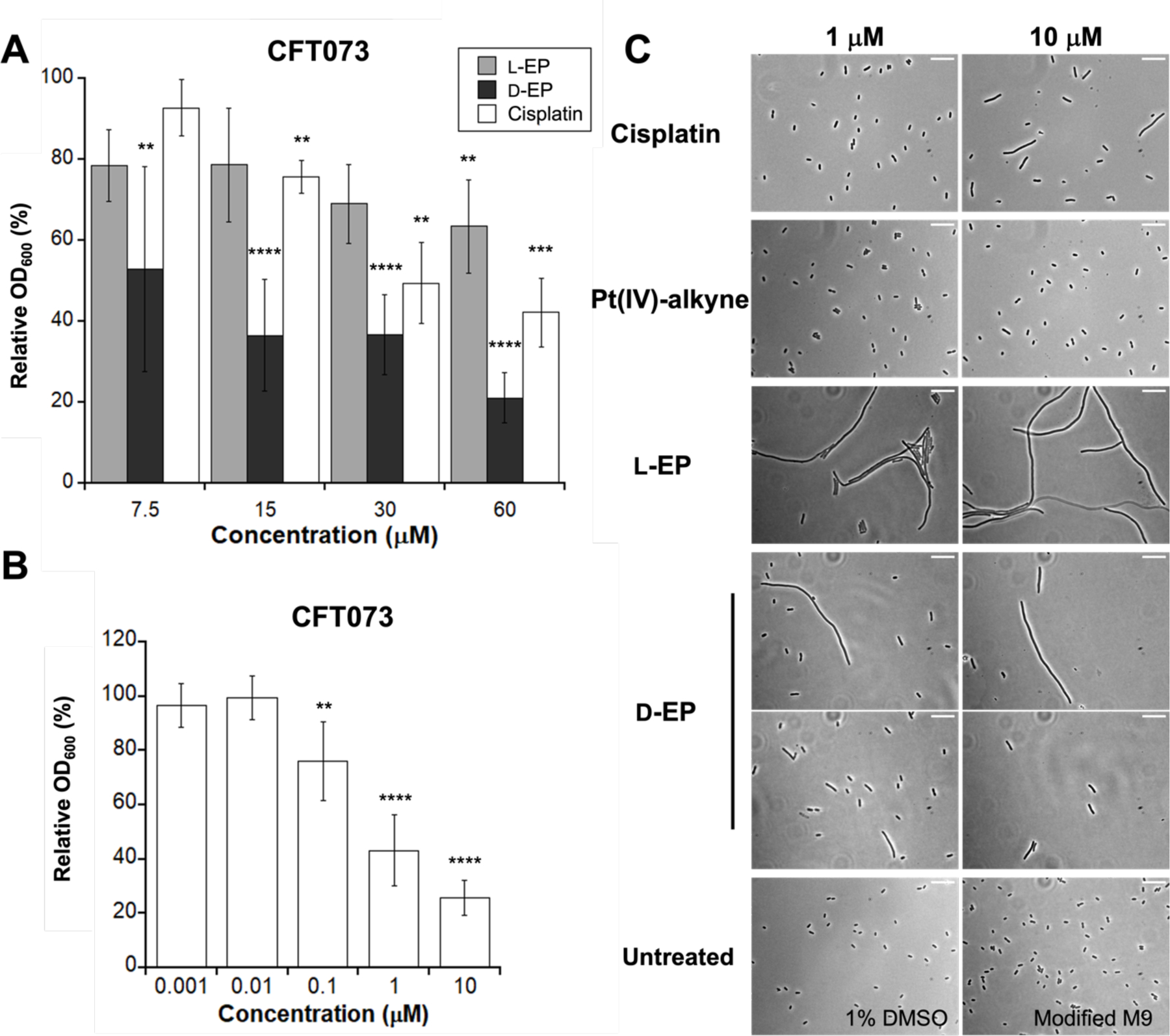
Comparison of activity of l-EP, d-EP and other Pt species. (A) Antibacterial activity of l-EP, d-EP and cisplatin against E. coli CFT073 (0–60 µM). (B) Antibacterial activity of d-EP against E. coli CFT073 (0–10 µM); Relative OD600 refers to the OD600 value of the treated sample divided by that of the untreated control (mean ± standard deviation, n ≥ 6). Statistical differences compared to untreated controls were calculated using two-tailed student t test assuming unequal variances; ****P < 0.0001, ***P < 0.001, **P < 0.01. (C) Representative phase-contrast micrographs E. coli CFT073 wild-type treated with cisplatin, Pt(IV)-alkyne 6, l-EP 4 and d-EP 5 (Scale bar: 10 µm). All assays were performed in modified M9 medium at 30 °C for 11 h.
We also examined the morphologies of E. coli CFT073 treated with d-EP, l-EP, cisplatin and Pt(IV)-alkyne in this low concentration range (Figure 7C). Both d-EP and l-EP induced filamentation at 1 and 10 µM, and bacteria treated with l-EP and d-EP displayed different morphologies (Figure 7C). The cells treated with l-EP were mostly very long filaments, but those treated with d-EP were comparatively sparse and exhibited various morphologies, including filaments (relatively shorter than those caused by l-EP), elongated cells, and normal cells. By contrast, only slight elongation was induced by 10 µM cisplatin. Almost all bacterial cells treated with Pt(IV)-alkyne were normal-sized and rod-shaped, presumably a consequence of poor penetration of the molecule across cell membrane. Thus, by leveraging the chiral recognition of Ent esterases, we synthesized the d-enantiomer of Ent-Pt(IV), which showed enhanced potency against E. coli compared to l-EP and cisplatin.
Ent conjugation affords enhanced Pt uptake by bacterial cells.
To examine whether Ent conjugation affects Pt uptake by bacterial cells, we treated E. coli CFT073 with 1 µM of l/d-EP, cisplatin, or Pt(IV)-alkyne in modified M9 medium at 30 °C for 30 min, and then quantified cell-associated Pt levels by ICP-MS. We found that Pt uptake was markedly enhanced for the Ent conjugates compared to cisplatin and Pt(IV)-alkyne. In particular, Pt uptake was ≥10-fold greater for E. coli CFT073 treated with l-EP or d-EP (8–10%) compared to cells treated with cisplatin or Pt(IV)-alkyne (Figure 8A), which showed negligible Pt uptake (<1%). This result indicates that Ent conjugation facilitates Pt uptake, which presumably results from the active transport of l/d-EP by the Ent transport system compared to passive diffusion of cisplatin and Pt(IV)-alkyne.
Figure 8.
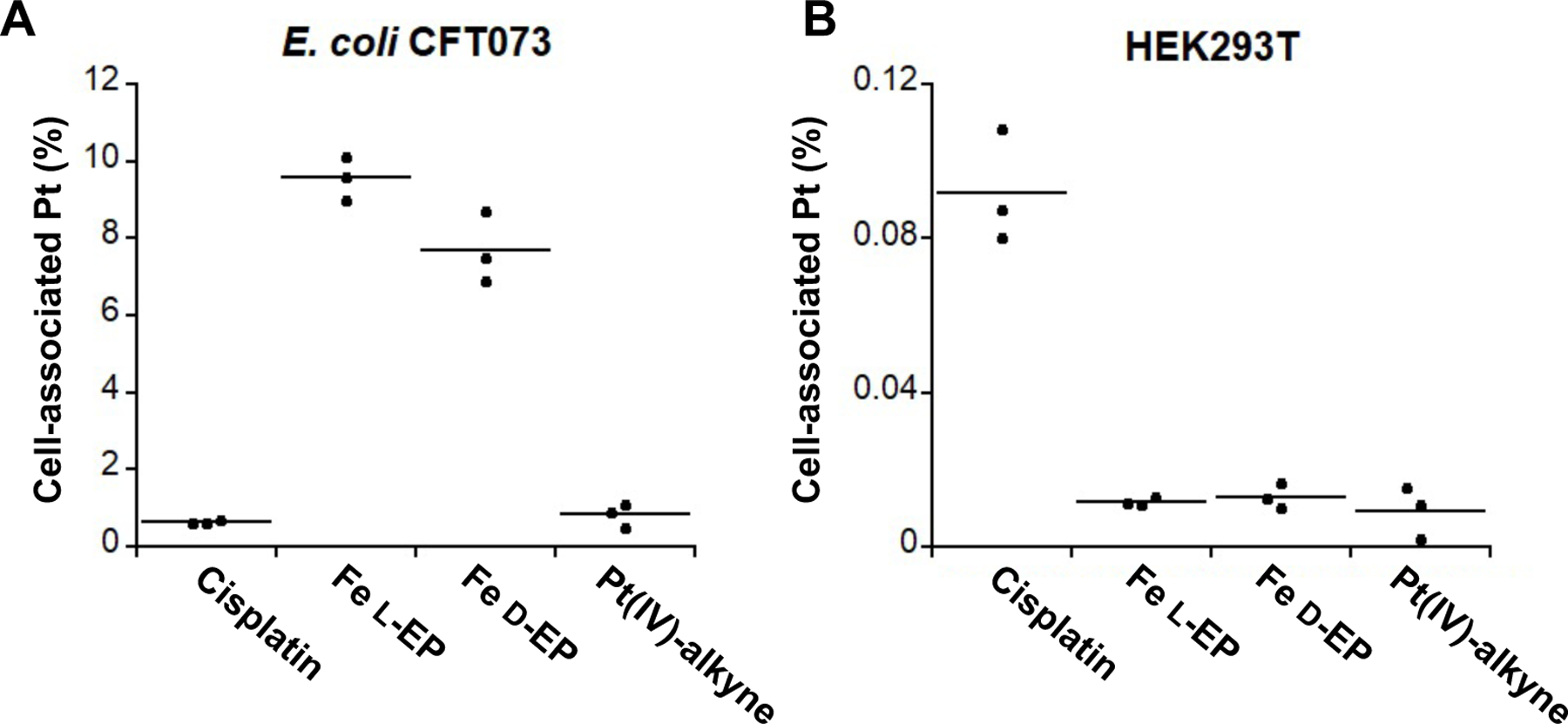
Pt uptake of E. coli CFT073 (A) and HEK293T cells (B) treated with 1 µM cisplatin, l-EP, d-EP, or Pt(IV)-alkyne 6, respectively. E. coli CFT073 cells were treated for 30 min in modified M9 medium at 30 °C, and HEK293T cells were treated for 6 h in DMEM+1% penicillin/streptomycin at 37 °C, 5% CO2 (n = 3).
Ent conjugation reduces Pt uptake by human cells.
Lastly, we examined whether Ent conjugation affects Pt uptake by human cells. In contrast to bacterial cells, to our knowledge human cells do not have an Ent receptor. We reasoned that the l/d-EP conjugates would not enter human cells by passive diffusion because they are relatively large (1369 Da). We performed a Pt uptake assay with HEK293T cells, which were treated with 1 µM of l/d-EP, cisplatin, or Pt(IV)-alkyne in DMEM supplemented with 1% penicillin/streptomycin at 37 °C for 6 h. The cell-associated Pt levels were then quantified by ICP-MS.66 Under these conditions, the HEK293T cells showed ~9-fold greater Pt uptake for cisplatin (~0.09%) than for l/d-Ent or Pt(IV) alkyne (~0.01%) (Figure 8B). The negligible uptake of Pt(IV) alkyne is presumably attributed to its poor lipophilicity due to its short carbon chain axial ligands.67–68 Because higher cellular Pt accumulation is accompanied by greater DNA platination and therefore cell growth inhibition,69–70 our Pt uptake assay suggests that Ent modification affords decreased cytotoxicity towards human cells compared to cisplatin.
Conclusion
In this work, we used l-EP as a proof-of-concept to show that Ent conjugation repurposes cisplatin as an antibiotic and mediates its delivery to the cytoplasm of E. coli. Then, guided by fundamental studies on chiral recognition of Ent by cytoplasmic esterases, we synthesized the enantiomer d-EP, which displays enhanced antibacterial activity against E. coli. Lastly, we demonstrated that Ent conjugation facilitates Pt uptake in bacteria and reduces Pt uptake in HEK293T cells. We propose the following mechanism for l/d-EP (Figure 9): upon Ent-mediated delivery into the E. coli cytoplasm, l/d-EP undergoes intracellular reduction and thereby release one equivalent of cisplatin, acetate and the l-/d-Ent-containing ligand. Cisplatin further undergoes aquation, and the resulting electrophilic complexes ([Pt(NH3)2Cl(H2O)]+ and [Pt(NH3)2(H2O)2]2+) readily bind to the purine bases of DNA, resulting in cross-links which distort and bend the DNA structure.30, 53 In response, bacteria initiate the SOS response, which mediates the inhibition of cell division while damaged DNA is repaired.53, 71 With l-EP treatment, esterase-mediated l-Ent cleavage releases bound Fe, which increases bacterial fitness and leads to the formation of extreme filaments. Such extreme filaments are not observed for d-EP because the d-Ent scaffold is not a substrate for Ent esterases. Moreover, d-EP exhibits significantly enhanced the antibacterial activity compared to l-EP. This outcome provides a foundation for further leveraging d-Ent in siderophore-based delivery. More broadly, the current work also provides a modular synthetic approach for repurposing other Pt-based anticancer agents and metallopharmaceuticals as targeted antibiotics against bacterial pathogens using siderophores.
Figure 9.
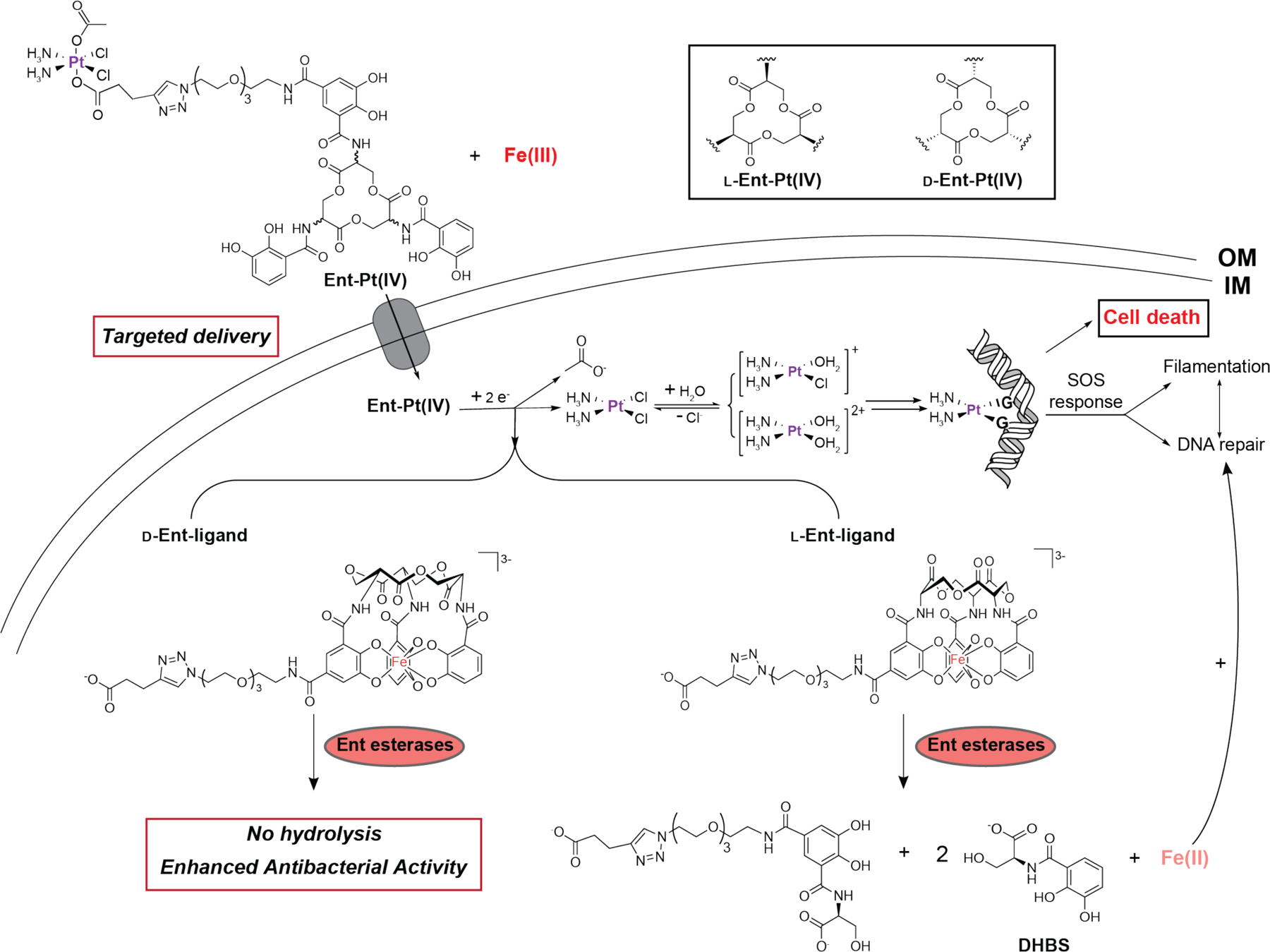
Proposed model for the activity for l- and d-EP against E. coli. DHBS, 2,3-dihydroxybenzoyl serine; G, a guanine base.
Along with our prior work on the Ent-Cipro conjugate,20 the present work also deepens our appreciation of the role of esterase-catalyzed Ent hydrolysis in the activity of Ent conjugates with cytoplasmic targets. In the case of Ent-Cipro, the pathogen-associated esterase IroD was required for antibacterial activity, indicating that hydrolysis of the Fe-Ent moiety by IroD was an essential processing step for Ent-Cipro activation. By contrast, Fes and IroD compromised the potency of l-EP, indicating that hydrolysis of the Fe-Ent moiety by these enzymes provides E. coli with a fitness advantage. The different outcomes for Ent-Cipro and l-EP may result, at least in part, to the relatively potencies of the two cargos. Ciprofloxacin is approximately 104-fold more potent than cisplatin. Thus, for Ent-Cipro, less conjugate and therefore less Fe needs to be delivered to the cell for cell death. Regardless, the current results highlight that it is necessary to think about the delivery of nutrient Fe bound to the siderophore moiety, especially for cargos with cytoplasmic targets. In addition to our current examination of d-Ent as a strategy to avoid Fe(III) release, Ga(III)-loaded SACs have been studied to avoid Fe co-delivery and thereby afford enhanced antibacterial activity compared to the corresponding Fe(III)-loaded SACs.72–74 In a preliminary investigation, we observed negligible antibacterial activity when treating E. coli K12 and CFT073 with Ga(III)-bound l-EP. This observation is consistent with a prior report that Ga(III)-Ent is a poor transport substrate for the Ent uptake machinery.75
In closing, despite advances in drug repurposing, no drugs have been repurposed as antibiotics to date.76 Studies of SACs have mainly focused on modifying classic antibiotics.18 To our knowledge, only one example of a SAC designed for drug repurposing has been reported, a compound containing a mycobacterial siderophore linked to the anti-malarial drug artemisinin that targets Mycobacterium tuberculosis.77 We anticipate that the current exploration of Ent-directed cisplatin delivery will motivate further efforts towards leveraging the siderophore-based “Trojan-horse” strategy for repurposing a broad scope of non-antibiotic toxic drugs into targeted antibacterials.
Supplementary Material
Acknowledgments
This work was supported by the 2018 Professor Amar G. Bose Research Grant. NMR and MS instrumentation is housed by the MIT DCIF. The ICP-MS instrument at MIT is maintained by the MIT CEHS (NIH P30-ES002109). Instrumentation for phase-contrast and fluorescence microscopy instrumentation is housed by the W. M. Keck Microscopy Facility at Whitehead Institute. We thank Phoom Chairatana, Timothy Johnstone and Artur Sargun for technical assistance during the early stage of this project. We thank Jessica Patrick in the Shoulders lab at MIT for assistance with tissue culture.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org. Complete experimental methods, supporting discussion l-EP stability in growth medium, Tables S1–S3, Figures S1–S23, and NMR spectroscopic data.
Conflicts of interest
None
References
- 1.Brown ED; Wright GD, Antibacterial drug discovery in the resistance era. Nature 2016, 529, 336–343. [DOI] [PubMed] [Google Scholar]
- 2.Blaser M, Stop the killing of beneficial bacteria. Nature 2011, 476, 393–394. [DOI] [PubMed] [Google Scholar]
- 3.Cho I; Blaser MJ, The human microbiome: at the interface of health and disease. Nat. Rev. Genet 2012, 13, 260–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Farha MA; Brown ED, Drug repurposing for antimicrobial discovery. Nat. Microbiol 2019, 4, 565–577. [DOI] [PubMed] [Google Scholar]
- 5.Czech T; Lalani R; Oyewumi MO, Delivery systems as vital tools in drug repurposing. AAPS PharmSciTech 2019, 20, 116. [DOI] [PubMed] [Google Scholar]
- 6.Hider RC; Kong X, Chemistry and biology of siderophores. Nat. Prod. Rep 2010, 27, 637–657. [DOI] [PubMed] [Google Scholar]
- 7.Pantopoulos K; Porwal SK; Tartakoff A; Devireddy L, Mechanisms of mammalian iron homeostasis. Biochemistry 2012, 51, 5705–5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Braun V; Hantke K, Recent insights into iron import by bacteria. Curr. Opin. Chem. Biol 2011, 15, 328–334. [DOI] [PubMed] [Google Scholar]
- 9.Hood MI; Skaar EP, Nutritional immunity: transition metals at the pathogen-host interface. Nat. Rev. Microbiol 2012, 10, 525–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weinberg ED, Nutritional immunity. Host’s attempt to withold iron from microbial invaders. JAMA 1975, 231, 39–41. [DOI] [PubMed] [Google Scholar]
- 11.McPherson CJ; Aschenbrenner LM; Lacey BM; Fahnoe KC; Lemmon MM; Finegan SM; Tadakamalla B; O’Donnell JP; Mueller JP; Tomaras AP, Clinically relevant Gram-negative resistance mechanisms have no effect on the efficacy of MC-1, a novel siderophore-conjugated monocarbam. Antimicrob Agents Chemother 2012, 56, 6334–6342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Braun V; Pramanik A; Gwinner T; Köberle M; Bohn E, Sideromycins: tools and antibiotics. Biometals 2009, 22, 3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nolan EM; Fischbach MA; Koglin A; Walsh CT, Biosynthetic tailoring of microcin E492m: post-translational modification affords an antibacterial siderophore–peptide conjugate. J. Am. Chem. Soc 2007, 129, 14336–14347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sassone-Corsi M; Nuccio S-P; Liu H; Hernandez D; Vu CT; Takahashi AA; Edwards RA; Raffatellu M, Microcins mediate competition among Enterobacteriaceae in the inflamed gut. Nature 2016, 540, 280–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vassiliadis G; Destoumieux-Garzón D; Lombard C; Rebuffat S; Peduzzi J, Isolation and characterization of two members of the siderophore-microcin family, microcins M and H47. Antimicrob Agents Chemother 2010, 54, 288–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Palmer JD; Mortzfeld BM; Piattelli E; Silby MW; McCormick BA; Bucci V, Microcin H47: a class IIb microcin with potent activity against multidrug resistant Enterobacteriaceae. ACS Infect. Dis 2020, 6, 672–679. [DOI] [PubMed] [Google Scholar]
- 17.Roosenberg JM II; Lin Y-M; Lu Y; Miller MJ, Studies and syntheses of siderophores, microbial iron chelators, and analogs as potential drug delivery agents. Curr. Med. Chem 2000, 7, 159–197. [DOI] [PubMed] [Google Scholar]
- 18.Lin YM; Ghosh M; Miller PA; Möllmann U; Miller MJ, Synthetic sideromycins (skepticism and optimism): selective generation of either broad or narrow spectrum Gram-negative antibiotics. Biometals 2019, 32, 425–451. [DOI] [PubMed] [Google Scholar]
- 19.Zheng T; Nolan EM, Enterobactin-mediated delivery of β-lactam antibiotics enhances antibacterial activity against pathogenic Escherichia coli. J. Am. Chem. Soc 2014, 136, 9677–9691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Neumann W; Sassone-Corsi M; Raffatellu M; Nolan EM, Esterase-catalyzed siderophore hydrolysis activates an enterobactin-ciprofloxacin conjugate and confers targeted antibacterial activity. J. Am. Chem. Soc 2018, 140, 5193–5201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sargun A; Johnstone TC; Zhi H; Raffatellu M; Nolan EM, Enterobactin- and salmochelin-β-lactam conjugates induce cell morphologies consistent with inhibition of penicillin-binding proteins in uropathogenic Escherichia coli CFT073. Chem. Sci 2021, 12, 4041–4056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sargun A; Sassone-Corsi M; Zheng T; Raffatellu M; Nolan EM, Conjugation to enterobactin and salmochelin S4 enhances the antimicrobial activity and selectivity of β-lactam antibiotics against nontyphoidal Salmonella. ACS Infect. Dis 2021, 7, 1248–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raymond KN; Dertz EA; Kim SS, Enterobactin: an archetype for microbial iron transport. Proc. Natl. Acad. Sci. U. S. A 2003, 100, 3584–3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lewis K, Platforms for antibiotic discovery. Nat. Rev. Drug Discov 2013, 12, 371–387. [DOI] [PubMed] [Google Scholar]
- 25.Rosenberg B; Van Camp L; Krigas T, Inhibition of cell division in Escherichia coli by electrolysis products from a platinum electrode. Nature 1965, 205, 698–699. [DOI] [PubMed] [Google Scholar]
- 26.Rosenberg B; Van Camp L; Grimley EB; Thomson AJ, The inhibition of growth or cell division in Escherichia coli by different ionic species of platinum(IV) complexes. J. Biol. Chem 1967, 242, 1347–1352. [PubMed] [Google Scholar]
- 27.Johnstone TC; Suntharalingam K; Lippard SJ, The next generation of platinum drugs: targeted Pt(II) agents, nanoparticle delivery, and Pt(IV) prodrugs. Chem. Rev 2016, 116, 3436–3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dhar S; Kolishetti N; Lippard SJ; Farokhzad OC, Targeted delivery of a cisplatin prodrug for safer and more effective prostate cancer therapy in vivo. Proc. Natl. Acad. Sci. U.S.A 2011, 108, 1850–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chairatana P; Zheng T; Nolan EM, Targeting virulence: salmochelin modification tunes the antibacterial activity spectrum of β-lactams for pathogen-selective killing of Escherichia coli. Chem. Sci 2015, 6, 4458–4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilson JJ; Lippard SJ, Synthetic methods for the preparation of platinum anticancer complexes. Chem. Rev 2014, 114, 4470–4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hambley TW; Battle AR; Deacon GB; Lawrenz ET; Fallon GD; Gatehouse BM; Webster LK; Rainone S, Modifying the properties of platinum(IV) complexes in order to increase biological effectiveness. J. Inorg. Biochem 1999, 77, 3–12. [DOI] [PubMed] [Google Scholar]
- 32.Reithofer MR; Bytzek AK; Valiahdi SM; Kowol CR; Groessl M; Hartinger CG; Jakupec MA; Galanski M; Keppler BK, Tuning of lipophilicity and cytotoxic potency by structural variation of anticancer platinum (IV) complexes. J. Inorg. Biochem 2011, 105, 46–51. [DOI] [PubMed] [Google Scholar]
- 33.Wexselblatt E; Yavin E; Gibson D, Platinum(IV) prodrugs with haloacetato ligands in the axial positions can undergo hydrolysis under biologically relevant conditions. Angew. Chem. Int. Ed 2013, 52, 6059–6062. [DOI] [PubMed] [Google Scholar]
- 34.Chin CF; Tian Q; Setyawati MI; Fang W; Tan ESQ; Leong DT; Ang WH, Tuning the activity of platinum(IV) anticancer complexes through asymmetric acylation. J. Med. Chem 2012, 55, 7571–7582. [DOI] [PubMed] [Google Scholar]
- 35.Wexselblatt E; Gibson D, What do we know about the reduction of Pt(IV) pro-drugs? J. Inorg. Biochem 2012, 117, 220–229. [DOI] [PubMed] [Google Scholar]
- 36.Ong JX; Yap SQ; Wong DYQ; Chin CF; Ang WH, Platinum(IV) carboxylate prodrug complexes as versatile platforms for targeted chemotherapy. Chimia 2015, 69, 100–103. [DOI] [PubMed] [Google Scholar]
- 37.Chen S; Yao H; Zhou Q; Tse MK; Gunawan YF; Zhu G, Stability, reduction, and cytotoxicity of platinum(IV) anticancer prodrugs bearing carbamate axial ligands: comparison with their carboxylate analogues. Inorg. Chem 2020, 59, 11676–11687. [DOI] [PubMed] [Google Scholar]
- 38.Lemma K; Berglund J; Farrell N; Elding LI, Kinetics and mechanism for reduction of anticancer-active tetrachloroam(m)ine platinum(IV) compounds by glutathione. J. Biol. Inorg. Chem 2000, 5, 300–306. [DOI] [PubMed] [Google Scholar]
- 39.Lemma K; Sargeson AM; Elding LI, Kinetics and mechanism for reduction of oral anticancer platinum(IV) dicarboxylate compounds by L-ascorbate ions. J. Chem. Soc., Dalton Trans. 2000, 1167–1172. [Google Scholar]
- 40.Hall MD; Hambley TW, Platinum(IV) antitumour compounds: their bioinorganic chemistry. Coord. Chem. Rev. 2002, 232, 49–67. [Google Scholar]
- 41.Johnstone TC; Alexander SM; Lin W; Lippard SJ, Effects of monofunctional platinum agents on bacterial growth: a retrospective study. J. Am. Chem. Soc 2014, 136, 116–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang JZ; Bonnitcha P; Wexselblatt E; Klein AV; Najajreh Y; Gibson D; Hambley TW, Facile preparation of mono-, di- and mixed-carboxylato platinum(IV) complexes for versatile anticancer prodrug design. Chemistry 2013, 19, 1672–1676. [DOI] [PubMed] [Google Scholar]
- 43.Zheng T; Bullock JL; Nolan EM, Siderophore-mediated cargo delivery to the cytoplasm of Escherichia coli and Pseudomonas aeruginosa: syntheses of monofunctionalized enterobactin scaffolds and evaluation of enterobactin–cargo conjugate uptake. J. Am. Chem. Soc 2012, 134, 18388–18400. [DOI] [PubMed] [Google Scholar]
- 44.Kubas G; Monzyk B; Crumbliss A, Tetrakis(acetonitrile) copper(I) hexafluorophosphate. Inorg Synth 1979, 19, 90–92. [Google Scholar]
- 45.Hall MD; Telma KA; Chang K-E; Lee TD; Madigan JP; Lloyd JR; Goldlust IS; Hoeschele JD; Gottesman MM, Say no to DMSO: dimethylsulfoxide inactivates cisplatin, carboplatin, and other platinum complexes. Cancer Res 2014, 74, 3913–3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chan TR; Hilgraf R; Sharpless KB; Fokin VV, Polytriazoles as copper(I)-stabilizing ligands in catalysis. Org. Lett 2004, 6, 2853–2855. [DOI] [PubMed] [Google Scholar]
- 47.Blango MG; Ott EM; Erman A; Veranic P; Mulvey MA, Forced resurgence and targeting of intracellular uropathogenic Escherichia coli reservoirs. PLoS One 2014, 9, e93327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lin H; Fischbach MA; Liu DR; Walsh CT, In vitro characterization of salmochelin and enterobactin trilactone hydrolases IroD, IroE, and Fes. J. Am. Chem. Soc 2005, 127, 11075–11084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fischbach MA; Lin H; Liu DR; Walsh CT, In vitro characterization of IroB, a pathogen-associated C-glycosyltransferase. Proc. Natl. Acad. Sci. U.S.A 2005, 102, 571–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fischbach MA; Lin H; Liu DR; Walsh CT, How pathogenic bacteria evade mammalian sabotage in the battle for iron. Nat. Chem. Biol 2006, 2, 132–138. [DOI] [PubMed] [Google Scholar]
- 51.Hantke K; Nicholson G; Rabsch W; Winkelmann G, Salmochelins, siderophores of Salmonella enterica and uropathogenic Escherichia coli strains, are recognized by the outer membrane receptor IroN. Proc. Natl. Acad. Sci. U.S.A 2003, 100, 3677–3682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Léveillé S; Caza M; Johnson JR; Clabots C; Sabri M; Dozois CM, Iha from an Escherichia coli urinary tract infection outbreak clonal group A strain is expressed in vivo in the mouse urinary tract and functions as a catecholate siderophore receptor. Infect. Immun 2006, 74, 3427–3436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jamieson ER; Lippard SJ, Structure, recognition, and processing of cisplatin−DNA adducts. Chem. Rev 1999, 99, 2467–2498. [DOI] [PubMed] [Google Scholar]
- 54.Witkin EM, The radiation sensitivity of Escherichia coli B: a hypothesis relating filament formation and prophage induction. Proc. Natl. Acad. Sci. U. S. A 1967, 57, 1275–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Adler HI; Hardigree AA, Postirradiation growth, division, and recovery in bacteria. Radiat. Res 1965, 25, 92–102. [PubMed] [Google Scholar]
- 56.Rosenkranz HS; Garro AJ; Levy JA; Carr HS, Studies with hydroxyurea I. The reversible inhibition of bacterial DNA synthesis and the effect of hydroxyurea on the bactericidal action of streptomycin. Biochim. Biophys. Acta 1966, 114, 501–515. [PubMed] [Google Scholar]
- 57.Lippert B, Cisplatin: chemistry and biochemistry of a leading anticancer drug John Wiley & Sons, 1999. DOI: 10.1002/9783906390420. [DOI] [Google Scholar]
- 58.Reslova S, The induction of lysogenic strains of Escherichia coli by cis-dichlorodiammineplatinum (II). Chem-Biol. Interact 1971, 4, 66–70. [DOI] [PubMed] [Google Scholar]
- 59.Baba T; Ara T; Hasegawa M; Takai Y; Okumura Y; Baba M; Datsenko KA; Tomita M; Wanner BL; Mori H, Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol 2006, 2, 2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zobell CE; Cobet AB, Filament formation by Escherichia coli at increased hydrostatic pressures. J. Bacteriol 1964, 87, 710–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.ThermoFischer Scientific LIVE/DEAD BacLight Bacterial Viability Kits https://www.thermofisher.com/document-connect/document-connect.html?url=https://assets.thermofisher.com/TFS-Assets/LSG/manuals/mp07007.pdf&title=TElWRSYjNDc7REVBRCAmbHQ7aSZndDtCYWMmbHQ7L2kmZ3Q7TGlnaHQgQmFjdGVyaWFsIFZpYWJpbGl0eSBLaXRz (accessed 2020-06-19).
- 62.Hagan EC; Mobley HLT, Uropathogenic Escherichia coli outer membrane antigens expressed during urinary tract infection. Infect. Immun 2007, 75, 3941–3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Puig S; Ramos-Alonso L; Romero AM; Martínez-Pastor MT, The elemental role of iron in DNA synthesis and repair. Metallomics 2017, 9, 1483–1500. [DOI] [PubMed] [Google Scholar]
- 64.Abergel RJ; Zawadzka AM; Hoette TM; Raymond KN, Enzymatic hydrolysis of trilactone siderophores: where chiral recognition occurs in enterobactin and bacillibactin iron transport. J. Am. Chem. Soc 2009, 131, 12682–12692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Larsen NA; Lin H; Wei R; Fischbach MA; Walsh CT, Structural characterization of enterobactin hydrolase IroE. Biochemistry 2006, 45, 10184–10190. [DOI] [PubMed] [Google Scholar]
- 66.Fadzen CM; Wolfe JM; Zhou W; Cho C-F; von Spreckelsen N; Hutchinson KT; Lee Y-C; Chiocca EA; Lawler SE; Yilmaz OH; Lippard SJ; Pentelute BL, A platinum(IV) prodrug—perfluoroaryl macrocyclic peptide conjugate enhances platinum uptake in the brain. J. Med. Chem 2020, 63, 6741–6747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gabano E; Ravera M; Colangelo D; Osella D, Bioinorganic chemistry: the study of the fate of platinum-based antitumour drugs. Curr. Chem. Biol 2007, 1, 278–289. [Google Scholar]
- 68.Alessio M; Zanellato I; Bonarrigo I; Gabano E; Ravera M; Osella D, Antiproliferative activity of Pt(IV)-bis(carboxylato) conjugates on malignant pleural mesothelioma cells. J. Inorg. Biochem 2013, 129, 52–57. [DOI] [PubMed] [Google Scholar]
- 69.Ravera M; Gabano E; Zanellato I; Bonarrigo I; Alessio M; Arnesano F; Galliani A; Natile G; Osella D, Cellular trafficking, accumulation and DNA platination of a series of cisplatin-based dicarboxylato Pt(IV) prodrugs. J. Inorg. Biochem 2015, 150, 1–8. [DOI] [PubMed] [Google Scholar]
- 70.Raveendran R; Braude JP; Wexselblatt E; Novohradsky V; Stuchlikova O; Brabec V; Gandin V; Gibson D, Pt(IV) derivatives of cisplatin and oxaliplatin with phenylbutyrate axial ligands are potent cytotoxic agents that act by several mechanisms of action. Chem. Sci 2016, 7, 2381–2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Justice SS; Hunstad DA; Seed PC; Hultgren SJ, Filamentation by Escherichia coli subverts innate defenses during urinary tract infection. Proc. Natl. Acad. Sci. U.S.A 2006, 103, 19884–19889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tsionou MI; Knapp CE; Foley CA; Munteanu CR; Cakebread A; Imberti C; Eykyn TR; Young JD; Paterson BM; Blower PJ; Ma MT, Comparison of macrocyclic and acyclic chelators for gallium-68 radiolabelling. RSC Adv 2017, 7, 49586–49599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Emery T; Hoffer PB, Siderophore-mediated mechanism of gallium uptake demonstrated in the microorganism Ustilago sphaerogena. J. Nucl. Med 1980, 21, 935–939. [PubMed] [Google Scholar]
- 74.Pandey A; Savino C; Ahn SH; Yang Z; Van Lanen SG; Boros E, Theranostic gallium siderophore ciprofloxacin conjugate with broad spectrum antibiotic potency. J. Med. Chem 2019, 62, 9947–9960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ecker DJ; Matzanke BF; Raymond KN, Recognition and transport of ferric enterobactin in Escherichia coli. J. Bacteriol 1986, 167, 666–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Boyd NK; Teng C; Frei CR, Brief overview of approaches and challenges in new antibiotic development:a focus on drug repurposing. Front. Cell. Infect. Microbiol 2021, 11, 684515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Miller MJ; Walz AJ; Zhu H; Wu C; Moraski G; Möllmann U; Tristani EM; Crumbliss AL; Ferdig MT; Checkley L; Edwards RL; Boshoff HI, Design, synthesis, and study of a mycobactin−artemisinin conjugate that has selective and potent activity against tuberculosis and malaria. J. Am. Chem. Soc 2011, 133, 2076–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.