

Abstract
Introduction:
Recurrent and metastatic pheochromocytoma (PCC) are rare advanced endocrine neoplasms with limited treatment options. Insight into the pathogenic molecular alterations in patients with advanced PCC can provide therapeutic options for precisely targeting dysregulated pathways.
Objective:
We report the discovery and characterization of a novel BRAF-containing fusion transcript and its downstream molecular alterations in a patient with recurrent pheochromocytoma with peritoneal seeding (pheochromocytomatosis).
Methods:
We reviewed the medical record of a patient with pheochromocytomatosis. A comprehensive pan-cancer molecular profiling using next-generation sequencing (NGS) as well as confirmatory RT-qPCR were performed on surgical specimens. BRAF rearrangement and downstream molecular changes were assayed using fluorescence in situ hybridization (FISH) and immunohistochemistry (IHC), respectively. Western blot was used to assess the in vitro activation of the MAPK signaling pathway and the EMT markers in transfected HEK-293 cells.
Results:
The NGS analysis of a specimen from a 72-year-old female patient with pheochromocytomatosis showed an in-frame fusion of exon 3 of Glucocorticoid Induced 1 (GLCCI1) to exon 9 of BRAF. The upstream auto-inhibitory domain of BRAF was excluded from the GLCCI1-BRAF fusion; however, the downstream BRAF kinase domain was intact. A BRAF rearrangement was confirmed via a BRAF-specific break-apart FISH assay. Four separate tumor foci harbored GLCCI1-BRAF fusion. IHC demonstrated increased phosphorylated MEK. HEK-293 cells transfected with the GLCCI1-BRAF fusion demonstrated increased phosphorylated MEK as well as higher expression of EMT markers SNAI1 and ZEB1 in vitro.
Conclusion:
We demonstrate a novel pathogenic gene fusion of GLCCI1 with the oncogenic kinase domain of BRAF, resulting in an activation of the MAPK signaling pathway and EMT markers. Thus, this patient may benefit from clinically available MEK and/or BRAF inhibitors when systemic therapy is indicated.
Keywords: BRAF, GLCCI1, pheochromocytoma, pheochromocytomatosis
Summary
This report is the first of GLCCI1 fused to BRAF in a human neoplasm and only the second BRAF-containing fusion transcript in PCC. Detailed molecular characterization of PCC can be a valuable tool in managing patients with recurrent PCC and pheochromocytomatosis that represents a significant clinical challenge.
Introduction
Pheochromocytoma (PCC) is a rare neuroendocrine tumor originating from medullary chromaffin cells of the adrenal glands (1, 2). PCCs secrete bioactive amines (catecholamines) that cause episodic symptoms, elevate blood pressure, and lead to cardiovascular morbidity and mortality (3). Surgical resection is the recommended first-line treatment and can be curative for localized disease. Metastasis is seen in about 5–26% of patients with PCC, most commonly to the liver (4, 5). Locoregional and systemic therapies have been used for advanced PCC but are rarely curative. Advanced PCC can cause devastating complications via acute and chronic blood pressure elevation, including heart failure and stroke. One of the important risk factors for local recurrence is tumor spillage by violating the tumor capsule during PCC resection (6). In pheochromocytomatosis, spilled PCC cells grow into numerous tumor deposits that can be functionally active and induce cardiovascular and other end-organ complications (7). Complete surgical clearance of pheochromocytomatosis is technically challenging and may require a multi-visceral resection (8). Thus, there is a need to identify additional therapies for PCC that are not amenable to surgical management.
The molecular pathogenesis of PCC is incompletely understood. All known somatic and germline driver mutations in PCC are clustered in one of three main molecular signatures described in a landmark study from the Cancer Genome Atlas (TCGA) cohort: kinase signaling, pseudohypoxia, and Wnt-altered (9). Over 50% of the tumors in the TCGA cohort were driven by dysregulation of the kinase signaling pathway. Within this kinase signaling subgroup, fusion transcripts involving NF1, NGFR, and BRAF were detected. The BRAF fusion gene detected in one case was RUNDC1-BRAF and resulted in 5.2-fold BRAF overexpression (9). No other BRAF-containing fusions have been described in PCC.
In this report, we describe a patient with pheochromocytomatosis found to have a novel fusion of GLCCI1 and BRAF. Further study showed activated downstream signaling molecules in the mitogen-activated protein kinase (MAPK) pathway and the upregulation of SNAI1 and ZEB1, suggesting a mechanism of pathogenicity of this gene fusion.
Methods
Patient enrollment
The studies were approved by the Office of Human Subject Research, National Institutes of Health (NIH). The patients or guardians provided written informed consent and were enrolled in two protocols: Prospective Comprehensive Molecular Analysis of Endocrine Neoplasms and Diagnosis, Pathophysiology, and Molecular Biology of Pheochromocytoma and Paraganglioma (Clinical Trial Registration no. NCT01005654 and NCT00004847, NIH protocols 09-C-0242 and 00-CH-0093). All patients with PCC had germline genetic testing performed. This patient underwent the Invitae Hereditary Paraganglioma-Pheochromocytoma 14-gene Panel testing (Test #01302, Invitae, San Francisco, CA) (Supplemental Table 1). To demonstrate the activation of the MAPK signaling pathway downstream to BRAF in this patient’s tumor sample, we identified another female patient with no germline mutations associated with the MAPK signaling pathway from our prospectively maintained institutional database.
TruSight Oncology 500 assay
A tissue sample from a perisplenic nodule of recurrent PCC was formalin-fixed and paraffin-embedded (FFPE). The tissue was confirmed as an extra-adrenal PCC by histopathologic examination. RNA was extracted using the RNeasy FFPE kit (Qiagen, Germantown, MD). The hybrid capture-based assay TruSight Oncology 500 (TSO-500) and library preparation kit (# 20028214, TruSight Oncology 500 DNA Kit, Illumina, San Diego, CA) proceeded following the manufacturer’s instructions.
BRAF break apart assay
The BRAF-specific fluorescence in situ hybridization (FISH) assay was performed on FFPE tissue by NeoGenomics Laboratories, Fort Myers, FL.
Immunohistochemistry
Immunohistochemistry (IHC) was performed on 5-μm sections from FFPE tumor samples as previously reported (10). Briefly, the sections were incubated in a 1:50 dilution of anti-phosphorylated MEK (166F8, Cell Signaling Technology, Danvers, MA) overnight at 4oC followed by 1-hour incubation in biotinylated secondary antibody at room temperature. Slides were developed in SignalStain® Boost IHC Detection Reagent (#8114S, Cell Signaling Technology, Danvers, MA). We captured the representative images with a Zeiss Axio Observer D1 microscope (Carl Zeiss, Inc., Oberkochen, Germany). Hematoxylin and eosin (H&E) staining was performed on sections from the specimens.
Plasmid construction
The plasmid expressing FLAG-tagged BRAF was a gift from Dr. Dustin Maly (Addgene plasmid # 40775; http://n2t.net/addgene:40775; RRID: Addgene_40775). Plasmid integrity was verified by restriction digestion, and insert identity was confirmed by Sanger sequencing prior to downstream cloning operations. For the construction of pcDNA3.1 FLAG-GLCCI1-BRAF, the coding region for human GLCCI1 exon 1 (aa 2–157) was synthesized as three overlapping DNA oligonucleotides (Ultramers, IDT DNA) which were annealed and amplified with linker-containing primers using PCR. This fragment was combined with a synthetic DNA fragment (gBLOCK, IDT DNA) encoding GLCCI1 exons 2 and 3 (aa 158–232) and BRAF (aa 381–618) and used to replace the existing BRAF sequence (aa 2–618) in pcDNA3.1 FLAG-BRAF between the EcoRI and PflMI restriction sites. For the construction of pcDNA3.1 FLAG-only, the coding region for the start codon, the FLAG tag, and the eight amino acid spacer [M-DYKDDDDK-GILYFQSN] was amplified from the pcDNA3.1 FLAG-BRAF plasmid with linker-containing primers containing a stop codon using PCR. This fragment was used to replace the existing FLAG-BRAF sequence in pcDNA3.1 FLAG-BRAF between the EcoRI and XhoI restriction sites. For the construction of pcDNA3.1 FLAG-BRAF (V600E), the coding region FLAG-BRAF plasmid between the EcoRI and PflMI restriction sites was amplified in two segments (1.9 kb and 86 bp) with mutagenic primers encoding the BRAF(V600E) substitution using PCR. These fragments were used to replace the existing BRAF sequence (aa 2–618) in pcDNA3.1 FLAG-BRAF between the EcoRI and PflMI restriction sites. All PCR amplification were performed with Q5 High-Fidelity DNA polymerase (New England Biolabs). All cloning reactions were performed using ligation-independent cloning (In-Fusion, Takara, Inc) and transformed into NEB Stable cells (New England Biolabs). Insert-containing clones were verified by restriction digestion and Sanger sequencing prior to transfection testing.
Cell Culture
HEK-293 cells were purchased from American Type Culture Collection™ (CRL-1573™ Manassas, VA). HEK-293 cells were cultured in Minimum Essential Medium Eagle (MEM) with Earle’s salts and L-glutamine (Corning, Tewksbury, MA) with 10% FBS and maintained in 5% CO2 atmosphere at 37°C. Routine subculture was performed as required depending on the degree of confluence.
Transient Transfection
BRAF, GLCCI1-BRAF fusion, BRAF V600E, and empty plasmid were each transfected into HEK-293 cells using Lipofectamine® 3000 Transfection Kit (Invitrogen, Life Technologies Corporation, Carlsbad, CA) per manufacturers protocol. Briefly, plasmid DNA was incubated with Lipofectamine® 3000 reagent, and P3000® reagent in OPTI-MEM® reduced serum medium with HEPES, sodium bicarbonate, and L-glutamine (Thermo Fisher Scientific) for 15–30 minutes at room temperature and combined with cells in culture medium.
Western Blot Analysis
48 hours after transfection, cell lysates were prepared from HEK-293 cells, and protein was quantified using a Pierce™ BCA assay kit (Thermo Fisher Scientific). An equal amount of protein was used from each transfection condition for western blot and separated by electrophoresis on 4–15% gradient gels. After electrophoresis, the protein was transferred to a PVDF membrane, blocked with 5% milk in TBS-Tween buffer (ThermoFisher), then incubated overnight at 4°C with primary antibodies diluted 1:1000 in 5% bovine serum albumin (BSA). Primary antibodies used were anti-FLAG™ (F3165 Sigma-Aldrich, St. Louis, MO), anti-phospho-MEK (166F8 Cell Signaling Technology, Danvers, MA [CST]), anti-MEK (L38C12, CST), anti-SNAI1 (3879S, CST), and anti-ZEB1 (3396T, CST). Equal loading was confirmed by incubating with either anti-p84 (GTX7002 GeneTex, Irvine, CA) or horseradish peroxidase (HRP) conjugated anti-GAPDH (D16H11, CST) or anti-β-actin (12262S, CST). Next, PVDF membranes were washed with TBS-Tween for 30 minutes and incubated at room temperature for 1 hour with either anti-rabbit (7074S, CST) or anti-mouse (7076S, CST) IgG HRP conjugated secondary antibodies for all except HRP-anti-GAPDH and HRP-anti-β-actin. After secondary antibody incubation, membranes were washed again and analyzed using an enhanced chemiluminescence reagent (Thermo Fisher Scientific, Pierce™).
Real-Time-quantitative PCR (RT-qPCR) and sequencing
Primers and probes were synthesized by Integrated DNA Technologies (Coralville, Iowa). RNA was extracted using the RNeasy FFPE kit (Qiagen, Germantown, MD) and reverse transcribed to cDNA using Superscript IV First strand synthesis system (Thermo Fisher, Waltham, MA). RT-qPCR assays were performed using TaqMan Fast Advanced Master Mix (Thermo Fisher) on a ViiA™ 7 RT-qPCR System (Applied Biosystem, Carlsbad, CA). Cycling conditions consisted of initial denaturation at 95oC for 20 sec followed by 45 cycles of 95oC for 1 sec and 60oC for 20 sec. Expression of the GLCCI1-BRAF fusion was assayed with primers GLCCI1 e3A and BRAF e10A along with probe GLCCI1 e3B (Table 1). As an internal control, commercially available primers and probes were used to assay the expression of GAPDH (Life Technologies, Carlsbad, CA). As a negative control specimen, RNA was isolated from a FFPE xenograft of the RH30 rhabdomyosarcoma cell line (11) and assayed for GLCCI1-BRAF and GAPDH expression. Data analysis was performed with QuantStudio Real-Time PCR Software (Applied Biosystems).
Table 1:
Primers for PCR assays
| Name | Sequence (5’-3’) | Modification |
|---|---|---|
| BRAF e10A | CCACGAAATCCTTGGTCTCTAA | None |
| BRAF e10B | TCCTTTCTCGCTGAGGTCCT | None |
| GLCCI1 e3A | GCAGAAGAGGGTGCAGAAA | None |
| GLCCI1 e3B | ATGACGCAGAACGCTGATGTGACC | 6-FAM/TAMRA |
| GLCCI1 e3C | CATGGGGGAGTGCTGATCAA | None |
To isolate a portion of the fusion transcript for sequencing, RNA from the perisplenic specimen was reverse transcribed and amplified using primer GLCCI1 e3C and BRAF e10B (Table 1) with MyTaq DNA polymerase (Meridian Biosciences, Cincinnati, OH). An aliquot of the PCR product was cloned into the pCR2.1 vector using the TOPO TA Cloning kit (Thermo Fisher) and transformed in 5-alpha competent E. coli (New England Biolabs, Ipswich, MA). Clones were screened by EcoR1 digestion and agarose gel electrophoresis, and selected inserts were analyzed by standard Sanger sequencing (Eurofins Genomics, Louisville, KY).
Case report
A 72-year-old female presented to the NIH Warren Magnuson Clinical Center after the discovery of recurrent PCC. There was no family history of PCC or other phenotypes of PCC-related syndromes. She had previously undergone a laparoscopic transperitoneal left adrenalectomy at an outside institution over ten years before being evaluated at NIH. The pathology report from this initial operation noted a 2.5 cm PCC with extensive laceration, indicating tumor rupture during surgery. She did not have subsequent surveillance. One year before establishing care at the NIH, she had a hypertensive ischemic stroke, leaving her with persistent slurred speech secondary to right-sided cranial nerve XII palsy. Workup led to the discovery of recurrent PCC. The hypertensive episodes secondary to her recurrent PCC were managed with doxazosin and propranolol. Preoperative workup demonstrated elevated plasma metanephrine of 390 pg/mL (reference range 12–61 pg/mL), normetanephrine of 742 pg/mL (reference range 18–112 pg/mL), epinephrine of 125 pg/mL (reference range 0–50 pg/mL), and norepinephrine of 2904 pg/mL (reference range 112 – 750 pg/mL) – levels consistent with PCC. Twenty-four-hour urine studies demonstrated markedly elevated normetanephrine of 3210 mcg/24 hours (reference range <900 mcg/24 hours) and metanephrine of 2330 mcg/24 hours (reference range <400 pg/mL). Genetic testing did not demonstrate any pathogenic germline mutations in genes known to cause hereditary PCC syndromes. A contrast-enhanced CT scan and a gadolinium-enhanced MRI of the abdomen showed numerous hypervascular lesions near the left diaphragm, kidney, paracolic gutter, and spleen (Figure 1a and b). 68-Gallium DOTATATE PET/CT and 18-FDOPA PET/CT revealed radioavid tumors, consistent with recurrent PCC in the left adrenal bed (Figure 1c and d). The consensus reached at a multidisciplinary tumor board was to offer a surgical resection to control her hypertension and to reduce the risk of catastrophic cerebrovascular or cardiovascular events. She was admitted preoperatively for blood pressure control via alpha adrenoceptor blockade with phenoxybenzamine.
Figure 1:
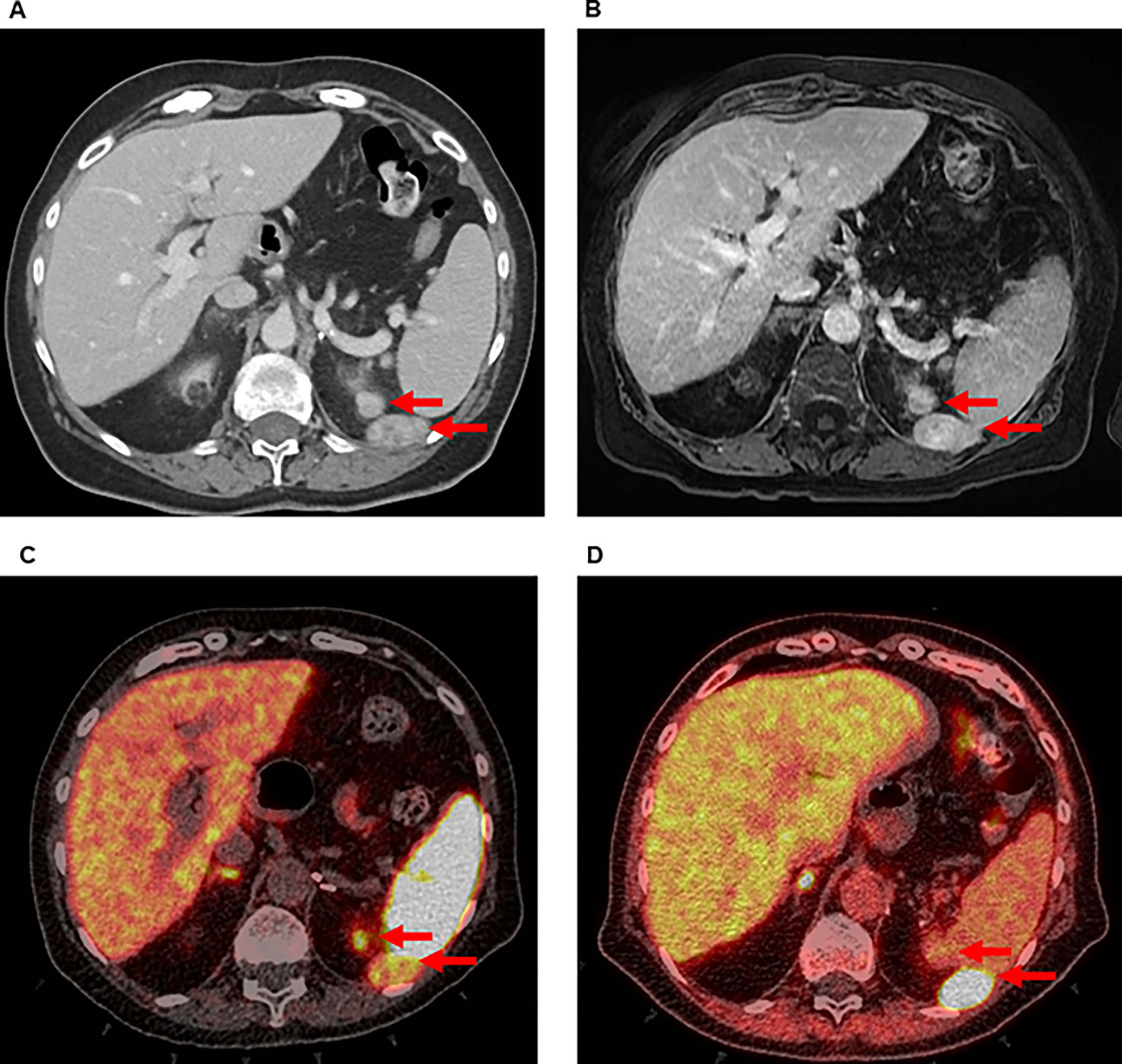
Preoperative imaging of a patient with recurrent PCC in left upper quadrant of the abdomen by (A) CT, (B) MRI, (C) 68-Ga DOTATATE PET/CT, and (D) 18-FDOPA PET/CT. Arrows indicate radiographically detectable lesions.
On exploratory laparotomy, multiple purple nodules in the left upper quadrant were visualized, consistent with pheochromocytomatosis. An extensive cytoreduction was performed, including resection of peritoneal and retroperitoneal nodules, distal pancreatectomy, splenectomy, diaphragm resection, and partial nephrectomy. Intraoperatively, the patient had labile blood pressure during tumor manipulation that required esmolol and sodium nitroprusside administration, as well as hypotension briefly requiring phenylephrine. At the conclusion of the case, there was no visible evidence of disease.
The histopathologic assessment of these nodules showed neuroendocrine cells in nests, consistent with PCC, as well as confirmation of clear margins. Additional IHC staining showed intact expression of SDHB and SDHA, and MIB1 staining showed a low proliferative index of 1–2% (data not shown). The patient underwent a surveillance CT scan at three months postoperatively with no evidence of disease. The postoperative biochemical testing at this time demonstrated normalized plasma metanephrine (44 pg/mL) and decreased normetanephrine (346 pg/mL). At 1 year following cytoreduction, the patient’s radiologic surveillance demonstrated no evidence of disease recurrence.
Results
Detection of a novel GLCCI1-BRAF fusion transcript using next-generation sequencing
To better elucidate any actionable pathogenic mutations in this recurrent PCC, we performed NGS with the TSO-500 panel. The NGS analysis revealed a novel transcript with an in-frame fusion between GLCCI1 exon 3 and BRAF exon 9 (Figure 2a). This fusion transcript is postulated to result from breaks in GLCCI1 intron 3 and BRAF intron 8 followed by joining of the breaks to give a GLCCI1-BRAF fusion gene with a chimeric intron consisting of parts of GLCCI1 intron 3 and BRAF intron 8 (Figures 2b–c). The GLCCI1-BRAF fusion transcript was detected in 579 unique reads (criterion for positive ≥ 6 reads). The location of the BRAF fusion junction in the predicted GLCCI1-BRAF protein product excluded the upstream autoinhibitory region that regulates BRAF downstream signaling (Figure 2d). Given its structural similarity to other known pathogenic BRAF fusions (12), this fusion was determined to be likely pathogenic. N-of-One, the Catalogue of Somatic Mutations in Cancer (COSMIC), and cBioportal databases were queried, with no prior entrees of this fusion listed. No other pathogenic or likely pathogenic variants were detected. Further analysis of the NGS data revealed the tumor to have a low mutational burden (0.786 Mutations/Megabase), no reportable copy number variations, and a stable microsatellite status. No pathogenic variants previously associated with PCC or paraganglioma were identified. No copy number variations were detected in the 92 genes tested by TSO-500 panel.
Figure 2:

Fusion between GLCCI1 exon 3 and BRAF exon 9. (A) NGS data aligned to genomic DNA reference. (B) Diagram of a portion of the fusion gene and the resulting fusion transcript. In the fusion gene, GLCCI1 exon 3 and part of GLCCI1 intron 3 are joined to part of BRAF intron 8 and BRAF exon 9. This fusion gene is transcribed and processed into a fusion transcript in which GLCCI1 exon 3 is joined to BRAF exon 9. The boxes represent exons, and the horizontal lines represent introns. (C) Nucleotide sequence of the corresponding regions of the fusion gene and fusion transcript. Exons and introns are shown as upper case and lower-case letters, respectively. The amino acid sequence is shown with single letter abbreviations below the sequence. (D) Map of the predicted protein products of the wild-type GLCCI1, GLCCI1-BRAF fusion and wild-type BRAF genes. The blue rectangles represent functional domains: FAM117, FAM117 family homology domain; RBD, Ras-binding domain; C1D, Phorbol ester/diacylglycerol-binding zinc finger motif; KD, serine-threonine-specific protein kinase domain. The vertical dashed line represents the fusion point between the GLCCI1 and BRAF protein products.
Next, we assessed whether the novel fusion oncogene was present in other recurrent tumor foci. We isolated RNA from four other tumor specimens obtained during the same cytoreductive surgery and performed RT-qPCR to amplify GLCCI1-BRAF. All four specimens did contain evidence of the novel fusion transcript, as did RNA taken from the first sample on which TSO-500 was performed (Supplemental Figure 1a). Further RT-qPCR analysis of the perisplenic specimen revealed a PCR product for which the predicted size of 168 bp was confirmed by agarose gel electrophoresis. After subcloning the PCR product into a plasmid, standard sequence analysis identified the predicted fusion junction (Supplemental Figure 1b). Taken together, the GLCCI1-BRAF fusion was present in multiple separate tumor foci suggesting that the fusion was not an isolated subclonal event.
Confirmation of BRAF gene rearrangement
To generate the fusion gene, a rearrangement is predicted to generate a break within the BRAF locus and thereby separate the 5’ and 3’ ends of the BRAF gene. To confirm the presence of BRAF rearrangement at the genomic DNA level, we tested a sample of the patient’s recurrent PCC tissue using a BRAF-specific FISH break-apart assay. An isolated 3’ signal was present in 46% (criterion for positive ≥ 9%) of nuclei analyzed (Figure 3). Coupled with the GLCCI1-BRAF fusion transcript identified by the NGS, the discontinuity of BRAF seen via FISH was consistent with rearrangements of GLCCI1 and BRAF to form a fusion gene. Since both GLCCI1 and BRAF are located on chromosome 7, we hypothesize that the fusion gene can be formed by an inversion or translocation mechanism (Figures 4a–c).
Figure 3:

FISH assay confirming BRAF gene rearrangement. The break-apart assay was positive for an isolated BRAF 3’ signal in 46% of the analyzed nuclei (criterion for positive ≥ 9%). Probes: BRAF 5’ end – green, BRAF 3’ end – red (indicated by red arrows).
Figure 4:

Diagrams of normal and rearranged chromosome 7. (A) Normal chromosome 7 with the locations and transcriptional directions of the GLCCI1 and BRAF genes. (B), (C) Two possible chromosomal rearrangement mechanisms (inversion (B) or translocation (C)) that can generate the GLCCI1-BRAF fusion gene.
Activated MAPK signaling pathway in tumor bearing GLCCI1-BRAF fusion
We sought to confirm the BRAF/MAPK pathway activation in the specimen containing the novel fusion. For comparison, we queried a prospectively maintained database of PCC specimens resected at our institution that had NGS performed using the TSO-500 panel. We selected one of these PCCs for further analysis, resected from a female patient with a germline Von Hippel-Lindau (VHL) mutation 499C>T, as well as three somatic variants of unknown significance (VUS) that are associated with the MAPK signaling pathway (MAP3K1 1415T>G, NOTCH3 3314G>C, and PDGFRA 2411G>A). Microscopic examination of H&E stained sections of the BRAF fusion-positive specimen demonstrated polygonal neoplastic cells with abundant eosinophilic cytoplasm and conspicuous nucleoli consistent with PCC cells (Figure 5). IHC was performed to determine if the MAPK signaling pathway was upregulated in the recurrent PCC tissue with GLCCI1-BRAF fusion. Compared to tumor samples from the patient with VHL, the PCC with GLCCI1-BRAF fusion had an increased level of phosphorylated MEK (pMEK), consistent with constitutive BRAF activation resulting from loss of the BRAF autoinhibitory domain in the GLCCI1-BRAF gene product.
Figure 5:

Immunohistochemical staining of pMEK demonstrates MAPK pathway activation in BRAF fusion compared with a VHL-driven PCC. Representative photomicrographs of sections stained with H&E at 10x and pMEK IHC at 10x and 40x magnification are shown for each specimen.
GLCCI1-BRAF pathogenically signals through the MAPK pathway in vitro
To further elucidate the effect of the GLCCI1-BRAF fusion protein on cell signaling, we generated the novel fusion oncogene in a plasmid and transiently transfected it into HEK-293 cells. Western blotting confirmed the expression of the FLAG-tagged protein. The fusion GLCCI1-BRAF protein had a lower molecular weight than WT and V600E BRAF, matching with the predicted molecular weight based on the DNA sequence (Figure 6a). The transfection of the fusion construct resulted in increased levels of phosphorylated MEK after 48 hours relative to a negative control plasmid, comparable to that of the BRAF V600E positive control. To identify the molecular process associated with multifocal intraperitoneal recurrence, we studied the EMT markers. The transfection of HEK-293 cells with the fusion-containing plasmid increased the expression of SNAI1 and ZEB1 relative to control plasmid (Figure 6b). The results suggested that GLCCI1-BRAF fusion confers pathogenicity through the MAPK pathway and the EMT process in a manner similar to other known BRAF mutant cancers.
Figure 6:

Downstream changes resulting from GLCCI1-BRAF fusion in vitro. Western blots of HEK-293 cells transfected with plasmids containing either WT BRAF, GLCCI1-BRAF, BRAFV600E, or empty vector. (A) Top panels: confirmation of BRAF expression. Bottom panels: phosphorylated MEK and total MEK expression. (B) Top panels: SNAI1 expression. Bottom panels: ZEB1 expression.
Discussion
Primary PCC occurs at an incidence of 2–8 per million people (13–16). Metastatic disease is found in 5–26% of cases and can occur at any time, both synchronously and years after initial resection (5, 17). While surgery can be curative for primary PCC, functional metastases confer significant cardiovascular morbidity and reduces 5-year overall survival from 89% to as low as 37% (18, 19). Clinical risk factors associated with metastasis include male sex, older age at diagnosis, larger tumor size, elevated dopamine or norepinephrine, SDHB mutation, and failure to undergo resection of a primary tumor (14, 20–23). Pathologic risk factors used to predict survival have been included in scoring systems such as Pheochromocytoma of the Adrenal Gland Scaled Score (PASS) and the Grading System for Adrenal Pheochromocytoma and Paraganglioma (GAPP) (23–26). Treatment of metastatic PCC is generally palliative, with cytoreductive procedures, when possible, demonstrating improvement of symptoms (17, 27). Less is known about the etiology of pheochromocytomatosis. While it is thought to arise iatrogenically from spilled tumor cells during surgery, patient and genetic factors that lead to symptomatic local recurrence are not specifically described (7, 8). In this case report, the authors believe that technical error with primary tumor spillage and the GLCCI1-BRAF fusion causing tumor cells to invade contributed to recurrent pheochromocytomatosis. Due to the paucity of data on management, pheochromocytomatosis generally follows the same treatment algorithms as metastatic PCC. Thus, there is a need to develop additional therapeutic options for metastatic PCC and pheochromocytomatosis.
Knowledge of PCC genetics may be useful to guide the choice of targeted therapy for unresectable disease (1, 28). Pathogenic germline mutations identified by TCGA include SDHB, RET, VHL, NF1, SDHD, MAX, EGLN1, and TMEM127 (9). Of the germline mutations that drive PCC, SDHB has the highest association with metastasis (29–31). For somatic genes identified by TCGA, the most frequently mutated in PCC was HRAS, found upstream of BRAF in signal transduction. Somatic alterations associated with aggressive disease include MAML3 fusions, mutations in SETD2, ATRX, genes involved in the Wnt and pseudohypoxia pathways, as well as a high total number of somatic mutations (9, 32). Currently, no targeted molecular agents are approved for metastatic PCC (33, 34). The treatment with somatostatin receptor analogs (SSAs) with or without radioisotope conjugates can be considered in a clinical trial.
Fusion transcripts are a significant source of pathogenesis in cancer (35) and may generate tumor-specific proteins potentially targetable by small molecules and/or immunotherapy (36). Of the ten previously reported fusion transcripts in PCC by TCGA analysis, seven contained MAML3 and were implicated in the Wnt-altered oncogenic pathway. In contrast, the remaining three fusion transcripts were classified in the kinase pathway, including a RUNDC1-BRAF fusion found in one case (9). Together with our report on the novel GLCCI1-BRAF fusion, these are the only two known BRAF-containing fusion transcripts in PCC to date. Other BRAF fusions have been found in melanoma, pancreatic adenocarcinoma, thyroid carcinoma, prostate carcinoma, gastric adenocarcinoma, and the majority of pilocytic astrocytomas (12, 37, 38). In the patient described above with resected pheochromocytomatosis, selection of a BRAF inhibitor (BRAFi) and/or a systemic agent targeting downstream molecules of the MAPK signaling pathway may become relevant should an unresectable recurrence arise.
The BRAF isoform of the RAF family of proteins is one of the most commonly mutated oncoproteins in solid tumors (39, 40). Functionally, BRAF is a cytoplasmic serine/threonine kinase known to be involved in the RAS-RAF-MEK-ERK-MAPK signal transduction pathway (41). Structurally, BRAF contains an upstream autoinhibitory domain, a linker region, and a downstream kinase domain (12). Of the known activating mutations in BRAF, point mutations within the kinase domain at V600 are the most common (39). Several V600 substitutions in particular, are targetable with the FDA-approved small molecule inhibitors vemurafenib, dabrafenib, and encorafenib (42). There is one report of PCC harboring a BRAF V600E mutation (43). Compared to activating point mutations, fusion transcripts containing BRAF are relatively rare in cancer. Many of the known BRAF fusions are thought to be oncogenic due to an exclusion of the upstream autoinhibitory domain found in wild-type BRAF (12). Targeted therapies for fusions involving BRAF are described in other cancers but not in PCC.
BRAF mutations have three mechanistic classes relevant for pharmacological inhibition (39). Class I mutant BRAF is defined as V600 substitution that constitutively activates the MAPK signaling pathway as a monomer. The mechanism of monomer signaling is salt bridge formation with the K507 residue that structurally mimics the activated kinase conformation typical of dimerized wild-type BRAF. In contrast, non-V600 mutated BRAF functions as a dimer, either with high kinase activity (class II) or reduced “kinase-dead” activity through alternative signaling involving a second mechanism (class III). The majority of BRAF fusions with an intact kinase domain are considered to function comparably to class II point mutations (44). We predict that GLCCI1-BRAF is a class II mutation, based on (1) the upregulation of phosphorylated MEK in transfected cells and (2) the deletion of the autoregulatory region of the BRAF gene in a manner similar to other known pathogenic BRAF fusions. The overexpression of EMT markers such as SNAI1 and ZEB1 caused by the GLCCI1-BRAF fusion is consistent with invasive phenotypes described previously in BRAF-mutant thyroid cancer and melanoma (45).
BRAFi show a range of efficacy based on mutation class. For class I mutations, the first-generation BRAFi dabrafenib (46), encorafenib (47), and vemurafenib (48) have all been FDA-approved for cutaneous melanoma and have shown experimental promise for multiple solid tumor types containing V600 mutations (49, 50). Due to their specificity for activated mutant BRAF monomers, first-generation BRAFi are generally less effective against class II and III mutant BRAF. Second generation BRAFi are under investigation as dual pathway inhibitors, or “paradox-breakers,” that limit MEK, ERK, and MAPK paradoxical upregulation. Many paradox-breakers also function as pan-RAF inhibitors by blocking dimerization-dependent signaling that is seen in class II and fusion BRAF. PLX8394 is one such second-generation BRAFi that has shown in vitro efficacy against class II and fusion BRAF-driven cell lines as an ERKi and MAPKi (51), in addition to blockade of BRAF dimer-dependent signaling (52). A clinical trial investigating PLX8394 in advanced solid tumors is currently enrolling patients (NCT02428712).
Like other reported pathogenic BRAF fusions, GLCCI1-BRAF excludes the upstream autoinhibitory domain but preserves the wild-type kinase functional domain. BRAF fusions have been demonstrated to exhibit BRAFi resistance similar to class II mutations, with paradoxical upregulation of MAPK signaling that is sensitive to paradox-breaker therapies (53). In addition, most fusions with wild-type kinase domains signal in a dimer-dependent mechanism. Therefore, the selection of a second-generation agent such as PLX8394 would be a rational strategy for systemic therapy should the patient’s pheochromocytoma recur without options for surgical management. Additionally, because these tumors expressed somatostatin receptors and were radioavid on 68-Ga DOTATATE PET/CT, this patient may be eligible for clinical trials of 177-Lu DOTATATE or systemic SSA therapy. Given the advanced nature of the patient’s disease, lifelong surveillance is appropriate in order to rapidly initiate treatment and avoid cardiovascular complications.
While BRAF fusions display active kinase signal transduction in a dimer-dependent mechanism (44, 54), the 5’ partner may exert some influence over this interaction (55). GLCCI1 is a glucocorticoid inducible gene product found in multiple tissues. It is known for modulating the asthmatic response to inhaled glucocorticoids, as polymorphisms of GLCCI1 were shown to correlate with reduced FEV1 improvement in a genome-wide association study (56). While GLCCI1 has also been implicated in normal renal glomerulus function (57) and steroid-induced thymocyte apoptosis regulation (58, 59), the functional role of GLCCI1 in cancer is not well established. GLCCI1 has been found in several fusion proteins in cancer, including ST3GAL3-GLCCI1 in paraganglioma (36). Though we cannot exclude a functional role of GLCCI1 exons 1–3, we hypothesize that oncogenesis in GLCCI1-BRAF occurs primarily through mechanisms similar to that of other BRAF fusion proteins (12). Further studies would be required to show this mechanism definitively.
There are several limitations to this study. First, due to the remote timeline of the patient’s index operation at an outside hospital, we did not have access to the patient’s primary tumor. It is therefore unclear whether the GLCCI1-BRAF fusion event was present as the oncogenic driver of the primary tumor. In theory, the fusion could have existed in or developed from a subclone of pre-established PCC tumor deposits, accelerating the growth of the recurrent disease. Nevertheless, the presence of the GLCCI1-BRAF fusion in multiple metastatic tumor foci, along with the upregulation of MAPK signaling and the EMT markers, suggest the pathogenicity of this gene fusion. Second, our study does not characterize the mechanism of the GLCCI1-BRAF fusion event. Both BRAF and GLCCI1 are located on chromosome 7, suggesting the possibility of an inversion event following two double-strand breaks in one copy of chromosome 7, or a translocation event following one double-strand break on each of the two copies of chromosome 7.
Supplementary Material
Supplemental Figure 1: (A) RT-qPCR demonstrating presence of GLCCI1-BRAF within RNA isolated from indicated specimens from the patient’s cytoreductive operation. Negative control: RH30 xenograft rhabdomyosarcoma. Positive control: perisplenic nodule used for TSO-500. (B) Electrophoresis gel of RT-qPCR products from perisplenic specimen, and raw data of subsequent sequencing.
Supplemental Table 1: List of genes tested in the Invitae Hereditary Paraganglioma-Pheochromocytoma 14-gene Panel.
Funding
This research was supported by the Intramural Research Program of the National Cancer Institute and Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health.
Footnotes
Declaration of competing interest
Disclosure Statement: The authors have nothing to disclose.
References
- 1.Favier J, Amar L, Gimenez-Roqueplo AP. Paraganglioma and phaeochromocytoma: from genetics to personalized medicine. Nat Rev Endocrinol. 2015;11(2):101–11. [DOI] [PubMed] [Google Scholar]
- 2.Turchini J, Cheung VKY, Tischler AS, De Krijger RR, Gill AJ. Pathology and genetics of phaeochromocytoma and paraganglioma. Histopathology. 2018;72(1):97–105. [DOI] [PubMed] [Google Scholar]
- 3.Lenders JWM, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. The Lancet. 2005;366(9486):665–75. [DOI] [PubMed] [Google Scholar]
- 4.Ayala-Ramirez M, Feng L, Johnson MM, Ejaz S, Habra MA, Rich T, et al. Clinical risk factors for malignancy and overall survival in patients with pheochromocytomas and sympathetic paragangliomas: primary tumor size and primary tumor location as prognostic indicators. J Clin Endocrinol Metab. 2011;96(3):717–25. [DOI] [PubMed] [Google Scholar]
- 5.Eisenhofer G, Bornstein SR, Brouwers FM, Cheung NK, Dahia PL, de Krijger RR, et al. Malignant pheochromocytoma: current status and initiatives for future progress. Endocr Relat Cancer. 2004;11(3):423–36. [DOI] [PubMed] [Google Scholar]
- 6.Rafat C, Zinzindohoue F, Hernigou A, Hignette C, Favier J, Tenenbaum F, et al. Peritoneal implantation of pheochromocytoma following tumor capsule rupture during surgery. J Clin Endocrinol Metab. 2014;99(12):E2681–5. [DOI] [PubMed] [Google Scholar]
- 7.Robledo AB, Marco JLP, Ibáñez TB, Anastasio MFM, Gavara CG. Pheochromocytomatosis: A Risk after Pheochromocytoma Surgery. The American Surgeon. 2010;76(8):122–4. [PubMed] [Google Scholar]
- 8.Li ML, Fitzgerald PA, Price DC, Norton JA. Iatrogenic pheochromocytomatosis: a previously unreported result of laparoscopic adrenalectomy. Surgery. 2001;130(6):1072–7. [DOI] [PubMed] [Google Scholar]
- 9.Fishbein L, Leshchiner I, Walter V, Danilova L, Robertson AG, Johnson AR, et al. Comprehensive Molecular Characterization of Pheochromocytoma and Paraganglioma. Cancer Cell. 2017;31(2):181–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nilubol N, Boufraqech M, Zhang L, Gaskins K, Shen M, Zhang YQ, et al. Synergistic combination of flavopiridol and carfilzomib targets commonly dysregulated pathways in adrenocortical carcinoma and has biomarkers of response. Oncotarget. 2018;9(68):33030–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Olanich ME, Sun W, Hewitt SM, Abdullaev Z, Pack SD, Barr FG. CDK4 Amplification Reduces Sensitivity to CDK4/6 Inhibition in Fusion-Positive Rhabdomyosarcoma. Clin Cancer Res. 2015;21(21):4947–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ross JS, Wang K, Chmielecki J, Gay L, Johnson A, Chudnovsky J, et al. The distribution of BRAF gene fusions in solid tumors and response to targeted therapy. Int J Cancer. 2016;138(4):881–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lowery AJ, Walsh S, McDermott EW, Prichard RS. Molecular and therapeutic advances in the diagnosis and management of malignant pheochromocytomas and paragangliomas. Oncologist. 2013;18(4):391–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hamidi O, Young WF Jr., Iniguez-Ariza NM, Kittah NE, Gruber L, Bancos C, et al. Malignant Pheochromocytoma and Paraganglioma: 272 Patients Over 55 Years. J Clin Endocrinol Metab. 2017;102(9):3296–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berends AMA, Buitenwerf E, de Krijger RR, Veeger N, van der Horst-Schrivers ANA, Links TP, et al. Incidence of pheochromocytoma and sympathetic paraganglioma in the Netherlands: A nationwide study and systematic review. Eur J Intern Med. 2018;51:68–73. [DOI] [PubMed] [Google Scholar]
- 16.Crona J, Taieb D, Pacak K. New Perspectives on Pheochromocytoma and Paraganglioma: Toward a Molecular Classification. Endocr Rev. 2017;38(6):489–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Filpo G, Maggi M, Mannelli M, Canu L. Management and outcome of metastatic pheochromocytomas/paragangliomas: an overview. J Endocrinol Invest. 2021;44(1):15–25. [DOI] [PubMed] [Google Scholar]
- 18.Zarnegar R, Kebebew E, Duh QY, Clark OH. Malignant pheochromocytoma. Surg Oncol Clin N Am. 2006;15(3):555–71. [DOI] [PubMed] [Google Scholar]
- 19.Hamidi O, Young WF Jr., Gruber L, Smestad J, Yan Q, Ponce OJ, et al. Outcomes of patients with metastatic phaeochromocytoma and paraganglioma: A systematic review and meta-analysis. Clin Endocrinol (Oxf). 2017;87(5):440–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roman-Gonzalez A, Zhou S, Ayala-Ramirez M, Shen C, Waguespack SG, Habra MA, et al. Impact of Surgical Resection of the Primary Tumor on Overall Survival in Patients With Metastatic Pheochromocytoma or Sympathetic Paraganglioma. Ann Surg. 2018;268(1):172–8. [DOI] [PubMed] [Google Scholar]
- 21.Hescot S, Curras-Freixes M, Deutschbein T, van Berkel A, Vezzosi D, Amar L, et al. Prognosis of Malignant Pheochromocytoma and Paraganglioma (MAPP-Prono Study): A European Network for the Study of Adrenal Tumors Retrospective Study. J Clin Endocrinol Metab. 2019;104(6):2367–74. [DOI] [PubMed] [Google Scholar]
- 22.Crona J, Lamarca A, Ghosal S, Welin S, Skogseid B, Pacak K. Genotype-phenotype correlations in pheochromocytoma and paraganglioma: a systematic review and individual patient meta-analysis. Endocr Relat Cancer. 2019;26(5):539–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mei L, Khurana A, Al-Juhaishi T, Faber A, Celi F, Smith S, et al. Prognostic Factors of Malignant Pheochromocytoma and Paraganglioma: A Combined SEER and TCGA Databases Review. Horm Metab Res. 2019;51(7):451–7. [DOI] [PubMed] [Google Scholar]
- 24.Wachtel H, Hutchens T, Baraban E, Schwartz LE, Montone K, Baloch Z, et al. Predicting Metastatic Potential in Pheochromocytoma and Paraganglioma: A Comparison of PASS and GAPP Scoring Systems. J Clin Endocrinol Metab. 2020;105(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kimura N, Takayanagi R, Takizawa N, Itagaki E, Katabami T, Kakoi N, et al. Pathological grading for predicting metastasis in phaeochromocytoma and paraganglioma. Endocr Relat Cancer. 2014;21(3):405–14. [DOI] [PubMed] [Google Scholar]
- 26.Thompson LD. Pheochromocytoma of the Adrenal gland Scaled Score (PASS) to separate benign from malignant neoplasms: a clinicopathologic and immunophenotypic study of 100 cases. Am J Surg Pathol. 2002;26(5):551–66. [DOI] [PubMed] [Google Scholar]
- 27.Nolting S, Grossman A, Pacak K. Metastatic Phaeochromocytoma: Spinning Towards More Promising Treatment Options. Exp Clin Endocrinol Diabetes. 2019;127(2–03):117–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nolting S, Ullrich M, Pietzsch J, Ziegler CG, Eisenhofer G, Grossman A, et al. Current Management of Pheochromocytoma/Paraganglioma: A Guide for the Practicing Clinician in the Era of Precision Medicine. Cancers (Basel). 2019;11(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Amar L, Baudin E, Burnichon N, Peyrard S, Silvera S, Bertherat J, et al. Succinate dehydrogenase B gene mutations predict survival in patients with malignant pheochromocytomas or paragangliomas. J Clin Endocrinol Metab. 2007;92(10):3822–8. [DOI] [PubMed] [Google Scholar]
- 30.Turkova H, Prodanov T, Maly M, Martucci V, Adams K, Widimsky J Jr., et al. Characteristics and Outcomes of Metastatic Sdhb and Sporadic Pheochromocytoma/Paraganglioma: An National Institutes of Health Study. Endocr Pract. 2016;22(3):302–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Benn DE, Gimenez-Roqueplo AP, Reilly JR, Bertherat J, Burgess J, Byth K, et al. Clinical presentation and penetrance of pheochromocytoma/paraganglioma syndromes. J Clin Endocrinol Metab. 2006;91(3):827–36. [DOI] [PubMed] [Google Scholar]
- 32.Fishbein L, Khare S, Wubbenhorst B, DeSloover D, D’Andrea K, Merrill S, et al. Whole-exome sequencing identifies somatic ATRX mutations in pheochromocytomas and paragangliomas. Nat Commun. 2015;6:6140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pang Y, Liu Y, Pacak K, Yang C. Pheochromocytomas and Paragangliomas: From Genetic Diversity to Targeted Therapies. Cancers (Basel). 2019;11(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Y, Liu L, Zhu F. Therapies targeting the signal pathways of pheochromocytoma and paraganglioma. Onco Targets Ther. 2019;12:7227–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Salokas K, Weldatsadik RG, Varjosalo M. Human transcription factor and protein kinase gene fusions in human cancer. Sci Rep. 2020;10(1):14169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gao Q, Liang WW, Foltz SM, Mutharasu G, Jayasinghe RG, Cao S, et al. Driver Fusions and Their Implications in the Development and Treatment of Human Cancers. Cell Rep. 2018;23(1):227–38 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jones DT, Hutter B, Jager N, Korshunov A, Kool M, Warnatz HJ, et al. Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet. 2013;45(8):927–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Palanisamy N, Ateeq B, Kalyana-Sundaram S, Pflueger D, Ramnarayanan K, Shankar S, et al. Rearrangements of the RAF kinase pathway in prostate cancer, gastric cancer and melanoma. Nat Med. 2010;16(7):793–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dankner M, Rose AAN, Rajkumar S, Siegel PM, Watson IR. Classifying BRAF alterations in cancer: new rational therapeutic strategies for actionable mutations. Oncogene. 2018;37(24):3183–99. [DOI] [PubMed] [Google Scholar]
- 40.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–54. [DOI] [PubMed] [Google Scholar]
- 41.Lavoie H, Therrien M. Regulation of RAF protein kinases in ERK signalling. Nature Reviews Molecular Cell Biology. 2015;16(5):281–98. [DOI] [PubMed] [Google Scholar]
- 42.Karoulia Z, Gavathiotis E, Poulikakos PI. New perspectives for targeting RAF kinase in human cancer. Nat Rev Cancer. 2017;17(11):676–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Luchetti A, Walsh D, Rodger F, Clark G, Martin T, Irving R, et al. Profiling of somatic mutations in phaeochromocytoma and paraganglioma by targeted next generation sequencing analysis. Int J Endocrinol. 2015;2015:138573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schreck KC, Grossman SA, Pratilas CA. BRAF Mutations and the Utility of RAF and MEK Inhibitors in Primary Brain Tumors. Cancers (Basel). 2019;11(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mitchell B, Dhingra JK, Mahalingam M. BRAF and Epithelial-Mesenchymal Transition: Lessons From Papillary Thyroid Carcinoma and Primary Cutaneous Melanoma. Adv Anat Pathol. 2016;23(4):244–71. [DOI] [PubMed] [Google Scholar]
- 46.Hauschild A, Grob J-J, Demidov LV, Jouary T, Gutzmer R, Millward M, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. The Lancet. 2012;380(9839):358–65. [DOI] [PubMed] [Google Scholar]
- 47.Dummer R, Ascierto PA, Gogas HJ, Arance A, Mandala M, Liszkay G, et al. Overall survival in patients with BRAF-mutant melanoma receiving encorafenib plus binimetinib versus vemurafenib or encorafenib (COLUMBUS): a multicentre, open-label, randomised, phase 3 trial. The Lancet Oncology. 2018;19(10):1315–27. [DOI] [PubMed] [Google Scholar]
- 48.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Subbiah V, Puzanov I, Blay JY, Chau I, Lockhart AC, Raje NS, et al. Pan-Cancer Efficacy of Vemurafenib in BRAF (V600)-Mutant Non-Melanoma Cancers. Cancer Discov. 2020;10(5):657–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Salama AKS, Li S, Macrae ER, Park JI, Mitchell EP, Zwiebel JA, et al. Dabrafenib and Trametinib in Patients With Tumors With BRAF(V600E) Mutations: Results of the NCI-MATCH Trial Subprotocol H. J Clin Oncol. 2020;38(33):3895–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tutuka CSA, Andrews MC, Mariadason JM, Ioannidis P, Hudson C, Cebon J, et al. PLX8394, a new generation BRAF inhibitor, selectively inhibits BRAF in colonic adenocarcinoma cells and prevents paradoxical MAPK pathway activation. Mol Cancer. 2017;16(1):112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yao Z, Gao Y, Su W, Yaeger R, Tao J, Na N, et al. RAF inhibitor PLX8394 selectively disrupts BRAF dimers and RAS-independent BRAF-mutant-driven signaling. Nat Med. 2019;25(2):284–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sievert AJ, Lang SS, Boucher KL, Madsen PJ, Slaunwhite E, Choudhari N, et al. Paradoxical activation and RAF inhibitor resistance of BRAF protein kinase fusions characterizing pediatric astrocytomas. Proc Natl Acad Sci U S A. 2013;110(15):5957–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yao Z, Torres NM, Tao A, Gao Y, Luo L, Li Q, et al. BRAF Mutants Evade ERK-Dependent Feedback by Different Mechanisms that Determine Their Sensitivity to Pharmacologic Inhibition. Cancer Cell. 2015;28(3):370–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Botton T, Talevich E, Mishra VK, Zhang T, Shain AH, Berquet C, et al. Genetic Heterogeneity of BRAF Fusion Kinases in Melanoma Affects Drug Responses. Cell Rep. 2019;29(3):573–88 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tantisira KG, Lasky-Su J, Harada M, Murphy A, Litonjua AA, Himes BE, et al. Genomewide association between GLCCI1 and response to glucocorticoid therapy in asthma. N Engl J Med. 2011;365(13):1173–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nishibori Y, Katayama K, Parikka M, Oddsson A, Nukui M, Hultenby K, et al. Glcci1 deficiency leads to proteinuria. J Am Soc Nephrol. 2011;22(11):2037–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chapman MS, Askew DJ, Kuscuoglu U, Miesfeld RL. Transcriptional control of steroid-regulated apoptosis in murine thymoma cells. Mol Endocrinol. 1996;10(8):967–78. [DOI] [PubMed] [Google Scholar]
- 59.Kiuchi Z, Nishibori Y, Kutsuna S, Kotani M, Hada I, Kimura T, et al. GLCCI1 is a novel protector against glucocorticoid-induced apoptosis in T cells. FASEB J. 2019;33(6):7387–402. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1: (A) RT-qPCR demonstrating presence of GLCCI1-BRAF within RNA isolated from indicated specimens from the patient’s cytoreductive operation. Negative control: RH30 xenograft rhabdomyosarcoma. Positive control: perisplenic nodule used for TSO-500. (B) Electrophoresis gel of RT-qPCR products from perisplenic specimen, and raw data of subsequent sequencing.
Supplemental Table 1: List of genes tested in the Invitae Hereditary Paraganglioma-Pheochromocytoma 14-gene Panel.