

Abstract
Background:
Humanized 3F8-bispecific antibody (hu3F8-BsAb) using the IgG(L)-scFv format, where the anti-CD3 huOKT3 scFv is fused with the carboxyl end of the hu3F8 light chain, has potent anti-tumor cytotoxicity against GD2(+) tumors. To overcome the insufficient number and function of T cells in cancer patients, they can be rejuvenated and expanded ex vivo before arming with hu3F8-BsAb for adoptive transfer, potentially reducing toxic side effects from direct BsAb administration.
Procedure:
T cells from normal volunteers were expanded and activated ex vivo using CD3/CD28 beads for 8 days. Activated T cells (ATCs) were harvested and co-incubated with a GMP grade hu3F8-BsAb at room temperature for 20 minutes. These armed ATCs were tested for cytotoxicity in vitro and in vivo against human GD2(+) cell lines and PDXs xenografts in BALB-Rag2−/−IL-2R-γc-KO (BRG) mice.
Results:
Hu3F8-BsAb armed ATCs showed robust antigen-specific tumor cytotoxicity against GD2(+) tumors in vitro. In vivo, T-cells armed with hu3F8-BsAb were highly cytotoxic against GD2(+) melanoma and neuroblastoma xenografts in mice, accompanied by T-cell infiltration without significant side effects. Only zeptomole (10−21) quantities of BsAb per T-cell was required for maximal antitumor effects. Tumor response was a function of T-cell dose.
Conclusion:
BsAb armed T cells may have clinical utility as the next generation of cytotherapy combined with recombinant BsAb against human tumors for both adult and pediatrics, if autologous T-cells can be activated and expanded ex vivo.
Keywords: GD2, bispecific antibody, neuroblastoma, melanoma, cytotherapy, armed T cells
Introduction:
Disialoganglioside GD2 is a tumor associated antigen (TAA) expressed on neuroectodermal derived tumors in both pediatric and adult cancers such as neuroblastoma, melanoma, retinoblastoma, as well as sarcomas including osteosarcoma, rhabdomyosarcoma and Ewing sarcoma.1 GD2 is a proven target for antibody-based immunotherapy given its relative antigen homogeneity, membrane proximity and infrequent antigen loss.2–4 Numerous efforts have been made in the last two decades to improve on anti-GD2 therapeutics, including novel antibodies, radiolabel conjugates, drug conjugates, immunocytokines, and CAR-Ts.5 Dinutuximab was the first anti-GD2 antibody to be FDA approved and together with dinutuximab-β, have become the standard consolidation therapy for neuroblastoma in the U.S and Europe.6,7 Evolving from the era of murine or chimeric anti-GD2 monoclonal antibodies (mAb), hu14.18-K322A and hu3F8 (naxitamab) were developed to further reduce the immunogenicity and anti-drug antibodies.8,9,10 The toxicity profile of hu3F8 even at 2.5x the antibody dose of mouse 3F8 or ch14.18 appeared easier to manage as outpatient treatments.10
Among pediatric cancers, neuroblastoma is the first to demonstrate the benefit of anti-GD2 monoclonal antibody in controlling chemoresistant microscopic marrow disease,11 where negative MRD by qRT-PCR measurement after 2 mAb cycles was predictive of survival.12 Yet, anti-GD2 mAb by itself is relatively ineffective against bulky disease. Several strategies have been tested to overcome this limitation. Chemoimmunotherapy was introduced with the intention to change the microenvironment, modify vascularity, to create neoantigen and to induce immunogenic cell death.13 Since neuroblastoma expresses PD-L1, which could inhibit T-cells and NK cells leading to poor treatment outcomes14, combination with immune checkpoint inhibitors (ICI) have also been investigated (NCT02914405).15
Cytotherapy using chimeric antigen receptor (CAR) T cells has shown excellent clinical responses against relapsed and refractory lymphoblastic leukemia.16 However, the success of CAR-Ts in solid tumors such as neuroblastoma and sarcomas has not been as robust.17 Anti-GD2 CAR-Ts have been generated and tested against neuroblastoma in phase1 clinical trials,18–20 showing safety and activity against minimal residual disease. To improve anti-tumor response, modification continues to be explored such as combinations with radiation, chemotherapy or ICIs.21
CAR-Ts are personalized medicine products, which require T-cells to be genetically engineered for individual patients. As a living drug, the persistence of CAR-Ts is required for clinical benefit; yet, its life-long presence could have severe on target off tumor sequelae (e.g. CD19(+) B cell aplasia). The cost and complexity in the manufacture and distribution of CAR-Ts are well known.22 An alternative method of redirecting polyclonal T-cells to the tumor is to attach bispecific antibodies onto their cell surface without the need of genetic modification.23 Anti-GD2 T cell engaging BsAb was previously reported, showing potent anti-tumor cytotoxicity in vitro and in vivo.24 Unlike CAR-Ts, T-cells activated by BsAb do not undergo activation induced cell death (AICD) or exhaustion.25 With Fc silencing that removed Fc-receptor binding and complement activation, cytokine storm was substantially reduced.24,26
T-cell extraction and expansion ex vivo is a well-established process both as reagents in research and as drugs in the clinic. T-cells armed with chemically conjugated BsAbs showed efficient killing of GD2(+) neuroblastoma cell lines.27 A phase 1 clinical trial of such armed T-cells is ongoing in children and young adults with neuroblastoma and osteosarcoma (NCT02173093).28 A similar strategy was used to arm T-cells with chemically conjugated EGFRxCD3 BsAb against glioblastoma and gastrointestinal cancers, 29 with HER2xCD3 BsAb against Breast cancer (NCT03661424),30 and in combination with immune checkpoint inhibitors (NCT03406858). The excess antibodies and released cytokines during arming were removed before reinfusion to reduce cytokine release syndrome.
Neuroblastoma is the most common cancer in the first year of life,31 and the third most common pediatric malignancy with an incidence of 750 new cases a year in the United States. Despite increasing intensity of chemotherapy combined with surgery and radiation, cure rate of high-risk metastatic neuroblastoma is only 50% despite anti-GD2 mAb therapy.7 While anti-GD2 mAb works through Fc dependent cytotoxicity that exploit NK cells, myeloid cells and complement cascade, T cells are under-utilized even though they are well-established professional killers. With the downregulation of HLA on neuroblastoma and scarcity of tumor infiltrating lymphocytes, classic T cell immunity is rare. We now report ex vivo arming of polyclonal T-cells using recombinant T-BsAb specific for GD2, to demonstrate their anti-tumor properties in vitro and in vivo against melanoma and neuroblastoma (NB) cell lines and PDX model.
Materials and Methods:
Cell lines
The cell lines M14 was obtained from the University of California, Los Angeles (Los Angeles, CA) and IMR32 was purchased from the ATCC. They were confirmed by short-tandem repeat profiling using the PowerPlex 1.2 System (Promega) and were periodically tested for Mycoplasma using a commercial kit (Lonza). For the study to confirm its specificity against GD2 antigen, GD2 negative acute myeloid leukemia cell line MOLM13 purchased from the ATCC was used as a negative control.
T-cell activation
Human T-cells were isolated from the whole blood of healthy donors (New York Blood Center) by using Ficoll-Hypaque density gradient centrifugation (Corning™), isolated T-cells by using human Pan T-cell isolation kit (Miltenyi Biotec) and stored at −80°C till use. For the experiments, frozen T-cells were thawed and seeded into culture flasks, activated with CD3/CD28 Dynabeads beads (Invitrogen) according to the manufacturer’s protocol, and expanded for 8 days in the presence of 30 IU/ul of interleukin 2 (Prometheus Laboratories Inc.) in RPMI1640 medium (Corning) supplemented with 10% heat inactivated FBS (SIGMA) and antibiotics (Corning™ ). All T-cells were analyzed for CD3 (BD bioscience), CD4 (BD bioscience), and CD8 (BD bioscience) by flow cytometry which showed more than >90% CD3 positive population with CD4 and CD8, approximately 50% and 50%, respectively after activation process.
Arming procedures and binding assay
The hu3F8-BsAb has been reported previously.24 For arming, activated T-cells were harvested and the beads were removed by a DynaMag™−15 magnet (Thermofisher). Appropriate number of activated T-cells were divided in conical tubes and mixed with hu3F8 BsAb at the concentration of 0.05, 0.5, 5ug/106 T-cell at room temperature for 20 minutes. After washing twice, the cells were resuspended in culture medium at a concentration of 106/10ul. Each time after arming, we checked the antibodies bound to T-cells by flow cytometry using A1G4 anti-3F8 idiotype antibody labeled with Alexa Fluor 647 (Life Technologies Corporation).
Cell cytotoxicity (chromium-51 release assay)
To assess cell cytotoxicity, chromium-51 release assay was used as previously described.32 Isolated T-cells were activated, expanded, and armed with hu3F8 BsAbs at 0.05, 0.5, 5ug/106 T-cells. Unarmed T-cells from same donor was used as controls.
Washing-off experiment
To quantify the amount of antibody fallen off from T-cells during washing process of arming, we tested the concentration of antibodies in supernatant while arming T-cells with hu3F8 BsAbs. We analyzed the Abs concentration of supernatant after 1st wash and 2nd wash by ELISA assay.
Xenograft studies
All animal procedures were performed in compliance with the MSK Institutional Animal Care and Use Committee guidelines. For all in vivo study, immunodeficient mice BALB-Rag2/IL-2R-γc-KO (BRG) from Taconics were used. The mice were maintained under clean conditions and fed with Sulfatrim food for infection prophylaxis. The mice at the start of experiment were 6–10 week-old and each treatment group included 5 mice per group.
The mice were treated with the schedule as follows. Tumor cell lines or patient derived xenografts were implanted subcutaneously on the flank on day1 of the experiment. On day 8, mice were treated with armed T cells, administered intravenously by retro-orbital injection. IL 2 (1000 U/dose/mouse) was administered subcutaneously twice a week to maintain T cell survival. The tumor was measured by the Peira TM900 imaging device (PEIRA) twice a week. For Neuroblastoma and Melanoma cell line studies, IMR-32-Luc cells or M14 Luc cells (2 million cells/ mouse) in Matrigel (BD Bioscience) were used. For Neuroblastoma PDX model, fresh surgical specimens and early passage in NSG mice were used. The Piro20-lung PDX which was established from a fresh surgical specimen collected from the lung metastasis from a neuroblastoma patient, provided by Dr. Piro Lito at MSK was implanted subcutaneously and when tumors became palpable, they were randomly assigned to each experimental group.
Flow cytometry analysis
On day 36 of the experiment, tumors were harvested, and processed to obtain single cell preparations to study infiltrating lymphocytes using human CD45 antibody (BD LSRFortessa flow cytometer, Biolegend) using appropriate isotype matched control antibodies, and analyzed by FlowJo software.
Immunohistochemistry staining
The immunohistochemistry staining was performed by MSK Molecular Cytology Core Facility using a Discovery XT processor (Ventana Medical Systems). The details were described as previous published.22
Statistical analysis
Statistical analysis for all experiment were done using GraphPad Prism version 8 (GraphPad Software, San Diego, CA). Differences between samples were tested for statistical significance using Student t test, and P<0.05 was considered statistically significant.
Result:
Hu3F8-BsAb binds to ex vivo expanded and activated T-cells
We used the hu3F8-BsAb as detailed previously.24 It was built using the platform IgG(L)-scFv format in which the heavy chain was derived from that of hu3F8 IgG18 except for an N297A mutation to remove glycosylation to silence Fc functions, and N322A to remove binding to C1q.26 The light chain was extended with a C-terminal (G4S)3 linker followed by huOKT3 scFv. Ex vivo arming process was summarized in Figure 1A, at increasing concentrations of hu3F8-BsAbs per million T-cells (0.05, 0.5, 5ug/106 T-cells). By 48 hours of incubation at 37°C (Figure 1B), ~25 % of T-cells remained positive for surface bound hu3F8bsAb. By 72 hours surface hu3F8-BsAb was no longer detectable. (Figure 1B)
Figure 1. Characteristics of bead activated T cells (ATCs) armed with hu3F8 BsAbs.
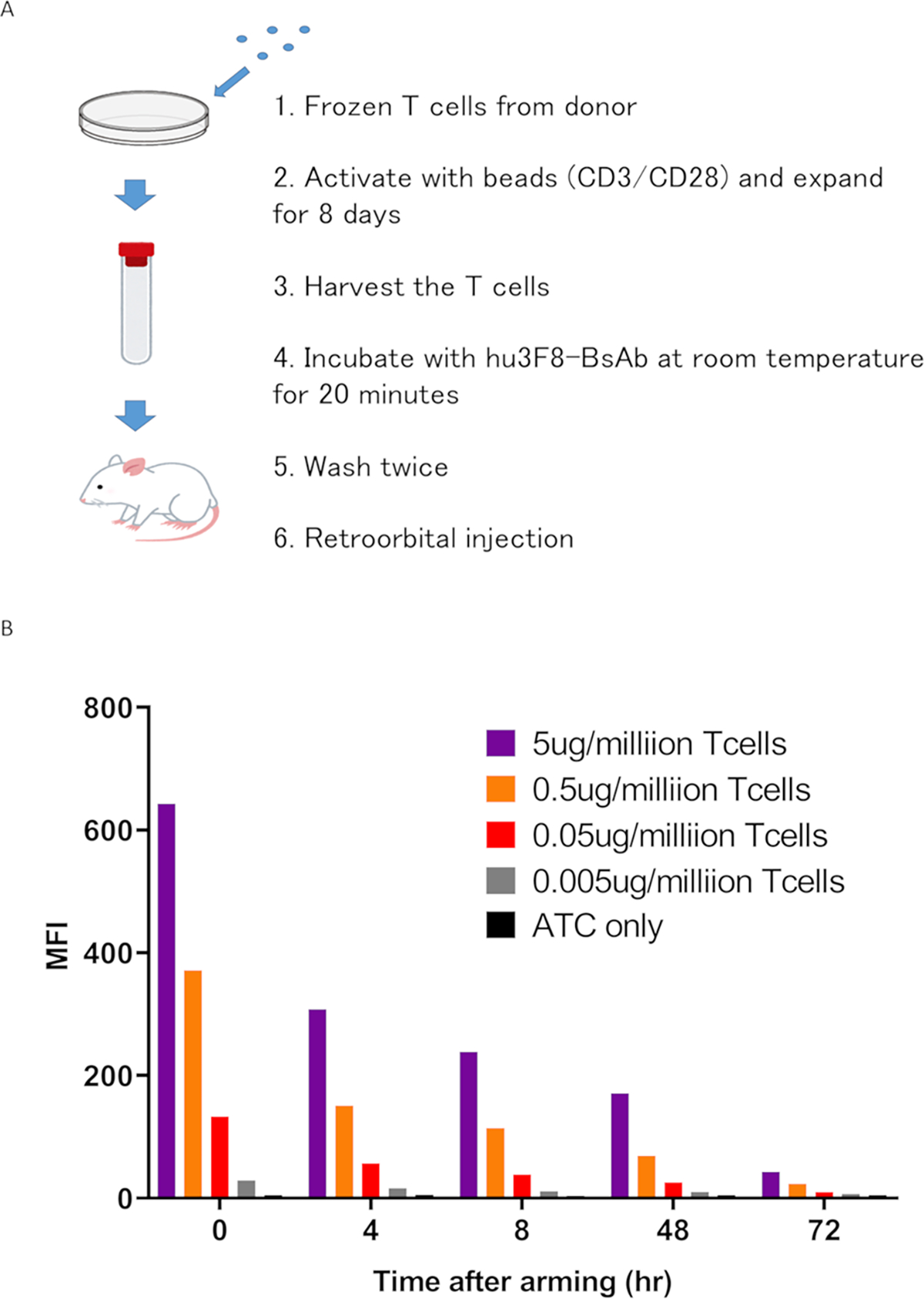
(A) The process of expanding T cells ex vivo and arming them with hu3F8 BsAb. (B) Surface hu3F8-BsAb on ATC was assayed using anti-hu3F8 anti-idiotypic antibody A1G4 (labeled with Alexa Fluor 647). Armed ATC was cultured in the absence of hu3F8 BsAb for 4, 8, 24, 48 and 72 hours, washed and stained with A1G4. Surface BsAb was detectable at least through 24 hours after arming. By 72 hours, BsAb has substantially decreased.
MFI, mean fluorescence intensity
T-cell armed with hu3F8-BsAb killed human tumor cell lines in vitro
By 4-hour 51Cr release assay hu3F8-BsAb armed T-cells showed potent cytotoxicity against Melanoma cell line M14 Luc. Compared to continual presence of hu3F8-BsAB during the cytotoxicity assay, once BsAb was washed off, ex vivo armed T-cells were less cytotoxic as expected (Figure 2A). For armed T-cells, there was a hu3F8-BsAb dose dependent increase in cytotoxicity which plateaued at 0.5ug/106 T-cells (Figure 2B).
Figure 2. ATCs armed with hu3F8BsAbs killed the GD2 positive tumor in vitro. In vitro killing was not improved by increasing the arming antibody dose above 0.05ug/million T cells.
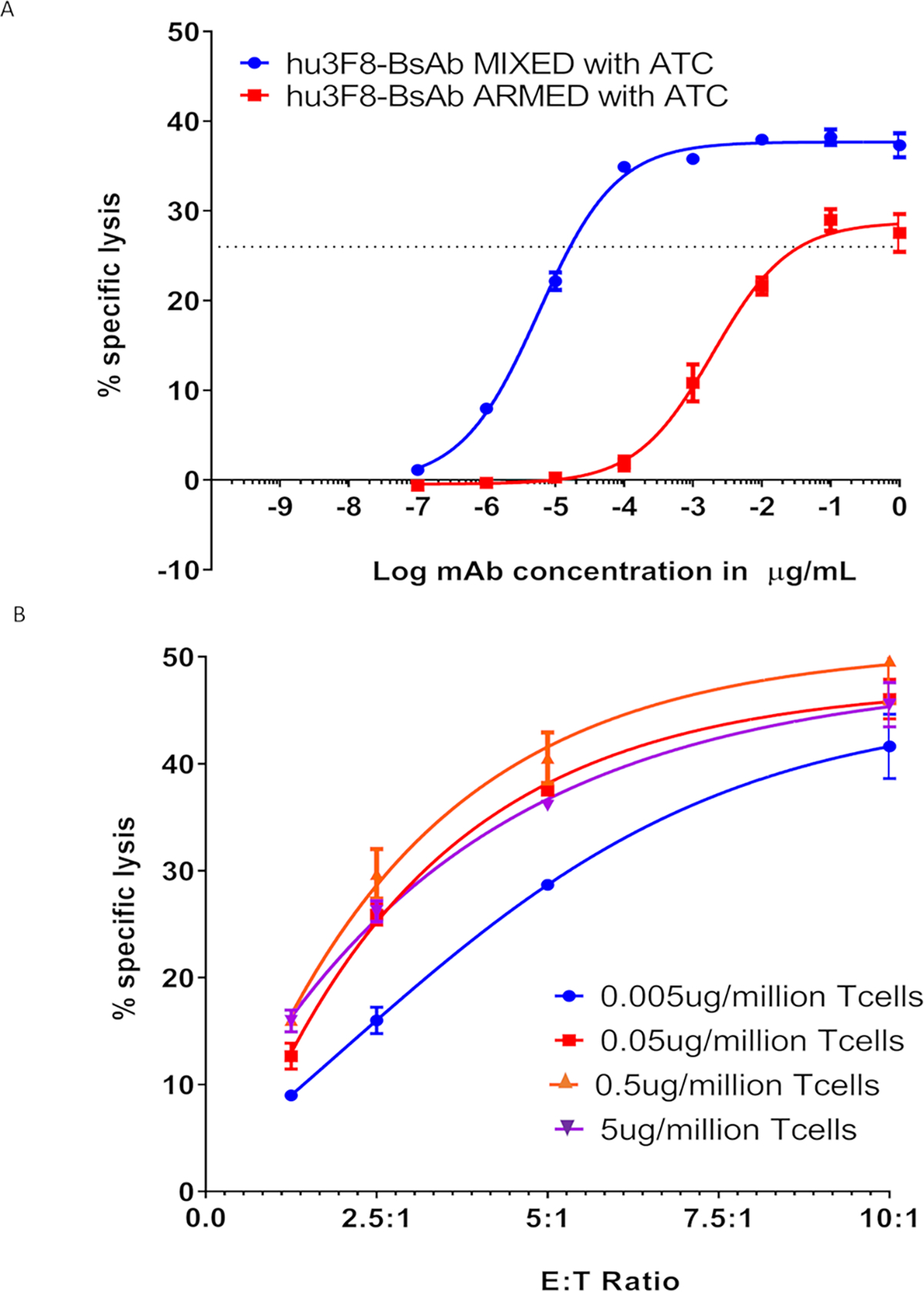
(A) Armed T cells killed the M14 melanoma cells in vitro. (B) Hu3F8-BsAb armed with T cells killing by 51Cr assay M14 cells showing % specific lysis based on different ATC to target cell ratio.
Efficacy of hu3F8-BsAb against Melanoma xenografts model
We tested the in vivo anti-tumor activity of hu3F8-BsAb against M14 melanoma and IMR32 (MYCN amplified) neuroblastoma xenografts in BRG mice as previously described.33 Xenografts were planted subcutaneously (sc) and both T-cell and hu3F8-BsAb were given intravenously (iv). Decreasing the arming dose of hu3F8-BsAb (from 5 to 0.5 to 0.05 ug/106 T-cell) did not change the anti-tumor response, consistent with the in vitro findings. At 0.05 ug/106 arming dose, where 10% of the antibody became T cell bound, the estimated surface density of BsAb was in the zepto molar range. When the frequency of armed T-cells was increased from once a week to twice a week, there was a consistent improvement in anti-tumor response (Figure 3A). When the dose of injected T-cells was increased (5, 10, 20 to 40 million per dose), there was also an improvement in response (Figure 3B).
Figure 3. ATCs armed with hu3F8 BsAbs suppressed GD2(+) xenograft in vivo.
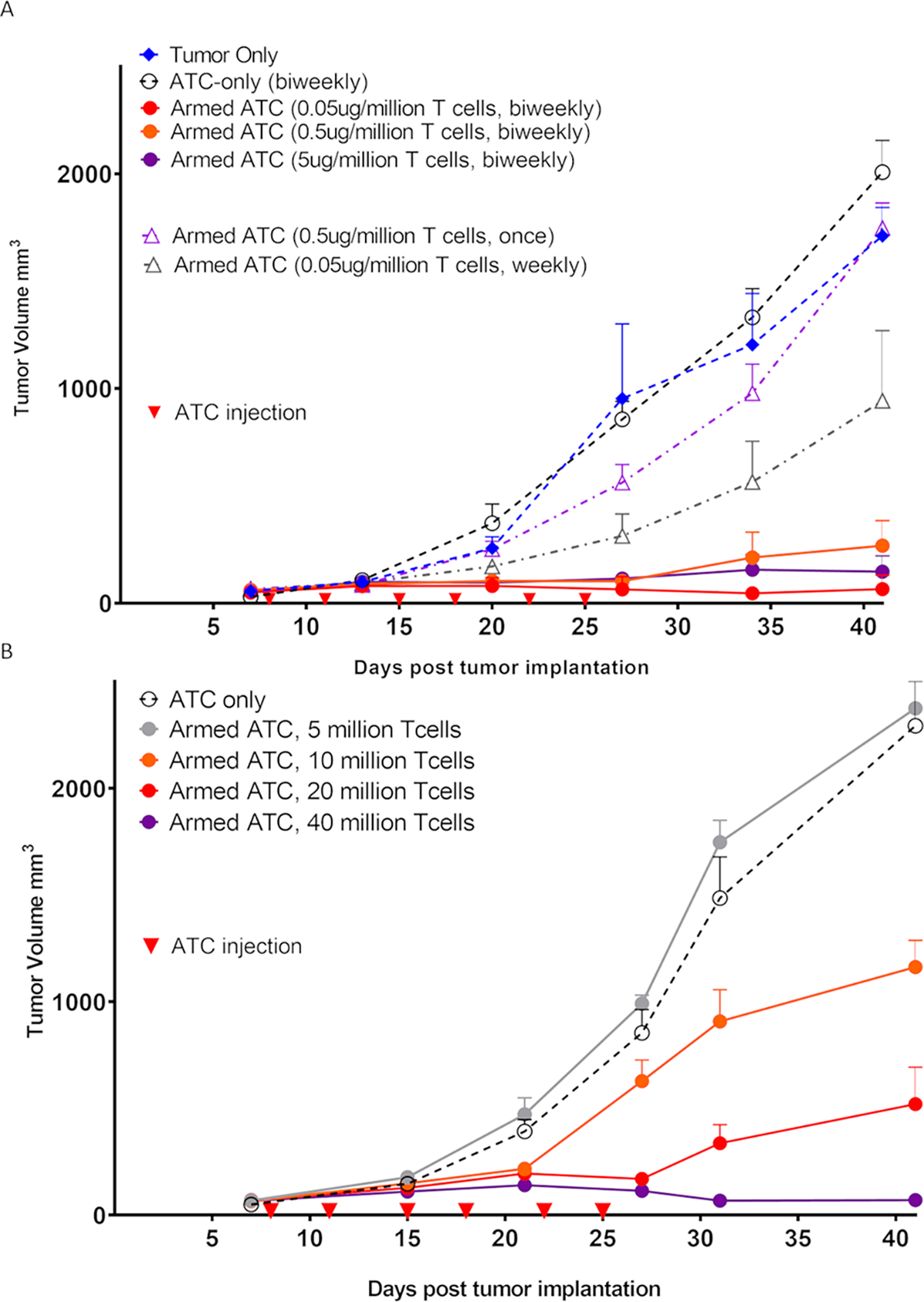
(A) ATCs armed with increasing arming dose (0.05, 0.5 or 5ug/106 T-cell) of hu3F8-BsAb were used to treat GD2(+) M14 melanoma xenografts in BRG mice. The tumor response was near identical at all three arming dose levels. (n=5 mice/group) (B) Increasing biweekly doses of armed ATCs improved tumor response. (n=5 mice/group). Once: only one time injection, Weekly: weekly injection for total 3 times, Biweekly: biweekly injection for total 6 times.
T-cells armed with hu3F8-BsAb infiltrated M14 xenograft
After treatment with hu3F8-BsAb armed T-cells, tumors were harvested for human tumor infiltrating lymphocytes (TILs) analysis. By FACS using anti-huCD45 antibody (Figure 4A) and by immunohistochemistry using anti-CD3 antibody (Figure 4B), there was significant infiltration of huCD45(+) or huCD3(+) T-cells into tumor when armed with hu3F8-BsAb; no TILS were seen with unarmed T-cells.
Figure 4. ATCs armed with hu3F8 BsAb infiltrated into the xenograft.
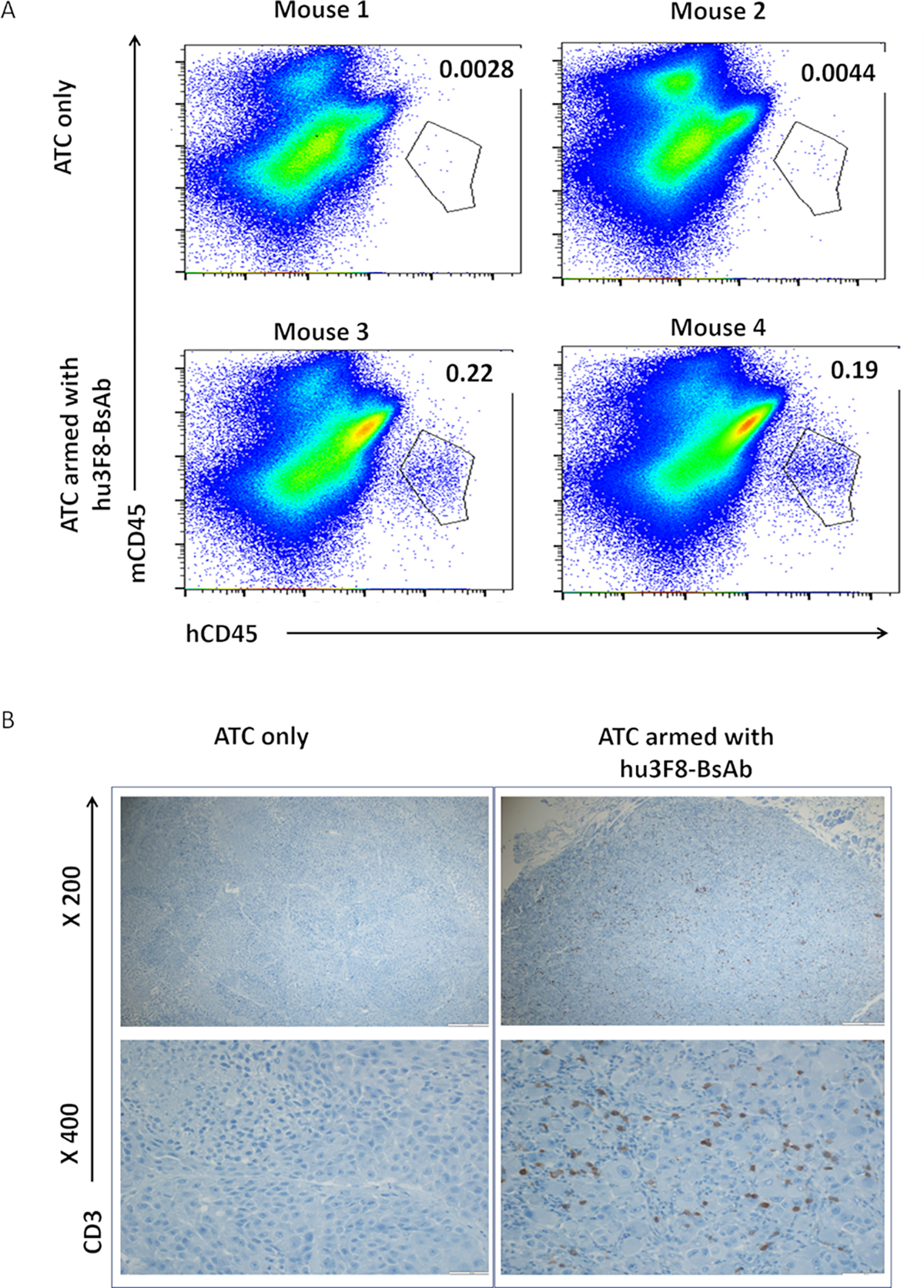
Tumors were harvested and analyzed for human tumor infiltrating lymphocytes (TILs), by (A) FACS analysis for huCD45(+) cells and (B) by immunohistochemistry for huCD3(+) cells. There were two mice per group.
T cells armed with hu3F8-BsAb suppressed growth of NB cell line and patient derived xenografts
We next tested the in vivo anti-tumor effect of T-cells armed with hu3F8-BsAb against neuroblastoma xenografts at arming doses of 0.5 ug/106 and 0.05 ug/106 T-cell, at 2×107 T-cells/injection. Tumor growth was suppressed by hu3F8-BsAb armed T cells, but not by T-cells armed with control BsAbs both for IMR32 (Figure 5A) and for Piro20-lung NB PDX (Figure 5B) where response was durable for 70–80 days. Throughout the treatment and follow-up, there was no significant weight loss (>10%) or clinical toxicities (Figure 5C). Overall survival was also significantly prolonged in the group treated with T cells armed with 3F8-BsAbs compared to control groups (Figure 5D). When the same T cells armed with hu3F8-BaAb were used against a GD2-negative tumor (MOLM13, human AML), no anti-tumor effect was seen (data not shown).
Figure 5. ATCs armed with hu3F8-BsAb suppressed growth of NB cell line and patient derived xenografts.
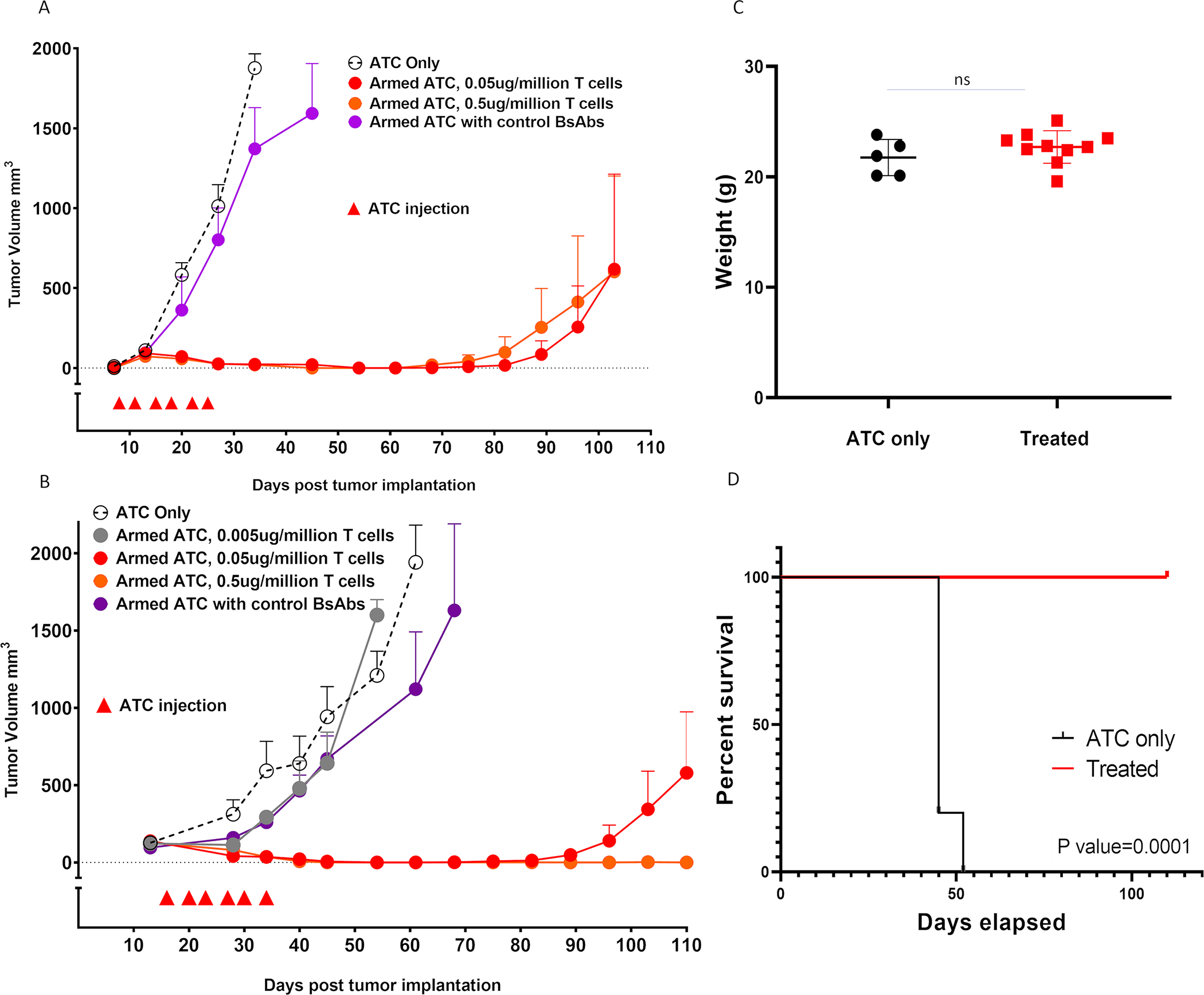
(A) Anti-tumor response among IMR32 xenografts (n=5 mice/group), (B) Anti-tumor response among patient derived xenografts (n=5 mice/group), (C) Body weight during the first 50 days of treatment in B, and (D) Improvement of survival after armed ATC treatment in B. All treatments were given biweekly for total 6 times, with 20 million armed ATCs. Control BsAbs were given at BsAb concentration of 0.05 ug/million T cells.
Discussion:
In this study, we described the consistent antitumor effect of T-cells armed with hu3F8-BsAb, a T cell bispecific antibody previously described.24 We showed that these ex vivo armed T cells retained the BsAb on the cell surface for sufficient length of time to mediate efficient killing of tumor cell lines in vitro and in vivo. They readily infiltrated cold tumors and showed substantial anti-tumor activity against melanoma and neuroblastoma xenografts without observable clinical toxicities by their healthy appearance and no weight loss. Akin to CAR-Ts, these polyclonal T cells are no longer restricted by HLA in order to recognize the target. Unlike CAR-Ts, they do not undergo clonal expansion in vivo and have minimal cytokine release during their circulation. Most importantly, by not persisting, permanent autoimmune process that followed living CAR-Ts could be avoided. In addition, ex vivo arming is a less complicated procedure and might be less costly for production and distribution.
Our studies showed that there is dependency on T cell dose. While 10 million armed T-cells/dose could suppress tumor growth, the response was not consistent. This is likely because of T cell engraftment efficiency in the BRG mice, a genetic background known to resist human cells more than NSG mice. One key benefit for ex vivo arming is to eliminate the surge of cytokines when T cells are engaged by anti-CD3 scFv, being washed off before infusion into the patient, a major dose limiting side effect of current CAR-Ts and bispecific antibodies.
Another concerning side effect of CAR-Ts is neurotoxicity. Richman et al. recently published the data of severe neurotoxicities occurring in mice treated with anti-GD2 CAR-Ts and its severity was correlated with the efficacy of CAR-Ts.34 In our studies, ex vivo armed T cells or hu3F8-BsAb did not cause any neurotoxicity, in contrast to CAR-Ts (Cheung, unpublished results). This is partly a characteristic of BsAb or BsAb armed T cells that do not cross the blood brain barrier. The downside is leaving the CNS at risk as sanctuaries for some tumors. But for target antigens expressed in the normal brain (e.g. GD2), this is probably the less of the two evils.
BsAbs armed with T-cells have previously been tested in patients against several antigens, such as HER2,35 CD20,36 EGFR,29 and GD2.37 More recently, the combination of BsAb armed T cells with the checkpoint inhibitor Pembrolizumab in metastatic castration resistant prostate cancer was initiated. It is expected that future studies using recombinant BsAb will yield more favorable responses than previous attempts with ex vivo armed T cells. For neuroblastoma, current standard therapy includes induction chemotherapy with local control with surgery and radiation followed by high dose chemotherapy (HDC) and stem cell rescue before consolidation therapy with immunotherapy using anti-GD2 monoclonal antibodies. It is conceivable that T cells harvested early in treatment could be rejuvenated ex vivo, armed with BsAb before reinfusion to patients to control minimal residual disease.
Limitations of our study is that we used immunodeficient mice for our experiments which might underestimate side effect of immunotherapies as well as the fact that we used subcutaneous models instead of orthotopic modes. Syngeneic models using immunocompetent mice should provide more supportive data, although immunogeneicity of humanized proteins in these models has been a major limitation.
In summary, our study demonstrated the potential of ex vivo armed T cells using anti-GD2 BsAb, built on the IgG(L)-scFv platform using the hu3F8 anti-GD2 backbone and huOKT3 anti-CD3 scFv. This should be applicable to other anti-GD2 antibodies or anti-tumor antibodies. This approach may offer a potential alternative to CAR-T approach.
Acknowledgement:
This work was supported by funds from Enid A. Haupt Endowed Chair, the Robert Steel Foundation, Kids Walk for Kids with Cancer, as well as sponsored research fund from Y-mAbs Therapeutics. Technical service provided by the MSK Animal Imaging Core Facility, Antitumor Assessment Core Facility, and Molecular Cytology Core Facility were supported in part by the NCI Cancer Center Support Grant P30 CA008748.
Abbreviation
- hu3F8-BsAb
humanized 3F8-bispecific antibody
- ATC
activated T cell
- BsAb
Bispecific antibody
- GMP
Good Manufacturing Practice
- TAA
tumor associated antigen
- CAR
chimeric antigen receptor
- FDA
The Food and Drug Administration
- mAb
monoclonal antibody
- MRD
minimal residual disease
- qRT-PCR
quantitative Reverse Transcription PCR
- PD-L1
Programmed cell death 1-Ligand1
- ICI
immune checkpoint inhibitor
- CD
cluster of differentiation
- AICD
activation induced cell death
- EGFR
epidermal growth factor receptor
- HER2
human epidermal growth factor receptor 2
- NK cells
natural killer cells
- HLA
human leukocyte antigen
- PDX
patient derived xenograft
- ATCC
American Type Culture Collection
- MSK
Memorial Sloan Kettering Cancer Center
- NSG
NOD scid gamma mouse
- MYCN
N-myc
- TILs
tumor infiltrating lymphocytes
- FACS
fluorescence-activated cell sorting
- AML
acute myelogenous leukemia
- BRG
BALB-Rag2/IL-2R-γc-KO
- scFv
single-chain variable fragment
Footnotes
Conflict of Interest statement:
Both MSK and NKC have financial interest in Y-mAbs, Abpro-Labs and Eureka Therapeutics. NKC reports receiving commercial research grants from Y-mabs Therapeutics and Abpro-Labs Inc. NKC was named as inventor on multiple patents filed by MSK, including those licensed to Ymabs Therapeutics, Biotec Pharmacon, and Abpro-labs. NKC is a SAB member for Abpro-Labs and Eureka Therapeutics. HX was named as co-inventor in a patent on hu3F8 bispecific antibody filed by MSK. The other authors have no disclosures to report.
Data availability statement:
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References:
- 1.Dobrenkov K, Cheung NK: GD2-targeted immunotherapy and radioimmunotherapy. Semin Oncol 41:589–612, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cheung NK, Dyer MA: Neuroblastoma: developmental biology, cancer genomics and immunotherapy. Nat Rev Cancer 13:397–411, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kramer K, Gerald WL, Kushner BH, et al. : Disialoganglioside G(D2) loss following monoclonal antibody therapy is rare in neuroblastoma. Clin Cancer Res 4:2135–9, 1998 [PubMed] [Google Scholar]
- 4.Schumacher-Kuckelkorn R, Volland R, Gradehandt A, et al. : Lack of immunocytological GD2 expression on neuroblastoma cells in bone marrow at diagnosis, during treatment, and at recurrence. Pediatr Blood Cancer 64:46–56, 2017 [DOI] [PubMed] [Google Scholar]
- 5.Ahmed M, Cheung NK: Engineering anti-GD2 monoclonal antibodies for cancer immunotherapy. FEBS Lett 588:288–97, 2014 [DOI] [PubMed] [Google Scholar]
- 6.Cheung NK, Lazarus H, Miraldi FD, et al. : Ganglioside GD2 specific monoclonal antibody 3F8: a phase I study in patients with neuroblastoma and malignant melanoma. J Clin Oncol 5:1430–40, 1987 [DOI] [PubMed] [Google Scholar]
- 7.Yu AL, Gilman AL, Ozkaynak MF, et al. : Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med 363:1324–34, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheung NK, Guo H, Hu J, et al. : Humanizing murine IgG3 anti-GD2 antibody m3F8 substantially improves antibody-dependent cell-mediated cytotoxicity while retaining targeting in vivo. OncoImmunology 1:477–486, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Navid F, Sondel PM, Barfield R, et al. : Phase I Trial of a Novel Anti-GD2 Monoclonal Antibody, Hu14.18K322A, Designed to Decrease Toxicity in Children With Refractory or Recurrent Neuroblastoma. J Clin Oncol 32:1445–52, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kushner BH, Cheung IY, Modak S, et al. : Humanized 3F8 Anti-GD2 Monoclonal Antibody Dosing With Granulocyte-Macrophage Colony-Stimulating Factor in Patients With Resistant Neuroblastoma: A Phase 1 Clinical Trial. JAMA Oncol 4:1729–1735, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheung NK, Cheung IY, Kushner BH, et al. : Murine Anti-GD2 Monoclonal Antibody 3F8 Combined With Granulocyte-Macrophage Colony-Stimulating Factor and 13-Cis-Retinoic Acid in High-Risk Patients With Stage 4 Neuroblastoma in First Remission. J Clin Oncol 30:3264–70, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kushner BH, Ostrovnaya I, Cheung IY, et al. : Prolonged progression-free survival after consolidating second or later remissions of neuroblastoma with Anti-GD2 immunotherapy and isotretinoin: a prospective Phase II study. Oncoimmunology 4:e1016704, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mody R, Naranjo A, Van Ryn C, et al. : Irinotecan-temozolomide with temsirolimus or dinutuximab in children with refractory or relapsed neuroblastoma (COG ANBL1221): an open-label, randomised, phase 2 trial. Lancet Oncol 18:946–957, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dondero A, Pastorino F, Della Chiesa M, et al. : PD-L1 expression in metastatic neuroblastoma as an additional mechanism for limiting immune surveillance. Oncoimmunology 5:e1064578, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Siebert N, Zumpe M, Juttner M, et al. : PD-1 blockade augments anti-neuroblastoma immune response induced by anti-GD2 antibody ch14.18/CHO. Oncoimmunology 6:e1343775, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grupp SA, Kalos M, Barrett D, et al. : Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med 368:1509–18, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mirzaei HR, Rodriguez A, Shepphird J, et al. : Chimeric Antigen Receptors T Cell Therapy in Solid Tumor: Challenges and Clinical Applications. Front Immunol 8:1850, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park JR, Digiusto DL, Slovak M, et al. : Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol Ther 15:825–33, 2007 [DOI] [PubMed] [Google Scholar]
- 19.Pule MA, Savoldo B, Myers GD, et al. : Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med 14:1264–70, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Louis CU, Savoldo B, Dotti G, et al. : Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 118:6050–6, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schultz LM, Majzner R, Davis KL, et al. : New developments in immunotherapy for pediatric solid tumors. Curr Opin Pediatr 30:30–39, 2018 [DOI] [PubMed] [Google Scholar]
- 22.Salmikangas P, Kinsella N, Chamberlain P: Chimeric Antigen Receptor T-Cells (CAR T-Cells) for Cancer Immunotherapy - Moving Target for Industry? Pharm Res 35:152, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kontermann RE, Brinkmann U: Bispecific antibodies. Drug Discov Today 20:838–47, 2015 [DOI] [PubMed] [Google Scholar]
- 24.Xu H, Cheng M, Guo H, et al. : Retargeting T cells to GD2 pentasaccharide on human tumors using Bispecific humanized antibody. Cancer Immunol Res 3:266–77, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoseini SS, Dobrenkov K, Pankov D, et al. : Bispecific antibody does not induce T-cell death mediated by chimeric antigen receptor against disialoganglioside GD2. Oncoimmunology 6:e1320625, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang LL, Hoseini SS, Xu H, et al. : Silencing Fc in T cell Engaging Bispecific Antibodies is Critical for T cell Trafficking and Anti-Tumor Potency Cancer Immunology Research:(in press), 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yankelevich M, Kondadasula SV, Thakur A, et al. : Anti-CD3 x anti-GD2 bispecific antibody redirects T-cell cytolytic activity to neuroblastoma targets. Pediatr Blood Cancer 59:1198–205, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yankelevich M, Modak S, Chu R, et al. : Phase I study of OKT3 x hu3F8 bispecific antibody (GD2Bi) armed T cells (GD2BATs) in GD2-positive tumors. Journal of Clinical Oncology 37:2533–2533, 2019 [Google Scholar]
- 29.Zitron IM, Thakur A, Norkina O, et al. : Targeting and killing of glioblastoma with activated T cells armed with bispecific antibodies. BMC Cancer 13:83, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lum LG, Sen M: Activated T-cell and bispecific antibody immunotherapy for high-risk breast cancer. Bench to bedside. Acta Haematol 105:130–6, 2001 [DOI] [PubMed] [Google Scholar]
- 31.Maris JM: Recent advances in neuroblastoma. N Engl J Med 362:2202–11, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheng M, Ahmed M, Xu H, et al. : Structural design of disialoganglioside GD2 and CD3-bispecific antibodies to redirect T cells for tumor therapy. Int J Cancer 136:476–86, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu H, Cheng M, Guo H, et al. : Retargeting T cells to GD2 pentasaccharide on human tumors using bispecific humanized antibody. Cancer immunology research 3:266–277, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Richman SA, Nunez-Cruz S, Moghimi B, et al. : High-Affinity GD2-Specific CAR T Cells Induce Fatal Encephalitis in a Preclinical Neuroblastoma Model. Cancer Immunol Res 6:36–46, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lum LG, Thakur A, Al-Kadhimi Z, et al. : Targeted T-cell Therapy in Stage IV Breast Cancer: A Phase I Clinical Trial. Clin Cancer Res 21:2305–14, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lum LG, Thakur A, Pray C, et al. : Multiple infusions of CD20-targeted T cells and low-dose IL-2 after SCT for high-risk non-Hodgkin’s lymphoma: a pilot study. Bone Marrow Transplant 49:73–9, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yankelevich M, Modak S, Lee DT, et al. : Phase I study of OKT3 x hu3F8 bispecific antibody armed T cells (GD2BATs) in NB and GD2+ tumors, ASCO 2019 Annual Meeting. Chicago, IL, Journal of Clinical Oncology, 2018, pp 2533 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.