

Abstract
Adenine base editors (ABEs) have been subjected to multiple rounds of mutagenesis with the goal of optimizing their function as efficient and precise genome editing agents. Despite an ever-expanding data set of ABE mutants and their corresponding DNA or RNA-editing activity, the molecular mechanisms defining these changes remain to be elucidated. In this study, we provide a systematic interpretation of the nature of these mutations using an entropy-based classification model that relies on evolutionary data from extant protein sequences. Using this model in conjunction with experimental analyses, we identify two previously reported mutations that form an epistatic pair in the RNA-editing functional landscape of ABEs. Molecular dynamics simulations reveal the atomistic details of how these two mutations affect substrate-binding and catalytic activity, via both individual and cooperative effects, hence providing insights into the mechanisms through which these two mutations are epistatically coupled.
Introduction
The ability to introduce A•T to G•C base pair conversion in the genetic code of an organism, in a precise, efficient, and programmable manner, has the potential to correct almost 60% of known pathogenic point mutations in human beings.1 Targeted A•T to G•C conversions have recently been realized through the development of adenine base editors (ABEs).2
ABEs consist of two subunits: a catalytically impaired Cas9 (Cas9n), which serves as a programmable DNA-targeting module, and an engineered variant of a tRNA adenosine deaminase enzyme TadA3 (hereafter referred to as TadA*, where * indicates the incorporation of mutations in the natural enzyme), which serves as the single-stranded DNA (ssDNA)-editing module and enables the hydrolytic deamination of targeted adenosines (A) into inosines (I). Inosine is subsequently converted into guanosine (G) by the DNA repair and replication machinery, completing the A•T to G•C base pair conversion by ABEs (Fig. 1A).
FIG. 1.

(A) Schematic representation of base editing by adenine base editors (ABEs; PDB ID: 6VPC).56 The binding of Cas9n to the target genomic locus unwinds the DNA double helix and exposes a small region of single-stranded DNA. TadA* hydrolytically deaminates the adenine (A) to form inosine (I), which is subsequently converted to guanine (G) by cellular DNA repair and replication machinery. (B) Engineering efforts in the field to generate and improve upon ABEs, starting from Escherichia coli wtTadA. (C) Primary and secondary structure of E. coli wtTadA with key mutations indicated. The line colors correspond to colors shown in (B), indicating the ABE version in which these mutations were identified. Solid lines are mutations that were incorporated into final ABEs, while dashed lines are mutations that were experimentally tested in previous work but not incorporated into final ABEs. Color images are available online.
ABEs continue to remain a focal point of interest for the genome editing community, not only because of their potential as therapeutic agents4–9 but also because of the remarkable scientific effort that went into their development. Extensive protein engineering and evolutionary methods were employed to impart ssDNA-editing capabilities onto an RNA-editing enzyme, the wild-type Escherichia coli TadA (wtTadA), resulting in the seminal ABE7.10 base editor.2
Although the mutations that gave rise to the original ABE7.10 construct successfully imparted ssDNA-editing capability onto TadA*, they did not suppress the inherent RNA-editing activity of TadA*. It was subsequently demonstrated that ABE7.10 induces considerable gRNA-independent off-target RNA editing throughout the transcriptome.10–12
Since the development of ABE7.10, major efforts have been devoted to its further mutagenesis on two separate fronts (Fig. 1B). First, additional rounds of directed evolution were employed to increase the on-target ssDNA-editing activity by TadA, resulting in ABE8.2013 and ABE8e.14 Second, structural analyses of the TadA–RNA complex followed by rational engineering was employed to decrease the off-target RNA-editing activity by TadA, resulting in ABE7.10F148A,15 ABE7.10V106W,16 and SECURE-ABEs.17
Due to the lack of naturally occurring ssDNA-editing enzymes (cytosine deaminases are a rare exception18), the expansion of the existing base editing repertoire will inevitably require evolution and engineering strategies on new enzymes analogous to those used in the development of ABEs. The success of structure-based protein engineering efforts is highly dependent on the availability of appropriate X-ray or cryo-EM structures of the protein–nucleic acid complexes. Even when structural data are available to guide this process, most mutations, especially those concentrated near the active site of the enzyme, are likely to have detrimental effects on the enzyme's function.16,17,19
Thus, to facilitate the design of future base editors, especially with reduced off-target RNA editing activities, it is important to understand fully the atomistic features that are essential for the native RNA-editing function of TadA*, and how specific mutations can affect changes to its substrate binding and catalytic mechanism.
To date, many studies have mutated ABEs to manipulate its DNA- and RNA-editing abilities, producing a large amount of experimental data associated with mutations at 46 of the 167 residue sites of TadA* (Fig. 1B and C). To gain fundamental insights into ABE's editing activity from this ever-expanding pool of mutational information and to guide future efforts in the development of new base editors, we carried out a systematic data-driven computational study combined with experimental assays to understand better, in atomistic detail, the effects of individual mutations on the activity of TadA*.
Methods
Data curation and sequence entropy
Extant homologs were obtained with the BLAST program20 using E. coli wtTadA as the initial query sequence, with an e-value cutoff of 0.1 in the SWISSPROT database.21 We further filtered the data set by removing sequences with >40% gap percentage and to minimize redundant sequences with >95% identity to the query sequence. The final filtered data set is composed of 35 homologs. The resultant data set was used to calculate the sequence entropy score, defined as:
| (1) |
where refers to the statistical probability of having a particular amino acid n at site i, and N is the total number of amino acids. Further details regarding the data set and entropy calculation can be found in the Supplementary Materials and Methods.
Experimental methods
Cloning
All plasmids used in this study were produced using USER cloning with ABE0.122 as a template, using Phusion U Hot Start DNA Polymerase (Thermo Fisher Scientific). All DNA vector amplification was carried out using NEB 10-beta competent cells (New England Biolabs). All plasmids were purified using the ZymoPURE II Plasmid Midiprep Kit (Zymo Research).
Cell culture
HEK293T (ATCC CRL-3126) cells were cultured in DMEM-GlutaMAX (Gibco) media supplemented with 10% (vol./vol.) fetal bovine serum (FBS; Gibco) and 100 IU/mL penicillin-streptomycin (Gibco) at 37°C with 5% CO2. Prior to transfection, the media was replaced with antibiotic-free media.
Transfections
DNA samples for transfections were prepared with 750 ng of a base editor plasmid and 250 ng of a nontargeting sgRNA plasmid. Each sample was diluted to 12.5 μL with Opti-MEM™ (Gibco). Lipofectamine 2000 (L2000; Invitrogen) was diluted with Opti-MEM at a ratio of 1.5 μL L2000 to 11 μL Opti-MEM, with 12.5 μL of this mixture being added to each DNA sample. After 15 min of incubation at room temperature, each transfection sample was added to a well containing 9:5 × 105 HEK293T cells suspended in 250 μL DMEM-GlutaMAX (Gibco) supplemented with 10% FBS.
High-throughput RNA sequencing
Cells were lysed 36 h after transfection with 300 μL RNA lysis buffer (Zymo Research), and RNA was extracted with either a Zymo Quick-RNA Miniprep Kit or a Qiagen RNeasy Mini Kit following the manufacturer's instructions. RNA was reverse transcribed to produce cDNA using the SuperScript III First-Strand Synthesis System (Invitrogen) following the manufacturer's instructions. Target sites were amplified from cDNA using two rounds of polymerase chain reaction (PCR). The first round used site-specific primers (Supplementary Tables S1 and S2) to amplify the target sequence from cDNA.
Target amplification was confirmed with gel electrophoresis. The product from the first round of PCR was used as a template in the second round of PCR, which added unique sets of p5/p7 Illumina barcodes to each sample. Amplification was confirmed with gel electrophoresis. Then, amplicons of similar size were pooled, and gel purified on a 2% agarose gel. The target amplicon was excised from the gel and dissolved in three volumes QG buffer by incubating at 42°C for 10 min. After chilling on ice for 5 min, 1/3 volume of 100% isopropanol was added, and the sample was run through a Qiaquick PCR purification column on a vacuum manifold. The column was washed with 750 μL of PE wash buffer, and residual ethanol was removed by spinning the column at 16,000 g for 3 min.
PCR products were eluted with 30 μL HyClone water. Pooled libraries were quantified by Qubit and sequenced on an Illumina MiniSeq according to the manufacturer's protocol.
HTS data analysis
Sequencing reads were demultiplexed in MiniSeq Reporter (Illumina), and individual FASTQ files were analyzed using CRISPResso2.23
Computer simulations
The TadA*0.1 model was built using the crystal structure of E. coli TadA (PDB ID: 1Z3A).24 Given the sequence homology between Staphylococcus aureus TadA and E. coli TadA, we combined the saTadA-RNA structure (PDB ID: 2B3J) with the TadA*0.1 model to build the TadA*0.1–RNA model.25 The TadA*0.1 was transformed into the various ABE mutants using the swapaa command in Chimera.26 For both apo-TadA* and TadA*–RNA models, all crystallographic water molecules within 3 Å distance of the surface of the protein or the RNA were preserved during the modeling procedure. All titratable residues were protonated using the H++ server employing the default settings.27,28
The protein was represented using Amber ff14SB,29 and the RNA was represented using RNA.OL3 force field.30–32 The metal-containing active site of TadA* was represented with custom force field parameters obtained using the MCPB.py approach at B3LYP/6-31G* level of theory.33 LEap tool from AmberTools was used to immerse the apo-TadA* and TadA*–RNA complexes into a pre-equilibrated truncated octahedron box of explicit TIP3P water, with a 15 Å buffer distance. Varying numbers of Na+ ions were added to each of the systems to maintain electroneutrality, and the simulation cell was then replicated infinitely in three dimensions to impose periodic boundary conditions.
All MD simulations were performed under periodic boundary conditions using the CUDA accelerated version of PMEMD implemented in the Amber18 suite of programs.34–37 The structures were first relaxed using a combination of steepest descent and conjugate gradient minimization. This was followed by 1 ns heating to 298.15 K and multistep equilibration under progressively decreasing harmonic restraints for 40 ns. Subsequently, we removed all restraints and carried out ∼1 μs unbiased MD simulations for the four TadA* mutants and corresponding TadA*–RNA complexes.
We calculated the free-energy binding profiles of the TadA*–RNA complexes along the collective variable corresponding to the distance between the centers of mass of the protein and the RNA substrate. For each TadA*–RNA complex, the PMF along this collective variable was calculated using umbrella sampling (US) simulations.38 Starting from the equilibrated TadA*–RNA structures, we conducted four independent sets of US simulations for all the four TadA*–RNA complexes, and the final PMFs were reconstructed using the weighted histogram analysis method (WHAM) algorithm.39,40 Additional error analysis was carried out using a custom block averaging script based on the method described by Zhu and Hummer.41
The free energy changes for the deprotonation of the activated water molecule by the Glu59 residue for the TadA* (and TadA*–RNA) models were computed for the various systems through a hybrid quantum mechanical/molecular mechanical (QM/MM) approach. The QM subsystem consisted of the side chains of the active site residues (His57, Glu59, Cys87, and Cys90), the Zn+2 ion, and the activated water for both the apo-TadA* and TadA*–RNA models. These QM atoms were treated using self-consistent charge density functional tight binding (DFTB) method implemented within Amber18.42 The atoms beyond this active site cluster were represented the MM subsystem and were treated using the force fields as in the unbiased MD.
The difference of the distances between the active site water oxygen atom and shared proton and the Glu59 O and shared proton was chosen as the collective variable to monitor the deprotonation reaction. For both the apo TadA* and TadA*–RNA complexes, the reaction profile along this collective variable was calculated using US simulations following a procedure similar to the one employed for the calculation of the TadA*–RNA binding profiles as summarized above.
The CPPTRAJ module implemented within Amber18 was used to analyze all the MD trajectories.43,44 The visualization of the MD trajectories was rendered using Chimera, and data were plotted using Matplotlib.45
Additional details for all simulation protocols can be found in the Supplementary Materials and Methods. Supplementary Table S3 summarizes all the simulations that were carried out during this study.
Results
Sequence entropy calculation
The principal tenet of biochemistry is that the primary sequence of amino acids comprising a protein dictates its three-dimensional molecular structure, which then determines its biological function.46 To date, most ABE engineering efforts have relied on the second and third tiers of this tenet: structural analyses15–17 followed by site-directed mutagenesis and experimental measurements of the resulting functional properties of TadA (second tier), or directed evolution where TadA is randomly mutated and functional variants are identified through a selection scheme (third tier)2,13,14 (Fig. 1B).
Due to the time- and resource-intensive nature of these second- and third-tier methods, we decided to begin our investigations by focusing instead on the first tier of this tenet—that is, investigating how the primary sequence of TadA can be used to rationalize the effects that individual mutations, identified experimentally, have on the native function of TadA* (i.e., RNA-editing activity; Fig. 1C).
With the expansion of reliable protein sequence databases,21,47 the statistical analyses of protein homologs have already enabled the successful prediction of mutational effects on the function of several enzymes,48 including cytosine base editors.49
For our sequence-based analyses of the ABE mutations, we used the amino acid sequence of E. coli wtTadA3 as our query for a BLAST search20 of its extant homologs in the SWISSPROT database,47 which generated a data set of 75 homologs. However, as our primary focus is to identify residues essential for the function of TadA on its native RNA substrate, we filtered out distant homologs using stringent percentage identity and coverage length cutoff values (Fig. 2A). This filtering resulted in a more focused data set, as it removed functionally distinct and redundant sequences from our initial BLAST search. Despite reducing the size of the data set considerably (to 35 homologs), this filtered data set still captures the diversity of our initial unfiltered data set (Fig. 2A).
FIG. 2.

(A) The top two principal components of the pairwise sequence distance matrix of extant homologs comprising the filtered (indicated with circles and dots) and unfiltered data sets. Based on the similarity of the sequences, the data set is clustered into four separate sets, colored purple, brown, red, and blue. The sequences in the filtered data set are highlighted in each cluster. (B) Multiple sequence alignment of extant homologs of wtTadA to calculate the statistical probability of occurrence of individual amino acids at residue site i . This is subsequently used to assign a conservation score to site i, using Shannon's definition of information entropy equation 1. (C) Information entropy of individual residue sites of the wtTadA query, with its secondary structure elements mapped below in (D). I Entropy values mapped on to the three-dimensional structure of E. coli TadA using a color gradient to signify conserved residues and mutational hotspots. Color images are available online.
To visualize the sequence space captured by our unfiltered and filtered data sets and to highlight relationships among these wtTadA homologs, we performed a dimensionality reduction of the data set using principal component analysis. This allowed us to project the hyperdimensional sequence space associated with the homologs onto two dimensions, while still preserving the relationships among the various homologs. By partitioning the data set into four representative clusters (Supplementary Fig. S1), the outcome of filtering becomes more apparent, as each cluster consists of functionally similar homologs. These clusters are represented by different colors (purple, brown, red, and blue) in Figure 2A.
From this analysis, it can be observed that this filtered data set indeed captures the overall diversity of the unfiltered data set, as three of the four clusters are represented. The purple cluster (containing the query sequence) consists primarily of TadAs and their eukaryotic equivalents (ADAT2s), and hence has the greatest number of filtered sequences (26 of the 35 filtered sequences).
The next most populated cluster, the brown cluster, consists of 22 sequences in the unfiltered set, and six sequences after filtering. Only 3 out of 11 sequences were selected from the red cluster, with two of these sequences belonging to the cytidine deaminase superfamily (and having 50% similarity to the query sequence) and the third sequence corresponding to a guanine deaminase.
Given the distance between the blue cluster and the query sequence (i.e., the lack of similarity between these sequences, as they represent the catalytically inactive Tad3 and ADAT3 proteins), no sequences were selected from this cluster upon the implementation of our filters.
It is important to note that our filtered data set consists entirely of RNA-editing enzymes, demonstrating the effectiveness of our filters. We therefore reasoned that any primary sequence analyses of our filtered data set would be highly biased toward illuminating aspects of RNA-editing activity by the wtTadA (Supplementary Fig. S1B).
Having obtained a reliable data set of extant TadA homologs, we next sought to quantify the evolutionary conservation and functional importance of individual residues of wtTadA. An extensively studied and widely used approach to address this problem is the evaluation of information theory-inspired sequence entropy (Hi) scores (Fig. 2B).50–53 The value of Hi ranges between 0 and 1, with an entropy value of 0 indicating that the site has only one unique amino acid represented within the data set (suggesting that the site is highly conserved from an evolutionary standpoint), and an entropy value of 1 indicating that the site has every possible amino acid represented within the data set (suggesting that such a site is naturally more tolerant to mutations).
Applying equation 1 to the filtered data set of TadA homologs (Fig. 2A), we calculated the site entropy for the entire wtTadA sequence (Fig. 2C). The active site of wtTadA consists of a zinc ion tetrahedrally coordinated by Cys87, Cys90, His57, and a water molecule. This water molecule is activated for deamination reaction by the highly conserved Glu59 residue. Consistent with the importance of these residues for the canonical RNA-editing activity of TadA, we observed for these four active site residues. This active site is further stabilized by a β-sheet core, and the entropy values for 24 of these 38 core residues are also low 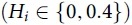 . The surface-exposed residues have relatively higher values of Hi, with the C- and N-terminal residues having the highest values (Hi > 0.4; Fig. 2D).
. The surface-exposed residues have relatively higher values of Hi, with the C- and N-terminal residues having the highest values (Hi > 0.4; Fig. 2D).
By mapping these entropy scores onto the structure of wtTadA24 (Fig. 2E), the correlation between the entropy values and the three-dimensional structure of TadA is clearly apparent. Thus, this sequence-based entropy model is capable of representing the structural information encoded by the amino acid sequence of wtTadA.
Sequence entropy as a binary classifier of TadA* RNA-editing activity
Building upon these results, we used the sequence-based entropy model to rationalize the role played by different amino-acid mutations that have been experimentally shown to modulate the function (i.e., RNA-editing activity) of ABEs (Fig. 1B and C). Based on the biochemical interpretation of the two extreme entropy values, we chose as an initial cutoff value to distinguish the functional implications of the entropy data obtained for the wtTadA sequence (Fig. 2C and E) in the context of all mutations reported for the ABEs (Fig. 1B and C).54
Within this model, we hypothesize that residue sites having will either induce no change in the activity of wtTadA or, if mutated appropriately, can have a favorable impact on the native activity (i.e., RNA-editing activity) of wtTadA. Conversely, sites with are predicted to have adverse effects on the canonical RNA-editing activity of wtTadA.15–17 It should be noted that since our data set comprises of only RNA-editing enzymes, we are primarily referring to the impact that individual mutations have on the native function of the wtTadA sequence (SI sequences and Supplementary Fig. S1B).
To quantify the performance of the sequence-based entropy model, we computed a confusion matrix where each prediction (Fig. 3A and B) is validated against the corresponding experimental editing outcome for 46 total mutations2,13–17 (Fig. 3C). By construction, the main diagonal elements (running from top left to bottom right) of the confusion matrix thus correspond to correct predictions, while the off-diagonal elements indicate incorrect predictions.
FIG. 3.

(A) and (D) Mutations reported to have beneficial or neutral effects on the RNA and DNA editing activity of the ABEs, respectively. (B) and (C) Mutations reported to have detrimental effects on the RNA and DNA editing activity of the ABEs, respectively. (C) and (F) Confusion matrix of the experimental data and the entropy-based classifier. (G) Local environment of 84 and 108 residues in the wtTadA structure. (H) Binned entropy values and distribution of amino acids at sites 84 and 108. Color images are available online.
We found that our sequence-based entropy model exhibits an accuracy of 89.1% and an F1 score of 0.92 (Fig. 3C). Specifically, the model correctly predicts the impact of all the mutations that reduced the RNA-editing activity of TadA* for which there are data, but incorrectly predicts the impact of five low-entropy mutations, which increased the RNA-editing activity of TadA*.
Sequence entropy as a binary classifier of TadA* DNA-editing activity
Given the vast amount of experimental data available regarding the mutations that impact the ssDNA-editing efficiency of TadA*, we were additionally interested in assessing the performance of the entropy-based model on these mutations. Hence, mutations that increase the ssDNA-editing ability of ABEs (as discovered using either directed evolution2,13,14 or site-directed mutagenesis17) are deemed to be correctly classified using our information entropy-based model if their Hi value is greater than 0.5 (Fig. 3D).
Despite being entirely derived from the information content of amino-acid sequences contained in a highly biased RNA-editing data set, the sequence-based entropy model applied to all the reported ABE mutations that have data regarding ssDNA-editing activity exhibits a remarkable accuracy of 86.9% and an F1 score of 0.91 (Fig. 3F). In certain cases, residues have been or can be mutated in multiple different ways, which may lead to conflicting editing outcomes (Supplementary Table S4). To resolve these conflicts and classify such sites, precedence was given to the editing outcome produced by the most chemically conserved mutation at such sites.
Here, we found our model to predict the effects of six mutations incorrectly, all of which correspond to residues with low entropy values that were experimentally found to increase ssDNA-editing activity. Specifically, we found that site 82 (Val in the wild-type enzyme), which has a low entropy value (), results in enhanced ssDNA editing in ABE8s when mutated to Ser.13 Notably, mutation of this residue to Gly abrogates just the RNA-editing activity of TadA* in SECURE-ABEs, while its mutation to Trp abrogates both the RNA as well as ssDNA editing by the ABEs,17 which was correctly predicted by our RNA-editing data set (Supplementary Table S4). This suggests higher predictability of the entropy classifier regarding the native RNA-editing activity of TadA* than its ssDNA-editing activity, as expected.
Furthermore, both sites 84 (Leu in wtTadA) and 108 (Asp in wtTadA) are associated with low entropy values but were mutated to enhance ssDNA-editing activity during the development of the foundational ABE7.10.2 Similarly, a low entropy value is found for site 149 (phenylalanine in wtTadA, ), which was mutated to enhance DNA-editing activity in ABE8e14 and was found to have no significant impact on the RNA-editing activity in the SECURE-ABEs.17
Overall, these misclassifications are restricted to mutations that impact the ssDNA-editing efficiency of TadA*, thus highlighting that the entropy model, just like other sequence-based coevolutionary methods, is limited by quality of the sequence data set.55
Binned evaluation of sequence entropy by considering the chemical nature of sidechains
To understand these six misclassified residues better and to identify possible deficiencies of our model and refine our classification scheme, we sought to analyze the amino acid distribution at these residue sites further (Fig. 3A and D).
The Asp108Asn mutation was the critical first mutation that led to the onset of ssDNA-editing activity of TadA*.2 Moreover, this residue is part of a surface-exposed loop in the structure of TadA* (Fig. 3G). Hence, we would expect this residue to display high entropy. To dissect the anomalous misclassification of site 108 through our entropy-based model further, we analyzed the distribution of various amino acids at this site within our data set (Fig. 3H). We observed that although the mutational entropy of this site is marginally low, approximately 36% of the data set sequences record an Asn at this site, making it the second most probable amino acid at site 108 (Supplementary Table S5).
This observation is particularly striking, given the importance of the Asp108Asn mutation. It was observed during the first round of directed evolution of the foundational ABE7.10,2 and we recently discovered that reversion of this mutation in the ABE7.10 construct resulted in complete loss of ssDNA-editing activity by TadA*.22 It is therefore quite significant that a mutation that is so critical for imparting novel ssDNA-editing functionality to an RNA-editing enzyme has such a high incidence in naturally occurring TadA homologs (Fig. 3G). Additionally, this suggests that while the TadA* enzyme developed activity toward DNA substrates, it retained activity toward its native RNA targets.
Upon conducting a similar distribution analysis for site 84, which is also a low entropy site that favorably affects ssDNA editing, we found that while this core residue has a low sequence entropy of as defined by Shannon's entropy, 88.6% of sequences had an aliphatic amino acid (Leu, Val, or Ile) at this position (Supplementary Table S5). In direct contrast, it was mutated to Phe in ABE7.10 (Fig. 3G). Thus, unlike the D108N mutation, the L84F mutation is a novel mutation that was not explored by natural protein evolution.
In the case of the highly conserved phenylalanine at site 149 , which has been mutated to Ala without abrogating either the ssDNA or the RNA-editing activity of ABEs17 and also mutated to Tyr through the directed evolution in ABE8e,14 the binned entropy analysis revealed that the Tyr is the second most preferred amino acid (19.4%) at site 149 in the naturally occurring TadA homologs. Hence, analogous to the analysis of site 108, the distribution of amino acids at site 149 also highlights that directed evolution explores sequence space previously accessed by naturally evolved homologs.
The misclassification of site 82 () also adheres to this trend as the second most prevalent amino acid at this position in the naturally occurring TadA homologs is Thr, whose chemical properties are similar to Ser, which has been shown to enhance the DNA-editing activity of TadA* in the evolved ABE8s.
This analysis of the distribution of the possible amino acids based on their chemical nature helps identify the types of mutations that are tolerated at various sites of the TadA* sequence (Supplementary Fig. S2A). Hence, we recalculated the entropy values for wtTadA by binning amino acid residues according to their side-chain classifications: polar uncharged, positively charged, negatively charged, hydrophobic-aliphatic, hydrophobic-aromatic, and special (Gly, Pro). The resulting binned entropy values (Supplementary Fig. S2B–D) are greater than 0.5 for site 108 while remaining lower than 0.5 for site 84 (Fig. 3G and H).
These results thus indicate that the entropy-based analysis allows not only for the quantification of the mutational propensity of individual wtTadA sites but also for the characterization of the chemical properties that make mutations to a specific class of amino acids relatively more favorable (Supplementary Table S5). Moreover, we also speculate that residue sites having marginally low Hi values can in fact be mutated based on the amino acid distribution observed in the extant homologs to confer novel functionality to the enzyme (as seen for D108N mutation) or to disrupt native functionality (as seen for L84F).
Experimental analyses
We next sought to test experimentally our hypothesis that the conservation scores and amino acid distributions derived from the entropy-based model could be used to predict the effects of mutations on the RNA-editing activity of TadA*. It is well known that later-generation ABEs induce transcriptome-wide RNA editing, but it is unknown if this is a “carryover” activity from wtTadA being able to edit RNA sequences other than its native tRNA substrate, or if the various mutations identified through directed evolution enhanced not only the ssDNA-editing activity of TadA*, but also its nonspecific RNA-editing activity.
We first tested the ability of ABE0.1 (as both monomeric and dimeric wtTadA fused to Cas9n) to introduce A-to-I edits in mRNA in a gRNA-independent manner. We transfected HEK293T cells with constructs encoding monomeric ABE0.1, dimeric ABE0.1, or heterodimeric ABE7.10 (wtTadA-TadA*-Cas9n), extracted mRNA after 36 h, and used high-throughput sequencing to quantify A-to-I editing at six different sites throughout the transcriptome that had previously been shown to be edited by ABE7.10 in a gRNA-independent manner.17 We observed >50% A-to-I RNA-editing efficiencies at all six sites by both wild-type constructs.
Moreover, consistent with the recent report comparing the kinetics of ABEs on RNA substrates,56 the RNA-editing activity of dimeric ABE0.1 was on average 21% higher than ABE7.10, highlighting the remarkable shift in substrate preference of wtTadA enzyme due to the many mutations that were found through directed evolution for ABE7.10.
Our entropy-based analysis suggests that non-aliphatic mutations at site 84 would diminish the RNA-editing activity of wtTadA, while certain mutations at site 108 would retain (or even enhance) the RNA-editing activity of wtTadA (Fig. 3 and Supplementary Fig. S2). To test this hypothesis, we generated six different monomeric ABE variants (i.e., TadA*-Cas9n)—ABE1.1 (where ABE1.1 = ABE0.1[D108N]2), ABE0.1(L84F), and ABE1.1(L84F)—and their corresponding heterodimeric constructs (i.e., wtTadA-TadA*-Cas9n), and compared their RNA-editing activities with ABE0.1 and ABE7.10 at the same six sites.
Each variant was tested as a monomer and a heterodimer, as this has been shown to have drastic effects on the off-target RNA editing activities of ABEs.13,17 While the aim of this was to observe differences due to dimerization, monomeric ABE8e has been shown to spontaneously dimerize in trans.56
Consistent with our hypothesis and the entropy-based classification model, the D108N mutation leads to a modest 11.2% (range 5.7–21.8%) increase in the A-to-I RNA-editing activity of ABE1.1 compared to ABE0.1. Moreover, the L84F mutation leads to a 25% (range 19.8–31.7%) or almost 1.7-fold decrease in RNA-editing efficiency of the enzyme compared to ABE0.1 across the six different RNA sites that were analyzed (Fig. 4). These editing patterns were also observed at an additional UACG motif within RNA site 1, although the editing levels here were much lower than in the other six sites (Supplementary Note 1 and Supplementary Fig. S3).
FIG. 4.

A-to-I base editing efficiencies in HEK293T cells by various ABE mutants at six different gRNA-independent RNA off-target sites. Fold-decrease values associated with the reduction in the RNA editing upon incorporation of the L84F mutation in ABE0.1 are indicated. Values and error bars reflect the mean and standard deviation of three independent biological replicates performed on different days. Color images are available online.
This loss of function due to the L84F mutation can be restored either by dimerizing the protein with wtTadA (as ABE0.1[L84F, heterodimer]) or by adding the D108N mutation (as ABE1.1[L84F]). We speculate that in the case of ABE0.1(L84F, heterodimer), the observed increase in RNA editing is due to the addition of the wtTadA subunit, which is capable of efficient RNA editing on its own (as in the ABE0.1 monomeric construct). In the case of ABE1.1(L84F), whose activity is comparable to ABE1.1, we observed a modest 4.3% (range 1.6–13%) increase over ABE0.1.
This restoration of the RNA-editing efficiency upon the combination of D108N and L84F mutations is particularly interesting, as it highlights the nonadditive and epistatic effect that mutations can have on enzyme function. Thus, upon combining a high entropy mutation with a low entropy mutation, the resultant double mutant exhibits high activity rather than an average of the two activities. Furthermore, this double mutant exhibits increased activity toward a different substrate (ssDNA).
Intriguingly, the RNA-editing activity of ABE7.10, which has 12 other mutations in addition to D108N and L84F, is slightly lower than that of ABE0.1 by 8.9% (range 2.5–13.8%) or 1.2-fold (Fig. 4). This observation further reinforces the nonadditivity of the TadA* mutations identified using directed evolution of ABEs. The early mutations led to a broadening of the substrate specificity of TadA* (i.e., imparting ssDNA-editing capabilities on to TadA) and later mutations enhanced the ssDNA-editing activity while potentially suppressing the RNA-editing activity (as with the L84F mutation).
Interaction and binding of RNA with TadA*
In a previous study,22 we demonstrated that the effects of individual mutations on the ssDNA-editing activity of ABEs can be studied using a minimalistic model of the system, composed of the TadA* mutants and the nucleic acid substrates, while ignoring Cas9, which acts as a mere carrier of the nucleotide editing module to its target genomic locus. Moreover, it has been experimentally proven that the off-target RNA editing by ABEs occurs in a Cas9 (or gRNA)-independent manner, which reinforces the notion that only the TadA* portion of the ABEs act on the RNA off-target substrates.10,13–16,57
To understand the complex epistatic relationship between the L84F and the D108N mutations in the context of the RNA-editing activity of ABEs (Figs. 3 and 4), we modeled the ABE–RNA systems by combining the experimentally resolved structure of wild-type E. coli TadA (PDB :1Z3A)24 and its native 14-mer RNA-hairpin substrate (5′-UUGACUACGAUCAA-3′) (PDB :2B3J).25
The RNA sequence in our simulation models, as well as the off-target RNA sites that we tested experimentally (Fig. 4), have the same consensus sequence (-UACG-) as that reported previously10,17 (Supplementary Fig. S4). Moreover, the secondary structures for three out of six RNA editing sites (sites 1, 2, and 4) are predicted to resemble the hairpin loop structure of the native target of TadA* that we simulated, indicating the strong preference of TadA* for its native substrate (Supplementary Fig. S5).
Having generated these models, we then carried out molecular dynamics (MD) simulations for each of the four TadA*–RNA complexes for 1 μs and examined the trajectories for changes in interactions between individual TadA* residues and the nucleic acid substrate. Since the mutations we are interested in lie near the active site of the TadA*, we focused predominantly on the interactions between the nucleotide bases splayed in the active site, that is, the target adenine and its 5′ and 3′ flanking bases (UACG) and neighboring TadA* residues. To home in on the amino acids in direct contact with these nucleotides, we carved out a 4 Å search radius around these bases, and then projected the residues that lie within this sphere onto asteroid plots (Fig. 5A–D and Supplementary Fig. S6).
FIG. 5.

Asteroid plots for the analysis of the interaction of (A) TadA*0.1, (B) TadA*1.1, (C) TadA*0.1(L84F), and (D) TadA*1.1(L84F) with substrate RNA. I Binding affinity comparisons for the various TadA*–RNA complexes. (F) The collective variable (ξ) used to monitor the binding/unbinding of the TadA*–RNA complexes. (G) Parameters associated to harmonic functions fitted to binding energy curves shown in I. Color images are available online.
In these plots, the nucleotides in the active site are represented collectively as the central node, and the peripheral nodes correspond to all amino acids within the first interaction shell of the nucleotides in the active site. The size of the encircling nodes is proportional to the time that the corresponding residues spend within the first interaction shell of the RNA bases throughout the entire MD trajectory. The hydrogen bonds (H-bonds) between these residues and the RNA bases are depicted as arrows connecting the relevant nodes in each asteroid plot, with the thickness of the arrows being proportional to the stability of the H-bond itself, which is defined as the frequency of appearance of that H-bond during the simulation.
The comparisons between the TadA*0.1/TadA*0.1(L84F), TadA*0.1/TadA*1.1, and TadA*1.1/TadA*1.1(L84F) mutants indicate that the D108N mutation leads to the formation of a favorable H-bond between the Asn108 residue and the U base flanking the target A. In fact, the TadA*1.1(L84F) mutant has the strongest interaction with RNA, as the D108N mutation, when combined with the L84F mutation, causes additional structural rearrangements surrounding the active site, resulting in a double H-bond interaction with the RNA substrate through residue 152.
The weak H-bond between D108 and the 2′-OH group of the flanking U base predicted by our simulations is also found in the crystallographic structure of the wtTadA-tRNA complex (PDB ID: 2B3J25). However, this weak H-bond does not appear in the TadA*0.1(L84F) mutant and is replaced by a much stronger H-bond in the TadA*1.1 mutant upon mutation of glutamate to asparagine at site 108. The formation of this stronger H-bond also leads to an increase in interactions between some of the peripheral residues (57, 59, 82, 85, 86, and 87) and the RNA bases in the active site, indicating a more stable conformation of the target adenine.
A similar increase in the interaction induced by the H-bond formed by the D108N residue was also observed in our MD simulations of the TadA*–ssDNA complex.22 In the context of ssDNA editing by ABEs, the D108N mutation in ABE1.1 leads to the onset of activity on DNA via the formation of this H-bond donation.2,22 However, in the context of RNA-editing efficiency, the D108N mutation, and consequent formation of the H-bond with RNA, only amounts to a slight increase in the activity due to wtTadA (ABE0.1), being already highly proficient in editing its native RNA substrate as well as ssRNA in general (Fig. 4).
Although the L84F mutation, unlike the D108N mutation, is accompanied by a more pronounced effect on the RNA-editing activity of wtTadA (Fig. 4) and is in fact a novel mutation in the first interaction shell of the RNA bases (Fig. 3G), the comparison of the asteroid plots corresponding to TadA*0.1 and TadA*0.1(L84F) shows less drastic changes than those observed in the TadA*1.1 asteroid plot. Specifically, the L84F mutation leads to the elimination of the weak H-bonds established by the 107 and 108 residues in TadA*0.1.
To quantify these differences, we performed US simulations to determine local changes in the free energies of the various TadA*–RNA complexes about the active site. Starting from the equilibrated structure of each TadA*–RNA complex, we modeled the binding process using a collective variable (ξ) defined as the distance between the TadA* and RNA centers of mass, which was varied from 17 to 37 Å. We successfully used the same collective variable to characterize the binding process of the analogous TadA*–DNA complexes in our previous study.22
The free-energy changes along this collective variable were calculated for each of the four TadA* mutants using the WHAM39,40 (see Supplementary Figs. S7 and S8 for convergence analysis of these US simulations). Figure 5E shows that the TadA*1.1–RNA and TadA*1.1(L84F)–RNA complexes are more tightly bound than the TadA*0.1–RNA complex. While these trends help explain the experimental RNA-editing efficiencies of these D108N mutants when compared to ABE0.1, they do not apply to the TadA*0.1 and TadA*0.1(L84F) mutants, which exhibit similar local free-energy changes as RNA is pulled out of the active site.
This observation reciprocates the results of our previous study of the TadA*–ssDNA complex, showing that mutations installed at later stages of the directed evolution process do not further enhance the binding strength relative to TadA1.1 but instead most likely impact the catalytic activity of TadA*.
As the subtle conformational changes that we observe between the asteroid plots of TadA*0.1 and TadA*0.1(L84F) (Fig. 5A and C) do not result in significant changes in the binding strength between these two mutants, we thus sought to quantify the effects of these conformational changes on the catalysis.
Water in the active site and implications for catalysis
The hydrolytic deamination reaction catalyzed by TadA involves a zinc-coordinated water molecule (hereafter referred to as the activated water molecule) that is deprotonated by the active site Glu59 residue (a highly conserved residue, see Fig. 2) during the first step of the reaction. In addition to this activated water molecule, the active site also includes another structurally important water molecule (hereafter referred to as the bridging water), which acts as a bridge between Glu59 and the carbonyl backbone of Leu84 (Fig. 6A and B). Both water molecules are resolved in several high-resolution crystal structures of TadA homologs (Supplementary Fig. S9), which further reinforces their importance in the stabilization of the active site cavity.
FIG. 6.

(A) Side view of the of the TadA*–RNA system highlighting the location of the catalytically relevant residues. The Zn+2 ion is coordinated by His57, Cys87, and Cys90 (not shown here for clarity) and a water molecule. This water molecule is activated by Glu59, which is also connected to another water molecule. This second water acts as a bridge between the Glu59 and the carbonyl backbone of residue 84. The target adenine is deep within the active site, and residue 108 is farther away from the active site waters. (B) Simplified flat lay representation to highlight the interactions of active site waters. Modified chord diagrams to demonstrate the persistence of the active site waters for (C) TadA*0.1– RNA, (D) TadA*1.1–RNII TadA*0.1(L84F)–RNA, and (F) TadA*1.1(L84F)–RNA. The red chords connecting Glu59 with Zn+2 depict the stability of the activated water molecule. Similarly, the blue chords connecting Glu59 with residue 84 depict the stability of the bridging water molecule. Different colors signify unique water molecules, with the thickness of individual chords being directly proportional to the total time these water molecules interact with the active site of TadA*–RNA during the simulation. (G) Reaction profile for the deprotonation of the activated water molecule the various TadA*–RNA systems. (H) Conformation of the TadA*0.1–RNA when the proton resides on Glu59. (I) Conformation of the TadA*0.1(L84F)–RNA when the proton resides on stability on the Glu59. The target A has moved back into the active site toward the Phe84 and is separated from the active site residues by an additional water molecule—the mediating water. Color images are available online.
To characterize the role played by these two water molecules in the active site of the various TadA*–RNA mutant systems, we analyzed the data from our MD simulations in the form of modified chord diagrams in (Fig. 6C–F). The persistence of the activated water molecule is depicted in red in the left partitions, while that of the bridging water is depicted in blue in the right partitions of the four panels. The thickness of each chord is proportional to the time spent by the corresponding water molecules in the active site (Supplementary Fig. S10 and Supplementary Table S6).
For the TadA*0.1–RNA and TadA*1.1–RNA systems, we found that these two water molecules are highly stable in their respective positions and do not undergo any diffusion throughout the entirety of our MD simulations (1 μs). In contrast, for the TadA*0.1(L84F)–RNA system, both water molecules exhibit higher mobility and are exchanged several times with water molecules initially located in the bulk solution at the beginning of the simulation. We speculate that the hydrophobic-aromatic nature of the phenylalanine residue may be responsible for the decreased stability of both water molecules in the active site (Supplementary Figs. S10 and S11).
The stability of the two water molecules is restored in the TadA*1.1(L84F)–RNA complex. This implies that the D108N mutation can cancel out the destabilizing effects of the L84F mutation and effectively modulate the hydration of the active site, despite not engaging in any direct contact with either water molecules.
We observe similar trends when comparing these mutations in the apo-TadA* simulations. Specifically, the apo-TadA*0.1(L84F) system shows an analogous increased flux of the two water molecules in the active site, which is again suppressed after the installation of the D108N mutation (Supplementary Fig. S12). Importantly, the changes in the persistence of these catalytically relevant water molecules in the active site of the TadA*–RNA/TadA* systems (Fig. 6C–F and Supplementary Figs. S10–S12, and Supplementary Table S6) mirrors the changes in RNA-editing activity measured for the ABEs (Fig. 4).
Since the first step of the adenine deamination reaction involves the deprotonation of the activated water molecule by the Glu59 residue, we speculate that the changes we observe in the stability of the active site water molecules may lead to changes in the reaction rates in the four TadA* mutants. Hence, for a more explicit comparison with the experimental catalytic data of these four TadA* mutants, we performed QM/MM simulations to investigate the first step of the reaction mechanism.
Owing to the high computational cost of simulating the entire system at the QM level, QM/MM simulations offer an optimal trade-off between accuracy and computational efficiency by simulating the reaction centers with QM accuracy, while the remaining system is treated at the MM level of theory.
In our QM/MM simulations, the QM region encompasses the side chains of the active site residues (His57, Glu59, Cys87, Cys90), Zn+2, and the activated water molecule, which treated at the DFTB3 level with 3OB parameterization,58–60 while all other atoms of the system are included in the MM region. Similar DFTB approaches have been successfully employed in the past to study several zinc-containing enzymes,42,61–63 including deaminases, which are homologous to TadA.64–66
All QM/MM simulations were initiated from configurations taken from the US windows corresponding to the PMF minima shown in Figure 5. In modeling the first step of the deamination reaction, configurations with undissociated activated water molecules define the reactant state, while configurations with the protonated Glu59 residue define the product state. In the transition state, the proton is equally shared by the activated water molecule and Glu59.
To determine the energetics associated with this proton-transfer reaction, QM/MM US simulations were carried out along the proton-transfer coordinate, which is defined as the difference between the distances of the shared proton from the activated water and Glu59, as used in the study by Zhang et al.67 The PMFs calculated using the WHAM for all four TadA* mutants are shown in Figure 6G and are consistent with the energetics reported for other zinc-containing deaminases (E. coli CDA,68 yeast CDA,67,69 and guanine deaminase70).
The PMF profiles indicate that only the TadA*0.1(L84F)–RNA complex is associated with a weakly stable product state (i.e., a protonated Glu59). At first glance, these results seem to be counterintuitive and contrary to the experimental observation of a lower RNA-editing activity for the ABE0.1(L84F) mutant. However, upon further examination of the product state in the TadA*0.1(L84F)–RNA complex, we observed that proton transfer from the activated water to Glu59 is accompanied by the concomitant movement of the target adenine base away from the active site residue and toward the aromatic phenylalanine ring deeper into the active site, forming a staggered pi-stack with L84F residue (Supplementary Table S7).
The cavity formed as a result of this conformational change of the target adenine is filled by another water molecule (hereafter referred to as the mediating water) that may contribute to the following reaction steps, thereby altering the reaction mechanism for TadA*0.1(L84F). In this context, it should be noted that a deamination mechanism involving extra water molecules was characterized for cytosine deaminase in the study by Matsubara et al.71 In the case of the TadA*0.1 (or TadA*1.1 and TadA*1.1[L84F]) system, our simulations predict that the active site retains its configuration, with the adenine base primed for the next steps of the reaction (Fig. 6H).
We thus hypothesize that the proximity of the adenine base prevents the transfer of the proton from the activated water molecule to Glu59. QM/MM US simulations carried out for the apo-TadA* mutants provide support for this hypothesis, showing the formation of a stable product state for all the TadA* mutants due to the lack of the adenine base (Supplementary Figs. S12G, S13 and S14).
We thus conclude that the L84F mutation, a novel mutation at a low entropy residue site, affects the decrease in the activity of TadA* on the native RNA substrate through two key changes in its deamination chemistry. First, this mutation destabilizes the two water molecules in the active site, which are both structurally and functionally crucial for the initiation of the deamination reaction. Second, it pulls the target adenine base away from the protonated Glu59, thereby making the subsequent reaction steps less feasible or leading to an alternate reaction pathway involving additional steps (e.g., through the mediating water).
Our simulations indicate that the combination of the D108N mutation, which increases the RNA binding affinity, with the L84F mutation conserves the integrity of the active site by both stabilizing the two water molecules and positioning the target adenine appropriately for subsequent reaction steps, thereby rescuing the catalytic activity of the ABE1.1(L84F) mutant (Fig. 4).
Discussion
Through a systematic investigation of the various mutations that have been thus far identified in TadA*, our study retraces the evolutionary trajectory followed by this enzyme using a data-driven approach that combined statistical models, MD simulations, and experimental assays.
The information contained in the naturally evolved TadA homologs aids in rationalizing the effects of the mutations that have accumulated in the laboratory engineered TadA* (Fig. 2). We have demonstrated that mutations with a favorable impact on the RNA-editing activity of TadA* occur at residue sites having higher entropy, whereas mutations with an unfavorable impact on the RNA-editing activity occur at residue sites with lower entropy (Fig. 3). Moreover, these low entropy sites when mutated to previously unvisited amino acids in the sequence space, such as the L84F mutation, can also have an adverse impact on the native function of the enzyme.
Our experimental analyses also reveal that ABE0.1 has remarkably high gRNA-independent off-target RNA editing and is even higher than the evolved ABE7.10 variant (Fig. 4).13,14,17,18 These results indicate that such entropy-based scores, albeit being extracted from a highly RNA-biased data set, can serve as a preliminary screen for site-directed mutagenesis and guide the library preparation for evolving future base editors with reduced transcriptome-wide off-target editing activity.
The most reliable inferences that can be derived from such biased data sets are related to the native RNA-editing functionality of the query sequence. Hence, we propose that this entropy-based tool be preferentially applied for the search of mutations that can suppress the inherent RNA-editing activity of potential base editors, a problem that cannot be solved at present using the traditional directed evolution methods.
Despite having reasonable success at predicting sites with low and high functionality for TadA*, the entropy-based tool has some inherent drawbacks.
First, the assumption of using 0.5 as the dividing value for this classifier may be too simplistic. For future studies, instead of defining a single dividing value, we speculate that using a range of values (e.g., ) may lead to improvements in the predictive power of this classifier.
Second, the number of sites predicted to have the desirable mutational outcome using the entropy-based model can exceed the available design budget for a novel target protein. However, information from the entropy model can be combined with structural insights to conduct focused mutagenesis of residues that are in close proximity to the substrate and have entropy greater than 0.4. This would ensure that the resultant library spans the beneficial mutants needed to improve the functionality of the enzyme while reducing the size of the designed library (Supplementary Fig. S15).
Third, the entropy-based model proves that laboratory evolution plays by the rules set by natural evolution and that learning these rules from extant enzyme homologs can help guide future protein engineering endeavors. However, it is by construction, not a generative model. That is, this model cannot be extended beyond the sequences in the data set. It is limited by the diversity in the training data set, which in our case is highly biased toward RNA editing. Hence, we observed that directed evolution does explore regions of the sequence space that have not been explored by natural protein evolution (Supplementary Table S4).
Lastly, statistical models for protein sequence analysis, such as the entropy-based model used here, are known to perform poorly on predicting the relationship between co-evolving residues.72 This is apparent in our mutual entropy analysis of 84 and 108 residue sites, where the entropy-based model is unable to decipher the correlations between these two sites (Supplementary Note S2 and Supplementary Fig. S16), which can instead be understood through MD simulations of the TadA*–RNA complexes.
Using MD simulations, we observed that the L84F mutation decreases the experimental RNA-editing efficiency of TadA* by both destabilizing the active site water molecules and disrupting the conformation of the target A base. These effects are fundamentally different from those induced by the D108N mutation, which increases the experimental RNA editing of TadA*. The D108N mutation increases the strength of the interactions between the substrate RNA and TadA* by establishing favorable H-bonds—a phenomenon that we have previously observed in our TadA*–ssDNA simulations.22 These additional interactions due to the D108N mutation alleviate the destabilizing effects of the L84F mutation, thereby restoring the experimental efficiency of TadA*1.1(L84F) mutant.
Hence, the D108N and L84F mutations constitute an epistatic pair in the functional landscape of TadA*.73 The prevalence of such nonadditive and complex relationships between the mutating residues make the task of rational protein design notoriously challenging, especially in cases such as that of TadA*, where the native RNA-editing fitness landscape is strongly coupled with the ssDNA-editing fitness landscape.
Conclusions
We described here the use of a combination of theoretical, bioinfomatic, and experimental approaches to analyze retrospectively the mutations that have previously been reported to modulate the activity of the ABEs, and we uncovered a critical epistatic pair in its functional landscape. We anticipate that such synergistic combinations can stimulate the development and application of similar multilevel strategies (informed by a combination of sequence, structure, and function of enzymes) to tackle the complex problem of the prospective design of future base editors.
Supplementary Material
Acknowledgments
The authors thank M. Norman for a Director's Discretionary Allocation on the Comet GPU cluster at the San Diego Supercomputer Center. K.L.R. thanks C. Egan, E. Lambros, and S. Gu for helpful discussions. An earlier draft of this manuscript was posted as a preprint at bioRxiv (DOI: 10.1101/2020.12.24.424366).
Author Disclosure Statement
A.C.K. is a member of the SAB and a consultant of Pairwise Plants and is an equity holder for Pairwise Plants and Beam Therapeutics. A.C.K.'s interests have been reviewed and approved by the University of California, San Diego, in accordance with its conflict-of-interest policies. All other authors declare that they have no competing interests.
Funding Information
This research was supported by the University of California San Diego and the NIH through grants no. 1R21GM135736-01 and 1R35GM138317-01. All computer simulations used resources of the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by NSF through grant no. ACI-1548562. B.L.R. is supported by the Chemistry- Biology Interface (CBI) Training Program (NIGMS, 5T32GM112584).
Supplementary Material
References
- 1. Landrum MJ, Lee JM, Benson M, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res 2016;44:D862–D868. DOI: 10.1093/nar/gkv1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gaudelli NM, Komor AC, Rees HA, et al. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 2017;551:464–471. DOI: 10.1038/nature24644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wolf J, Gerber AP, Keller W. TadA, an essential tRNA-specific adenosine deaminase from Escherichia coli. EMBO J 2002;21:3841–3851. DOI: 10.1093/emboj/cdf362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zeng Y, Li J, Li G, et al. Correction of the Marfan syndrome pathogenic FBN1 mutation by base editing in human cells and heterozygous embryos. Mol Ther 2018;26:2631–2637. DOI: 10.1016/j.ymthe.2018.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Liu Z, Chen M, Chen S, et al. Highly efficient RNA-guided base editing in rabbit. Nat Commun 2018;9:1–10. DOI: 10.1038/s41467-018-05232-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Song CQ, Jiang T, Richter M, et al. Adenine base editing in an adult mouse model of tyrosinaemia. Nat Biomed Eng 2020;4:125–130. DOI: 10.1038/s41551-019-0357-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ryu SM, Koo T, Kim K, et al. Adenine base editing in mouse embryos and an adult mouse model of Duchenne muscular dystrophy. Nat Biotechnol 2018;36:536–539. DOI: 10.1038/s41551-019-0357-8. [DOI] [PubMed] [Google Scholar]
- 8. Hua K, Tao X, Yuan F, et al. Precise A•T to G•C base editing in the rice genome. Mol Plant 2018;11:627–630. DOI: 10.1016/j.molp.2018.02.007. [DOI] [PubMed] [Google Scholar]
- 9. Yan F, Kuang Y, Ren B, et al. Highly efficient A•T to G•C base editing by Cas9n-guided tRNA adenosine deaminase in rice. Mol Plant 2018;11:631–634. DOI: 10.1016/j.molp.2018.02.008. [DOI] [PubMed] [Google Scholar]
- 10. Grünewald J, Zhou R, Garcia SP, et al. Transcriptome-wide off-target RNA editing induced by CRISPR-guided DNA base editors. Nature 2019;569:433–437. DOI: 10.1038/s41586-019-1161-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jin S, Zong Y, Gao Q, et al. Cytosine, but not adenine, base editors induce genome-wide off-target mutations in rice. Science 2019;364:292–295. DOI: 10.1126/science.aaw7166. [DOI] [PubMed] [Google Scholar]
- 12. Zuo E, Sun Y, Wei W, et al. Cytosine base editor generates substantial off-target single-nucleotide variants in mouse embryos. Science 2019;364:289–292. DOI: 10.1126/science.aav9973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gaudelli NM, Lam DK, Rees HA, et al. Directed evolution of adenine base editors with increased activity and therapeutic application. Nat Biotechnol 2020;38:892–900. DOI: 10.1038/s41587-020-0491-6. [DOI] [PubMed] [Google Scholar]
- 14. Richter MF, Zhao KT, Eton E, et al. Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nat Biotechnol 2020;38:883–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhou C, Sun Y, Yan R, et al. Off-target RNA mutation induced by DNA base editing and its elimination by mutagenesis. Nature 2019;571:275–278. DOI: 10.1038/s41586-019-1314-0. [DOI] [PubMed] [Google Scholar]
- 16. Rees HA, Wilson C, Doman JL, et al. Analysis and minimization of cellular RNA editing by DNA adenine base editors. Sci Adv 2019;5:eaax5717. DOI: 10.1126/sciadv.aax5717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Grünewald J, Zhou R, Iyer S, et al. CRISPR DNA base editors with reduced RNA off-target and self-editing activities. Nat Biotechnol 2019;37:1041–1048. DOI: 10.1038/s41587-019-0236-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Komor AC, Kim YB, Packer MS, et al. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016;533:420–424. DOI: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Arnold FH. Unnatural selection: molecular sex for fun and profit. Eng Sci 1999;62:40–50. [Google Scholar]
- 20. Johnson M, Zaretskaya I, Raytselis Y, et al. NCBI BLAST: a better web interface. Nucleic Acids Res 2008;36:W5–W9. DOI: 10.1093/nar/gkn201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bairoch A, Apweiler R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res 2000;28:45–48. DOI: 10.1093/nar/28.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rallapalli KL, Komor AC, Paesani F. Computer simulations explain mutation induced effects on the DNA editing by adenine base editors. Sci Adv 2020;6:eaaz2309. DOI: 10.1126/sciadv.aaz2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Clement K, Rees H, Canver MC, et al. Accurate and rapid analysis of genome editing data from nucleases and base editors with CRISPResso2. Nat Biotechnol 2019;37:224–226. DOI: 10.1038/s41587-019-0032-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kim J, Malashkevich V, Roday S, et al. Structural and kinetic characterization of Escherichia coli TadA, the wobble-specific tRNA deaminase. Biochemistry 2006;45:6407–6416. DOI: 10.1021/bi0522394. [DOI] [PubMed] [Google Scholar]
- 25. Losey HC, Ruthenburg AJ, Verdine GL. Crystal structure of Staphylococcus aureus tRNA adenosine deaminase TadA in complex with RNA. Nat Struct Mol Biol 2006;13:153–159. DOI: 10.1038/nsmb1047. [DOI] [PubMed] [Google Scholar]
- 26. Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 2004;25:1605–1612. DOI: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 27. Gordon JC, Myers JB, Folta T, et al. H++: a server for estimating p K as and adding missing hydrogens to macromolecules Nucleic Acids Res 2005;33:W368–W371. DOI: 10.1093/nar/gki464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Anandakrishnan R, Aguila B, Onufriev AV. H++ 3.0: automating p K prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations Nucleic Acids Res 2012;40:W537–W541. DOI: 10.1093/nar/gks375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Maier JA, Martinez C, Kasavajhala K, et al. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput 2015;11:3696–3713. DOI: 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pérez A, Marchán I, Svozil D, et al. Refinement of the AMBER force field for nucleic acids: improving the description of α/γ conformers. Biophys J 2007;92:3817–3829. DOI: 10.1529/biophysj.106.097782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Banás P, Hollas D, Zgarbová M, et al. Performance of molecular mechanics force fields for RNA simulations: stability of UUCG and GNRA hairpins J Chem Theory Comput 2010;6:3836–3849. DOI: 10.1021/ct100481h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zgarbová M, Otyepka M, Šponer J, et al. Refinement of the Cornell et al. nucleic acids force field based on reference quantum chemical calculations of glycosidic torsion profiles. J Chem Theory Comput 2011;7:2886–2902. DOI: 10.1021/ct200162x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li P, Merz KM. MCPB.py: a Python based metal center parameter builder. J Chem Inf Model 2016;56:599–604. DOI: 10.1021/acs.jcim.5b00674. [DOI] [PubMed] [Google Scholar]
- 34. Salomon-Ferrer R, Case DA, Walker RC. An overview of the Amber biomolecular simulation package. WIRES Comput Mol Sci 2013;3:198–210. DOI: 10.1002/wcms.1121. [DOI] [Google Scholar]
- 35. Salomon-Ferrer R, Götz AW, Poole D, et al. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh Ewald J Chem Theory Comput 2013;9:3878–3888. DOI: 10.1021/ct400314y. [DOI] [PubMed] [Google Scholar]
- 36. Case DA, Cheatham III TE, Darden T, et al. The Amber biomolecular simulation programs. J Comput Chem 2005;26:1668–1688. DOI: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Case D, Ben-Shalom I, Brozell S, et al. AMBER 2018. San Francisco, CA: University of California, 2018. [Google Scholar]
- 38. Jarzynski C. Nonequilibrium equality for free energy differences. Phys Rev Lett 1997;78:2690. DOI: 10.1103/PhysRevLett.78.2690. [DOI] [Google Scholar]
- 39. Kumar S, Rosenberg JM, Bouzida D, et al. The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J Comput Chem 1992;13:1011–1021. DOI: 10.1002/jcc.540130812. [DOI] [Google Scholar]
- 40. Grossfield A. WHAM: the weighted histogram analysis method. Available online at: http://membrane.urmc.rochester.edu/wordpress/?page_id=126 (last accessed June 9, 2019).
- 41. Zhu F, Hummer G. Convergence and error estimation in free energy calculations using the weighted histogram analysis method J Comput Chem 2012;33:453–465. DOI: 10.1002/jcc.21989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Elstner M, Frauenheim T, Suhai S. An approximate DFT method for QM/MM simulations of biological structures and processes. J Mol Struct THEOCHEM 2003;632:29–41. DOI: 10.1016/S0166-1280(03)00286-0. [DOI] [Google Scholar]
- 43. Roe DR, Cheatham III TE. PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J Chem Theory Comput 2013;9:3084–3095. DOI: 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- 44. Roe DR, Cheatham III TE. Parallelization of CPPTRAJ enables large scale analysis of molecular dynamics trajectory data. J Comput Chem 2018;39:2110–2117. DOI: 10.1002/jcc.25382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hunter JD. Matplotlib: A 2D graphics environment. Comput Sci Eng 2007;9:90–95. DOI: 10.1109/MCSE.2007.55. [DOI] [Google Scholar]
- 46. Anfinsen CB. Principles that govern the folding of protein chains. Science 1973;181:223–230. DOI: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 47. UniProt Consortium. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res 2019;47:D506–D515. DOI: 10.1093/nar/gky1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chaparro-Riggers JF, Polizzi KM, Bommarius AS. Better library design: data driven protein engineering. Biotechnol J 2007;2:180–191. DOI: 10.1002/biot.200600170. [DOI] [PubMed] [Google Scholar]
- 49. Yu Y, Leete TC, Born DA, et al. Cytosine base editors with minimized unguided DNA and RNA off-target events and high on-target activity. Nat Commun 2020;11:1–10. DOI: 10.1038/s41467-020-15887-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shannon CE. A mathematical theory of communication. Bell Syst Tech J 1948;27:379–423. [Google Scholar]
- 51. Sander C, Schneider R. Database of homology-derived protein structures and the structural meaning of sequence alignment. Proteins Struct Funct Bioinf 1991;9:56–68. DOI: 10.1002/prot.340090107. [DOI] [PubMed] [Google Scholar]
- 52. Valdar W. Scoring residue conservation. Proteins Struct Funct Bioinf 2002;48:227. DOI: 10.1002/prot.10146. [DOI] [PubMed] [Google Scholar]
- 53. Capra JA, Singh M. Predicting functionally important residues from sequence conservation. Bioinformatics 2007;23:1875–1882. DOI: 10.1093/bioinformatics/btm270. [DOI] [PubMed] [Google Scholar]
- 54. Voigt CA, Mayo SL, Arnold FH, et al. Computationally focusing the directed evolution of proteins. J Cell Biochem 2001;84:58–63. DOI: 10.1002/jcb.10066. [DOI] [PubMed] [Google Scholar]
- 55. Fodor AA, Aldrich RW. Influence of conservation on calculations of amino acid covariance in multiple sequence alignments. Proteins 2004;56:211–221. DOI: 10.1002/prot.20098. [DOI] [PubMed] [Google Scholar]
- 56. Lapinaite A, Knott GJ, Palumbo CM, et al. DNA capture by a CRISPR-Cas9–guided adenine base editor. Science 2020;369:566–571. DOI: 10.1126/science.abb1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Park S, Beal PA. Off-target editing by CRISPR-guided DNA base editors. Biochemistry 2019;58:3727–3734. DOI: 10.1021/acs.biochem.9b00573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Walker RC, Crowley MF, Case DA. The implementation of a fast and accurate QM/MM potential method in Amber. J Comput Chem 2008;29:1019–1031. DOI: 10.1002/jcc.20857. [DOI] [PubMed] [Google Scholar]
- 59. Gaus M, Cui Q, Elstner M. DFTB3: extension of the self-consistent-charge density functional tight-binding method (SCC-DFTB). J Chem Theory Comput 2011;7:931–948. DOI: 10.1021/ct100684s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lu X, Gaus M, Elstner M, Cui Q. Parametrization of DFTB3/3OB for magnesium and zinc for chemical and biological applications. J Phys Chem B 2015;119:1062–1082. DOI: 10.1021/jp506557r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Xu D, Cui Q, Guo H. Quantum mechanical/molecular mechanical studies of zinc hydrolases. Int Rev Phys Chem 2014;33:1–41. DOI: 10.1080/0144235X.2014.889378. [DOI] [Google Scholar]
- 62. Chakravorty DK, Wang B, Lee CW, et al. Simulations of allosteric motions in the zinc sensor CzrA. J Am Chem Soc 2012;134:3367–3376. DOI: 10.1021/ja208047b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Pecina A, Haldar S, Fanfrlik J, et al. SQM/COSMO scoring function at the DFTB3-D3H4 level: unique identification of native protein–ligand poses. J Chem Inform Model 2017;57:127–132. DOI: 10.1021/acs.jcim.6b00513. [DOI] [PubMed] [Google Scholar]
- 64. Xu Q, Guo H. Quantum mechanical/molecular mechanical molecular dynamics simulations of cytidine deaminase: from stabilization of transition state analogues to catalytic mechanisms. J Phys Chem B 2004;108:2477–2483. DOI: 10.1021/jp037529d. [DOI] [Google Scholar]
- 65. Xu Q, Guo H, Gorin A, Guo H. Stabilization of a transition-state analogue at the active site of yeast cytosine deaminase: importance of proton transfers. J Phys Chem B 2007;111:6501–6506. DOI: 10.1021/jp0670743. [DOI] [PubMed] [Google Scholar]
- 66. Guo H, Rao N, Xu Q, Guo H. Origin of tight binding of a near-perfect transition state analogue by cytidine deaminase: implications for enzyme catalysis. J Am Chem Soc 2005;127:3191–3197. DOI: 10.1021/ja0439625. [DOI] [PubMed] [Google Scholar]
- 67. Zhang X, Zhao Y, Yan H, et al. Combined QM (DFT)/MM molecular dynamics simulations of the deamination of cytosine by yeast cytosine deaminase (yCD). J Comput Chem 2016;37:1163–1174. DOI: 10.1002/jcc.24306. [DOI] [PubMed] [Google Scholar]
- 68. Manta B, Raushel FM, Himo F. Reaction mechanism of zinc-dependent cytosine deaminase from Escherichia coli: a quantum-chemical study. J Phys Chem B 2014;118:5644–5652. DOI: 10.1021/jp501228s. [DOI] [PubMed] [Google Scholar]
- 69. Sklenak S, Yao L, Cukier RI, et al. Catalytic mechanism of yeast cytosine deaminase: an ONIOM computational study. J Am Chem Soc 2004;126:14879–14889. DOI: 10.1021/ja046462k. [DOI] [PubMed] [Google Scholar]
- 70. Sen A, Gaded V, Jayapal P, et al. Insights into the dual shuttle catalytic mechanism of guanine deaminase. J Phys Chem B 2021;125:8814–8826. DOI: 10.1021/acs.jpcb.1c06127. [DOI] [PubMed] [Google Scholar]
- 71. Matsubara T, Ishikura M, Aida M. A quantum chemical study of the catalysis for cytidine deaminase: contribution of the extra water molecule. J Chem Inform Model 2006;46:1276–1285. DOI: 10.1021/ci050479k. [DOI] [PubMed] [Google Scholar]
- 72. Talavera D, Lovell SC, Whelan S. Covariation is a poor measure of molecular coevolution. Mol Biol Evol 2015;32:2456–2468. DOI: 10.1093/molbev/msv109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Starr TN, Thornton JW. Epistasis in protein evolution. Protein Sci 2016;25:1204–1218. DOI: 10.1002/pro.2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.