

Abstract
The massive sequencing of severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) and global genomic surveillance strategies allowed the detection of many variants of concern and interest. The variant of interest Lambda (C.37), which originated in South America, has been the most prevalent in Peru and Chile, but its dispersion in other continents still remains unknown. The current study aims to determine the phylogenetic relationship among C.37 isolates worldwide, focusing on spike mutations to understand the spread of Lambda in pandemics. A total of 7441 sequences identified as C.37 were downloaded from the GISAID database; local analysis was carried out to identify spike mutations and phylogenetic analysis was carried out to determine the rate of spread of the virus. Our results showed some spike mutations of Lambda that allowed us to detect small local outbreaks in different countries that occurred in the past and identify several clades that have not yet been designated. Although the lineage C.37 is not epidemiologically relevant in Europe or North America, the endemic behavior of this variant in Peru had a major impact on the second SARS‐CoV‐2 wave.
Keywords: genomic surveillance, lambda, local outbreaks., SARS‐CoV‐2, Spike mutations
1. INTRODUCTION
Genomic surveillance using next‐generation sequencing (NGS) is the gold standard tool for tracking the severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) pandemic in real time. Currently, more than six million genomes have been submitted to the GISAID database (https://www.gisaid.org/); nevertheless, the genome quantity reported by each country is very different. United States, Iceland, the Netherlands, the United Kingdom, and Australia are countries with massive genomic records, mainly due to better support in terms of sequencing technology, logistically, and financially. 1 At the beginning of 2021, the global genomic surveillance of SARS‐CoV‐2 allowed the identification of lineages thereafter considered as viral variants by WHO; these variants were the result of dispersion, geographically constrained, and natural mutations of the virus. 2 In this sense, currently, there are five lineages that are most important or called a variant of concern (VOC): the variant B.1.1.7 (Alpha), B.1.351 (Beta), P.1 (Gamma), B.1.617.2 (Delta), and the recently designed B.1.1.529 (Omicron). In addition, two variants of interest (VOI), C.37 (Lambda) and B.1.621 (Mu), are still under surveillance.
The C.37 lineage of SARS‐CoV‐2 was termed Lambda by the World Health Organization and considered as a VOI. In Peru, the emergence of Lambda was estimated to occur around October 2020. 3 C.37 was initially described to present seven nonsynonymous mutations in the S gene (Δ246–252, G75V, T76I, L452Q, F490S, D614G, T859N) and a deletion in the ORF1a gene (Δ3675–3677), similar to 19 other mutations. 4 Two mutations (L452Q, F490S) are present in the receptor‐binding protein, and one of them (F490S) has shown reduced susceptibility to antibody neutralization. 5 Although it is only categorized as a variant of interest, many countries with more efficient public health systems were concerned about its transmission and infectivity due to the rapid increase in the number of cases in South American countries. One study established that the Lambda (C.37) variant has more infectivity and reduced susceptibility to neutralization in comparison with other variants like Alfa (B.1.1.7) and Gamma (P1). 6 Lambda mainly spread in Peru, Chile, and Argentina. Other countries in the region like Ecuador, Colombia, Brazil, Venezuela, and Bolivia detected only a minimal number of cases, possibly due to greater spread of other variants like Gamma, Alpha, and Mu.
The rapid dispersion of lambda through South America has been well documented. Nevertheless, knowledge of its dispersion in other continents is still unclear. The description of spike mutations in terms of their spread and outbreaks in other countries could be important to understand the global behavior of pandemics. Here, the main focus of the research is to discuss the diversity and spread of the lambda variant (C.37) over time, focusing on genomic spike mutations and distribution in the early stages.
2. MATERIALS AND METHODS
Data of the genomic surveillance project used in the present study were collected in Peru from March 2020 to September 2021. A total of 9877 samples were collected gradually from all departments of the country and sent to the National Institute of Health in Peru. A random selection of samples with C t < 30 was used for RNA isolation using the Quick‐DNA/RNA Viral MagBead Kit (Zymo Research) in the automated platform Opentrons OT‐2. The library preparation was performed using the Illumina COVIDSeq Kit and sequenced in the NextSeq. 550 (Illumina®) according to the manufacturer's instructions.
The quality of reads was assessed and removal of reads contamination was performed using FastQC v0.11.9 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and Kraken2 v2.0.8, respectively. 7 The filter reads were mapped to the NCBI reference sequence (NC_045512) isolated from Wuhan using the software package BWA v.0.7.17. 8 The consensus sequence was obtained using Samtools v.1.9 and IVAR v.1. 9 , 10 Finally, the annotation of consensus sequences was performed using NextClade (https://clades.nextstrain.org/), and lineage designation was defined using the software Pangolin v.3.1.2 (https://pangolin.cog-uk.io/).
For rapid detection and visualization of new mutations related to spike protein, a Python script was developed (GitHub). A maximum‐likelihood phylogenetic analysis was conducted using IQ‐TREE software with 30 independent searches and 1000‐Lart replicates to assess for node support. 11 Data selected for phylogeny were composed of the reference sequence Wuhan‐Hu‐1 (NC_045512) and all the sequences available in the GISAID database (https://www.gisaid.org/) until September 26, 2021 corresponding to the C.37 Pango lineage. In our research, viral genomes with poor‐quality sequences were not included in the downstream analysis. Data were filtered based on the N number (<10%) and a minimum length of 28 000 bp.
3. RESULTS
We collected 7374 genomes from the GISAID database, filtering 170 genomes according to the parameters described previously. Here, we describe the results of analysis of a total of 7204 complete genomes of the Lambda variant from a wide variety of geographical sites. We found that South American countries (3054 Peru and 1607 Chile) and North American (1081, United States) countries have the highest numbers of records. Of the total Peruvian sequences of C.37, 2791 were identified at our institution. In contrast, European countries have reported lower cases (Spain, Germany, and France), and Oceania, Africa, and Asia countries have shared only a few reports (Figure 1). Epidemiological data available on the GISAID database indicate that the first genome of Lambda was reported in South America and collected on November 8, 2020 from Argentina (EPI_ISL_2158693). Afterward, Peru and Chile reported cases of the first SARS‐CoV‐2 genome sequence of the Lambda variant in April 2021.
Figure 1.

Global distribution of the SARS‐CoV‐2 Lambda variant. Countries with a greater number of cases are represented in color: Peru and France (red); Chile and Italy (blue); Argentina and Germany (light blue); Ecuador (yellow); Colombia (pink); Mexico and Spain (Orange); United States and United Kingdom (Green); and Switzerland (purple). Other countries are shown in gray. SARS‐CoV2, severe acute respiratory syndrome coronavirus 2.
We found 8027 nucleotide mutations in all genomes analyzed; after applying a filter, we obtained 177 mutations (2.2%) shared by more than 40 genomes (Figure 2). These mutations were present in nine genes of the virus: ORF1a (58), ORF1b (21), Spike (30), ORF3a (12), ORF6 (6), ORF7 a (6), ORF8 (9), gen M (6), and gen N (17), and intergenic regions (12). In phylogenetic analysis, we focus on spike mutations and some nucleotide sharing in phylogenetic clades. We detected eight mutations in the spike gene as the most frequent (>6200 genomes) and emerged at the same time: G21786T, C21789T, T22917A, the 22 299–22 319 deletion, T23031C, A23403G, C23731T, and C24138A. Likewise, spike mutations with less frequency are listed in Table 1. The emergence and disappearance of some C.37 mutations over time are shown in Figure 2, and it is clear that most of them were widely spread and conserved since the appearance of the Lambda variant. More details of the emergence of new mutations over time are provided in Supporting Information: Data 1 and 2).
Figure 2.
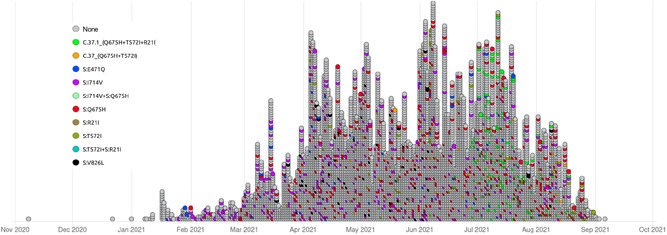
Mutations of the SARS‐CoV‐2 Lambda variant from November (2020) to September (2021) from all samples available on the GISAID database. Each circle represents a sample. SARS‐CoV2, severe acute respiratory syndrome coronavirus 2.
Table 1.
Spike mutation frequency with <6200 genomes
| Mutations | ||
|---|---|---|
| Nucleotide | Amino acidic | Frequency |
| G23587T | Q675H | 596 |
| C21727T | ‐ | 408 |
| A23702G | I714V | 355 |
| A23203G | ‐ | 224 |
| C23277T | T572I | 212 |
| 21 749–21 787 | ‐ | 176 |
| C21575T | L5F | 151 |
| G21777A | S71− | 137 |
| G21624T | R21I | 132 |
| C21691T | ‐ | 98 |
| C21621G | ‐ | 93 |
| G22111A | ‐ | 87 |
| 22 301–22 321 | ‐ | 83 |
| C21676T | ‐ | 70 |
| G22973C | ‐ | 60 |
| A25336C | ‐ | 59 |
| G22346T | ‐ | 54 |
| G24038T | V826L | 54 |
| G22319A | ‐ | 51 |
| G23593T | ‐ | 49 |
| C21614T | ‐ | 43 |
| G22801T | ‐ | 42 |
C.37 phylogenetics revealed that this lineage is widely present in South American samples. Within the phylogenetic tree (Figure 3), we can find the C.37.1 subgroup, the only sublineage described in C.37. Our results show that the C.37.1 sublineage is defined mainly by the presence of North American and European samples, with R21I, T572I, Q675H, and D253N, and other mutations in ORF1ab, ORF1b, ORF3a, and N genes. From the phylogenetic tree, we infer that some emergency mutations could help to delimit new sublineages over the course of time. Among these mutations, we found E471Q, T572I, Q675H, and I714V as possible emerging sublineages.
Figure 3.

(A) The C.37 phylogenetic tree showing the C.37.1 sublinage and potentially new subclades with E471Q, T572I, Q675H, and I714V mutations. (B) Genomic representation highlighting relevant mutations by each gene.
Mutation Q675H was found to be present in many emerging clades in the phylogenetic analysis, the most representative clade is composed of 430 genomes (Figure 3). It is also defined by the presence of P51L and D253N mutations in ORF9b and Spike genes, respectively. Other mutations like E471Q, T572I, and I714V emerged in one clade, each composed of 58, 30, and 355 genomes, respectively. The D253N mutation is of concern due to its constant prevalence. More details about these new possible emerging clades are shown in Figure 3.
Our analysis also found local outbreaks in Peru (Arequipa and Northern Peru), Germany (North Rhine‐Westphalia), and some European countries. All these outbreaks are defined by spike gene mutations (Figure 4).
Figure 4.

Distribution of local outbreaks of the C.37 lineage worldwide. (A) Local outbreak in Northern Peru. (B) Local outbreak in Peru (Arequipa) and Germany (North Rhine‐Westphalia). (C) Outbreak in Europe, emergence of the C.37.1 sublineage. (D) Number of genomes by country.
The proposed and designated C.37.1 sublineage of lambda emerged in June and its dispersion continued up to August 2021. It mainly spread in Europe, in Spain (64%), and Switzerland (16%). Two main outbreaks of Lambda emerged in Peru, one of them occurring in the Northern region (Piura and La Libertad city), delimited by the presence of the T572I mutation, from July to September 2021. Another one emerged in the Southern region (Arequipa), with the Q675H mutation, from April to August 2021. Simultaneously, we found the emergence of another outbreak in the North Rhine‐Westphalia city (Germany) from February to June 2021, with the Q675H mutation predominating.
4. DISCUSSION
Since the first report of the Lambda variant (C.37 lineage) in April 2021, several cases across all continents have been reported. Information related to this VOI transmission and dispersion is still scarce. Nevertheless, it is important to constantly track the new mutations of the Lambda variant that are currently spreading despite the emergence of variants of concern.
The prevalence of Lambda was high in two South American countries: Peru and Chile (>1000 genomes). Nevertheless, lambda variant remains less epidemiological important in other countries like United States compared to high records of Alfa, Gamma, 12 Delta, and the recent introduction of Omicron. 13 Novel reports of the spread of this VOI to Europe 14 led to the creation of a new sublineage called C.37.1, but the spread of cases reported is very slow compared to other VOCs circulating in Europe. 15 In this sense, the emergence of the Lambda variant was probably endemic and represented the principal lineage prevalent during the second wave in Peru. 3
Mutations detected in the Lambda variant show high individual diversity, but there are 26 shared mutations among all populations of the virus located in the genes Spike, ORF1a, ORF1b, and N, and the intergenic region. The main mutations that characterize the Lambda Spike gene are G75V, T76I, R246N, a deletion of seven amino acids SYLTPGD in positions 247–253, L452Q, F490S, D614G, and T859N, 3 but we found possible additional emerging mutations such as Q675H, V826L, I714V, L5F, R21I, T572I, and S71del. The mutation Q675H appears in two clades: the first clade includes the C.37.1 sublineage with additional spike mutations R21I and T572I. The C.37.1 sublineage is currently spreading in Europe and America (82 Spain, 59 USA, 20 Switzerland, 17 Germany, 8 Denmark, 7 Netherlands, 7 Belgium, 5 Dominican Republic, 4 Ireland, 3 Sweden, 3 Italy, 2 France, 1 Portugal, 1 Norway, 1 the United Kingdom, 1 Mexico, and 1 Aruba). Nevertheless, our phylogenetic analyses reveal that Q675H is not responsible for the monophyletic clade; the spike mutations R21I and T572I cluster into a real monophyletic clade. In a sense, we suggest withdrawing the mutation Q675H as a biomarker to classify C.37.1 sublineage samples. The second clade is monophyletic and carries the mutation Q675H in genomes from Germany, Chile, Peru, and the United States. The emergence of the same mutation in different contexts and countries is probably related to gain transmission; genomes with this mutation are currently active in Peru and Chile, and the spread of clade C.37.1 is slow in Spain and Switzerland.
The mutation V826L was associated with outbreaks in Europe and North America; phylogenetic relations are very close to the sublineage C.37.1. The T572I mutation that was found in La Libertad city as a local outbreak (Figure 4) was previously demonstrated to cause the shift of the coiled region to a helix. 16 Nowadays, genomes with V826L and some Q675H are misclassified as C.37.1 using the Pango algorithm (https://cov-lineages.org/). Therefore, we consider that it is crucial to reanalyze and focus on local spike diversity due to possible limitations present in the PANGO lineage related to identifying clades or convergent occurrence of mutations. 17
In conclusion, by tracking the emergence of spike mutations of Lambda, it is possible to detect past outbreaks in different countries. Although the C.37 lineage is not currently epidemiologically relevant in Europe or North America, due to the endemic behavior of this variant in South America (Peru and Chile), it became the most prevalent from February to July (2021) in the second wave. It is essential to continue local genomic surveillance of Lambda spike mutations, especially considering the late emergence of the Omicron variant in South America.
AUTHOR CONTRIBUTIONS
Orson Mestanza and Wendy Lizarraga designed and performed all experiments. Víctor Jimenez‐Vasquez, Verónica Hurtado, Iris S. Molina, Luis Barcena, Steve Acedo, Alicia Nuñez, Sara Gordillo, Nieves Sevilla, and Princesa Medrano collaborated in the data collection, RNA extraction, library preparation, and sequencing. Carlos Padilla‐Rojas, Henri Bailon, Omar Cáceres, Marco Galarza, Nancy Rojas‐Serrano, Natalia Vargas‐Herrera, Priscila Lope‐Pari, Joseph Huayra, Roger V. Araujo‐Castillo, and Lely Solari designed and managed the genomic surveillance project. All authors read and approved the final manuscript.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
Supplementary information.
ACKNOWLEDGMENTS
We are grateful to the COVID‐19 diagnostic team of the National Institute of Health in Peru, laboratory staff, and many international research groups of COVID‐19 who have been contributing to this study by sharing genomic sequences in a free access database. This study was financially supported by the Genomic Surveillance Project of SARS‐CoV‐2 of the National Institute of Health in Peru.
Mestanza O, Lizarraga W, Padilla‐Rojas C, et al. Genomic surveillance of the Lambda SARS‐CoV‐2 variant in a global phylogenetic context. J Med Virol. 2022;94:4689‐4695. 10.1002/jmv.27889
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available in the GISAID database (EpiCoV™) at https://www.gisaid.org/.
REFERENCES
- 1. Furuse Y. Genomic sequencing effort for SARS‐CoV‐2 by country during the pandemic. Int J Infect Dis. 2021;103:305‐307. 10.1016/j.ijid.2020.12.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Poterico JA, Mestanza O. Genetic variants and source of introduction of SARS‐CoV‐2 in South America. J Med Virol. 2020. 10.1002/jmv.26001 [DOI] [PMC free article] [PubMed]
- 3. Padilla‐Rojas C, Jimenez‐Vasquez V, Hurtado V, et al. Genomic analysis reveals a rapid spread and predominance of lambda (C.37) SARS‐COV‐2 lineage in Peru despite circulation of variants of concern. J Med Virol. 2021. 10.1002/jmv.27261 [DOI] [PMC free article] [PubMed]
- 4. Romero PE, Dávila‐Barclay A, Salvatierra G, et al. The emergence of SARS‐CoV‐2 variant lambda (C.37) in South America [WWW document]. medRxiv. 2021. https://www.medrxiv.org/content/10.1101/2021.06.26.21259487v1 [DOI] [PMC free article] [PubMed]
- 5. Liu Z, VanBlargan LA, Bloyet L‐M, et al. Identification of SARS‐CoV‐2 spike mutations that attenuate monoclonal and serum antibody neutralization. Cell Host Microbe. 2021;29:477‐488 . e4. 10.1016/j.chom.2021.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Acevedo ML, Alonso‐Palomares L, Bustamante A, et al. Infectivity and immune escape of the new SARS‐CoV‐2 variant of interest lambda [WWW document]. medRxiv. 2021. https://www.medrxiv.org/content/10.1101/2021.06.28.21259673v1.
- 7. Wood DE, Lu J, Langmead B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019;20:257. 10.1186/s13059-019-1891-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754‐1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Danecek P, Bonfield JK, Liddle J, et al. Twelve years of SAMtools and BCFtools. Gigascience. 2021;10(2):giab008. 10.1093/gigascience/giab008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Grubaugh ND, Gangavarapu K, Quick J, et al. An amplicon‐based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol. 2019;20:8. 10.1186/s13059-018-1618-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nguyen L‐T, Schmidt HA, von Haeseler A, Minh BQ. IQ‐TREE: a fast and effective stochastic algorithm for estimating maximum‐likelihood phylogenies. Mol Biol Evol. 2015;32:268‐274. 10.1093/molbev/msu300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Paul P, France AM, Aoki Y, et al. Genomic surveillance for SARS‐CoV‐2 variants circulating in the United States, December 2020–May 2021. Morb Mortal Wkly Rep. 2021;70:846‐850. 10.15585/mmwr.mm7023a3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Herlihy R, Bamberg W, Burakoff A, et al. Rapid increase in circulation of the SARS‐CoV‐2 B.1.617.2 (Delta) variant—Mesa County, Colorado, April–June 2021. Morb Mortal Wkly Rep. 2021;70:1084‐1087. 10.15585/mmwr.mm7032e2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Baj A, Novazzi F, Ferrante FD, et al. Introduction of SARS‐COV‐2 C.37 (WHO VOI lambda) from Peru to Italy. J Med Virol. 2021. 10.1002/jmv.27235 [DOI] [PMC free article] [PubMed]
- 15. Mishra S, Mindermann S, Sharma M, et al. Changing composition of SARS‐CoV‐2 lineages and rise of delta variant in england. EClinicalMedicine. 2021;39:101064. 10.1016/j.eclinm.2021.101064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chand GB, Banerjee A, Azad GK. Identification of twenty‐five mutations in surface glycoprotein (Spike) of SARS‐CoV‐2 among Indian isolates and their impact on protein dynamics. Gene Rep. 2020;21:100891. 10.1016/j.genrep.2020.100891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. O'Toole Á, Scher E, Underwood A, et al. Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool. Virus Evol. 2021;7(2):veab064. 10.1093/ve/veab064 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information.
Data Availability Statement
The data that support the findings of this study are available in the GISAID database (EpiCoV™) at https://www.gisaid.org/.