

SUMMARY
Chemical modifications of mRNA, the so-called epitranscriptome, represent an additional layer of post-transcriptional regulation of gene expression. The most common epitranscriptomic modification, N6-methyladenosine (m6A), is generated by a multi-subunit methyltransferase complex. We show that alphaherpesvirus kinases trigger phosphorylation of several components of the m6A methyltransferase complex, including METTL3, METTL14, and WTAP, which correlates with inhibition of the complex and a near complete loss of m6A levels in mRNA of virus-infected cells. Expression of the viral US3 protein is necessary and sufficient for phosphorylation and inhibition of the m6A methyltransferase complex. Although m6A methyltransferase complex inactivation is not essential for virus replication in cell culture, the consensus m6A methylation motif is under-represented in alphaherpesvirus genomes, suggesting evolutionary pressure against methylation of viral transcripts. Together, these findings reveal that phosphorylation can be associated with inactivation of the m6A methyltransferase complex, in this case mediated by the viral US3 protein.
Graphical Abstract

In brief
Jansens et al. demonstrate that phosphorylation can be associated with inactivation of the m6A methyltransferase complex, in this case mediated by the alphaherpesviral US3 protein.
INTRODUCTION
Post-transcriptional modification of transcripts by N6-methyladenosine (m6A) is involved in several aspects of mRNA biology, and dysregulation is associated with various diseases, including cancer and many different viral infections (Williams et al., 2019; Barbieri and Kouzarides, 2020). m6A in mRNA is deposited by a multi-subunit complex called the m6A writer complex, in which METTL3 is the catalytic subunit (Bokar et al., 1994; Wang et al., 2014, 2016a; Śledź and Jinek, 2016; Wang et al., 2016b). Despite the identification of the components of the m6A writer complex and a wide array of studies into the role of m6A methylation, the regulation of the complex is still poorly understood. Studies showing differences in methylation levels in transcripts are typically associated with up- or downregulation of components of the complex (Pinello et al., 2018; Peng et al., 2019). However, since the discovery of the m6A writer complex, there has been speculation regarding potential post-translational regulation mechanisms (Bokar et al., 1994). More recently, SUMOylation of METTL3 has been shown to suppress m6A methyltransferase activity (Du et al., 2018), while WTAP and METTL3 phosphorylation moderately increased methyltransferase activity (Sun et al., 2020). These studies provided the first evidence for post-translational regulation of the m6A methyltransferase complex, despite the relatively small effects on its activity.
A potential strategy for uncovering mechanisms that regulate the m6A writer complex is to examine virus-induced regulation of m6A levels. Studies from the 1970s showed that m6A levels in viral and host poly(A) RNA were markedly reduced after infection with the alphaherpesvirus herpes simplex virus 1 (HSV-1) (Bartkoski and Roizman, 1976, 1978). A recent report also showed that infection of cells with HSV-1 reduces m6A levels (Srinivas et al., 2021). This effect depended on the viral ICP27 transcription regulator (Srinivas et al., 2021). However, the early studies on the interaction of alphaherpesviruses with m6A showed that the reduction in m6A methylation depends on an early or catalytic late viral protein, while ICP27 is a non-catalytic immediate-early protein (Bartkoski and Roizman, 1976, 1978, Tombácz et al., 2009). In addition, it is currently unknown if expression of ICP27 alone, in the absence of virus infection, affects m6A methylation. Therefore, it is unclear if ICP27 directly inhibits the m6A methylation complex or indirectly contributes to this inhibition by affecting expression of an early or late protein that inhibits the m6A writer complex.
Here, we report that infection of cells with the alphaherpesviruses pseudorabies virus (PRV) or HSV-1 results in a near complete loss of m6A-methylated transcripts. By detecting m6A levels in nascent mRNA, we demonstrate that the loss of m6A after PRV infection is due to inactivation of the m6A writer complex. In addition, we report that PRV infection results in phosphorylation of different components of the m6A writer complex, including METTL3, METTL14, and WTAP. Phosphorylation of METTL3 and METTL14 critically depended on the conserved alphaherpesvirus serine/threonine protein kinase US3 but was independent of its kinase activity. Expression of the US3 protein alone was sufficient to reduce m6A levels in the absence of infection. Our results indicate that the m6A writer complex remains intact in the nucleus during PRV infection but dissociates from chromatin, suggesting that in PRV-infected cells, the m6A writer complex may no longer be attached to nascent mRNA. Importantly, we show that the viral US3 protein is required for this dissociation of the m6A writer complex from chromatin. As a consequence, levels of m6A-methylated transcripts are restored in cells infected with US3null PRV. Together, these results reveal mechanistic insights in virus-induced inactivation of the m6A writer complex. In addition, the findings highlight phosphorylation as an important post-translational regulatory mechanism of the m6A methyltransferase complex.
RESULTS
Infection with the alphaherpesviruses PRV or HSV-1 triggers a near complete loss in m6A-methylated mRNA and inhibition of the m6A methyltransferase complex
Early studies on m6A methylation indicated that m6A levels are reduced during infection of cells with the alphaherpesvirus HSV-1 (Bartkoski and Roizman, 1976, 1978). At the time, the proteins involved in the regulation of m6A methylation were unknown. To further investigate this, we used a state-of-the-art mass spectrometry approach to assess the impact of PRV or HSV-1 infection on the levels of m6A in mRNA in infected cells. The method relies on the specific digestion of m6A in the DRACH (in which D can be A, G, or U; R can be A and G; and H can be A, C, or U) context, which eliminates m6A signal from rRNA (Salisbury et al., 2021; Mirza et al., 2022). Figures 1A and 1B show that both alphaherpesviruses trigger a dramatic decrease in m6A-methylated mRNA. A time-course assay of PRV infection showed that a substantial reduction in m6A-methylated mRNA can be observed from 6 h post-inoculation (hpi) onward and is virtually complete at 9 hpi (Figure 1C).
Figure 1. Alphaherpesvirus infection results in a decrease in m6A-methylated mRNA.
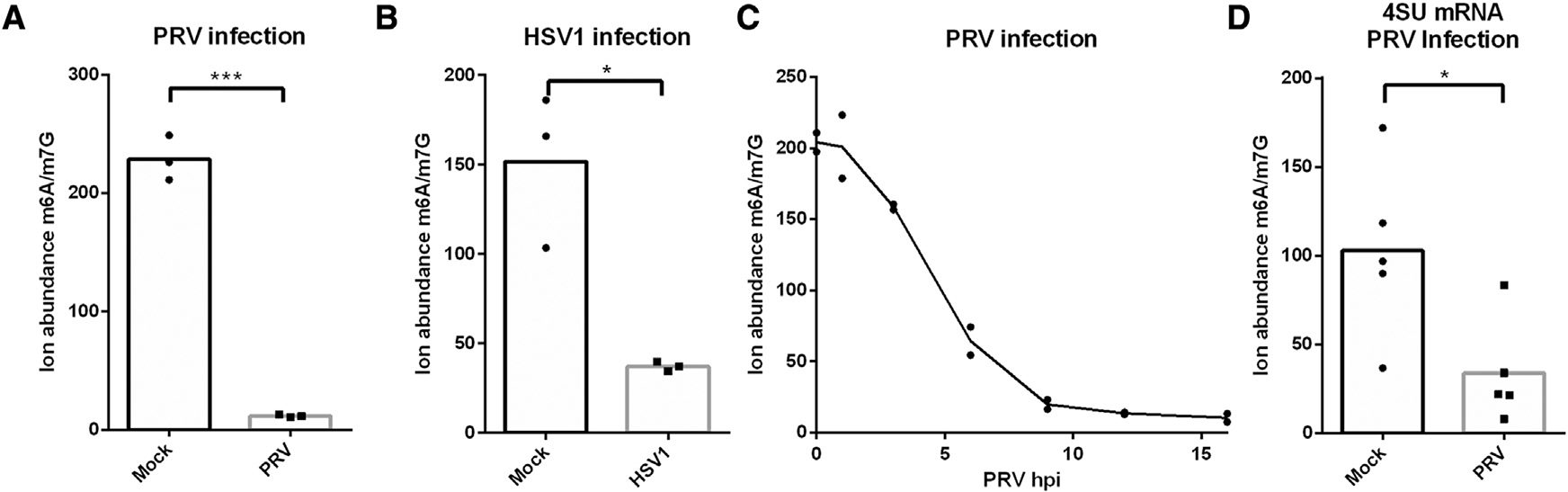
Quantification of m6A levels using mass spectrometry in (A) mRNA from mock-infected or PRV-infected ST cells at 16 hpi (n = 3 biological replicates), (B) mRNA from mock-infected or HSV-1-infected HEK293-T cells at 16 hpi (n = 3 biological replicates), (C) mRNA from PRV-infected ST cells at different time points post-inoculation (n = 2 biological replicates), and (D) 4SU-labeled mRNA produced between 7 and 9 hpi of mock-infected and PRV-infected ST cells (n = 5 biological replicates). Statistical significance was calculated by unpaired Student’s t test. *p < 0.05; ***p < 0.001.
To assess whether the m6A writer complex is inactivated in infected cells, we determined m6A levels in newly formed mRNA in mock- or PRV-infected cells. We treated PRV- or mock-infected cells with 4-thiouridine (4SU) from 7 to 9 hpi to label RNA that was produced during this time period. We then purified the 4SU-labeled RNA and quantified the m6A levels in the mRNA fraction using mass spectrometry (Figure 1D). By determining m6A levels in newly formed mRNA, we can largely exclude the possibility of increased degradation of methylated transcripts and thus measure the action of the m6A writer complex. These assays show that newly formed mRNA also contain decreased levels of m6A in PRV-infected cells, indicating that indeed alphaherpesvirus infection results in inhibition of m6A methylation.
PRV infection results in phosphorylation of several m6A methyltransferase complex proteins via the viral US3 and UL13 protein kinases
In order to determine whether alterations in one or more of the components of the writer complex might explain inhibition of m6A methylation, we performed western blotting of several of the components of the m6A writer complex. All components of the writer complex appeared to be slowly and modestly downregulated during the course of PRV infection (Figure 2A), arguing against a rapid virus-induced degradation of the m6A writer complex. However, WTAP showed a prominent upshift in the apparent molecular weight on western blot from 6 hpi onward, suggesting virus-induced post-translational modifications of the m6A writer complex. Phosphatase treatment showed that the virus-induced upshift in the apparent molecular weight of WTAP is caused by phosphorylation (Figure 2B). Alphaherpesviruses encode two viral serine/threonine protein kinases, US3 and UL13. To assess whether these viral kinases are involved in PRV-induced phosphorylation of WTAP, assays were done using wild-type or isogenic US3null and UL13null PRV strains. Western blotting of cells infected with mutants of either of the two viral kinases revealed that the PRV-induced phosphorylation of WTAP is partially dependent on the viral UL13 kinase (Figure 2C). Transfection assays showed that the active UL13 kinase, but not the kinase-dead (KD) version, induces a major upshift in the apparent molecular weight of WTAP. The US3 kinase on the other hand, induces a minor upshift of the apparent molecular weight of WTAP (Figure 2D). Together these data show that both of the viral kinases trigger phosphorylation of WTAP.
Figure 2. PRV infection triggers phosphorylation of the m6A writer complex.
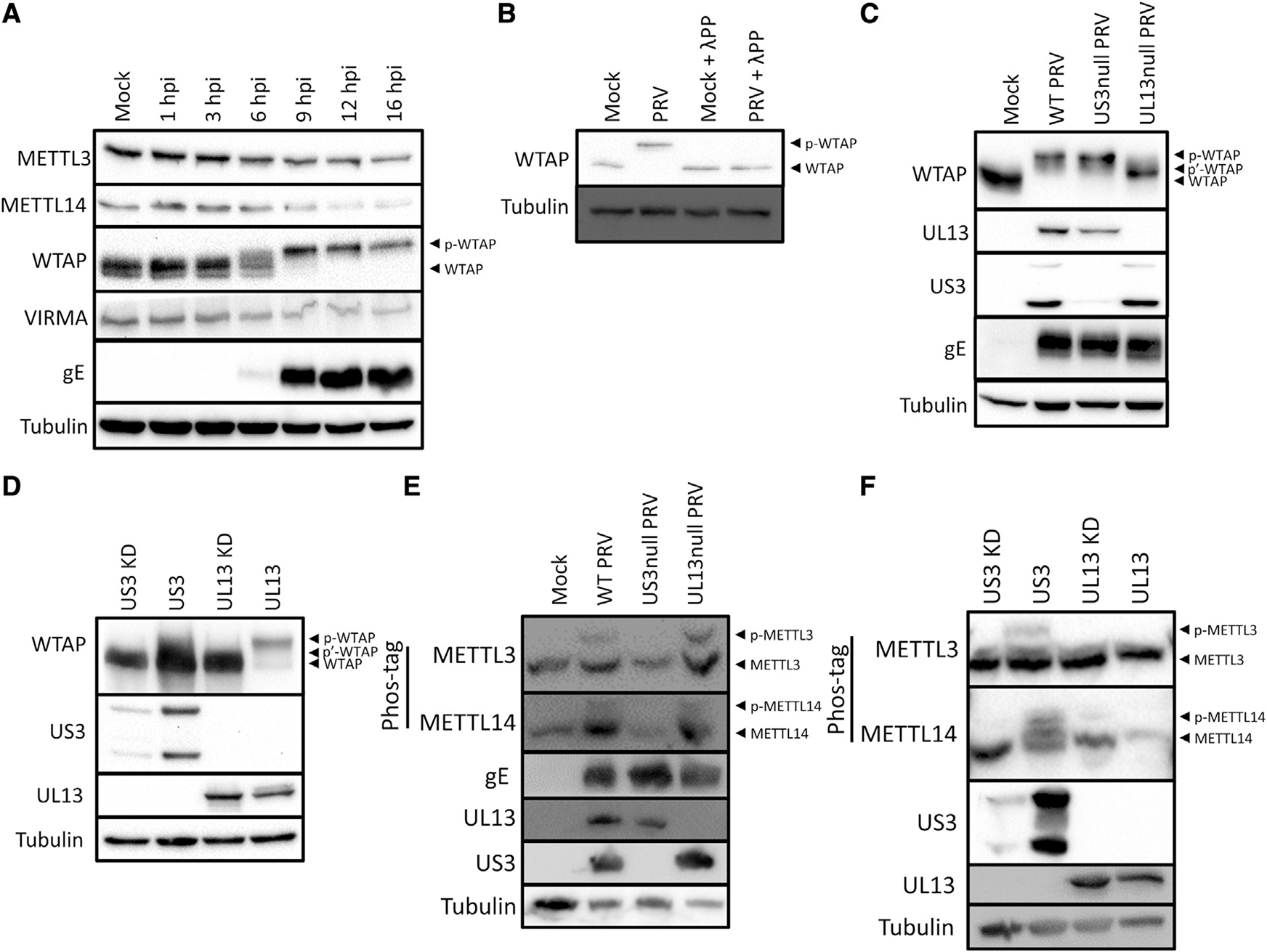
(A) Western blotting of a time-course experiment of different components of the m6A writer complex upon infection of ST cells with PRV at an MOI of 10 (n = 2 biological replicates).
(B) Western blotting of WTAP in mock- or PRV-infected ST cells at 16 hpi that were either or not treated with λ-phosphatase (n = 3 biological replicates).
(C) Western blotting of ST cells infected with wild-type (WT) PRV or isogenic PRV strains lacking expression of either of the viral protein kinases at 16 hpi (n = 3 biological replicates).
(D) Western blotting of HEK293-T cells transfected with either viral protein kinase at 48 h post-transfection (hpt) (n = 3 biological replicates). Kinase-dead (KD) US3 and UL13 contain a point mutation in the ATP binding site or the catalytic site, respectively.
(E) Phos-tag assays of ST cells mock-infected or infected with WT PRV or isogenic PRV strains lacking expression of US3 or UL13 at 16 hpi.
(F) Phos-tag assays of HEK293-T cells transfected with either of the viral protein kinases at 48 hpt.
See also Figure S1.
Not all protein phosphorylation events result in a visually detectable upshift in molecular weight on SDS-PAGE. To more sensitively assess whether other components of the m6A writer complex may also be phosphorylated in PRV-infected cells, we used Phos-tag gels, which increase differences in the migration speed of phosphorylated proteins. Figure 2E shows that PRV infection results in phosphorylation of both METTL3 and METTL14. Phosphorylation of METTL3 and METTL14 was still observed upon infection with UL13null virus, but not upon infection with US3null virus, showing that these phosphorylation events depend on the US3 protein kinase. We next performed a time-course assay, which showed that phosphorylation of METTL3 and WTAP can be clearly observed at 6 hpi, correlating well with expression of US3 and UL13 (Figure S1A). Furthermore, we confirmed that at 6 hpi, METTL3 phosphorylation in PRV-infected cells also depends on expression of US3, while both US3 and UL13 contribute substantially to WTAP phosphorylation at that time point (Figure S1B). To determine whether the sole expression of the US3 protein is sufficient to trigger phosphorylation of METTL3 and METTL14, in the absence of viral infection, transfection assays were performed. Expression of the US3 kinase, but not the UL13 kinase, triggered phosphorylation of both METTL3 and METTL14 (Figure 2F). Note that the poor expression of the kinase-inactive version of the US3 kinase, in line with earlier reports (Jansens et al., 2020), prevented conclusions regarding the kinase dependency of this phosphorylation.
In conclusion, PRV infection results in phosphorylation of several components of the m6A writer complex, including WTAP, METTL3, and METTL14. Phosphorylation of METTL3 and METTL14 depends on the viral US3 protein kinase, whereas both US3 and UL13 trigger phosphorylation of WTAP.
The m6A methyltransferase complex is inhibited in a US3-dependent manner
We next investigated whether the viral US3 and/or UL13 kinases that drive phosphorylation of the m6A writer complex components are involved in modulating its activity. We determined m6A levels in mRNA from cells infected with wild-type PRV or PRV that lacks either the US3 or the UL13 kinase. We observed a clear decrease of m6A levels in mRNA of wildtype and UL13null PRV-infected cells compared with mock-infected cells (Figure 3A). Cells infected with US3null PRV did not show this decrease in m6A levels compared with mock-infected cells (Figure 3A). Similar results were obtained using 4SU-labeled mRNA produced between 7 and 9 hpi, confirming that the inhibition of the m6A methyltransferase complex during PRV infection is US3 dependent (Figure 3B). In line with these results in PRV, cells infected with US3null HSV-1 did not show reduced m6A levels in mRNA compared with mock-infected cells, in contrast to cells infected with wild-type HSV-1 (Figure S2).
Figure 3. PRV infection leads to US3-dependent inactivation of the m6A writer complex.

(A) Mass spectrometry-based quantification of m6A levels in total mRNA from ST cells infected with wild-type PRV strain NIA3 or isogenic PRV lacking either of the viral protein kinases at 9 hpi (n = 5 biological replicates).
(B) 4SU-labeled nascent mRNA produced between 7 and 9 hpi from mock-infected ST cells or ST cells infected with wild-type PRV strain NIA3 or isogenic US3null or UL13null PRV (n = 5 biological replicates).
(C) Total mRNA from HEK293-T cells transfected with an empty vector or either of the active or kinase-dead viral kinases (n = 3 biological replicates).
(D) Total mRNA from ST cells infected with wild-type PRV strain Becker or isogenic PRV lacking the US3 protein or PRV expressing a kinase-inactive US3 protein (KD) at 9 hpi (n = 3 biological replicates).
(E) Phos-tag assays of mock-infected ST cells or ST cells infected with wild-type PRV strain Becker or isogenic PRV lacking the US3 protein or expressing a kinase-inactive US3 protein at 6 hpi.
See also Figure S2. Statistical significance was calculated by one-way ANOVA. *p < 0.05; **p < 0.01; ***p < 0.001.
To determine whether the expression of US3, in the absence of virus infection, is sufficient to inactivate the m6A writer complex, transfection assays were performed. Expression of the US3 protein led to a dramatic reduction of m6A in mRNA (Figure 3C), showing that expression of this viral protein is sufficient to inactivate the m6A methyltransferase complex. Kinase-dead US3 and intact UL13 (but not KD UL13) also triggered a reduction of m6A levels, albeit not to the same degree as observed for intact US3 (Figure 3C).
The partial reduction of m6A levels after expression of inactive US3, combined with the poor expression of the kinase-inactive protein (Figure 2F), prompted us to investigate if the impact of US3 on phosphorylation and activity of the m6A writer complex are independent of its kinase activity. To this end, we used a PRV mutant that expresses a kinase-inactive version of the US3 protein. We found that although infection with an isogenic PRV mutant deleted for the US3 protein did not lead to decreased m6A levels, infection with a mutant expressing a kinase-inactive version of the US3 protein led to a decrease in m6A levels similar to wild-type PRV (Figure 3D). Interestingly, and in line with these results, phosphorylation of METTL3 and WTAP in infected cells at 6 hpi was also observed when using the PRV mutant expressing kinase-inactive US3, but not after infection with US3null PRV (Figure 3D), indicating that US3-induced phosphorylation of METTL3 occurs indirectly, likely through activation of (a) cellular kinase(s).
Overall, these data indicate that expression of the viral US3 protein kinase is necessary and sufficient to inactivate the m6A methyltransferase complex and is independent of its kinase activity.
PRV infection results in US3-dependent dissociation of the m6A writer complex from chromatin
Next, we investigated the mechanism through which the writer complex is inactivated. METTL3 function requires its association with chromatin to mediate its co-transcriptional methylation of nascent mRNA (Ke et al., 2017; Slobodin et al., 2017). In several instances, loss of m6A formation has been linked to delocalization from chromatin and relocalization either to the nucleoplasm or cytosol. This has been seen in experiments in which m6A writer components WTAP, VIRMA, and ZC3H13 have been depleted, the writer complex was delocalized, and m6A levels in mRNA dropped (Ping et al., 2014; Wen et al., 2018; Yue et al., 2018). Similarly, the writer complex was recently reported to relocalize from the nucleus to the cytoplasm in HSV-1-infected cells (Srinivas et al., 2021). In contrast to the latter report, immunofluorescence assays indicated that the m6A methyltransferase complex subunits METTL3 and WTAP predominantly localize to the nucleus in both PRV-infected and mock-infected cells and co-localize with US3 (Figures 4A and S3A). However, in PRV-infected cells, components of the writer complex appear to be more homogeneously distributed in the nucleus compared with mock-infected cells. Similarly, we also did not observe a relocalization of the components of the m6A writer complex to the cytoplasm upon infection of HeLa cells with HSV-1 (Figure S3B).
Figure 4. Chromatin dissociation of the m6A writer complex upon PRV infection.

(A) Immunofluorescence imaging of METTL3 and WTAP in ST cells infected with wild-type PRV- or mock-infected for 8 h (n = 3 biological replicates). Scale bar, 5 μm.
(B and C) ST cells were mock infected or infected with wild-type or isogenic US3null PRV for 16 h, after which WTAP (B) or METTL3 (C) was immunoprecipitated. The different components of the m6A writer complex were then detected using western blotting and quantified using densitometry (n = 2 biological replicates).
(D) ST cells were mock infected or infected with wild-type PRV or isogenic US3null or UL13null PRV and at 16 hpi fractioned in soluble and chromatin-bound lysates, followed using Western blotting for several components of the writer complex.
See also Figure S3.
To determine whether the m6A writer complex disassembles upon PRV infection, we performed co-immunoprecipitation assays in which immunoprecipitates of WTAP or METTL3 were analyzed for the presence of other components of the complex (Figures 4B and 4C). METTL3, METTL14, and VIRMA were found in similar or even higher amounts in WTAP immunoprecipitates derived from infected cells compared with uninfected cells, indicating that the composition of the m6A methyltransferase complex remains intact during PRV infection. In line with this, we could detect METTL14, WTAP, and VIRMA in METTL3 immunoprecipitates of both infected and uninfected cells.
Although we could not detect PRV infection-induced disassembly of the m6A writer complex and did not observe cytoplasmic relocation of METTL3 or WTAP, we still considered the possibility that the writer complex does not associate with chromatin after infection. To test this idea, cell lysates were separated into a fraction containing both cytoplasmic (i.e., tubulin) and soluble nuclear proteins (i.e., PCIF1) and a fraction containing chromatin-associated nuclear proteins (i.e., histone H3) (Figure 4D). Interestingly, Figure 4D shows that PRV infection results in a marked dissociation of the m6A methyltransferase complex from the chromatin-bound fraction, as all components of the writer complex relocate from the chromatin-associated fraction to the soluble fraction. Importantly, infection with US3null PRV did not cause this chromatin dissociation of the m6A methyltransferase complex, suggesting that US3-triggered phosphorylation of the m6A methyltransferase complex leads to its dissociation from nascent mRNA.
Taken together, these data show that PRV infection triggers a US3-dependent disruption of m6A methylation; phosphorylation of WTAP, METTL3, and METTL14; and dissociation of the m6A methyltransferase complex from the chromatin-bound fraction of cells.
Inactivation of the m6A writer complex is not required for PRV replication in cell culture
We next assessed if inactivation of the m6A writer complex by the US3 protein affects viral replication in cell culture. We first compared viral protein expression in ST cells upon infection with wild-type or US3null PRV using western blot (Figure 5A). We probed for viral proteins that are expressed at different times in the replication cycle of PRV: the immediate-early protein IE180, the early protein UL13, and the late protein gD. Each of these proteins was expressed similarly in cells infected with wild-type or US3null PRV (Figure 5A), arguing against a role for m6A writer complex inhibition in viral protein production in cell culture. To confirm this, we investigated viral RNA expression using qPCR, again analyzing transcripts from different kinetic classes. Although viral transcript levels were overall slightly lower in US3null-infected cells, we did not observe any statistically significant differences between the expression of viral transcripts in wild-type- or US3null-infected cells (Figure 5B). We also assessed the production of infectious particles by quantifying infectious virus particle production in cells infected with wild-type or US3-null PRV (Figure 5C). In line with previous reports, US3null PRV showed an only slightly reduced production of infectious virus compared with wild-type PRV (Figure 5C; De Wind et al, 1992).
Figure 5. Inactivation of the m6A writer complex is not essential for viral replication, but m6A consensus sites are significantly underrepresented in alphaherpesvirus genomes.
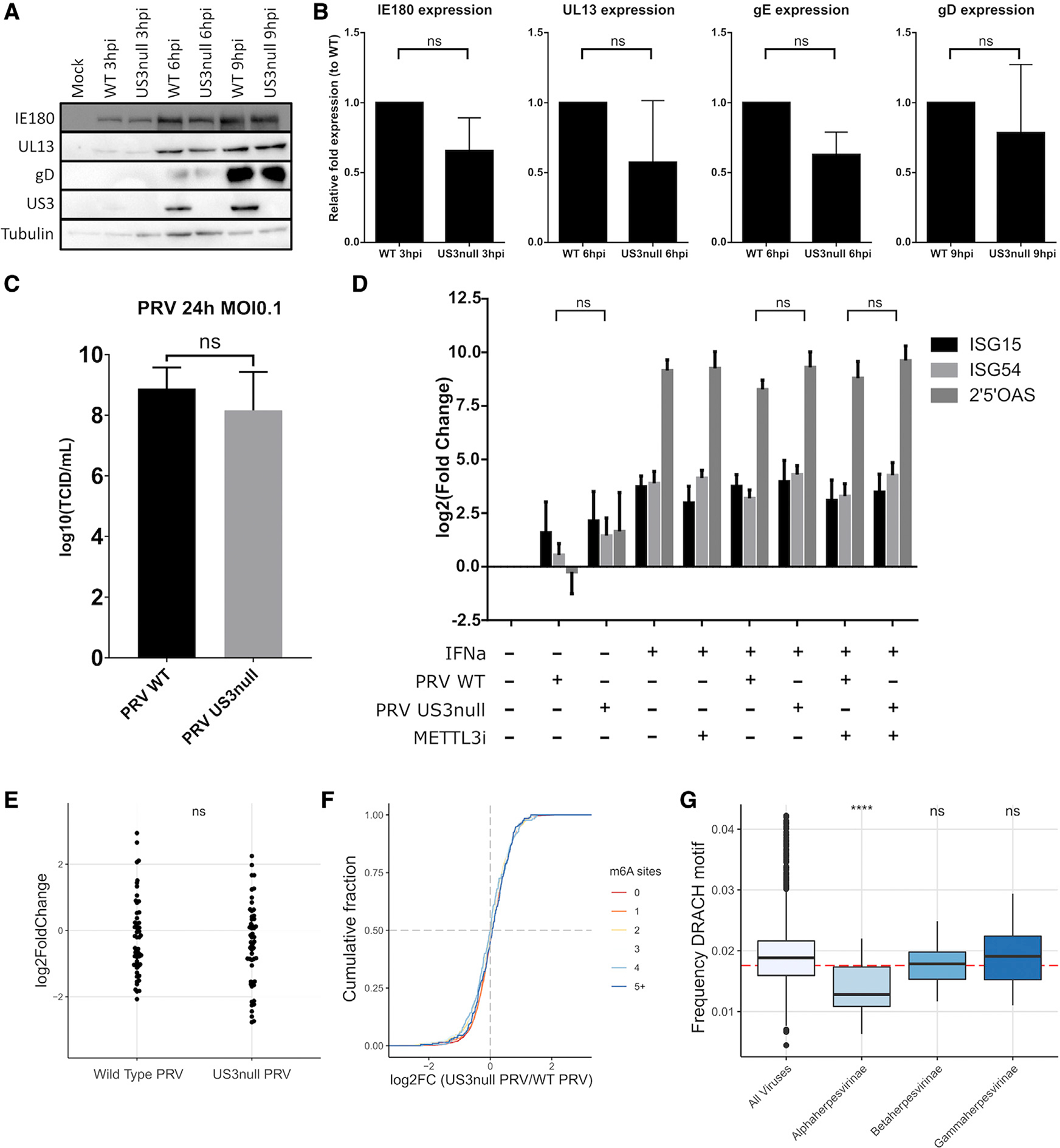
(A) ST cells were infected with wild-type or isogenic US3null PRV for different times, and viral protein levels were determined using western blotting.
(B) ST cells were infected with wild-type or isogenic US3null PRV for different times, and viral transcript levels were determined using qPCR. Data are represented as mean ± SEM (n = 3 biological replicates).
(C) ST cells were infected with wild-type or isogenic US3null PRV at an MOI of 0.1 for 24 h, and infectious viral particle counts were determined by titrations. Data are represented as mean ± SEM (n = 3 biological replicates).
(D) ST cells were infected with wild-type or isogenic US3null PRV for 8 h and treated with MG132 from 2 hpi. At 4 hpi, cells were either or not treated with 300 ng/mL IFNa. ISG transcript levels were determined using qPCR. Data are represented as mean ± SEM (n = 3 biological replicates).
(E) Fold changes of all porcine ISGs were determined using RNA-seq in wild-type or US3null-infected ST cells compared with mock-infected ST cells (n = 2 biological replicates).
(F) Cumulative fold change of m6A-methylated or non-methylated host transcripts as measured using RNA-seq comparing wild-type PRV-infected and isogenic US3null PRV-infected ST cells. ST cells were harvest at 16 h after infection. Host transcripts were binned on the basis of the number of m6A sites, with the red line representing unmethylated transcripts (n = 2 biological replicates).
(G) All available eukaryotic virus genomes were downloaded (n = 5,516 genomes), and the frequency of the DRACH motif (in which D can be A, G, or U; R can be A or G; and H can be A, C, or U) was analyzed and compared with the subfamilies alphaherpesvirinae (n = 43 genomes), betaherpesvirinae (n = 23 genomes), and gammaherpesvirinae (n = 39 genomes). The red dotted line represents the theoretical random frequency of the DRACH motif.
See also Figures S4 and S5. Statistical significance was calculated by unpaired Student’s t test or one-way ANOVA. ****p < 0.0001.
m6A methylation has been described to be a major regulator of the type I interferon (IFN) signaling pathway (Rubio et al., 2018; Winkler et al., 2019), which affects the replication of several viruses (Kennedy et al., 2016; Courtney et al., 2017; Tsai et al., 2018; Kim et al., 2020; Lu et al., 2020). To determine whether the inhibition of the m6A writer complex by the US3 protein constitutes an IFN evasion mechanism, we infected ST cells with either wild-type or US3null PRV and determined the expression of different interferon-stimulated genes (ISGs). We performed these experiments either with or without addition of interferon alpha (IFNa) and with or without treatment with a newly developed METTL3 inhibitor (Yankova et al., 2021). Treatment with this inhibitor was confirmed to reduce m6A levels by more than 75% (Figure S4). We reported earlier that PRV suppresses IFN signaling by degrading Janus kinases in a proteasome-dependent manner (Yin et al., 2021). Cells were therefore treated with the proteasome inhibitor MG132 to allow detectable expression of ISG transcripts. No difference was observed in the expression of ISG15, ISG54, or 2′5′OAS ISG transcripts after infection with either wild-type or US3null PRV (Figure 5D).
To assess whether there was an overall difference in the expression of all ISGs in US3null-infected cells compared with wild-type-infected cells, we performed RNA sequencing (RNA-seq) of ST cells that were either mock infected or infected with wild-type or US3null virus. We then determined the fold changes of porcine ISG transcript between mock-infected cells and wildtype- or US3null-infected cells. Overall, there was no statistical difference between expression of ISGs in wild-type- and US3null-infected cells (Figure 5E). In addition, we used the RNA-seq dataset to assess more generally whether host transcripts that are known to be m6A methylated (or unmethylated transcripts) show differences in stability in ST cells infected with either wild-type or US3null PRV. The data showed that there is no difference in stability of methylated cellular transcripts in US3null-infected cells compared with wild-type PRV-infected cells (Figure 5F).
Finally, we wondered whether the PRV-induced inactivation of the m6A writer complex may correlate with a selection pressure on the genome of PRV. We therefore quantified the frequency of the m6A consensus sequence (DRACH) in all known eukaryotic virus genomes. We discovered that the DRACH motif is significantly de-enriched in the genomes of alpha- but not beta- or gammaherpesviruses, suggesting that there is an evolutionary pressure against the incorporation of methylation sites in alphaherpesvirus genomes (Figure 5G). Interestingly, this effect can still be observed when accounting for the nucleotide composition of the genomes and hence taking into account the high GC content of some alphaherpesviruses, such as PRV and HSV-1 (Figure S5). Of additional interest, the latter analysis shows a significant enrichment of the DRACH motif in the genomes of gammaherpesviruses, in line with the notion that viral transcripts of different members of this herpesvirus subfamily are m6A methylated, which is thought to be important for different aspects of the biology of these viruses (Tan et al., 2017; Ye et al., 2017; Hesser et al., 2018; Baquero-Perez et al., 2019; Lang et al., 2019; Dai et al., 2021; Tang et al., 2021; Xia et al., 2021; Zheng et al., 2021; Macveigh-Fierro et al., 2022).
In conclusion, we show that the inactivation of the m6A writer complex is not essential for viral replication in the ST cell line and that no general differences on either ISG transcripts or cellular m6A-containing transcripts can be observed between wildtype- and US3null-infected ST cells. However, the data indicate a conserved evolutionary pressure against m6A methylation sites in the genomes of alphaherpesviruses.
DISCUSSION
The vast majority of m6A in mRNA is produced by a multi-subunit complex called the m6A writer complex (Bokar et al., 1994; Geula et al., 2015). The catalytic subunit METTL3, together with METTL14 and WTAP, make up the core of this enzymatic complex (Liu et al., 2014; Ping et al., 2014). Although phosphorylation of METTL3 and WTAP has been previously reported, this resulted in a relatively minor increase in methylation activity (Sun et al., 2020). Therefore, it was not clear if the m6A writer complex can be regulated by phosphorylation and how phosphorylation would affect the writer complex. Here, we describe an unprecedented inhibition of methylation activity mediated by an alphaherpesviral protein kinase, US3. M6A levels in cells infected with wild-type PRV expressing the US3 kinase are reduced up to 95%, while expression of the US3 kinase in the absence of an infection background reduces m6A levels by 85%, which are unprecedented reductions in m6A levels that have not been observed using any type of treatment or protein expression modulation.
Several components of the m6A writer complex were phosphorylated during PRV infection. The most apparent modification of the m6A writer complex upon infection was the phosphorylation of WTAP. WTAP phosphorylation is mediated by both the US3 and the UL13 viral kinases. Although transfection of the UL13 kinase resulted in a mild drop in m6A levels, a virus mutant lacking the UL13 protein kinase triggered a similar decrease in m6A methylation as wild-type virus, showing that the UL13 kinase is dispensable for inactivation of the writer complex during infection. Besides phosphorylation of WTAP, we also observed phosphorylation of METTL3 and METTL14. Although METTL3 and METTL14 phosphorylation was not observed by an upshift on standard SDS-PAGE, phosphorylation was readily detectable using Phos-tag gels. Unlike the phosphorylation of WTAP, these modifications were induced only by expression of the US3 protein kinase, and the emergence of METTL3 phosphorylation correlates well with the loss of m6A. We found that expression of the US3 protein kinase is necessary and sufficient for the phosphorylation of METTL3 and METTL14.
Our results are in accordance with early research from the 1970s that showed that HSV-1 infection leads to an inhibition of m6A methylation via an early and/or late viral protein, possibly with catalytic activity (Bartkoski and Roizman, 1976, 1978). US3, which is a viral early protein in both HSV-1 and PRV, matches these criteria. In line with our results on PRV, we found that inhibition of the m6A writer complex in HSV-1-infected HEK293T cells also depends on expression of US3. With regard to how expression of US3 leads to inhibition m6A methyltransferase activity, we found that the m6A writer complex is relocated from the chromatin-bound fraction to the soluble nuclear fraction upon PRV infection, suggesting that the writer complex detaches from nascent mRNA. The relocation is US3 dependent, therefore correlating with the inactivation of the m6A writer complex. Although we observed a US3-dependent relocation of the components of the m6A methyltransferase complex from the chromatin-bound fraction to the soluble fraction, immunofluorescence indicated that this was not associated with a relocation of METTL3 or WTAP from the nucleus to the cytoplasm. The latter is in contrast to a recent report showing that HSV-1 infection triggers a substantial relocation of the m6A methyltransferase complex to the cytoplasm (Srinivas et al., 2021). Although it is possible that HSV-1 and PRV interact with m6A methylation of transcripts differently, our assays on HSV-1-infected cells do not point to a cytoplasmic relocation of the m6A writer complex. Although speculative at this point, it is possible that these differences reflect differences in cell type or particular methodology, including fixation and permeabilization protocols.
The fact that US3 triggers phosphorylation of several critical components of the m6A methyltransferase complex complicates the identification of the exact phosphorylation sites and their contribution to inactivation of the complex. Interestingly, the inactivation of the m6A writer complex occurs independently of the kinase activity of the US3 protein. This indicates that the observed phosphorylations are driven by (a) cellular kinase(s) and that US3 co-opts an existing cellular pathway. As such, experiments aimed at further identifying the mechanism of US3-mediated inactivation of the writer complex may ultimately facilitate the detection of specific regulatory pathways that enable endogenous kinase pathways to inhibit m6A levels in mammalian cells.
Despite the near complete loss of m6A in infected cells, the role of the inactivation of the m6A writer complex during virus infection remains elusive. m6A methylation is important in the infection cycle of several different viruses, with direct effects of m6A in viral RNA, as seen in HIV-1, simian virus 40 (SV40), and influenza (Kennedy et al., 2016; Lichinchi et al., 2016; Tirumuru et al., 2016; Courtney et al., 2017; Tsai et al., 2018; Tsai et al., 2021). Also, a growing body of evidence indicates a role of m6A in type I IFN signaling in response to viral infections, as seen in human cytomegalovirus (HCMV), human metapneumovirus, and respiratory syncytial virus (RSV) (Rubio et al., 2018; Gokhale et al., 2019; Winkler et al., 2019; Lu et al., 2020; McFadden et al., 2021; Xue et al., 2021). A PRV mutant lacking the US3 kinase replicates at a similar efficiency as wild-type PRV in vitro, showing no major differences in either viral mRNA and protein expression or viral titers reached. We also did not observe significant differences in ISG expression after infection with wild-type or US3null PRV, nor did we observe a general effect on the expression of typically methylated transcripts compared with unmethylated transcripts. However, we do show that the DRACH motif, the consensus sequence for m6A methylation by METTL3, is significantly de-enriched in the genomes of alphaherpesviruses. This indicates a significant evolutionary pressure against m6A methylation of viral transcripts of alphaherpesviruses. Although our data are in line with other studies indicating that the US3 protein of alphaherpesviruses is dispensable for virus replication in cell culture, US3null mutants of alphaherpesviruses are severely attenuated in vivo (Deruelle and Favoreel, 2011). It is therefore likely that virus-induced inhibition of m6A methylation is not critical for virus replication in cell culture but may play an important role during in vivo infection of the host, which will be addressed in future assays.
In summary, the present study shows that expression of the alphaherpesvirus US3 protein kinase leads to phosphorylation and inactivation of the m6A writer complex. Inactivation of the m6A writer complex correlates with its phosphorylation and its dissociation from chromatin. These results contribute to our understanding of alphaherpesvirus biology as well as, more generally, the regulation of m6A methylation.
Limitations of the study
Our experiments show that m6A methylation can be regulated in an unprecedented manner and provide insights on how alphaherpesviruses interact with RNA processing pathways. However, there are some limitations to this study that we would like to discuss. The main limitation is that although we showed that phosphorylation of components of the writer complex correlates with the loss of m6A methylation, we were unable to identify the specific residues that are phosphorylated. As such, it is not yet clear if and which phosphorylation is causal for the inactivation of the writer complex. A second limitation is that although we show that there appears to be evolutionary pressure against methylation of alphaherpesvirus transcripts, it is still unclear how inhibition of m6A methylation contributes to the complex virus-host interplay of alphaherpesviruses.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Herman W. Favoreel (herman.favoreel@ugent.be).
Materials availability
Unique reagents generated in this study will be available upon MTA completion.
Data and code availability
The accession numbers for the RNA-seq data reported in this paper are Gene Expression Omnibus (GEO): GSE201012.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
ST cells
Swine testicle (ST) cells were cultured at 37°C in a humidified atmosphere at 5% CO2. Cells were cultured in Eagle’s Minimal Essential Medium (MEM, ThermoFisher) supplemented with 10% fetal calf serum (FCS), 1 mM sodium pyruvate, 105 U/L penicillin, 100 mg/L streptomycin and 50 mg/L gentamycin. Cells were obtained from the American Type Culture Collection (ATCC) and were not authenticated by authors. Cells were tested to be free of mycoplasma contamination via de LookOut Mycoplasma PCR detection kit (SigmaAldrich).
Human cell lines
HEK293-T and HeLa cells were cultured at 37°C in a humidified atmosphere at 5% CO2. Cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM, ThermoFisher) supplemented with 10% FCS, 105 U/L penicillin, 100 mg/L streptomycin and 50 mg/L gentamycin. Cell lines were kept at 37°C in a humidified atmosphere at 5% CO2. Cells were obtained from the ATCC and were not authenticated by authors. Cells were tested to be free of mycoplasma contamination via de LookOut Mycoplasma PCR detection kit (SigmaAldrich).
Viruses
Wild type HSV-1 F strain and its isogenic US3null mutant were described previously (Ejercito et al., 1968; Ryckman and Roller, 2004). Wild type PRV NIA3 and isogenic US3null and UL13null mutants were previously described (de Wind et al., 1990; De Wind et al, 1992; Kimman et al., 1992). Parental PRV Becker strain and its isogenic mutants that lack expression of US3 (US3null) or express a point mutated US3 in which a catalytic aspartate residue was replaced by an alanine residue (D223A, kinase inactive US3) were described earlier (Smith et al., 2004; Smith and Coller, 2008).
METHOD DETAILS
Infections
Confluent cells were inoculated at a multiplicity of infection (MOI) of 10 plaque-forming units (PFUs)/cell and analysed at 16 hpi unless indicated otherwise.
Transfections
HEK293-T cells were used for transfection assays to obtain a sufficiently high transfection efficiency. Cells were transfected with 3 μg of plasmid DNA and 5 μL PEI for each well of a 6 well plate using JetPEI (Poly-plus) according to the manufacturer’s instructions. The plasmids encoding wild type NIA3 US3 protein (pKG1) and kinase dead US3 protein with a K138Q mutation (pHF61) were described previously (Geenen et al., 2005; Deruelle et al., 2007). Transfected cells were lysed and analysed at 48 hpt.
Construction of expression vectors encoding wild type or kinase dead PRV UL13
Plasmids encoding wild type or kinase dead (KD) UL13 were generated by PCR amplification of UL13 from wild type NIA3 PRV or pGS1018 PRV encoding kinase inactive UL13 carrying a D194A alanine substitution for a catalytic aspartate residue (Coller and Smith, 2008), followed by ligation in the pcDNA3.1 backbone. In brief, UL13 was amplified with primers containing BamHI and XbaI restriction sites (forward: GGCGGACGGATCCCTGCTGACCCAATGGCTG reverse: CGCCCCGCTCTAGAACCGCAGGAAGGTGC) and using Herculase II Fusion DNA Polymerase (Agilent). The amplified fragment was purified and digested overnight. After purification, the digested fragment was ligated into a BamHI/XbaI digested pcDNA3.1 vector using the rapid DNA ligation kit (ThermoFisher). Ligated plasmids were electroporated into DH5alpha cells and positive colonies were checked by sequencing.
Cell treatments
The METTL3 inhibitor STM2457 was purchased from MedChemExpress and used at a concentration of 30 μM starting from 2 h before infection. The 26S proteasome inhibitor MG132 was purchased from Merck and treatment was performed 2 hpi at a concentration of 10 μM. A plasmid encoding recombinant porcine IFN-alpha (IFNa) was kindly provided by Simon Yongming (Kansas State University, USA). This plasmid was transfected into HEK293-T cells, and supernatant was collected at 48 hpt. The amount of IFNa secreted in the supernatant was measured by enzyme-linked immunosorbent assay (ELISA) as described before (Lamote et al., 2017). IFNa treatment was performed by treating the cells with 300 ng/mL IFNa at 4 hpi.
RNA isolation and m6A quantification
Total RNA was isolated using the RNeasy mini kit (Qiagen) according to the manufacturer’s instructions, including homogenization using the QIAshredder kit (Qiagen). DNase treatment was performed on-column to eliminate DNA contamination (Qiagen). Newly transcribed RNA was isolated following a protocol adapted from a method previously described (Garibaldi et al., 2017). Briefly, ST cells were infected with wild type, US3null, UL13null or mock infected. At 7 hpi, complete growth medium containing 500 μM 4-thiouridine (4SU) (Merck) was added to the cells. After 2 h of incubation, the cells were lysed using rlt buffer (Qiagen). 4SU labeled RNA was biotinylated using EZ-Link HPDP-Biotin (ThermoFisher). 100 μg of total RNA was incubated with 0.2 mg/mL biotin-HPDP in 1 mL of a buffer containing 10 mM Tris-HCl and 1 mM EDTA. After 90 min of incubation at room temperature, total biotinylated RNA was subjected to the RNA cleanup protocol of the RNeasy mini kit (Qiagen). The RNA was then heated to 65°C for 10 min, followed by 5 min on ice. 100 μL of streptavidin beads were added to the biotinylated RNA (Miltenyi Biotec). After 15 min of incubation at room temperature, biotinylated RNA was isolated on a μMACS column (Miltenyi Biotec). Finally, the biotinylated RNA was eluted via two elution steps using 100 mM DTT and the resulting RNA was precipitated in ethanol and resuspended in molecular grade water.
To specifically quantify m6A levels in mRNA, a sequential digestion protocol was used to only free m6A in the GAC context (Salisbury et al., 2021; Mirza et al., 2022). m6A in contaminating rRNA is not present in the GAC consensus sequence and is thus not digested. Cap m7G is first removed in order to normalize mRNA m6A to cap m7G by digesting the total RNA with 200 U of yDcpS (NEB) for 2 h at 37°C in a thermomixer. Next m6A in the GAC context is freed by digestion with 2 U RNAse T1 (Invitrogen) for 2 h at 37°C in a thermomixer, followed by digestion with 2 U of S1 nuclease (Invitrogen) for 1 h at 37°C in a thermomixer. The resulting product in then precipitated in methanol. The supernatant of the precipitation is used for LC-MS/MS using a platform comprised of an Agilent Model 1290 Infinity II liquid chromatography system coupled to an Agilent 6460 Triple Quadrupole mass spectrometer equipped with Agilent Jet Stream Technology. Chromatography of metabolites utilizes aqueous normal phase (ANP) chromatography on a Diamond Hydride column (Microsolv). Mobile phases consist of 50% isopropanol, containing 0.025% acetic acid (A), and 90% acetonitrile containing 5 mM ammonium acetate (B). To eliminate the interference of metal ions on chromatographic peak integrity and electrospray ionization, EDTA was added to the mobile phase at a final concentration of 5 μM. The following gradient was applied at the flow rate of 0.4mL/min: 0–1.0 min, 99% B; 1.0–5.0 min, to 30% B; 5 to 8min, to 0% B; 8 to 29min, 0% B, followed by a re-equilibration at 99% B for 5min. The column compartment temperature was set at 28°C. The injection volume was 2ul. MRM data was acquired in positive ion mode. Source parameters for m6A measurement were as follows: gas temperature, 230°C; gas flow, 6 L/min; nebulizer, 26 psi; sheath gas temperature, 400°C; sheath gas flow, 11 L/min; capillary voltage, 2600V; nozzle voltage, 300V; delta EMV, 200V. The source parameters for m7GMP were the same as for m6A except the capillary voltage was set at 1400V. m6A standard was purchased from Selleckchem (S3190). m7G standard was purchased from Jena Bioscience (NU-1135S).
Sequencing and analysis
RNA isolations were performed using the RNeasy mini kit (Qiagen) according to the manufacturer’s procedure, including homogenization using the QIAshredder kit (Qiagen). DNase treatment was performed on-column to eliminate DNA contamination (Qiagen). Concentration and quality of the total extracted RNA was checked via the Quant-it Ribogreen RNA assay (Life Technologies) and the RNA 6000 nano chip (Agilent Technologies). The QuantSeq 3′ mRNA library prep FWD kit (Lexogen) was used for library preparation. Library QC was performed using the high sensitivity DNA chip (Agilent technologies). Sequencing was performed on the NextSeq 500 SR 76 high output system (Illumina). All sequencing data was deposited in GEO under the following accession number: GSE201012.
To determine the difference in gene expression of m6A-methylated transcripts compared to unmethylated transcripts, differential expression was determined for transcripts binned by the amount of m6A residues they contain. Reads were aligned to the Sscrofa11.1 genome using STAR (Dobin et al., 2013). Differential expression was determined using DESeq2 (Love et al., 2014).
Western blotting
Unless indicated otherwise, cells were lysed at 48 hpt or 16 hpi in RIPA buffer (Abcam) with cOmplete mini EDTA free protease inhibitor cocktail (Roche) and PhosStop (Roche). Cell lysates were separated on a 10% polyacrylamide gel, followed by blotting on PVDF membrane (Amersham). Blots were blocked in 5% nonfat milk diluted in 0.1% Tween-20 in PBS for 1 h at room temperature. Primary antibodies were incubated overnight at 4°C. Following 3 consecutive 5 min washes in PBS-T, the membranes were incubated with the secondary antibody for 1 h at room temperature. Following 3 more 5 min washes, the blots were detected using Pierce enhanced chemiluminescence (ECL) substrate (Thermo Scientific), ECL Plus substrate (GE Healthcare), or SuperSignal West Femto maximum sensitivity substrate (Thermo Scientific) on a ChemiDoc MP imaging device (Bio-Rad). Phos-tag gels were analysed identically, except using a lysis buffer lacking EDTA and performing EDTA treatment of the gels before blotting to remove zinc ions for optimal transfer efficiency. Phos-tag gels were purchased from Fujifilm. Western blot assays were performed using primary antibodies against alpha-tubulin (Abcam ab40742, 1/1,000), METTL3 (Abcam ab1953521/1,000), METTL14 (Abcam ab252562, 1/500), WTAP (Cell Signaling Technologies 56501S, 1/1,000), VIRMA (Cell Signaling Technologies BET A302-124A, 1/1,000), PCIF1 (Proteintech 16082-1-AP,), histone H3 (Proteintech 17168-1-AP, 1/1,000), PRV US3 (Olsen et al., 2006, 1/100), PRV UL13 (Van Cleemput et al., 2021, 1/1,000), PRV gD (Nauwynck and Pensaert, 1995, 1/100), PRV gE (Nauwynck and Pensaert, 1995, 1/100) and PRV IE180 (Gomez-Sebastian and Tabares, 2004, 1/1,000).
Cell fractionation
Cells were collected by scraping followed by centrifugation at 700 g for 7 min at 4°C. The pellet was washed once with PBS. The cells were then lysed for 10 min on ice in lysis buffer containing 1% Nonidet P-40 in TNE with cOmplete mini EDTA free protease inhibitor cocktail (Roche). The insoluble fraction was then centrifuged at 10 000 g for 10 min. The supernatant was collected and contains the cytoplasm and soluble nuclear proteins. The pellet was first washed using Nonidet P-40 lysis buffer. The pellet was then lysed further for 30 min at 4°C using RIPA buffer (Abcam) with cOmplete mini EDTA free protease inhibitor cocktail (Roche) on a Vibrax shaker (IKA). The insoluble fraction was centrifuged at 10 000 g for 10 min. The supernatant was collected and contains chromatin bound proteins.
Immunoprecipitation
Cells intended for immunoprecipitation were lysed in lysis buffer containing 1% Triton-X in TBS with cOmplete mini EDTA free protease inhibitor cocktail (Roche). The lysate was diluted ½ in water before the addition of the IP antibody. WTAP was immunoprecipitated using sc-374280 antibody (Santa Cruz Biotechnology, Inc.), METTL3 was immunoprecipitated using ab195352 antibody (Abcam). After 4 h of incubation, the lysate/antibody mixture was added to magnetic protein A/G beads (ThermoFisher). After another 4 h of incubation, beads were washed 5 times using wash buffer containing 450 mM NaCl and 50 mM Tris pH 7.5, and boiled in Laemmli Buffer. Immunoprecipitates were analyzed using Western blotting as described above. If the detection antibody was raised in the same species as the immunoprecipitation antibody, blots were detected using Veriblot reagent (Abcam).
Immunofluorescence
Cells were fixed using 3% paraformaldehyde for 10 min after which they were permeabilized using methanol for 10 min. Primary antibodies were incubated overnight at 4°C. Following three washing steps with PBS, cells were incubated with secondary antibody for 1 h at 37°C at a 1/200 dilution. After three more washing steps with PBS, cells were mounted using glycerine-DABCO. Samples were imaged using a Leica SPE laser scanning confocal microscope (Leica). Immunofluorescence assays were performed using primary antibodies against METTL3 (Abcam ab195352, 1/100), WTAP (Cell Signaling Technologies 56501S, 1/100), METTL14 (Abcam ab98166, 1/100), PRV US3 (Olsen et al., 2006, 1/100) and HSV-1 ICP4 (Santa Cruz Biotechnology sc-56986, 1/100).
RNA isolation and real time quantitative PCR (RT-qPCR)
Total RNA isolations were performed using the RNeasy minikit (Qiagen) according to the manufacturer’s instructions. Purified RNA was treated with RNase free DNase I (New England Biolabs) at 37°C for 10 min to remove contaminating DNA. To stop DNase I activity, EDTA (Invitrogen) was added at a final concentration of 5 mM and samples were incubated at 75°C for 10 min. Reverse transcription was carried out with 500 ng RNA using an iScript cDNA synthesis kit (Bio-Rad) according to the manufacturer’s instructions. Quantitative PCR was performed using a StepOnePlus real-time PCR system (Applied Biosystems, Thermo Fisher Scientific) with SYBR green master mix (Applied Biosystems). The relative expression of each gene was analyzed by the double delta threshold cycle method and normalized to the level of expression of the 28S rRNA gene, which has been validated as a reference gene as previously described (Romero et al., 2020). Primers used for the different genes are listed in Table S1.
Virus titrations
Confluent ST cell monolayers were infected at a MOI of 0.1 for 24 h. Virus inoculum was washed away at 2 hpi, and the cells were washed twice with PBS. The cells were treated with sodium citrate buffer, pH 3.0 (40 mM sodium citrate, 10 mM KCl, 135 mM NaCl), for 2 min at room temperature to remove all remaining infectious virus from the input (Piret et al., 2002). Following two more washing steps with PBS, fresh ST medium was added.
Infectious virus in the supernatants was titrated by 1/10 serial dilution assays on ST cells and four experimental repeats were performed. The characteristic PRV-derived cytopathic effect served as a readout. Titers are expressed as the log10 of the TCID50/ml.
QUANTIFICATION AND STATISTICAL ANALYSIS
For the statistical analysis of the results either a two sided students t-test was used, or a one way ANOVA with a Dunnett test for multiple comparisons. p-values < 0.05 were considered to be significant. Analysis and visualization was performed in Graphpad Prism, except for Figures 5E and 5F, which were generated in R using ggplot2.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Alpha-tubulin | Abcam | ab40742 |
| METTL3 | Abcam | ab195352 |
| METTL14 (WB) | Abcam | ab252562 |
| METTL14 (IF) | Abcam | ab98166 |
| WTAP (WB) | Cell Signaling Technology | 56501S |
| WTAP (IP) | Santa Cruz Biotechnology | sc-374280 |
| VIRMA | Cell Signaling Technology | BET A302-124A |
| PRV US3 | Olsen et al., 2006 | N/A |
| PRVUL13 | Van Cleemput et al., 2021 | N/A |
| PRV gE | Nauwynck and Pensaert, 1995 | N/A |
| PRV gD | Nauwynck and Pensaert, 1995 | N/A |
| PRV IE180 | Gomez-Sebastian and Tabares, 2004 | N/A |
| HSV-1 ICP4 | Santa Cruz Biotechnology | sc-56986 |
| PCIF1 | Proteintech | 16082-1-AP |
| Histone H3 | Proteintech | 17168-1-AP |
| Anti-mouse HRP Secondary | Agilent | P0447 |
| Anti-rabbit HRP Secondary | Agilent | P0448 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| PRV NIA3 WT | de Wind et al., 1990 | N/A |
| PRV NIA3 US3null | De Wind et al, 1992 | N/A |
| PRV NIA3 UL13null | Kimman et al., 1992 | N/A |
| PRV Becker VP26-mRFP WT | Smith et al., 2004 | N/A |
| PRV Becker VP26-mRFP US3null | Coller and Smith, 2008 | N/A |
| PRV Becker VP26-mRFP US3 KD | Coller and Smith, 2008 | N/A |
| HSV1 F WT | Ejercito et al. (1968) | N/A |
| HSV1 F US3null | Ryckman and Roller, 2004 | N/A |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| STM2457 METTL3 inhibitor | Selleck Chemicals | HY-134836 |
| MG132 26S proteasome inhibitor | Merck | C2211 |
| 4-thiouridine | Merck | T4509-25MG |
| EZ-Link HPDP-Biotin | Thermo Fisher | 21341 |
| MEM | Gibco | 41090-028 |
| DMEM | Gibco | 61965-026 |
| Dithiothreitol (DTT) | Calbiochem | 3860-5GM |
| yDcpS | NEB | M0463S |
| Rnase T1 (1000 U/μl) | Thermo Fisher Scientific | EN0541 |
| S1 Nuclease (100 U/μl) | Thermo Fisher Scientific | EN0321 |
| Herculase II Fusion DNA Polymerase | Agilent | 600677 |
| m6A standard for LC-MS/MS | Selleckchem | S3190 |
| m7G standard for LC-MS/MS | Jena Bioscience | NU-1135S |
| RIPA buffer | Abcam | ab156034 |
| cOmplete mini EDTAfree protease inhibitor | Roche | 11836170001 |
| PhosStop | Roche | 4906845001 |
| PVDF membrane | Amersham | 10600023 |
| VeriBlot IP Detection Reagent (HRP) | Abcam | ab131366 |
| Pierce enhanced chemiluminescence (ECL) substrate | Thermo Scientific | 32106 |
| ECL Plus substrate | GE Healthcare | RPN2236 |
| SuperSignal West Femto maximum sensitivity substrate | Thermo Scientific | 54095 |
| BamHI-HF | New England Biolabs | R3136 |
| XbaI | New England Biolabs | R0145 |
| Nonidet P-40 lysis buffer | Merck | 11332473001 |
| Protein A/G beads | Thermo Fisher | 88802 |
| Hoechst 33342, Trihydrochloride, Trihydrate | Thermo Fisher Scientific | H1399 |
|
| ||
| Recombinant DNA | ||
|
| ||
| pKG1 plasmid (containing NIA3 US3 sequence) | Geenen et al., 2005 | N/A |
| pHF61 plasmid (containing kinase dead US3 K138Q) | Deruelle et al., 2007 | N/A |
| Plasmid containing NIA3 UL13 sequence | This paper | N/A |
| Plasmid containing kinase dead UL13 D194A | This paper | N/A |
| Plasmid containing porcine IFNa sequence | Yongming, Kansas State University | N/A |
|
| ||
| Critical commercial assays | ||
|
| ||
| Phostag gels | Fujifilm | 198-17981 |
| SYBR Green PCR Master Mix | Applied Biosystems | 4309155 |
| RNeasy Mini Kit | Qiagen | 74106 |
| mMACS Streptavidin Kit | Miltenyi Biotec | 130-074-101 |
| iScript cDNA Synthesis Kit | Bio-Rad | 1708891 |
| On-column RNase-Free DNase Set | Qiagen | 79254 |
| DNase I (RNase-free) | BioLabs | 79254 |
| Rapid DNA ligation kit | ThermoFisher | K1422 |
| JetPEI | Poly-plus | 101000053 |
| QIAshredder | Qiagen | 79654 |
|
| ||
| Deposited data | ||
|
| ||
| Raw data RNA-seq | This paper | Gene expression omnibus (GEO): GSE201012 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| ST cells | ATCC | RRID:CVCL_2204 |
| HEK293T cells | ATCC | RRID:CVCL_0063 |
| HeLa cells | ATCC | RRID:CVCL_0030 |
|
| ||
| Oligonucleotides | ||
|
| ||
| Oligonucleotides for RT-qPCR | Table S1 | N/A |
| UL13 forward primer for cloning GGCGG ACGGATCCCTGCTGACCCAATGGCTG | This paper | N/A |
| UL13 reverse primer for cloning CGCCC CGCTCTAGAACCGCAGGAAGGTGC | This paper | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| GraphPad Prism | GraphPad Software Inc | https://www.graphpad.com/ |
| RStudio 2021.09.2 | RStudio | www.rstudio.com |
| R-4.1.2 | R project | https://www.r-project.org/ |
| STAR 2.7.4a | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| DESeq2 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| ImageJ | NIH Image for the Macintosh | https://imagej.nih.gov/ij/ |
| Leica LAS X confocal microscopy software | Leica Microsystems | https://www.leica-microsystems.com/products/microscope-software/p/leica-las-x-ls/ |
Highlights.
Alphaherpesvirus infection leads to a near complete loss of m6A levels in mRNA
Viral US3 kinase triggers phosphorylation and inactivation of the m6A writer complex
m6A writer complex inactivation correlates with its dissociation from chromatin
ACKNOWLEDGMENTS
We would like to thank several researchers for their kind gift of reagents: Leigh Anne Olsen, Jolien Van Cleemput, and Lynn Enquist (Princeton University, Princeton, NJ) for the anti-PRV US3 and anti-PRV UL13 antibodies; Enrique Tabarés (Universidad Autonoma de Madrid, Madrid, Spain) for the anti-PRV IE180 antibody; Hans Nauwynck (Ghent University, Merelbeke, Belgium) for the anti-PRV gD and anti-PRV gE antibodies; the ID-DLO (the Netherlands) for the NIA3 PRV (mutant) strains; Greg Smith (Northwestern University, Chicago, IL) for the Becker PRV (mutant) strains; and Richard Roller (University of Iowa, Iowa City, IA) and Bernard Roizman (University of Chicago, Chicago, IL) for HSV-1 F (mutant) strains. We thank Nicolás Romero and Alexander Tishchenko for help with qPCR and western blot assays. This project was supported by grants from FWO-Vlaanderen (G019617N and G060119N) and the Special Research Fund of Ghent University (Concerted research grant GOA013-17) (H.W.F.), a grant from the Belgian American Education Foundation (BAEF) (R.J.J.J.), NIH grants R01NS064516 and R35NS111631 (S.R.J.), and NIH grants F32GM120987 and T32 CA062948 (A.O.-G.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111107.
DECLARATION OF INTERESTS
S.R.J. is an advisor to and owns equity in 858 Therapeutics and Lucerna Technologies.
REFERENCES
- Baquero-Perez B, Antanaviciute A, Yonchev ID, Carr IM, Wilson SA, and Whitehouse A (2019). The Tudor SND1 protein is an m6A RNA reader essential for replication of Kaposi’s sarcoma-associated herpesvirus. eLife 8, e47261. 10.7554/eLife.47261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri I, and Kouzarides T (2020). Role of RNA modifications in cancer. Nat. Rev. Cancer 20, 303–322. 10.1038/s41568-020-0253-2. [DOI] [PubMed] [Google Scholar]
- Bartkoski M, and Roizman B (1976). RNA synthesis in cells infected with herpes simple virus. XIII. Differences in the methylation patterns of viral RNA during the reproductive cycle. J. Virol. 20, 583–588. 10.1128/jvi.20.3.583-588.1976. http://www.ncbi.nlm.nih.gov/pubmed/186635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartkoski MJ, and Roizman B (1978). Regulation of herpesvirus macromolecular synthesis VII. Inhibition of internal methylation of mRNA late in infection. Virology 85, 146–156. 10.1016/0042-6822(78)90419-1. [DOI] [PubMed] [Google Scholar]
- Bokar JA, Rath-Shambaugh M, Ludwiczak R, Narayan P, and Rottman F (1994). Characterization and partial purification of mRNA N6-adenosine methyltransferase from HeLa cell nuclei. Internal mRNA methylation requires a multisubunit complex. J. Biol. Chem. 269, 17697–17704. 10.1016/s0021-9258(17)32497-3. [DOI] [PubMed] [Google Scholar]
- Coller KE, and Smith GA (2008). Two viral kinases are required for sustained long distance axon transport of a neuroinvasive herpesvirus. Traffic 9, 1458–1470. 10.1111/j.1600-0854.2008.00782.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtney DG, Kennedy EM, Dumm RE, Bogerd HP, Tsai K, Heaton NS, and Cullen BR (2017). Epitranscriptomic enhancement of influenza A virus gene expression and replication. Cell Host Microbe 22, 377–386.e5. 10.1016/j.chom.2017.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai DL, Li X, Wang L, Xie C, Jin Y, Zeng MS, Zuo Z, and Xia TL (2021). Identification of an N6-methyladenosine-mediated positive feedback loop that promotes Epstein-Barr virus infection. J. Biol. Chem. 296, 100547. 10.1016/j.jbc.2021.100547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wind N, Zijderveld A, Glazenburg K, Gielkens A, and Berns A (1990). Linker insertion mutagenesis of herpesviruses: inactivation of single genes within the Us region of pseudorabies virus. J. Virol. 64, 4691–4696. 10.1128/jvi.64.10.4691-4696.1990. http://www.ncbi.nlm.nih.gov/pubmed/2168958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Wind N, Domen J, and Berns A (1992). Herpesviruses encode an unusual protein-serine/threonine kinase which is nonessential for growth in cultured cells. J. Virol. 66, 5200–5209. 10.1128/jvi.66.9.5200-5209.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deruelle M, Geenen K, Nauwynck HJ, and Favoreel HW (2007). A point mutation in the putative ATP binding site of the pseudorabies virus US3 protein kinase prevents Bad phosphorylation and cell survival following apoptosis induction. Virus Res. 128, 65–70. 10.1016/j.virusres.2007.04.006. [DOI] [PubMed] [Google Scholar]
- Deruelle MJ, and Favoreel HW (2011). Keep it in the subfamily: the conserved alphaherpesvirus US3 protein kinase. J. Gen. Virol. 92, 18–30. 10.1099/vir.0.025593-0. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Hou G, Zhang H, Dou J, He J, Guo Y, Li L, Chen R, Wang Y, Deng R, et al. (2018). SUMOylation of the m6A-RNA methyltransferase METTL3 modulates its function. Nucleic Acids Res. 46, 5195–5208. 10.1093/nar/gky156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ejercito PM, Kieff ED, and Roizman B (1968). Characterization of herpes simplex virus strains differing in their effects on social behaviour of infected cells. J. Gen. Virol. 2, 357–364. 10.1099/0022-1317-2-3-357. [DOI] [PubMed] [Google Scholar]
- Garibaldi A, Carranza F, and Hertel KJ (2017). Isolation of Newly Transcribed RNA Using the Metabolic Label 4-Thiouridine. Methods Mol. Biol 1648, 169–176. 10.1007/978-1-4939-7204-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geenen K, Favoreel HW, Olsen L, Enquist LW, and Nauwynck HJ (2005). The pseudorabies virus US3 protein kinase possesses anti-apoptotic activity that protects cells from apoptosis during infection and after treatment with sorbitol or staurosporine. Virology 331, 144–150. 10.1016/j.virol.2004.10.027. [DOI] [PubMed] [Google Scholar]
- Geula S, Moshitch-Moshkovitz S, Dominissini D, Mansour AA, Kol N, Salmon-Divon M, Hershkovitz V, Peer E, Mor N, Manor YS, et al. (2015). m 6 A mRNA methylation facilitates resolution of naïve pluripotency toward differentiation. Science 347, 1002–1006. 10.1126/science.1261417. [DOI] [PubMed] [Google Scholar]
- Gokhale NS, McIntyre ABR, Mattocks MD, Holley CL, Lazear HM, Mason CE, and Horner SM (2019). Altered m6A modification of specific cellular transcripts affects flaviviridae infection. Mol. Cell 77, 542–555.e8. 10.1016/j.molcel.2019.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Sebastián S, and Tabarés E (2004). Negative regulation of herpes simplex virus type 1 ICP4 promoter by IE180 protein of pseudorabies virus. J. Gen. Virol. 85, 2125–2130. 10.1099/vir.0.80119-0. [DOI] [PubMed] [Google Scholar]
- Hesser CR, Karijolich J, Dominissini D, He C, and Glaunsinger BA (2018). N6-methyladenosine modification and the YTHDF2 reader protein play cell type specific roles in lytic viral gene expression during Kaposi’s sarcoma-associated herpesvirus infection. PLoS Pathog. 14, e1006995. 10.1371/journal.ppat.1006995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansens RJJ, Marmiroli S, and Favoreel HW (2020). An unbiased approach to mapping the signaling network of the pseudorabies virus US3 protein. Pathogens 9, 916. 10.3390/pathogens9110916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke S, Pandya-Jones A, Saito Y, Fak JJ, Vågbø CB, Geula S, Hanna JH, Black DL, Darnell JE, and Darnell RB (2017). m 6 A mRNA modifications are deposited in nascent pre-mRNA and are not required for splicing but do specify cytoplasmic turnover. Genes Dev. 31, 990–1006. 10.1101/gad.301036.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy E, Bogerd H, Kornepati A, Kang D, Ghoshal D, Marshall J, Poling B, Tsai K, Gokhale N, Horner S, and Cullen B (2016). Posttranscriptional m6A editing of HIV-1 mRNAs enhances viral gene expression. Cell Host Microbe 19, 675–685. 10.1016/j.chom.2016.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim G-W, Imam H, Khan M, and Siddiqui A (2020). N 6 -Methyladenosine modification of hepatitis B and C viral RNAs attenuates host innate immunity via RIG-I signaling. J. Biol. Chem. 295, 13123–13133. 10.1074/jbc.ra120.014260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimman TG, de Wind N, Oei-Lie N, Pol JMA, Berns AJM, and Gielkens ALJ (1992). Contribution of single genes within the unique short region of Aujeszky’s disease virus (suid herpesvirus type 1) to virulence, pathogenesis and immunogenicity. J. Gen. Virol. 73, 243–251. 10.1099/0022-1317-73-2-243. [DOI] [PubMed] [Google Scholar]
- Lamote JAS, Kestens M, Van Waesberghe C, Delva J, De Pelsmaeker S, Devriendt B, and Favoreel HW (2017). The pseudorabies virus glycoprotein gE/gI complex suppresses type I interferon production by plasmacytoid dendritic cells. J. Virol. 91, e02276–16. 10.1128/JVI.02276-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang F, Singh RK, Pei Y, Zhang S, Sun K, and Robertson ES (2019). EBV epitranscriptome reprogramming by METTL14 is critical for viral-associated tumorigenesis. PLoS Pathog. 15, e1007796. 10.1371/journal.ppat.1007796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichinchi G, Gao S, Saletore Y, Gonzalez GM, Bansal V, Wang Y, Mason CE, and Rana TM (2016). Dynamics of the human and viral m6A RNA methylomes during HIV-1 infection of T cells. Nat. Microbiol 1, 16011. 10.1038/nmicrobiol.2016.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, Jia G, Yu M, Lu Z, Deng X, et al. (2014). A METTL3–METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 10, 93–95. 10.1038/nchembio.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Zhang Z, Xue M, Zhao BS, Harder O, Li A, Liang X, Gao TZ, Xu Y, Zhou J, et al. (2020). N 6-methyladenosine modification enables viral RNA to escape recognition by RNA sensor RIG-I. Nat. Microbiol. 5, 584–598. 10.1038/s41564-019-0653-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macveigh-Fierro D, Cicerchia A, Cadorette A, Sharma V, and Muller M (2022). The m6A reader YTHDC2 is essential for escape from KSHV SOX-induced RNA decay. Proc. Natl. Acad. Sci. USA 119, e2116662119. 10.1073/pnas.2116662119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFadden MJ, McIntyre AB, Mourelatos H, Abell NS, Gokhale NS, Ipas H, Xhemalc e, B., Mason CE, and Horner SM (2021). Post-transcriptional regulation of antiviral gene expression by N6-methyladenosine. Cell Rep. 34, 108798. 10.1016/j.celrep.2021.108798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirza AH, Attarwala N, Gross SS, Chen Q, and Jaffrey SR (2022). Selective detection of m6A derived from mRNA using the Phospho-tag m6A assay. Preprint at bioRxiv. 10.1101/2022.05.23.493172. [DOI] [Google Scholar]
- Nauwynck HJ, and Pensaert MB (1995). Effect of specific antibodies on the cell-associated spread of pseudorabies virus in monolayers of different cell types. Arch. Virol. 140, 1137–1146. 10.1007/BF01315422. [DOI] [PubMed] [Google Scholar]
- Olsen LM, Ch’ng TH, Card JP, and Enquist LW (2006). Role of pseudorabies virus Us3 protein kinase during neuronal infection. J. Virol. 80, 6387–6398. 10.1128/JVI.00352-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng W, Li J, Chen R, Gu Q, Yang P, Qian W, Ji D, Wang Q, Zhang Z, Tang J, and Sun Y (2019). Upregulated METTL3 promotes metastasis of colorectal Cancer via miR-1246/SPRED2/MAPK signaling pathway. J. Exp. Clin. Cancer Res 38, 393. 10.1186/s13046-019-1408-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natalia P, Stephanie S, and Justin JLW (2018). Aberrant expression of enzymes regulating m 6 A mRNA methylation: implication in cancer. Cancer Biol. Med. 15, 323. 10.20892/j.issn.2095-3941.2018.0365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ping X-L, Sun BF, Wang L, Xiao W, Yang X, Wang WJ, Adhikari S, Shi Y, Lv Y, Chen YS, et al. (2014). Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 24, 177–189. 10.1038/cr.2014.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piret J, Roy S, Gagnon M, Landry S, Désormeaux A, Omar RF, and Bergeron MG (2002). Comparative study of mechanisms of herpes simplex virus inactivation by sodium lauryl sulfate and n-lauroylsarcosine. Antimicrob. Agents Chemother. 46, 2933–2942. 10.1128/AAC.46.9.2933-2942.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero N, Van Waesberghe C, and Favoreel HW (2020). Pseudorabies virus infection of epithelial cells leads to persistent but aberrant activation of the NF-kB pathway, inhibiting hallmark NF-κB-Induced proinflammatory gene expression. J. Virol. 94, e00196–20. 10.1128/JVI.00196-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio RM, Depledge DP, Bianco C, Thompson L, and Mohr I (2018). RNA m 6 A modification enzymes shape innate responses to DNA by regulating interferon β. Genes Dev. 32, 1472–1484. 10.1101/gad.319475.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryckman BJ, and Roller RJ (2004). Herpes simplex virus type 1 primary envelopment: UL34 protein modification and the US3-UL34 catalytic relationship. J. Virol. 78, 399–412. 10.1128/jvi.78.1.399-412.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salisbury DA, Casero D, Zhang Z, Wang D, Kim J, Wu X, Vergnes L, Mirza AH, Leon-Mimila P, Williams KJ, et al. (2021). Transcriptional regulation of N6-methyladenosine orchestrates sex-dimorphic metabolic traits. Nat. Metabol. 3, 940–953. 10.1038/s42255-021-00427-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Śledź P, and Jinek M (2016). Structural insights into the molecular mechanism of the m6A writer complex. Elife 5 (ptember), e18434. 10.7554/eLife.18434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slobodin B, Han R, Calderone V, Vrielink JAO, Loayza-Puch F, Elkon R, and Agami R (2017). Transcription impacts the efficiency of mRNA translation via Co-transcriptional N6-adenosine methylation. Cell 169, 326–337.e12. 10.1016/j.cell.2017.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GA, Pomeranz L, Gross SP, and Enquist LW (2004). Local modulation of plus-end transport targets herpesvirus entry and egress in sensory axons. Proc. Natl. Acad. Sci. USA 101, 16034–16039. 10.1073/pnas.0404686101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivas KP, Depledge DP, Abebe JS, Rice SA, Mohr I, and Wilson AC (2021). Widespread remodeling of the m 6 A RNA-modification landscape by a viral regulator of RNA processing and export. Proc. Natl. Acad. Sci. USA 118, e2104805118. 10.1073/pnas.2104805118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun HL, Zhu AC, Gao Y, Terajima H, Fei Q, Liu S, Zhang L, Zhang Z, Harada BT, He YY, et al. (2020). Stabilization of ERK-phosphorylated METTL3 by USP5 increases m6A methylation. Mol. Cell 80, 633–647.e7. 10.1016/j.molcel.2020.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan B, Liu H, Zhang S, da Silva SR, Zhang L, Meng J, Cui X, Yuan H, Sorel O, Zhang SW, et al. (2017). Viral and cellular N6-methyladenosine and N6, 2’-O-dimethyladenosine epitranscriptomes in the KSHV life cycle. Nat. Microbiol. 3, 108–120. 10.1038/s41564-017-0056-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Chen K, Song B, Ma J, Wu X, Xu Q, Wei Z, Su J, Liu G, Rong R, et al. (2021). m6A-Atlas: a comprehensive knowledgebase for unraveling the N6-methyladenosine (m6A) epitranscriptome. Nucleic Acids Res. 49, D134–D143. 10.1093/nar/gkaa692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirumuru N, Zhao BS, Lu W, Lu Z, He C, and Wu L (2016). N6-methyladenosine of HIV-1 RNA regulates viral infection and HIV-1 Gag protein expression. Elife 5, e15528. 10.7554/eLife.15528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tombácz D, Tóth JS, Petrovszki P, and Boldogkői Z (2009). Whole-genome analysis of pseudorabies virus gene expression by real-time quantitative RT-PCR assay. BMC Genom. 10. 10.1186/1471-2164-10-491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai K, Bogerd HP, Kennedy EM, Emery A, Swanstrom R, and Cullen BR (2021). Epitranscriptomic addition of m6A regulates HIV-1 RNA stability and alternative splicing. Preprint at bioRxiv, 2021.02.23.432449. 10.1101/gad.348508.121.been. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai K, Courtney DG, and Cullen BR (2018). Addition of m6A to SV40 late mRNAs enhances viral structural gene expression and replication. PLoS Pathog. 14, e1006919. 10.1371/journal.ppat.1006919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Cleemput J, Koyuncu OO, Laval K, Engel EA, and Enquist LW (2021). CRISPR/Cas9-Constructed pseudorabies virus mutants reveal the importance of UL13 in alphaherpesvirus escape from genome silencing. J. Virol. 95, e02286–20. 10.1128/JVI.02286-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li Y, Toth JI, Petroski MD, Zhang Z, and Zhao JC (2014). N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat. Cell Biol. 16, 191–198. 10.1038/ncb2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Doxtader K, and Nam Y (2016a). Structural basis for cooperative function of Mettl3 and Mettl14 methyltransferases. Mol. Cell 63, 306–317. 10.1016/j.molcel.2016.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Feng J, Xue Y, Guan Z, Zhang D, Liu Z, Gong Z, Wang Q, Huang J, Tang C, et al. (2016b). Structural basis of N6-adenosine methylation by the METTL3–METTL14 complex. Nature 534, 575–578. 10.1038/nature18298. [DOI] [PubMed] [Google Scholar]
- Wen J, Lv R, Ma H, Shen H, He C, Wang J, Jiao F, Liu H, Yang P, Tan L, et al. (2018). Zc3h13 regulates nuclear RNA m6A methylation and mouse embryonic stem cell self-renewal. Mol. Cell 69, 1028–1038.e6. 10.1016/j.molcel.2018.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams GD, Gokhale NS, and Horner SM (2019). Regulation of viral infection by the RNA modification N6 -methyladenosine. Annual Review of Virology 6, 235–253. 10.1146/annurev-virology-092818-015559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler R, Gillis E, Lasman L, Safra M, Geula S, Soyris C, Nachshon A, Tai-Schmiedel J, Friedman N, Le-Trilling VTK, et al. (2019). m6A modification controls the innate immune response to infection by targeting type I interferons. Nat. Immunol. 20, 173–182. 10.1038/s41590-018-0275-z. [DOI] [PubMed] [Google Scholar]
- Xia TL, Li X, Wang X, Zhu YJ, Zhang H, Cheng W, Chen ML, Ye Y, Li Y, Zhang A, et al. (2021). N(6)-methyladenosine-binding protein YTHDF1 suppresses EBV replication and promotes EBV RNA decay. EMBO (Eur. Mol. Biol. Organ.) J 22, e50128. 10.15252/embr.202050128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue M, Zhang Y, Wang H, Kairis EL, Lu M, Ahmad S, Attia Z, Harder O, Zhang Z, Wei J, et al. (2021). Viral RNA N6-methyladenosine modification modulates both innate and adaptive immune responses of human respiratory syncytial virus. PLoS Pathog. 17, e1010142. 10.1371/journal.ppat.1010142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yankova E, Blackaby W, Albertella M, Rak J, De Braekeleer E, Tsagkogeorga G, Pilka ES, Aspris D, Leggate D, Hendrick AG, et al. (2021). Small-molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature 593, 597–601. 10.1038/s41586-021-03536-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye F, Chen ER, and Nilsen TW (2017). Kaposi’s sarcoma-associated herpesvirus utilizes and manipulates RNA N6-adenosine methylation to promote lytic replication. J. Virol. 91, e00466–17. 10.1128/JVI.00466-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, Romero N, and Favoreel HW (2021). Pseudorabies virus inhibits type I and type III interferon-induced signaling via proteasomal degradation of Janus kinases. J. Virol. 95, e0079321. 10.1128/jvi.00793-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Y, Liu J, Cui X, Cao J, Luo G, Zhang Z, Cheng T, Gao M, Shu X, Ma H, et al. (2018). VIRMA mediates preferential m6A mRNA methylation in 3′UTR and near stop codon and associates with alternative polyadenylation. Cell Discov. 4, 10. 10.1038/s41421-018-0019-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Wang J, Zhang X, Fu Y, Peng Q, Lu J, Wei L, Li Z, Liu C, Wu Y, et al. (2021). RNA m6 A methylation regulates virus-host interaction and EBNA2 expression during Epstein-Barr virus infection. Immun. Inflamm. Dis 9, 351–362. 10.1002/iid3.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession numbers for the RNA-seq data reported in this paper are Gene Expression Omnibus (GEO): GSE201012.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.