

Summary
Since the beginning of the pandemic caused by the severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) the gastrointestinal (GI) tract has emerged as an important organ influencing the propensity to and potentially the severity of the related COVID‐19 disease. However, the contribution of the SARS‐CoV‐2 intestinal infection on COVID‐19 pathogenesis remains to be clarified. In this exploratory study, we highlighted a possible link between alterations in the composition of the gut microbiota and the levels of SARS‐CoV‐2 RNA in the gastrointestinal tract, which could be more important than the presence of SARS‐CoV‐2 in the respiratory tract, COVID‐19 severity and GI symptoms. As established by metaproteomics, altered molecular functions in the microbiota profiles of high SARS‐CoV‐2 RNA level faeces highlight mechanisms such as inflammation‐induced enterocyte damage, increased intestinal permeability and activation of immune response that may contribute to vicious cycles. Uncovering the role of this gut microbiota dysbiosis could drive the investigation of alternative therapeutic strategies to favour the clearance of the virus and potentially mitigate the effect of the SARS‐CoV‐2 infection.
Background
Novel coronavirus pneumonia, COVID‐19, is caused by severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2). The most common pathognomonic symptoms include cough, fever, and dyspnoea, but the heterogeneity of clinical manifestations is one of several intriguing facets of SARS‐CoV‐2 infection (Guan et al., 2020). Thus, even though COVID‐19 is mainly considered a respiratory disease, gastrointestinal symptoms have been described in COVID‐19 patients, the most recurrent of which are diarrhoea, vomiting, nausea and abdominal pain. These symptoms suggest that SARS‐CoV‐2 infection has a direct impact also on the gastrointestinal system (Wan et al., 2020).
At the very first stage of the pandemic, digestive symptoms were reported to be an early sign of COVID‐19 (Song et al., 2020). Various aetiopathogenetic hypotheses were subsequently advanced to explain the occurrence of diarrhoea in COVID‐19 patients, including loss of absorption capability by enterocytes (Guo et al., 2021), microscopic inflammatory damage to gastrointestinal mucosal and impaired angiotensin conversion enzyme 2 (ACE2) function. However, SARS‐CoV‐2 viral particles were also detectable in stool samples from patients without diarrhoea (Wang et al., 2020a). Other coronavirus diseases, such as SARS in 2002 and Middle East Respiratory Syndrome (MERS) in 2012 – with which SARS‐CoV‐2 shares 70% and 40% genomic sequence similarities respectively – were also reported to show some enteric involvement (Chu et al., 2020). Although the human intestinal tract was demonstrated to serve as an alternative infection route for MERS‐CoV (Zhou et al., 2017), the role of intestinal SARS‐CoV‐2 infection in the pathogenesis of COVID‐19 remains to be clarified.
A faecal–oral transmission route for SARS‐COV‐2 was proposed based on the evidence that viral RNA is detected in up to 50% of faecal samples from patients diagnosed with COVID‐19 despite a negative nasopharyngeal (NP) swab (Scaldaferri et al., 2020; Wang et al., 2020b). The persistent shedding of SARS‐CoV‐2 in stools from infected patients between 1 and 12 days after a negative NP test result (Xing et al., 2020; Xu et al., 2020; Xiao et al., 2020b) led to the hypothesis that infectious virions are secreted from infected gastrointestinal cells. The presence of SARS‐CoV‐2 RNA load in faecal samples calls for new research (Petrillo et al., 2021). Furthermore, re‐emergence of coronavirus disease in several regions was associated with the processing of frozen food products (Xie et al., 2021), and initial contamination via ingestion cannot be excluded. Evidence obtained using organoid models revealed SARS‐CoV‐2 to exclusively target the apical surface of mature villous enterocytes expressing high levels of three proteins enhancing virus entry into enterocytes: ACE2 and two related membrane‐bound serine proteases, TMPRSS2 and TMPRSS4 (Xiao et al., 2020a; Zhang et al., 2020a). Although the live virus can be detected by electron microscopy in SARS‐CoV‐2 positive faecal specimens (Hindson, 2020), virus isolation from faeces remains difficult (Wolfel et al., 2020; Zhang et al., 2020b). It is possible that SARS‐CoV‐2 may be in part inactivated in stool samples due to bioactive molecules present in stimulated low pH human colonic fluids (Zang et al., 2020).
Currently, little is known about the impact of the gastrointestinal presence of SARS‐CoV‐2 on the course of COVID‐19. Evidence that the gut microbiome influences ACE2 expression has led to several groups hypothesising a contribution of intestinal microbes to COVID‐19, but only very limited data are available on the profile of the gut microbiome in SARS‐CoV‐2‐infected patients (Segal et al., 2020; Villapol, 2020). Notably, microbiota alterations have mainly been discussed in relation to the detection of SARS‐CoV‐2 in the respiratory tract and COVID‐19 severity. Thus, Zuo et al., (2021) investigated the transcriptional activity of SARS‐CoV‐2 and temporal microbiome alterations in faecal samples from patients with COVID‐19. They observed that faecal samples showing high SARS‐CoV‐2 infectivity contained higher abundances of bacterial species Collinsella aerofaciens, Collinsella tanakaei, Streptococcus infantis, and Morganella morganii. Metatranscriptomics analysis revealed higher functional capacity for de novo nucleotide biosynthesis, amino acid biosynthesis and glycolysis in these samples. In contrast, faecal samples with low‐to‐no SARS‐CoV‐2 infectivity were associated with higher abundances of short‐chain fatty acid‐producing bacteria.
Nevertheless, transcript presence does not necessarily indicate protein synthesis and thus a functional impact. More precise functional information could be obtained by metaproteomics characterization (Van Den Bossche et al., 2021). Profiling the human gut microbiome via metaproteomics has proven its value in pathogenesis research in several diseases (Lai et al., 2019; Wang et al., 2020c). The potential of these techniques to help guide future clinical diagnosis has also been highlighted (Long et al., 2020).
In this exploratory study, we used a mass spectrometry‐based approach to profile the gut microbiota in terms of bacterial, archaeal, yeast and fungal content, and analysed the associated metaproteomic functions in patients with intestinal COVID‐19 infection. Altered microbiota compositions were found to be independent of the presence of SARS‐CoV‐2 in the respiratory tract, disease severity and gastro‐intestinal (GI) symptoms, but correlated with GI levels of SARS‐CoV‐2 RNA. Examination of the functional composition of the metaproteome provided a shortlist of both microbial and human biomarker candidates indicative of intestinal SARS‐CoV‐2 infection. These biomarkers could be used to monitor infection. Information on how intestinal SARS‐CoV‐2 affects the microbiota and the host could be useful in the search for alternative therapies promoting viral clearance, with a view to mitigating the impact of SARS‐CoV‐2 infection.
Results
Levels of SARS‐CoV‐2 RNA in the gut
A total of 39 faecal samples collected in April–May 2020 from 39 patients have been studied in this exploratory study (Table 1). Among them, 33 were included in the COVID group due to a positive RT‐PCR on NP swab and/or a positive CT scan (Fig. 1A). The remaining six patients were without COVID‐19 diagnosis (RT‐PCR and CT‐scan negative). The median ages and sex ratios (M/F) of patients with COVID‐19 and non‐COVID‐19 were 77 years (34–99) versus 76 (61–92) and 0.94 versus 1 respectively. To investigate the effect of intestinal SARS‐CoV‐2 infection on the composition of the gut microbiota, faecal samples were characterized by applying the pipeline presented in Fig. 1B. The presence of SARS‐CoV‐2 in the gut was analysed by subjecting stool samples to RT‐qPCR. Of the 39 patients, 10 (25.6%) presented a positive RT‐qPCR result. The median faecal viral load was 3.7 log10 copies per mg (IQR 4.5–2.7), as estimated by RT‐qPCR performed in parallel with standards. A viral load of 1.7 log10 copies per mg (50 copies mg−1) was arbitrarily chosen as the cut off value to group samples into high SARS‐CoV‐2 RNA levels, low SARS‐CoV‐2 RNA loads and negative samples (Fig. 2). No obvious association (Kruskal–Wallis test, p < 0.05) was found between plasma C‐reactive protein (CRP) levels and GI tract SARS‐CoV‐2 levels (p = 0.21). No correlation (Fisher exact test, p < 0.05) between the levels of SARS‐CoV‐2 RNA in the faecal samples and the positivity of NP swab tests on the first hand (p = 0.40), and with severity of COVID‐19 on the other (p = 1.00), were detected.
Table 1.
Clinical characteristics of subjects involved in this study.
| Patient | Sex | Age | RT‐PCR NP swab status | CT‐scan signs of COVID‐19 | SARS‐CoV‐2 in stool (copies mg−1) | Δ RT‐PCR NP swab and stool (days) | PlasmaCRP (mg L−1) | Severity of pulmonary signs | Comorbidities | Antibiotic treatment |
|---|---|---|---|---|---|---|---|---|---|---|
| C01P001 | M | 99 | (+) | No | 3.6E + 04 | 34 | 68.9 | No sign | 1, 2, 5 | Amoxicillin/Clavulanate |
| C01P002 | M | 66 | (−) | Yes | (−) | 5 | 64.5 | Mild | 2, 4 | |
| C01P003 | M | 79 | (+) | Yes | 1.4E + 03 | 8 | 32.4 | Moderate | 1, 6 | 3rd gen Cephalosporins, Metronidazole |
| C01P004 | F | 67 | (+) | Yes | (−) | 17 | 3.2 | Moderate | 0 | |
| C01P005 | F | 91 | (+) | No | (−) | 1 | 7.4 | No sign | 1, 2 | |
| C01P006 | F | 54 | (+) | Yes | (−) | 14 | 30.1 | Moderate | 0 | Vancomycin, Cephazolin Linezolid |
| C01P007 | F | 88 | (+) | No | 1.4E + 01 | 1 | 2.4 | No sign | 1, 2 | Amoxicillin/Clavulanate |
| C01P008 | M | 60 | (+) | Yes | (−) | 20 | 1.2 | Severe | 1 | |
| C01P009 | F | 83 | (+) | No | (−) | 4 | 12.3 | No sign | 0 | |
| C01P010 | F | 92 | (−) | No | (−) | – | 103.1 | No sign | 1, 2 | Amoxicillin/Clavulanate |
| C01P011 | M | 85 | (+) | No | (−) | 7 | 4.2 | No sign | 1, 2 | |
| C01P012 | F | 83 | (+) | Yes | (−) | 9 | 3.9 | Mild | 1, 2, 3, 5 | |
| C01P013 | F | 47 | (+) | Yes | (−) | 1 | 15.6 | Moderate | 0 | |
| C01P014 | M | 63 | (+) | Yes | (−) | 21 | 1.9 | Severe | 0 | |
| C01P015 | F | 54 | (−) | Yes | (−) | 8 | 7.6 | severe | 4 | |
| C01P016 | M | 82 | (+) | No | 6.8 | 5 | 48.9 | No sign | 1, 6 | |
| C01P017 | M | 61 | (−) | No | (−) | – | 23.3 | No sign | 2, 6 | |
| C01P018 | M | 48 | (+) | Yes | (−) | 11 | 65.6 | Mild | 4 | |
| C01P019 | F | 77 | (+) | No | (−) | 15 | 2.2 | Mild | 1 | |
| C01P020 | M | 96 | (+) | No | (−) | 3 | 37.9 | No sign | 1, 4 | |
| C01P021 | F | 56 | (+) | Yes | (−) | 6 | 30 | Moderate | 0 | |
| C01P022 | M | 76 | (+) | Yes | 2.5E + 01 | 10 | 251.3 | Severe | 1, 2 | |
| C01P023 | F | 82 | (−) | No | (−) | 3 | 70.9 | Mild | 1, 2, 3, 5 | Ceftriaxone |
| C01P024 | M | 60 | (−) | Yes | (−) | 30 | 3.9 | Severe | 0 | |
| C01P025 | M | 75 | (−) | No | (−) | – | 37.9 | No sign | 1, 2 | |
| C01P027 | M | 79 | (−) | No | (−) | – | 13 | No sign | 1, 6 | Trimethoprim/Sulfamethoxazole |
| C01P028 | F | 40 | (−) | Yes | (−) | – | 25.8 | Mild | 0 | |
| C01P029 | F | 84 | (+) | No | 3.6E + 04 | 4 | 133.7 | No sign | 1, 2, 4 | Amikin Imipenem/Cilastatin |
| C01P030 | F | 78 | (−) | Yes | (−) | – | 2.3 | No sign | 2, 4 | Cefotaxime, Spiramycin |
| C01P031 | F | 76 | (−) | No | (−) | – | 32.4 | No sign | 1, 3 | |
| C01P032 | M | 69 | (−) | Yes | (−) | 4 | 8 | Severe | 2 | Cefotaxime, Daptomycin |
| C01P033 | F | 95 | (+) | Yes | 9.6E − 01 | 2 | 14.1 | Mild | 1, 2 | |
| C01P035 | M | 34 | (+) | No | 1.5 | 3 | 1.6 | No sign | 4 | |
| C01P036 | F | 58 | (+) | Yes | 1.0E + 01 | – | 66.5 | Moderate | 2 | |
| C01P037 | M | 87 | (+) | No | (−) | 6 | 38.5 | No sign | 1, 2 | |
| C01P038 | M | 96 | (+) | No | (−) | 1 | 5 | No sign | 1, 2 | |
| C01P039 | F | 68 | (+) | Yes | (−) | 8 | 21.2 | Moderate | 2 | |
| C01P040 | F | 79 | (+) | No | (−) | 9 | 20.8 | Moderate | 1, 3 | Amoxicillin/Clavulanate |
| C01P041 | M | 91 | (+) | No | 6.9E + 01 | 3 | 85 | No sign | 1 |
Comorbidities: (0) Any, (1) Age > 70, (2) Antecedents of cardiovascular disease, (3) Uncontrolled diabetes with complications, (4) Chronic respiratory disease, (5) Chronic renal disease, (6) Cancer, (7) Obesity.
Fig. 1.
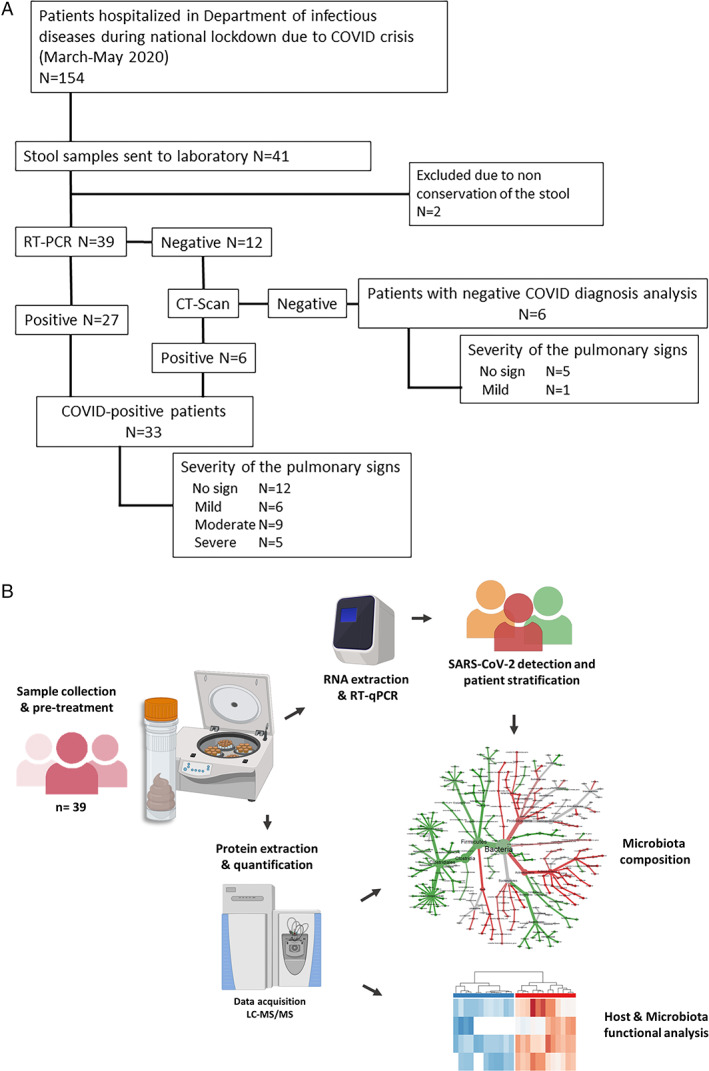
Microbiota profiling of SARS‐CoV‐2‐infected patients.
A. Flowchart of the study.
B. Schematic representation of the experimental analytical workflow. Stool samples were analysed in parallel by RT‐qPCR to detect the presence of SARS‐CoV‐2 RNA in the gut, and by shotgun tandem mass spectrometry to investigate the taxonomical and functional composition of the microbiota.
Fig. 2.
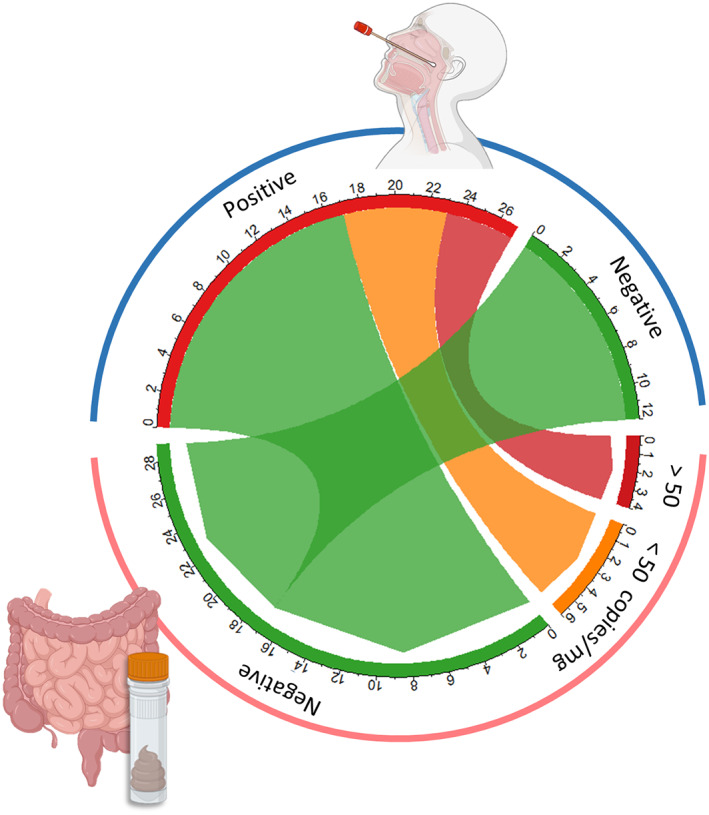
Presence of SARS‐CoV‐2 in the gut. Circos plot showing the relationship between RT‐PCR results from nasopharyngeal swab tests (blue arc) and RT‐qPCR results for stool samples (pink arc). The size of each arc section reflects the number of samples falling into each group. Red and orange indicate positive samples with high and low levels of SARS‐COV‐2 respectively; green indicates negative samples.
Significantly altered microbiota profile in patients with a high level of SARS‐CoV‐2 RNA in the GI tract
To gain insight into the relationship between SARS‐CoV‐2 infection, microbiota and host, we differentiated samples based on the levels of SARS‐CoV‐2 RNA detected in the GI tract. Each faecal sample was analysed three times by metaproteomics, carefully adjusting the peptide quantities to inject based on a pre‐screen by mass spectrometry. The total metaproteomic dataset from the 117 nanoLC–MS/MS runs comprised 7 761 229 MS/MS spectra. With the adjustment procedure, an overall average of 66 335 ± 3248 MS/MS was obtained per sample with low variation between samples. Peptides were identified by searching tandem mass spectra in a two‐step cascaded search, with the second search performed against a sample dedicated database. This strategy allowed 31 ± 6% of spectra to be assigned per sample. A total of 88 135 proteins and 60 179 protein groups were listed in the dataset. Overall, a higher number of proteins was identified in samples with lower levels of SARS‐CoV‐2 RNA or negative compared to positive samples with a viral load >50 copies mg−1 of faecal material (Supplementary Fig. S1).
For each sample, the distribution of the assigned Taxon‐Spectrum Matches (TSMs) as a function of their origin – microbial, host, or food‐related – (Supplementary Table S1) revealed a higher percentage of host signal in samples positive for SARS‐CoV‐2 RNA (Fig. 3A). The microbial component of the metaproteomes expressed as a proportion of the average protein biomass at the phylum level was dominated by bacteria (61 ± 19%; mean ± SD), followed by fungi and archaea, which represented 5 ± 2% and 3 ± 1% respectively. Firmicutes, Proteobacteria, Actinobacteria and Bacteroidetes were the predominant bacterial phyla, which combined accounted for an average of 48% of the biomass. No association (Kruskal–Wallis test, p < 0.05) was found between faecal SARS‐CoV‐2 viral load and the Firmicutes/Bacteroidetes ratio (p = 0.85). Among the archaeal signatures, the Euryarchaeota phylum was the most highly represented (3 ± 1%). A total of 201 genera were identified in microbiota profiles, with Clostridium, unclassified Lachnospiraceae, Bacteroides and Lachnoclostridium as dominant taxa in addition to the host. The relative biomass contributions for these groups ranged from <1% to 6% for bacterial genera and up to 58% for Homo sapiens. Among the 13 archaeal genera identified, Methanobrevibacter (28 792 TSMs) was the most abundant. Even though it remained a minor component of the microbiota, sequence coverage for this genus was high (6392 taxon‐specific peptides). Ascomycota and Streptophyta tended to be the most abundant Eukaryota phyla, with the notable exception of the Chordata host (Fig. 3B). After filtering out food‐related and host signals, dimension reduction by principal component analysis (PCA) revealed distinct microbiota profiles for patients with high intestinal levels of SARS‐CoV‐2 RNA (viral load >50 copies mg−1 of faecal material) compared to profiles for patients with low or no viral RNA (Fig. 3C). Two outliers were observed: sample C01P003 (faecal viral load 1.4 × 103 copies mg−1), clustered among negative samples; sample C01P033 (1 copy mg−1), which had a microbiota composition resembling high SARS‐CoV‐2 samples. The same unsupervised analysis failed to highlight clusters of microbiota profiles according to the RT‐PCR status of the NP swab, the severity of the pulmonary signs or the antibiotic treatments (Supplementary Fig. S2A–C).
Fig. 3.
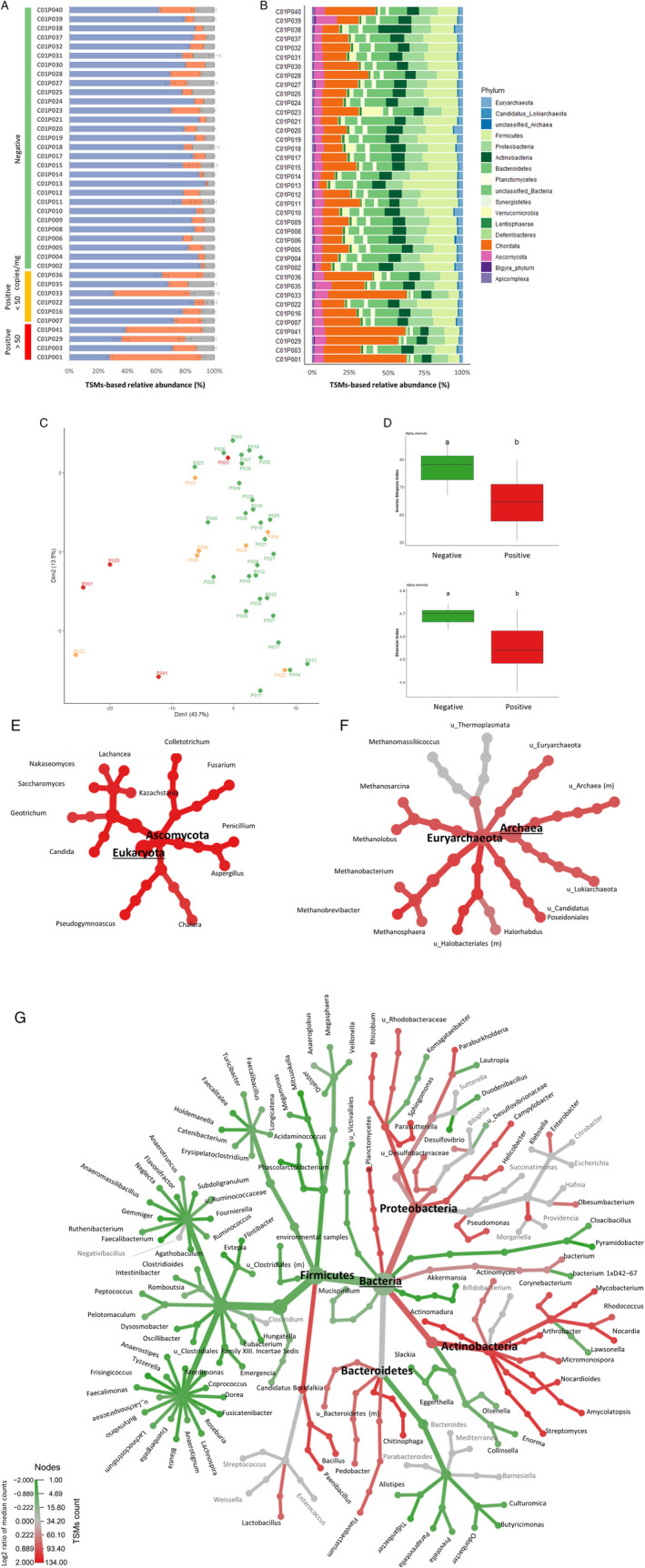
Metaproteomic profiling of gut microbiota.
A. Distribution of the assigned TSMs as a function of their origin, microbial (blue), host (orange) or food‐related (grey). Samples are grouped based on their SARS‐CoV‐2 RNA content.
B. Relative biomass contributions of the phyla identified for each sample. Each sample was analysed in three replicates, bars represent average relative abundances for each analysis.
C. PCA plot of the microbiota profiles. Red and orange indicate positive samples with high and low levels of SARS‐COV‐2 respectively; green indicates negative samples. Each point corresponds to one sample, represented as the sum of the contribution of its three corresponding replicates.
D. Box plots showing variation in alpha diversity across age‐matched samples from different groups based on the Inverse Simpson and Shannon indices. Red bars indicate positive samples with high levels of SARS‐COV‐2, green bars indicate negative samples. a, b: significant difference between groups [Tukey's Honest Significant Difference (HSD) test].
E. Differential heat tree for Eukaryota.
F. Differential heat tree for Archaea.
G. Differential heat tree for Bacteria. The trees show comparisons of microbiota compositions between samples containing high levels of SARS‐CoV‐2 RNA and negative samples. Phyla and genera are indicated. Taxa coloured red are more abundant in positive samples, green taxa are more abundant in negative samples.
Because age‐related changes to the microbiota have recently been reported (Nagpal et al., 2018), we further examined the alpha diversity of our samples after matching positive (viral load >50 copies mg−1 of faecal material) and negative patients by age (88.2 (±8.7) and 80.5 (±7.8) years old respectively). Both Inverse Simpson and Shannon indices indicated that microbial diversity was significantly decreased (Tukey's Honest Significant Difference) in samples containing SARS‐CoV‐2 RNA compared to negative samples (Fig. 3D). Overall comparison between microbiota determined for patients with a high SARS‐CoV‐2 load and negative samples (Wilcoxon rank‐sum test, p‐value <0.05) showed that patients with SARS‐CoV‐2 RNA in the intestine had a higher relative abundance (log2 FC > 1.6) of certain fungal genera, such as Candida, Fusarium, Penicillium, Aspergillus and Saccharomyces (Fig. 3E). For archaea, Methanosphaera and unclassified Halobacteriales were among the most enriched genera in samples containing SARS‐CoV‐2 (log2 FC = 1.4) (Fig. 3F). Among bacteria, several genera belonging to the Actinobacteria class (e.g. Streptomyces, Actinomadura, Amycolatopsis, Nocardia, Mycobacterium and Arthrobacter), as well as Paenibacillus, Chitinophaga and Sphingomonas were significantly enriched in SARS‐CoV‐2‐positive samples compared to controls (log2 FC > 1) (Fig. 3G). In contrast, significantly lower relative abundance was observed for genera belonging to the Ruminococcaceae and Lachnospiraceae families (log2 FC > −1), and more broadly to the Firmicutes phylum, except for members of the Bacilli class. Several genera from Odoribacteraceae, Prevotellaceae, Eggerthellaceae and Coriobacteriaceae families (log2 FC = −1), as well as Akkermansia (log2 FC = −1.7), were also detected at significantly lower abundance in virus‐positive samples (Fig. 3G). These differences were also observed when the comparative analysis was performed only on the most abundant microbial peptides identified in samples positive for SARS‐CoV‐2, demonstrating that the highly abundant human peptides identified in these samples do not impact taxa relative quantification (Supplementary Fig. S3).
Functional composition of the metaproteome reveals potential biomarkers of SARS‐CoV‐2 infection
To retrieve functional information from the metaproteome for different sample groups, microbiota and host proteins identified with an FDR of 1% were annotated and their functions were assigned to 887 KEGG Orthology (KO) entries (Supplementary Table S2).
The relative abundance of each functional term was calculated at phylum level to reduce loss of information when peptides were unambiguously assigned to the different taxa at a finer resolution. To investigate dissimilarities in microbiota‐derived KO functions (n = 664), unsupervised PCA was performed on age‐matched SARS‐CoV‐2‐positive (faecal viral load >50 copies mg−1) and negative samples (Fig. 4A). This PCA analysis revealed distinct clustering based on the presence of SARS‐CoV‐2, with only SARS‐CoV‐2‐positive C01P003 sitting closer to the negative samples. Comparative analysis of the functional profiles (Wilcoxon test, FDR‐adjusted p < 0.05) revealed 341 KOs significantly differentially abundant between the two groups of samples. Of these, 21 were increased in SARS‐CoV‐2‐positive samples (Fig. 4B/C, Supplementary Fig. S4). These KOs are included in 67 KEGG pathways, the most populated of which were metabolic pathways (10), biosynthesis of secondary metabolites (5), glycolysis/gluconeogenesis (3) and microbial metabolism in diverse environments (3). Interestingly, the identification of molecular functions related to citrulline flux (KO names OTC and arcA) suggested that Firmicutes species were still adapting to cope with stress and gain an energetic advantage. Evidence for this ongoing adaptation was also provided by the increase in abundance of polypeptides belonging to two‐component systems (mcp and yesN) and cobalamin production (cobS ‐ cobV, Firmicutes). Similarly, KOs for drug exporter pump (K06994), NADPH:quinone reductase (qor, CRYZ), and NTE family proteins associated with Actinobacteria suggest the implementation of mechanisms conferring a competitive advantage in stressful environments, such as the SARS‐CoV‐2‐infected gut. Modules involved in sulfur (soxD, Bacteroidetes) and glutathione metabolism (pepN, Actinobacteria) were also significantly increased. Another marker of the host response, K06856 (IGH, immunoglobulin heavy chain), was increased in association with both Actinobacteria and Firmicutes. Some fungi‐associated molecular functions were also altered (PDC, AdhP, SET2). Among these markers, AdhP is known to be involved in retinol metabolism, and the histone modification protein SET2 plays a key role in mucosal immune responses, and could be critically involved in integrating a variety of external signals driving fungal expansion. This fungal expansion would in turn influence the host immune response. Among the 21 altered KOs identified, nine (K00134, K00344, K01256, K07001, K01568, K22622, K07720, K11686, K16703) were linked to 14 pathways. Levels of these pathways were also significantly increased relative to age‐matched samples in which lower levels of SARS‐CoV‐2 RNA were detected (viral load <50 copies mg−1) (Fig. 4B/C).
Fig. 4.
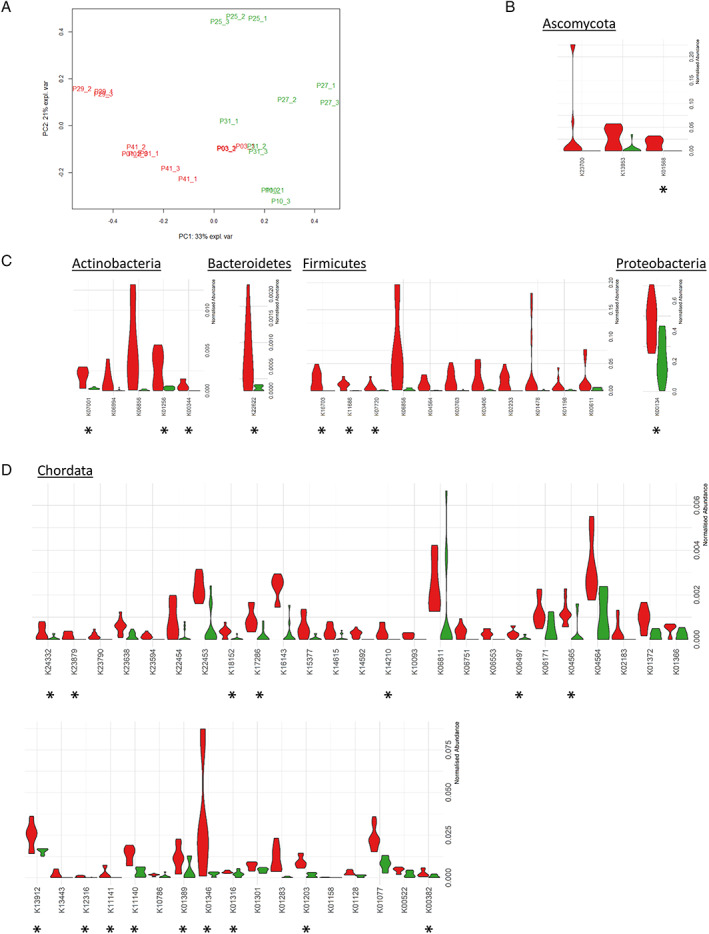
Comparative analysis of the functional composition of the metaproteome.
A. PCA analysis of functional microbiota profiles from aged‐matched samples containing high levels of SARS‐CoV‐2‐RNA, or negative for viral RNA.
B. Violin plots showing the relative abundance of the Ascomycota‐related KOs increased in SARS‐CoV‐2‐positive samples (red) or negative samples (green). Asterisks denote KOs that were also significantly increased when compared to levels detected in age‐matched samples containing lower levels of SARS‐CoV‐2 RNA.
C. Violin plots showing the relative abundance of the bacterial‐related KOs increased in SARS‐CoV‐2‐positive samples (red) or negative samples (green). Asterisks denote KOs that were also significantly increased when compared to levels detected in age‐matched samples containing lower levels of SARS‐CoV‐2 RNA.
D. Violin plots showing the relative abundance of host‐related KOs increased in SARS‐CoV‐2‐positive samples (red) compared to negative samples (green). Asterisks denote KOs that were also significantly increased when compared to levels detected in age‐matched samples containing lower levels of SARS‐CoV‐2 RNA.
To investigate alterations in host molecular functions, the relative abundance of host‐associated KOs was also analysed. The abundance of a total of 72 out of 187 KOs was altered in samples containing high levels of SARS‐CoV‐2‐RNA (viral load >50 copies mg−1) compared to negative samples (Supplementary Table S2). Of these KOs, 42, represented in 117 pathways, were increased in SARS‐CoV‐2‐positive samples with a high viral load (Fig. 4D). These KOs included host molecular functions involved in the ACE2 signalling network (Renin‐angiotensin system) such as peptidyl‐dipeptidase A (ACE), aminopeptidases (ANPEP/CD13), glutamyl aminopeptidase (ENPEP) and neprilysin (MME). In parallel, functions described by KOs such as latexin (LXN), sphingomyelinase‐related protein SMPDL3, dihydrolipoamide dehydrogenase (DLD), SOD2, Cu/Zn superoxide dismutase (SOD1), ferritin heavy chain (FTH1), tissue‐non‐specific alkaline phosphatase (K01077), antileukoproteinase (SLPI), bleomycin hydrolase (BLMH), aminopeptidase (NAALADL1), glutamate carboxypeptidase II (GCPII) and transmembrane serine protease 15 (TMPRSS15), proteins from the enterocyte brush‐border membrane – including sucrase‐isomaltase, calmodulin, cadherin‐17 and GCPII, trefoil factor family peptides (TFF2 and TFF3) were also significantly increased in faecal samples with high levels of SARS‐CoV‐2 RNA. Interestingly, faeces with high levels of SARS‐CoV‐2 contained a statistically higher abundance of proteins with CRP‐mediated functions: immunomodulatory Gal‐9, bone marrow proteoglycan (PRG2), glutathione S transferase, deleted in malignant brain tumour 1, gastric intrinsic factor, members of the lysosome pathway (DNASE2, CTSH, CD63 antigen, GAA and NPC2), l‐Tryptophan (SLC3A1) and choline (SLC44A2) transporters. These proteins are linked to functional pathways, and as such provide further insight into the host‐microbiota crosstalk in SARS‐CoV‐2‐positive GI tracts. In parallel, the increase in MHC class I antigen (MHC1), ADP‐Ribosyl Cyclase 2 (BST1), nicastrin (NCSTN), pre‐B lymphocyte gene (VPREB) and ferritin heavy chain (FTH1) suggests activation of immune processes. Sixteen of the altered KOs identified (K00382, K01203, K01316, K01346, K01389, K04565, K06497, K11140, K11141, K12316, K13912, K14210, K17286, K18152, K23879, K24332) were also specifically increased in samples containing <50 copies mg−1 viral RNA following comparison with their relative levels in SARS‐COV‐2 positive samples with high SARS‐CoV‐2 load.
Discussion
In the context of the SARS‐CoV‐2 infection, the GI tract has emerged as an organ significantly influencing the propensity to develop the ensuing disease COVID‐19, and potentially predict its severity (Albrich et al., 2021). Studies based on taxonomical molecular biology approaches have demonstrated that respiratory infections are associated with changes in the composition of the gut microbiota (Guo et al., 2021), but the correlation between respiratory disease and the amount of virions present in the gut has received less attention (Zuo et al., 2021). Here, we provide a broad profile of gut microbiota obtained by a mass spectrometry‐based approach detecting bacteria, archaea, yeasts and fungi. Organisms were proteotyped and their respective biomass contributions directly compared. We also investigated whether the presence of SARS‐CoV‐2 in the gastrointestinal tract could be associated with changes to the microbiota composition.
In this article, we report that the presence of SARS‐CoV‐2 RNA in the GI tract is not directly related to the detection of the virus in the respiratory tract or to COVID‐19 severity at the time of detection. This result is in agreement with the detection of SARS‐CoV‐2 in tissues throughout the GI tract, and virus shedding in stools in a significant proportion of patients. GI shedding often continues for prolonged periods following virus clearance from the respiratory tract (Segal et al., 2020). Although hampered by the limited number of patients analysed, the metaproteomics profiles obtained in this exploratory study support the hypothesis that the composition of the gut microbiota may be influenced by the abundance of SARS‐CoV‐2 RNA in the intestine more than by its presence in the respiratory tract, clinical symptoms or antibiotics exposure. In particular, albeit limited to a single time point analysis, our results show that the microbiota was significantly different in patients with a faecal viral load greater than 50 copies mg−1. In contrast, microbiota from patients with a lower or negative viral load tended to be more similar. Closer examination revealed a relatively high level of SARS‐CoV‐2 RNA to be associated with a significantly higher abundance of several genera belonging to the Euryarchaeota and Ascomycota phyla, as well as bacterial genera from the Actinobacteria order, Chitinophaga, Paenibacillus, Sphingomonas and Bacillus. The overgrowth of fungi is in itself indicative of a disruption of commensal communities. In addition, fungal overgrowth in the gut can cause macrophage polarization which has been linked to increased infiltration of inflammatory cells in allergic airways (Kim et al., 2014). Interestingly, bacterial genera from the Bacteroidetes phylum – known to be associated with suppression of colonic expression of ACE2 in the murine gut (Geva‐Zatorsky et al., 2015) – were significantly more abundant in SARS‐CoV‐2‐positive samples. Similarly, the relative abundance of bacterial genera belonging to the Firmicutes phylum, such as Faecalibacterium, Mitsuokella and Frisingicoccus, correlated well with the absence of SARS‐CoV‐2 in the gut. Interestingly, this class of bacteria are the main producer of butyrate in the intestinal lumen, which is extensively used by colonic epithelial cells as an energy source (Kalantar‐Zadeh et al., 2020). An in‐depth shotgun metagenomics analysis of samples from 15 patients hospitalized in Hong Kong (Zuo et al., 2020) revealed a similar shift in microbiota composition. Longitudinal studies on a larger number of patients could strengthen the observed changes and shed light on the cause or effect relationships of the microbial dysbiosis associated with COVID‐19 infection.
Our peptide‐based functional metaproteome analysis supported the differences observed between samples containing high levels of SARS‐CoV‐2 RNA and negative samples, and the existence of a complex interplay between the gut and SARS‐CoV‐2 infection. In particular, the alterations to host functions observed in faeces containing high levels of SARS‐CoV‐2 revealed an inflamed GI tract characterized by activation of the immune response, as reflected by the molecular alterations governing the host's antiviral defence system. In addition to serving as a receptor for SARS‐CoV‐2, ANPEP/CD13 expression is dysregulated in inflammatory diseases. Its detection in wide numbers of gut samples led to the persistent intestinal inflammation hypothesis (Bauvois and Dauzonne, 2006). In our samples, expression levels for this marker were consistent with increased LXN and SMPDL3, a sphingomyelinase‐related protein abundantly expressed on macrophages and dendritic cells (Heinz et al., 2015). Both of these proteins are upregulated by inflammatory stimuli. In parallel, functions described by KOs such as DLD, SOD2, Cu/Zn superoxide dismutase (SOD1) and ferritin heavy chain (FTH1) could reflect mitochondrial dysfunction and interplay between inflammation and oxidative stress. Interestingly, the expression of tissue‐nonspecific alkaline phosphatase in the colon was also previously reported to be upregulated during inflammatory episodes due to inflammation‐driven tissue infiltration by neutrophils (Lalles, 2014). The identification of elevated levels of KOs linked to functions located in the enterocyte brush border supports inflammation‐induced enterocyte damage and increased intestinal permeability. This possibility is supported by the over‐detection in faecal samples containing SARS‐CoV‐2 RNA of trefoil factor family peptides. These essential proteins are involved in the protection and repair of the gastrointestinal tract (Ghanemi et al., 2020). Similarly, the increased abundance of several leaky‐gut‐related functions like those associated with antileukoproteinase (SLPI) (Van Spaendonk et al., 2017); BLMH, a cytosolic aminopeptidase thought to contribute to MHC class I peptide presentation (Towne et al., 2007); aminopeptidase (NAALADL1), GCPII and transmembrane serine protease 15 (TMPRSS15) may contribute to the dysregulation of the protease/antiprotease balance and could potentially lead to epithelial damage and increased intestinal permeability. In parallel, the increase in MHC1, ADP‐Ribosyl Cyclase 2 (BST1), nicastrin (NCSTN), pre‐B lymphocyte gene (VPREB) and ferritin heavy chain (FTH1) suggests activation of immune processes that could be further explored in future studies to unravel the links between SARS‐CoV‐2 infection and gastrointestinal perturbations.
In this dysfunctional environment, individual members of the altered microbiota battle to gain an energetic advantage. Given the known regulatory role of H2S in mucosal inflammation and the involvement of glutathione in bio‐reduction of reactive oxygen species, the observed alterations suggest that the presence of SARS‐CoV‐2 triggers activation of microbial metabolic pathways that further fuel inflammation and related immune responses. An Ig‐like domain is frequently present in bacterial proteins that affect adhesion to host cells and tissues as part of the invasion or other steps in the infection process (Bodelon et al., 2013).
Our results corroborate the alterations recently reported in the faecal metabolome for COVID‐19 patients (Lv et al., 2021). The altered molecular functions described here suggest mechanisms feeding into vicious cycles. Furthermore, our results provide markers that could be valuable in monitoring the progression of SARS‐CoV‐2 infection and assessing therapeutic strategies to promote viral clearance and restore normal intestinal function. Among the protein functions described, some such as Galectin‐9 and CRP are already under investigation as biomarkers of disease severity (Ahnach et al., 2020; Gong et al., 2020; Moar and Tandon, 2021). Interestingly, elevated serum levels of CRP, especially in association with a high concentration of d‐dimers, are reported to be indicative of an increased risk of adverse outcomes in COVID‐19 (Smilowitz et al., 2021). Consequently, faecal CRP levels could represent a useful biomarker to stratify COVID‐19 patients as a function of the level of SARS‐CoV‐2 in the GI tract.
Although the COVID‐19 pandemic has led to the launch of a large number of clinical studies (Grenga and Armengaud, 2021), to our knowledge, this is the first time the composition and functionality of gut microbiota have been analysed by differential metaproteomics, with samples distinguished based on the presence of SARS‐CoV‐2 RNA in the intestinal tract. Interestingly, even in stool specimens containing high levels of viral genetic material, no SARS‐CoV‐2‐derived peptides were detected in our discovery metaproteomics approach despite preliminary evidence obtained by infecting Vero E6 cells with faecal filtrates supporting the presence of infectious SARS‐CoV‐2 particles (data not shown). This negative result could be ascribed to the complexity of the samples and a lack of sensitivity of our approach. Indeed, although targeted proteomics can successfully detect SARS‐CoV‐2 proteins in NP swabs and gargle samples (Ihling et al., 2020; Gouveia et al., 2020b; Saadi et al., 2021), SARS‐CoV‐2 viral proteins have never been detected by discovery proteomics approaches applied to complex matrices (Grenga and Armengaud, 2021). Notwithstanding the relatively modest number of samples analysed and the fact that extra‐pulmonary detection of viral RNA does not constitute proof that infectious virus particles are or were present, our results confirm the clinical relevance of testing for viral RNA in faeces because of its direct correlation with altered gut microbiota.
The association between COVID‐19 and the presence of SARS‐CoV‐2 in the intestine remains to be further explored. SARS‐CoV‐2 is the third highly pathogenic coronavirus (after SARS‐CoV and MERS‐CoV) to cross to humans within < 20 years, suggesting that new zoonotic coronavirus spill‐overs are likely to occur in the future. The potential role of the gut in the diseases induced by this family of viruses should be further explored in multiple cohorts and settings, longitudinally over the course of different stages of infection. ‘Long Covid’ describes the situation when the effects of COVID‐19 continue for weeks or months beyond the initial illness, and has now been reported for a significant number of patients (Norton et al., 2021). Some of these cases, especially those associated with abdominal pain, could be linked to continuous infection of the gut epithelium or severe alterations of the gut microbiota. Recently, Liu et al. (2022) provided observational evidence of compositional alterations of gut microbiome in patients with long‐term complications of COVID‐19. Examining how the microbiota composition affects how SARS‐CoV‐2 infects the GI tract is of utmost interest. Further mechanistic studies could help propose novel therapies to reduce the severity of COVID‐19.
Until now, the impact of SARS‐CoV‐2 infection on the gut microbiota has been scarcely studied. Here, we assessed on a cohort of 39 patients the gastrointestinal SARS‐CoV‐2 viral load and correlated it with their full‐range microbiota, including Bacteria, Archaea, Fungi and Moulds, accessible through metaproteomics. Our data show three key results that could be of major importance in the battle against COVID‐19: (i) faecal SARS‐CoV‐2 viral load is not correlated to symptoms, (ii) an important change in the microbial structure is observed for patients with high faecal SARS‐CoV‐2 viral load and (iii) a list of microbiome and human markers can be drawn from this study as possible candidates for diagnostic. These results should be an incentive for an extensive multi‐centric metaproteomics analysis of the gut microbiota of COVID‐19 patients.
Experimental procedures
Ethics approval, consent to participate and study population
This research has been performed in accordance with the Declaration of Helsinki. This retrospective study was approved by the local Institutional Review Board (IRB number: 20.05.01), ‘Comité d'éthique du CHU de Nîmes’. An informed letter was sent to patients to describe the study. Oral consent was obtained, witnessed and recorded for all participants. Stool samples received in the Department of Microbiology from the Department of Infectious Diseases (University Hospital Nîmes, France) from March 17, 2020, to May 11, 2020, were included. During this period, a nationwide lockdown was applied with an emergency state due to the context of the COVID‐19 health crisis. The hospital admitted exclusively patients with acute health problems.
The flowchart of the study is presented in Fig. 1A. Among the 41 stools received during the indicated period, two were excluded from the analysis due to the low quantity. For each patient, a NP swab was collected and sent to the Department of Microbiology which is accredited to perform RT‐PCR assay for SARS‐CoV‐2. RNA was extracted from clinical samples using the chemagic viral DNA/RNA kit special 96 on the chemagic platform (PerkinElmer, Waltham, MA, USA). The RT‐PCR was performed using the kit SARS‐CoV‐2 R‐Gene® (bioMérieux, Marcy‐l'Étoile, France) which targets three genes of the virus [the N gene of the nucleocapsid, the RNA‐dependent polymerase gene (RDRP) and the E gene which is common to all the SARS viruses], following the manufacturer's recommendations. Data including demographics, laboratory results and imaging results were extracted from the electronic medical records of the University Hospital management system (Clinicom, Intersystems SAS, France).
Collecting and processing faecal specimens
All the faecal samples received in the Department of Microbiology were routinely conserved at −80°C, according to the French microbiology standard (Rémic, 2018). The selected stools were transported in dry ice to the analytical facility following international guidelines (number UN3373, B category). Nucleic acid extraction and sample lysis performed on live SARS‐CoV‐2 samples prior to protein extraction were conducted in a level 3 biosafety facility.
Quantification of viral RNA by RT‐qPCR in faecal samples
SARS‐CoV‐2 RNA from stool samples was isolated using the NucleoSpin RNA Virus Mini kit (Macherey‐Nagel SAS, Hoerdt, France). Stools (200 mg) were suspended at 10% (vol./wt.) in PBS, homogenized vigorously with a vortex and then centrifuged for 3 min at 500g to remove large particles. As recommended by the manufacturer for the treatment of stool samples, proteinase K (20 μl at 20 mg ml−1) was added to the resulting supernatant (200 μl), incubated for 5 min at room temperature. Lysis was performed at 70°C and nucleic acid was extracted as recommended in the manufacturer's protocol for the treatment of viscous samples. Viral RNA was eluted in 50 μl RNase‐free H2O preheated to 70°C. RNA was quantified by RT‐qPCR using the SuperScript III Platinum One‐Step RT‐qPCR Kit (Thermo Fisher, Waltham, USA) and a CFX96 Touch Real‐Time PCR Detection System Thermal Cycler (Bio‐Rad, Marnes La Coquette, France). Primers targeting IP2 and IP4 (RdRp gene), 0.4 μM per reaction, were those recommended by Grenga et al., (2020): nCoV_IP2‐12669Fw (ATGAGCTTAGTCCTGTTG), nCoV_IP2‐12759Rv (CTCCCTTTGTTGTGTTGT), nCoV_IP2‐12696bProbe(+) (AGATGTCTTGTGCTGCCGGTA [5′]Hex [3′]BHQ‐1), nCoV_IP4‐14059Fw (GGTAACTGGTATGATTTCG), nCoV_IP4‐14146Rv (CTGGTCAAGGTTAATATAGG) and nCoV_IP4‐14084Probe(+) (TCATACAAACCACGCCAGG[5′]Fam [3′]BHQ‐1). Primers targeting E gene were: E_Sarbeco_Fw (ACAGGTACGTTAATAGTTAATAGCGT), E_Sarbeco_Rv (ATATTGCAGCAGTACGCACACA), and E_Sarbeco_Probe(+) (ACACTAGCCATCCTTACTGCGCTTCG[5′]Fam [3′]BHQ‐1). Standard curves were created using in vitro‐transcribed RNA derived from the BetaCoV_Wuhan_WIV04_2019 strain (EPI_ISL_402124), which contains the amplification regions of the RdRp and E gene as positive strand. Standard samples were prepared with 1011 copies of target sequences diluted in yeast tRNAs to facilitate recovery, and lyophilised.
Proteome extraction and digestion
Following sample homogenisation, three 50 mg aliquots of each faecal sample were transferred to fresh tubes to create three technical replicates for each sample. Aliquots were lysed in 100 μl LDS 1× (Lithium dodecyl sulfate) sample buffer (Invitrogen™, Thermo Fisher) supplemented with 5% beta‐mercaptoethanol (vol./vol.). Samples were sonicated for 5 min in an ultrasonic water bath (VWR ultrasonic cleaner) and incubated at 99°C for 5 min before transfer to 2‐ml Screw Cap microtubes (Sarstedt, Marnay, France) containing 200 mg ceramic beads, as previously described (Hayoun et al., 2019). Cell disruption was performed on a Precellys Evolution instrument (Bertin Technologies, Aix en Provence, France) operated at 10 000 rpm for 10 30‐s cycles, with 30 s rest between cycles. After lysis, samples were centrifuged at 16 000g for 3 min. For each sample, the resulting supernatant (25 μl) was loaded onto a NuPAGE 4%–12% Bis‐Tris gel, and proteins were subjected to short (5‐min) SDS‐PAGE migration. Proteins were stained for 5 min with Coomassie SimplyBlue SafeStain (Thermo Fisher) prior to in‐gel trypsin proteolysis with Trypsin Gold (Promega) using 0.011% ProteaseMAX surfactant (Promega, Madison, WI, USA), as described in Hartmann et al. (2014).
Preliminary quantitation of peptides extracted from faecal samples by high‐resolution mass spectrometry survey
For each faecal sample, 1 μl of the extracted peptide mixture was injected for analysis on an LTQ‐Orbitrap XL (Thermo Fisher Scientific, Waltham, USA) tandem mass spectrometer coupled to an Ultimate 3000 nano LC system (Thermo Fisher Scientific). The proteolysed products were desalted online on a reverse‐phase PepMap 100 C18 μ‐precolumn (5 μm, 100 Å, 300 μm id × 5 mm, Thermo Fisher) and resolved on a nanoscale PepMap 100 C18 nanoLC column (3 μm, 100 Å, 75 μm id × 50 cm, Thermo Fisher) at a flow rate of 0.3 μl min−1 prior to injection into the mass spectrometer. A linear chromatographic gradient of mobile phase A (0.1% HCOOH/100% H2O) and phase B (0.1% HCOOH/80% CH3CN) was applied from 5% to 40% B in 30 min. Full‐scan mass spectra were measured from m/z 350 to 1500 in data‐dependent mode using a Top5 strategy. Briefly, a scan cycle was initiated by a full high mass‐accuracy scan in the Orbitrap analyser, operated at 30 000 resolution, followed by MS/MS scans in the linear ion trap on the five most abundant precursor ions. A 10‐s dynamic‐exclusion window was applied to previously selected ions. Precursor ions were isolated using a 3‐m/z isolation window and activated with 35% normalized collision energy.
NanoLC–MS/MS characterization of peptides extracted from faecal samples
To normalize the peptide amount injected for each faecal sample, the total ion current (TIC) chromatogram obtained with the LTQ‐Orbitrap XL instrument was used to calculate the exact volume (μl) to be injected on a Q‐Exactive HF mass spectrometer (Thermo) by dividing 16.1E7 (the target optimal TIC previously established) by the TIC value obtained with the LTQ‐Orbitrap XL. The high‐field Orbitrap instrument was used in combination with an UltiMate 3000 LC system (Dionex‐LC) and operated in data‐dependent mode, as previously described (Klein et al., 2016). The appropriate volumes of peptides were injected for each aliquot (technical triplicate for each faecal sample), desalted on an Acclaim PepMap100 C18 precolumn (5 μm, 100 Å, 300 μm id × 5 mm), and then resolved on a nanoscale Acclaim PepMap 100 C18 column (3 μm, 100 Å, 75 μm id × 50 cm) with a 120‐min gradient at a flow rate of 0.2 μl min−1. The gradient was developed from 4% to 25% of (CH3CN, 0.1% formic acid) over 100 min, and then from 25% to 40% over 20 min. Peptides were analysed during scan cycles initiated by a full scan of peptide ions in the ultra‐high‐field Orbitrap analyser, followed by high‐energy collisional dissociation and MS/MS scans on the 20 most abundant precursor ions (Top20 method). Full‐scan mass spectra were acquired from m/z 350 to 1500 at a resolution of 60 000 with internal calibration activated on the m/z 445.12002 signal. During ion selection for MS/MS fragmentation and measurement, a 10‐s dynamic‐exclusion window was applied with an intensity threshold of 1.7 × 104. Only ions with positive charges 2+ and 3+ were considered.
Assigning peptides and analysing metaproteomics data
The Mascot Daemon 2.6.1 search engine (Matrix Science) was used to match MS/MS spectra to peptides in a multi‐round search process. The search parameters were as follows: full trypsin specificity, maximum of two missed cleavages, mass tolerances of 5 ppm on the parent ion and 0.02 Da on the MS/MS, carbamidomethylated cysteine (+57.0215) as fixed modification, and oxidized methionine (+15.9949) and deamidation of asparagine and glutamine (+0.9848) as variable modifications. The NCBInr database (National Center for Biotechnology Information, NIH, Bethesda) was downloaded as ftp://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/nr.gz (55 GB, downloaded on 2nd January 2020). Protein accession numbers were mapped to taxids using files from ftp://ftp.ncbi.nlm.nih.gov (downloaded on January 2020). Files included: assembly_summary_refseq.txt and assembly_summary_genbank.txt downloaded from ftp://ftp.ncbi.nlm.nih.gov/genomes/ASSEMBLY_REPORTS/ to map taxids to RefSeq (GCF) and GenBank assemblies (GCA), GCF/GCA *_assembly_report.txt files downloaded from ftp://ftp.ncbi.nlm.nih.gov/genomes/all/ to map GCF/GCA to nucleotides and the GCF/GCA *_genomic.gff.gz files downloaded from the same directory to map GCF/GCA to protein accessions. Python SQLite databases were created to perform fast protein‐to‐taxid matching. The file prot.accession2taxid.gz, downloaded from ftp://ftp.ncbi.nlm.nih.gov/pub/taxonomy/accession2taxid/ was used to supplement databases, especially for Eukaryota. Files containing the NCBI taxonomy, names.dmp and nodes.dmp were downloaded as ftp://ftp.ncbi.nlm.nih.gov/pub/taxonomy/taxdmp.zip to directly assign Peptide‐spectrum matches (PSMs) to the ‘canonical’ taxonomical levels (species, genus, family, order, class, phylum and superkingdom) of the NCBI taxonomic tree.
A first‐round Mascot search was performed on a reduced NCBInr‐based database, termed NCBInrS, containing 45 925 selected taxa. This database comprises 76 112 114 sequences and a total of 31 417 968 972 amino acids. This first round was performed with a selection of the 50 000 best MS/MS spectra, as determined using the open‐source algorithm ScanRanker. DirectTag version 1.4.66 (Ma et al., 2011) was used with the following parameters to apply the algorithm: PrecursorMzTolerance 0.1, FragmentMzTolerance 0.5, StaticMods ‘C 57.0215’, UseChargeStateFromMS true, NumChargeStates 4. Molecular ion peak lists were extracted with Proteome Discoverer daemon v1.4 software (Thermo scientific), applying the following parameters: 400 (minimum mass), 5000 (maximum mass), 0 (grouping tolerance), 0 (intermediate scans) and 1000 (threshold). Peptide‐to‐MS/MS spectrum assignments complied with the following constraints: full trypsin specificity, maximum of one missed cleavage, 2+ or 3+ peptide charges, mass tolerances on the parent ion of 3 ppm, and 0.02 Da on the MS/MS, static modification of carboxyamidomethylated cysteine (+57.0215), and oxidized methionine (+15.9949) as dynamic modification.
The Python version of Matrix Science's msparser (version 2.6.1) was used to parse the Mascot dat file, applying the ms_peptidesummary function. PSMs were validated when they had a Mascot expectation value below 0.3 according to their Mascot homology threshold; multiple PSMs were allowed per MS/MS spectrum. Each PSM associated with peptide sequences by Mascot was mapped to taxa using in‐house SQLite databases built from NCBI data, as described in Gouveia et al. (2020a).
The phylopeptidomics procedure (Gouveia et al., 2020a) was applied to identify taxonomies. The raw PSMs for each taxon (hereafter TSMs), the numbers of matching peptide sequences, specific peptide sequences and corresponding specific PSMs were determined for the species, genus, family, order, class, phylum and superkingdom ‘canonical’ taxonomical levels. Acceptance criteria for genera selection were adjusted for each individual dataset. Genera validated in the first‐round search, and all their descendants, were extracted from the full NCBInr database for a second Mascot search. All acquired spectra were used at this stage, and the Mascot settings were the same as for the first‐round search, except that the maximum number of missed cleavages was set to 2, and 5 ppm mass tolerance was allowed on the parent ion. Proteins validated with a Mascot p‐value of 0.05 during the second‐round search were extracted from NCBInr, and added to a selection of proteins specific to SARS‐CoV‐2, as detailed below, for a third‐round search. SARS‐CoV‐2 viral genomic sequences were downloaded (2020/05/11) from the GISAID database (Elbe and Buckland‐Merrett, 2017), and proteins were extracted after genome synchronization against the reference genome from the corresponding GFF file GCA_009858895.3_ASM985889v3_genomic.gff. Both these resources were downloaded from ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/009/858/895/GCA_009858895.3_ASM985889v3/. The 186 non‐redundant sequences from the N protein were added to the FASTA file used for the third‐round search. This final Mascot search was performed using the same spectra and settings as those used in the second‐round search. Peptides and proteins were identified with an FDR of 0.01 calculated from the relevant decoy database search. PSMs validated with a Mascot p‐value of 0.05 during the second‐round search were filtered using an FDR < 1% and subsequently used to infer peptide and protein identifications. Proteins were grouped if they shared at least one peptide. Label‐free quantification was performed based on PSM counts for each protein, applying the principle of parsimony. For each phylum, spectral counts for all the proteins in a group were summed to assign abundance values to each protein group. Proteins were KEGG‐annotated using the GhostKoala web service (https://www.kegg.jp/ghostkoala/) to match proteins to KO and KEGG pathway maps. The spectrum count values for peptides mapped to a KEGG through protein mapping were summed to attribute an abundance value to each functional term. Peptide‐to‐taxon mapping was also performed to allow taxon‐resolved functional quantification.
Statistical analyses
Count values from both taxonomic (number of TSMs) and functional data (spectrum counts) were scaled relative to their sum total in the sample. The categorical and continuous variables were compared among the patients using the Fisher exact test and the Kruskal–Wallis test respectively. PCA was performed as previously described (Gouveia et al., 2020a). The R package metacoder (Foster et al., 2017) was used to represent taxonomic abundance as a differential heat tree. Univariate differential analysis of taxon and KO abundances between conditions was performed by applying non‐parametric Wilcoxon tests corrected for multiple comparisons (Benjamini–Hochberg adjustment). Pairwise alpha‐diversity indices were calculated using the vegdist function from the vegan package in R.
Authors' Contributions
L.G., O.P., C.D.‐R., A.S., J.‐P.L. and J.A. conceived the project. L.G. and J.A. designed the overall experimental approach and analysed the data with O.P. L.G. drafted the manuscript with inputs from J.A. and other authors. G.M. performed the mass spectrometry measurements under the supervision of L.G. and J.A. S.R. and F.G. performed the RT‐qPCR. M.‐A.R. and L.G. performed sample inactivation and extraction. O.P. and K.C. contributed to data interpretation scripts. P.‐G.C., C.D.‐R., D.L., A.S. and J.‐P.L. were in charge of patient recruitment and diagnostics. All authors read and approved the final manuscript.
Supporting information
Fig. S1. Number of proteins identified per sample and relative abundance of broad taxonomic categories.
Fig. S2. Gut microbiota metaproteome analysis. PCA plot of the microbiota profiles based on results of nasopharyngeal PCR results (S2A), clinical symptoms (S2B) and antibiotic treatments (S2C). The letter in legend of Fig. S2C correspond to the following antibiotic treatments: (A) Amoxicillin/clavulanate, (B) 3rd gen cephalosporin plus metronidazole, (C) Vancomycin plus Cephazolin followed by Linezolid, (D) Ceftriaxone, (E) Trimethoprim/Sulfamethoxazole, (F) Amikin plus Imipenem/Cilastatin, (G) Cefotaxime plus Spiramycin, (H) Cefotaxime plus Daptomycin. Each point corresponds to one sample, represented as the sum of the contribution of its three corresponding replicates.
Fig. S3. Impact of highly abundant human peptides identified in samples positive for SARS‐CoV‐2 on microbial taxa relative quantification. Dot plot of the log2 median ratio and p‐value resulting from the comparative analysis performed before (A) and after (B) data transformation. In B, only on the most abundant microbial peptides identified in samples positive for SARS‐CoV‐2 were considered.
Fig. S4. Comparative analysis of the microbial functional profiles. Heat map of the relative abundance of the microbial‐related KOs in each one of the samples positive for SARS‐CoV‐2 (P01, P41, P03 and P29) and in the controls (P10, P25, P27 and P31) considered for the analysis described in Fig. 4B and C.
Table S1. Taxa identified and corresponding abundance based on the TSMs assigned at various taxonomical levels (superkingdom (Table S1a), phylum (Table S1b), class (Table S1c), order (Table S1d), family (Table S1e), and genus (Table S1f)).
Table S2. List of functions assigned grouped into 887 KO (KEGG Orthology) entries (Table S2a) and KO comparison between high viral load versus negative samples (Table S2b) and high versus low viral load samples (Table S2c).
Acknowledgements
We thank the Nîmes University hospital for its structural, human and financial support through the award obtained by our team during the internal call for tenders ‘Thématiques phares’. C.D.‐R., D.L., A.S. and J.‐P.L. belong to the FHU InCh (Federation Hospitalo Universitaire Infections Chroniques, Aviesan). We also express our gratitude to CEA for its long‐term support for developing the phylopeptidomics methodology, as well as GIS IBISA (‘Infrastructures en Biologie Santé et Agronomie’) and Région Occitanie for their support to the ‘ProGénoMix’ mass spectrometry facility. J.‐P.L. was supported by Montpellier University d'Excellence (MUSE) (MicroCOVID grant 2020).
Data Availability
The mass spectrometry and proteomics datasets acquired on faecal samples are available through the ProteomeXchange Consortium via the PRIDE partner repository (https://www.ebi.ac.uk/pride/), under dataset identifiers PXD024990 and 10.6019/PXD024990.
References
- Ahnach, M. , Zbiri, S. , Nejjari, S. , Ousti, F. , and Elkettani, C. (2020) C‐reactive protein as an early predictor of COVID‐19 severity. J Med Biochem 39: 500–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albrich, W.C. , Ghosh, T.S. , Ahearn‐Ford, S. , Mikaeloff, F. , Lunjani, N. , Forde, B. , et al. (2021) Excessive inflammatory and metabolic responses to acute SARS‐CoV‐2 infection are associated with a distinct gut microbiota composition. bioRxiv. doi: 10.1101/2021.10.26.465865. [DOI] [Google Scholar]
- Bauvois, B. , and Dauzonne, D. (2006) Aminopeptidase‐N/CD13 (EC 3.4.11.2) inhibitors: chemistry, biological evaluations, and therapeutic prospects. Med Res Rev 26: 88–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodelon, G. , Palomino, C. , and Fernandez, L.A. (2013) Immunoglobulin domains in Escherichia coli and other enterobacteria: from pathogenesis to applications in antibody technologies. FEMS Microbiol Rev 37: 204–250. [DOI] [PubMed] [Google Scholar]
- Chu, D.K.W. , Pan, Y. , Cheng, S.M.S. , Hui, K.P.Y. , Krishnan, P. , Liu, Y. , et al. (2020) Molecular diagnosis of a novel coronavirus (2019‐nCoV) causing an outbreak of pneumonia. Clin Chem 66: 549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbe, S. , and Buckland‐Merrett, G. (2017) Data, disease and diplomacy: GISAID's innovative contribution to global health. Glob Chall 1: 33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster, Z.S. , Sharpton, T.J. , and Grunwald, N.J. (2017) Metacoder: an R package for visualization and manipulation of community taxonomic diversity data. PLoS Comput Biol 13: e1005404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geva‐Zatorsky, N. , Alvarez, D. , Hudak, J.E. , Reading, N.C. , Erturk‐Hasdemir, D. , Dasgupta, S. , et al. (2015) In vivo imaging and tracking of host‐microbiota interactions via metabolic labeling of gut anaerobic bacteria. Nat Med 21: 1091–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghanemi, A. , Yoshioka, M. , and St‐Amand, J. (2020) Trefoil factor family member 2 (TFF2) as an inflammatory‐induced and anti‐inflammatory tissue repair factor. Animals (Basel) 10: 1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong, J. , Dong, H. , Xia, Q.S. , Huang, Z.Y. , Wang, D.K. , Zhao, Y. , et al. (2020) Correlation analysis between disease severity and inflammation‐related parameters in patients with COVID‐19: a retrospective study. BMC Infect Dis 20: 963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouveia, D. , Grenga, L. , Pible, O. , and Armengaud, J. (2020a) Quick microbial molecular phenotyping by differential shotgun proteomics. Environ Microbiol 22: 2996–3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouveia, D. , Miotello, G. , Gallais, F. , Gaillard, J.C. , Debroas, S. , Bellanger, L. , et al. (2020b) Proteotyping SARS‐CoV‐2 virus from nasopharyngeal swabs: a proof‐of‐concept focused on a 3 min mass spectrometry window. J Proteome Res 19: 4407–4416. [DOI] [PubMed] [Google Scholar]
- Grenga, L. , and Armengaud, J. (2021) Proteomics in the COVID‐19 battlefield: first semester check‐up. Proteomics 21: e2000198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grenga, L. , Gallais, F. , Pible, O. , Gaillard, J.C. , Gouveia, D. , Batina, H. , et al. (2020) Shotgun proteomics analysis of SARS‐CoV‐2‐infected cells and how it can optimize whole viral particle antigen production for vaccines. Emerg Microbes Infect 9: 1712–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan, W.J. , Ni, Z.Y. , Hu, Y. , Liang, W.H. , Ou, C.Q. , He, J.X. , et al. (2020) Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med 382: 1708–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, M. , Tao, W. , Flavell, R.A. , and Zhu, S. (2021) Potential intestinal infection and faecal‐oral transmission of SARS‐CoV‐2. Nat Rev Gastroenterol Hepatol 18: 269–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann, E.M. , Allain, F. , Gaillard, J.C. , Pible, O. , and Armengaud, J. (2014) Taking the shortcut for high‐throughput shotgun proteomic analysis of bacteria. Methods Mol Biol 1197: 275–285. [DOI] [PubMed] [Google Scholar]
- Hayoun, K. , Gouveia, D. , Grenga, L. , Pible, O. , Armengaud, J. , and Alpha‐Bazin, B. (2019) Evaluation of sample preparation methods for fast proteotyping of microorganisms by tandem mass spectrometry. Front Microbiol 10: 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz, L.X. , Baumann, C.L. , Koberlin, M.S. , Snijder, B. , Gawish, R. , Shui, G. , et al. (2015) The lipid‐modifying enzyme SMPDL3B negatively regulates innate immunity. Cell Rep 11: 1919–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindson, J. (2020) COVID‐19: faecal‐oral transmission? Nat Rev Gastroenterol Hepatol 17: 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihling, C. , Tanzler, D. , Hagemann, S. , Kehlen, A. , Huttelmaier, S. , Arlt, C. , and Sinz, A. (2020) Mass spectrometric identification of SARS‐CoV‐2 proteins from gargle solution samples of COVID‐19 patients. J Proteome Res 19: 4389–4392. [DOI] [PubMed] [Google Scholar]
- Kalantar‐Zadeh, K. , Ward, S.A. , Kalantar‐Zadeh, K. , and El‐Omar, E.M. (2020) Considering the effects of microbiome and diet on SARS‐CoV‐2 infection: nanotechnology roles. ACS Nano 14: 5179–5182. [DOI] [PubMed] [Google Scholar]
- Kim, Y.G. , Udayanga, K.G. , Totsuka, N. , Weinberg, J.B. , Nunez, G. , and Shibuya, A. (2014) Gut dysbiosis promotes M2 macrophage polarization and allergic airway inflammation via fungi‐induced PGE(2). Cell Host Microbe 15: 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein, G. , Mathe, C. , Biola‐Clier, M. , Devineau, S. , Drouineau, E. , Hatem, E. , et al. (2016) RNA‐binding proteins are a major target of silica nanoparticles in cell extracts. Nanotoxicology 10: 1555–1564. [DOI] [PubMed] [Google Scholar]
- Lai, L.A. , Tong, Z. , Chen, R. , and Pan, S. (2019) Metaproteomics study of the gut microbiome. Methods Mol Biol 1871: 123–132. [DOI] [PubMed] [Google Scholar]
- Lalles, J.P. (2014) Intestinal alkaline phosphatase: novel functions and protective effects. Nutr Rev 72: 82–94. [DOI] [PubMed] [Google Scholar]
- Liu, Q. , Mak, J.W.Y. , Su, Q. , Yeoh, Y.K. , Lui, G.C.‐Y. , Ng, S.S.S. , et al. (2022) Gut microbiota dynamics in a prospective cohort of patients with post‐acute COVID‐19 syndrome. Gut 71: 554–552. [DOI] [PubMed] [Google Scholar]
- Long, S. , Yang, Y. , Shen, C. , Wang, Y. , Deng, A. , Qin, Q. , and Qiao, L. (2020) Metaproteomics characterizes human gut microbiome function in colorectal cancer. NPJ Biofilms Microbiomes 6: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv, L. , Jiang, H. , Chen, Y. , Gu, S. , Xia, J. , Zhang, H. , et al. (2021) The faecal metabolome in COVID‐19 patients is altered and associated with clinical features and gut microbes. Anal Chim Acta 1152: 338267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, Z.Q. , Chambers, M.C. , Ham, A.J. , Cheek, K.L. , Whitwell, C.W. , Aerni, H.R. , et al. (2011) ScanRanker: quality assessment of tandem mass spectra via sequence tagging. J Proteome Res 10: 2896–2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moar, P. , and Tandon, R. (2021) Galectin‐9 as a biomarker of disease severity. Cell Immunol 361: 104287. [DOI] [PubMed] [Google Scholar]
- Nagpal, R. , Mainali, R. , Ahmadi, S. , Wang, S. , Singh, R. , Kavanagh, K. , et al. (2018) Gut microbiome and aging: physiological and mechanistic insights. Nutr Healthy Aging 4: 267–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norton, A. , Olliaro, P. , Sigfrid, L. , Carson, G. , Hastie, C. , Kaushic, C. , et al. (2021) Long COVID: tackling a multifaceted condition requires a multidisciplinary approach. Lancet Infect Dis 21: 601–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrillo, M. , Brogna, C. , Cristoni, S. , Querci, M. , Piazza, O. , and Van den Eede, G. (2021) Increase of SARS‐CoV‐2 RNA load in faecal samples prompts for rethinking of SARS‐CoV‐2 biology and COVID‐19 epidemiology. F1000Res 10: 370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rémic . (2018) Rémic: référentiel en microbiologie médicale 6.2. Paris, France: Société Française de Microbiologie (SFM). [Google Scholar]
- Saadi, J. , Oueslati, S. , Bellanger, L. , Gallais, F. , Dortet, L. , Roque‐Afonso, A.M. , et al. (2021) Quantitative assessment of SARS‐CoV‐2 virus in nasopharyngeal swabs stored in transport medium by a straightforward LC‐MS/MS assay targeting nucleocapsid, membrane, and spike proteins. J Proteome Res 20: 1434–1443. [DOI] [PubMed] [Google Scholar]
- Scaldaferri, F. , Ianiro, G. , Privitera, G. , Lopetuso, L.R. , Vetrone, L.M. , Petito, V. , et al. (2020) The thrilling journey of SARS‐CoV‐2 into the intestine: from pathogenesis to future clinical implications. Inflamm Bowel Dis 26: 1306–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal, J.P. , Mak, J.W.Y. , Mullish, B.H. , Alexander, J.L. , Ng, S.C. , and Marchesi, J.R. (2020) The gut microbiome: an under‐recognised contributor to the COVID‐19 pandemic? Therap Adv Gastroenterol 13: 1756284820974914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smilowitz, N.R. , Kunichoff, D. , Garshick, M. , Shah, B. , Pillinger, M. , Hochman, J.S. , and Berger, J.S. (2021) C‐reactive protein and clinical outcomes in patients with COVID‐19. Eur Heart J 42: 2270–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, Y. , Liu, P. , Shi, X.L. , Chu, Y.L. , Zhang, J. , Xia, J. , et al. (2020) SARS‐CoV‐2 induced diarrhoea as onset symptom in patient with COVID‐19. Gut 69: 1143–1144. [DOI] [PubMed] [Google Scholar]
- Towne, C.F. , York, I.A. , Watkin, L.B. , Lazo, J.S. , and Rock, K.L. (2007) Analysis of the role of bleomycin hydrolase in antigen presentation and the generation of CD8 T cell responses. J Immunol 178: 6923–6930. [DOI] [PubMed] [Google Scholar]
- Van Den Bossche, T. , Arntzen, M.Ø. , Becher, D. , Benndorf, D. , Eijsink, V.G.H. , Henry, C. , et al. (2021) The metaproteomics initiative: a coordinated approach for propelling the functional characterization of microbiomes. Microbiome 9: 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Spaendonk, H. , Ceuleers, H. , Witters, L. , Patteet, E. , Joossens, J. , Augustyns, K. , et al. (2017) Regulation of intestinal permeability: the role of proteases. World J Gastroenterol 23: 2106–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villapol, S. (2020) Gastrointestinal symptoms associated with COVID‐19: impact on the gut microbiome. Transl Res 226: 57–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan, Y. , Li, J. , Shen, L. , Zou, Y. , Hou, L. , Zhu, L. , et al. (2020) Enteric involvement in hospitalised patients with COVID‐19 outside Wuhan. Lancet Gastroenterol Hepatol 5: 534–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, D. , Hu, B. , Hu, C. , Zhu, F. , Liu, X. , Zhang, J. , et al. (2020a) Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus‐infected pneumonia in Wuhan, China. JAMA 323: 1061–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, W. , Xu, Y. , Gao, R. , Lu, R. , Han, K. , Wu, G. , and Tan, W. (2020b) Detection of SARS‐CoV‐2 in different types of clinical specimens. JAMA 323: 1843–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Zhou, Y. , Xiao, X. , Zheng, J. , and Zhou, H. (2020c) Metaproteomics: a strategy to study the taxonomy and functionality of the gut microbiota. J Proteomics 219: 103737. [DOI] [PubMed] [Google Scholar]
- Wolfel, R. , Corman, V.M. , Guggemos, W. , Seilmaier, M. , Zange, S. , Muller, M.A. , et al. (2020) Virological assessment of hospitalized patients with COVID‐2019. Nature 581: 465–469. [DOI] [PubMed] [Google Scholar]
- Xiao, F. , Sun, J. , Xu, Y. , Li, F. , Huang, X. , Li, H. , et al. (2020b) Infectious SARS‐CoV‐2 in feces of patient with severe COVID‐19. Emerg Infect Dis 26: 1920–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao, F. , Tang, M. , Zheng, X. , Liu, Y. , Li, X. , and Shan, H. (2020a) Evidence for gastrointestinal infection of SARS‐CoV‐2. Gastroenterology 158: e1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, Y. , Chen, Y. , Ma, M. , He, D. , and Yi, H. (2021) Re‐emergence of coronavirus disease in Chinese cities associated with chilled and frozen food products. J Infect 82: e18–e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing, Y.H. , Ni, W. , Wu, Q. , Li, W.J. , Li, G.J. , Wang, W.D. , et al. (2020) Prolonged viral shedding in feces of pediatric patients with coronavirus disease 2019. J Microbiol Immunol Infect 53: 473–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, Y. , Li, X. , Zhu, B. , Liang, H. , Fang, C. , Gong, Y. , et al. (2020) Characteristics of pediatric SARS‐CoV‐2 infection and potential evidence for persistent fecal viral shedding. Nat Med 26: 502–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zang, R. , Gomez Castro, M.F. , McCune, B.T. , Zeng, Q. , Rothlauf, P.W. , Sonnek, N.M. , et al. (2020) TMPRSS2 and TMPRSS4 promote SARS‐CoV‐2 infection of human small intestinal enterocytes. Sci Immunol 5: eabc3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, H. , Li, H.B. , Lyu, J.R. , Lei, X.M. , Li, W. , Wu, G. , et al. (2020a) Specific ACE2 expression in small intestinal enterocytes may cause gastrointestinal symptoms and injury after 2019‐nCoV infection. Int J Infect Dis 96: 19–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, W. , Du, R.H. , Li, B. , Zheng, X.S. , Yang, X.L. , Hu, B. , et al. (2020b) Molecular and serological investigation of 2019‐nCoV infected patients: implication of multiple shedding routes. Emerg Microbes Infect 9: 386–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, J. , Li, C. , Zhao, G. , Chu, H. , Wang, D. , Yan, H.H. , et al. (2017) Human intestinal tract serves as an alternative infection route for Middle East respiratory syndrome coronavirus. Sci Adv 3: eaao4966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo, T. , Liu, Q. , Zhang, F. , Lui, G.C. , Tso, E.Y. , Yeoh, Y.K. , et al. (2021) Depicting SARS‐CoV‐2 faecal viral activity in association with gut microbiota composition in patients with COVID‐19. Gut 70: 276–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo, T. , Zhan, H. , Zhang, F. , Liu, Q. , Tso, E.Y.K. , Lui, G.C.Y. , et al. (2020) Alterations in fecal fungal microbiome of patients with COVID‐19 during time of hospitalization until discharge. Gastroenterology 159: e1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Number of proteins identified per sample and relative abundance of broad taxonomic categories.
Fig. S2. Gut microbiota metaproteome analysis. PCA plot of the microbiota profiles based on results of nasopharyngeal PCR results (S2A), clinical symptoms (S2B) and antibiotic treatments (S2C). The letter in legend of Fig. S2C correspond to the following antibiotic treatments: (A) Amoxicillin/clavulanate, (B) 3rd gen cephalosporin plus metronidazole, (C) Vancomycin plus Cephazolin followed by Linezolid, (D) Ceftriaxone, (E) Trimethoprim/Sulfamethoxazole, (F) Amikin plus Imipenem/Cilastatin, (G) Cefotaxime plus Spiramycin, (H) Cefotaxime plus Daptomycin. Each point corresponds to one sample, represented as the sum of the contribution of its three corresponding replicates.
Fig. S3. Impact of highly abundant human peptides identified in samples positive for SARS‐CoV‐2 on microbial taxa relative quantification. Dot plot of the log2 median ratio and p‐value resulting from the comparative analysis performed before (A) and after (B) data transformation. In B, only on the most abundant microbial peptides identified in samples positive for SARS‐CoV‐2 were considered.
Fig. S4. Comparative analysis of the microbial functional profiles. Heat map of the relative abundance of the microbial‐related KOs in each one of the samples positive for SARS‐CoV‐2 (P01, P41, P03 and P29) and in the controls (P10, P25, P27 and P31) considered for the analysis described in Fig. 4B and C.
Table S1. Taxa identified and corresponding abundance based on the TSMs assigned at various taxonomical levels (superkingdom (Table S1a), phylum (Table S1b), class (Table S1c), order (Table S1d), family (Table S1e), and genus (Table S1f)).
Table S2. List of functions assigned grouped into 887 KO (KEGG Orthology) entries (Table S2a) and KO comparison between high viral load versus negative samples (Table S2b) and high versus low viral load samples (Table S2c).
Data Availability Statement
The mass spectrometry and proteomics datasets acquired on faecal samples are available through the ProteomeXchange Consortium via the PRIDE partner repository (https://www.ebi.ac.uk/pride/), under dataset identifiers PXD024990 and 10.6019/PXD024990.