

Abstract
Certain types of cationic lipids have shown promise in cancer immunotherapy, but their mechanism of action is poorly understood. In this study, we describe the properties of an immunotherapeutic consisting of the pure cationic lipid enantiomer R-1,2-dioleoyl-3-trimethyl-ammonium-propane (R-DOTAP) formulated with modified viral or self-peptide Ags. R-DOTAP formulations with peptide Ags stimulate strong cross-presentation and potent CD8 T cell responses associated with a high frequency of polyfunctional CD8 T cells. In a human papillomavirus tumor model system, a single s.c. injection of tumor-bearing mice with R-DOTAP plus human papillomavirus Ags induces complete regression of large tumors associated with an influx of Ag-specific CD8 T cells and a reduction of the ratio of regulatory/Ag-specific CD8 T cells. R-DOTAP also synergizes with an anti-PD1 checkpoint inhibitor, resulting in a significant inhibition of B16 melanoma tumor growth. We found that R-DOTAP stimulates type I IFN production by dendritic cells in vivo and in vitro. s.c. injection of R-DOTAP results in an IFN-dependent increase in draining lymph node size and a concomitant increase in CD69 expression. Using knockout mice, we show that type I IFN is required for the induction of CD8 T cell activity following administration of R-DOTAP plus Ag. This response requires Myd88 but not TRIF or STING. We also show that R-DOTAP stimulates both TLR7 and 9. Collectively, these studies reveal that R-DOTAP stimulates endosomal TLRs, resulting in a Myd88-dependent production of type I IFN. When administered with Ag, this results in potent Ag-specific CD8 T cell responses and antitumor activity.
Immunotherapy provides some of the more durable anticancer responses in advanced cancer patients. The most successful approaches to date include checkpoint blockade using Abs specific for CTLA-4 or programmed cell death protein 1 (PD1), both of which have FDA approval for multiple types of cancers (1, 2). The induction and infiltration of CD4 and CD8 tumor-specific T cells into the tumor microenvironment have also been demonstrated to be critical factors impacting successful treatment of cancers with immunotherapy (3, 4). In studies with checkpoint inhibitors, both PD-L1 expression and the presence of tumor-infiltrating T cells are found to correlate with responses to PD-1 blockade (4–6). The recent success of checkpoint inhibition in cancer immunotherapy relies on the release of inhibitory effects on preexisting tumor-specific CD8 T cells, allowing effective tumor infiltration and direct tumor lysis (4). However, only a subset of cancer patients will have significant numbers of tumor-specific CD8 T cells. Thus, one of the main goals of tumor immunotherapy is to increase the number and activity of tumor-specific CD8 T cells. There are two ways of accomplishing this: adoptive transfer of TCR or chimeric Ag receptor transduced T cells or in vivo priming of T cells against tumor-specific or tumor-associated Ags using immunotherapeutic vaccination. There are a variety of approaches toward Ag-specific tumor immunotherapy, each with advantages and drawbacks (7). A common goal among in vivo CD8 T cell priming approaches is to maximize cross-presentation of Ag and to deliver costimulation and cytokine signals optimized for differentiation of CD8 T cells into effector cells. Although viral vectors or nucleic acid vectors can accomplish this task, there are several limitations. Viral vectors can evoke an immune response, which limits viral gene expression in secondary administrations. Nucleic acid vectors require mechanisms for intracellular delivery (e.g., electroporation) and may not be specific in their targeting to the relevant cell types (e.g., APC). Finally, viral or nucleic acid vectors pose a risk of transforming recipient cells because of integration of vaccine DNA into recipient cell chromosomal DNA. An ideal in vivo CD8 T cell–activating immunotherapy will have a high safety profile, promote the delivery of high levels of CD8 peptide epitopes to APCs, and evoke a cytokine response known to enhance the development of effector CD8 T cells.
One very promising approach in the development of effective Ag-specific CD8 T cell immunotherapies uses nanoparticles composed of certain structurally specific cationic lipids (CLs). First described in the 1980s, synthetic CLs mediate efficient cellular uptake of nucleic acids and have been used extensively as transfection reagents (8). CLs also mediate efficient uptake of proteins (9). The mechanism of cellular uptake of nucleic acids has been extensively studied and involves 1) binding of anionic nucleic acid to CL-based particles (liposomes), 2) binding of the CL–nucleic acid liposomes to cell membranes and rapid uptake into endosomes, and 3) endosomal destabilization and delivery of cargo to the cytoplasm. Cationic liposomes bind nonspecifically to negatively charged cell membranes and appear to aggregate on cell membranes (10). Following binding, CL endocytosis occurs via a clathrin- but not caveolae-mediated mechanism (11–13). Following uptake, fusion occurs with the endosomal membrane, resulting in destabilization and delivery of cargo into the cytoplasm (14). Uptake and transport is an active process that is inhibited by ATP depletion, cytochalasin D, or 4°C (10, 15). The ability of CL liposomes to transport cargo into the cytoplasm results in strong cross-presentation of protein Ags into the MHC class I pathway (16–19). As a result of the unique delivery properties of CLs, a number of groups report promising results using CLs to deliver various protein Ags, including glycoprotein B of HSV (16), carcinoembryonic tumor Ag (20), soluble Leishmania Ags (21), influenza (22, 23), and human papillomavirus (HPV) Ags (24).
CLs represent a diverse group of molecules with different polar head groups and different lengths of carbon chains. In addition to the ability of CLs to promote cross-presentation, various CLs possess intrinsic immunostimulatory properties. Some CLs stimulate TLR2, TLR4, and NF-κB–dependent cytokines or NLRP3 inflammasomes and IL-1 production, depending on the nature of the polar head group and length of the carbon chain (25–28). The CL 1,2-dioleoyl-3-trimethyl-ammonium-propane (DOTAP) induces dendritic cell (DC) maturation and chemokine expression but does not appear to activate a classical NF-κB response, and no TLR stimulatory activity has been reported (29, 30). Instead, it has been suggested that DOTAP activates a unique response in DC involving reactive oxygen species induction and PI3 kinase/ERK pathways (29, 31). With respect to use in therapeutic T cell–activating immunotherapy, DOTAP formulated with long HPV peptides stimulates strong CD8 T cell responses and antitumor activity in a mouse model for HPV-induced cancer. Importantly, a single s.c. injection of tumor-bearing mice with a DOTAP–HPV peptide formulation consistently results in complete eradication of a TC-1 tumor expressing HPV E6 and E7 oncogenes (24, 32). DOTAP is a racemic mixture of two enantiomers, R and S. When the individual pure R- and S-enantiomers were independently synthesized, the vast majority of antitumor activity was observed in the product formulated with R-DOTAP and Ag, whereas formulations based on S-DOTAP had limited activity (33).
Despite the strong evidence that CLs such as R-DOTAP can promote a strong CD8 T cell response to protein and peptide Ags and antitumor activity, R-DOTAP’s mechanism of action as an immunotherapeutic agent has been only partially elucidated. We have prepared nanoparticles based on pure R-DOTAP lipid, named Versamune, and sought to better define its CD8 T cell–stimulatory ability by formulating the nanoparticles with a variety of short and long peptide Ags. Our results reveal that R-DOTAP is a potent activator of CD8 T cell responses to diverse protein and peptide Ags and stimulates robust antitumor immunity to large tumors when used as an Ag-specific immunotherapy. We show that R-DOTAP itself stimulates the type I IFN pathway and that both type I IFN responsiveness and Myd88, but not STING or TRIF, are required for the immunostimulatory activity of R-DOTAP. We also found that R-DOTAP stimulates endosomal TLR7 and TLR9, suggesting that endosomal TLR stimulation through Myd88 is the primary mechanism of type I IFN induction.
Materials and Methods
Animals
Six- to twelve-week-old C57BL6/J mice (B6 mice), B6.Cg-Tg(HLA-A/H2-D)2Enge/J transgenic breeder mice expressing a human HLA-A2 gene (AAD mice), and B6.129P2(SJL)-Myd88tm1.1defr/J (Myd88−/−), B6.129S2-Ifnar1tm1Agt/Mmjax (IFNAR−/−), C57BL/6J-Tmem173gt/J (STING−/−), C57BL/6J-Ticam1Lps2/J (Trif−/−), and C57BL/6-Tg(TcraTcrb)1100Mjb (OT1) breeder mice were obtained from The Jackson Laboratory (Bar Harbor, ME). All animals were housed and bred under specific pathogen–free conditions at the Division of Laboratory Animal Resources, University of Kentucky (UK) Medical Center, and all animal protocols were reviewed and approved by the UK Institutional Animal Care and Use Committee following the National Institutes of Health animal care guidelines.
Peptides and reagents
Current good manufacturing practice–grade (CGMP) R-DOTAP was provided by Merck & Cie (Shaffhausen, Switzerland). CGMP R-DOTAP liposomal nanoparticles were produced by Evonik (Vancouver, Canada) according to protocols described previously (32). CGMP peptide Ags (Table I) were provided by AmbioPharm (North Augusta, SC). Research-grade synthetic peptide Ags were synthesized and purified to >95% purity by GenScript (Piscataway, NJ). Fluorochrome-conjugated mouse monoclonal anti-mouse CD3 (clone: 145-2c11), CD4 (clone: GK1.5), CD8 (clone: YTS165.7.7), NKP36 (clone: 29A1.4), CD11b (clone: M1/70), IFN-γ (clone: XMG1.2), TNF-α (clone: MP6-XT22), and IL-2 (clone: JES6.5H4), were purchased from BioLegend (San Diego, CA). Fluorochrome-conjugated anti-mouse CD69 (clone: H1-2F3) and anti-mouse CD11c (clone: HL3), were purchased from BD Biosciences (San Jose, CA). Fluorochrome-conjugated anti-mouse monoclonal Foxp3 (clone: FJK-16s) and CD25 (clone: PC61.5) were purchased from eBioscience (San Diego, CA). Allophycocyanin-labeled H2-Db E749–57 (RAHYNIVTF) loaded MHC class I dextramers were purchased from Immudex (Fairfax, VA). Anti-mouse PD-1 (clone: RMP1-14) was purchased from Bio X Cell (West Lebanon, NH).
Table I.
Stimulatory peptides and tumor Ags used to evaluate vaccine efficacy
| Ag Identifier | Protein | Peptide Identifier | Sequence |
|---|---|---|---|
| RF9 | HPV-E7 | >E749–57 | RAHYNIVTF |
| KF18 | HPV-E7 | E743–57 | Palmitoyl-KSS-GQEPDRAHYNIVTF |
| V1A | Human MUC1 | MUC1150–158a | YLAPPAHGV |
| V2A | Human MUC1 | MUC1141–149a | YLDTRPAPV |
| C1A | Human MUC1 | MUC11172–1181a | YLAIVYLIAL |
| C2A | Human MUC1 | MUC11177–1186a | YLIALAVCQV |
| SL9 | Chicken OVA | OVA257–264 | SIINFEKL |
| Trp2 | Human tyrosinase related protein 2 | Trp2180–188 | SVYDFFVWL |
Modified sequences of human MUC1 protein. Modifications are shown in bold underlined letters.
Cell lines, tumor implantation, and tumor digestion
The TC-1 tumor cell line, originally produced by Dr. T.C. Wu (Johns Hopkins University, Baltimore, MD) was kindly provided by Dr. W.M. Kast (University of Southern California, Los Angeles, CA) with permission. TC-1 is a C57BL/6 mouse lung epithelial cell line transformed with HPV16 E6 and E7 oncogenes and activated H-Ras (34). TC-1 cells were cultured and maintained in RPMI media supplemented with 10% FBS, 1 mM L-glutamine, 1 mM sodium pyruvate, 1× MEM nonessential amino acids, 50 μM 2-ME, 100 U/ml penicillin, 100 U/ml streptomycin (complete RPMI 1640 [cRPMI] medium), and 0.4 mg/ml G418. B16F10 tumor cell lines were purchased from the American Type Culture Collection and were maintained in DMEM supplemented with 10% FBS, 100 U/ml penicillin, and 100 U/ml streptomycin. The B3Z CD8 T cell hybridoma cell line that recognizes the OVA257–339 peptide presented by H2Kb MHC class I was kindly provided by Dr. N. Shastri (University of California, Berkeley, CA). B16-Blue IFN-α/β cells used for detecting CL-induced type I IFN were purchased from InvivoGen (San Diego, CA), and cell cultures were maintained according to the manufacturer’s protocol. Tumors in mice were established by s.c. injecting 1 × 105 TC-1 or B16F10 tumor cells suspended in 50 μl of 50% Matrigel (Corning). Tumor growth was assessed by measuring the average diameter of the tumor using calipers and calculating the spherical tumor volume. For tumor-infiltrating lymphocyte analysis, s.c. tumors from euthanized mice were isolated, weighed, and digested enzymatically for 60 min at 37°C in a mixture containing 1 mg/ml collagenase (type IV), 0.1 mg/ml hyaluronidase, and 10 mg/ml DNase (type 1) in 0.2-μm filtered cRPMI media (cell dissociation mixture). Digested tumors were passed through a 100-μm cell strainer and washed twice with FACS buffer (1× PBS, 1% FBS, 0.1% NaN3, and 0.5 mM EDTA) to obtain single-cell suspensions for flow cytometer analysis.
R-DOTAP administration and immunotherapy
Mice were anesthetized using isoflurane for all injections and implants. The injection site was shaved and cleaned with 70% ethanol prior to s.c. injection of a formulation. The formulations were produced by mixing an equal volume of R-DOTAP nanoparticles (6 mg/ml in 280 mM sucrose) with the indicated proteins or peptides at 1 mg/ml, unless otherwise stated, immediately prior to treatment. No emulsion was created. For the mucin 1 (MUC1)–R-DOTAP; IFA, IL-12, GM-CSF, and HBV128–140 helper epitope (IFA-Cyt); or Montanide formulations, a proprietary multipeptide mixture of modified MUC1 peptides containing V1A, V2A, C1A, and C2A was used. For the “HPV mix” formulation, a proprietary multipeptide mixture of modified HPV peptides including KF18 and RF9 was used. For positive control or direct comparison, the lipid emulsion-based therapeutic vaccine formulation IFA-Cyt was prepared by emulsifying equal volumes of IFA and Ag–cytokine mixture containing peptide Ag at 1 mg/ml, HBV (128–140) core peptide (100 μg/dose; GenScript), recombinant mouse GM-CSF (5 μg/dose; BioLegend), and recombinant mouse IL-12 (3 μg/mouse; BioLegend) (35). The lipid emulsion vaccine formulation Montanide was prepared by emulsifying equal volumes of antigenic peptides and Montanide ISA-51 VG (Seppic). For tumor regression studies, mice were treated with one or multiple doses (100 μl) of the CL-based formulations delivered s.c. when tumors reached 4–9 mm in diameter. For assessing Ag-specific T cell responses using ELISPOT, mice were injected one or more times with 100 μl of formulation delivered s.c. at 7-d intervals, and the Ag-specific responses were measured 7 d after the last treatment.
In vitro and in vivo assessment of Ag uptake, processing, and endosomal accumulation
Ag uptake studies were performed by incubating bone marrow–derived DC (BMDCs) at 37°C with Alexa 647–conjugated whole OVA (Thermo Fisher Scientific), DQ-OVA (Molecular Probes), or peptide in the presence or absence of R-DOTAP for 10–60 min. Cells were then washed twice and analyzed several ways. Uptake of the fluorescent Alexa 647–OVA and DQ-OVA was measured using flow cytometry. Ag presentation was evaluated by measuring the ability of Ag-pulsed DC to activate the OVA257–264 (SL9)-specific T cell hybridoma B3Z as described (36). In vivo Ag presentation was assessed in an adoptive transfer system. CSFE-labeled spleen cells from the OT1 or DO11.10 TCR transgenic mouse were adoptively transferred into normal C57BL/6 mice at 5 × 106 CD8 T cells per mouse as described (37). After 24 h, adoptive transfer mice were injected with whole OVA alone or OVA plus R-DOTAP. After 3 d, draining lymph node (DLN) CD8 T cells were analyzed by flow cytometry for the extent of CFSE dilution as a measure of cell division.
RNA isolation and nanostring analysis
For gene expression analysis, mice were injected at the nape of the neck, and the draining axillary and brachial lymph nodes (LNs) were isolated and pooled separately from each of four mice. The LNs were enzymatically digested using cell dissociation mixture for 60 min at 37°C, and single-cell suspensions were used to sort CD11c+ cells using a Sony SY3200 cell sorter. Sort-purified CD11c+ cells (2 × 104 cells) were lysed in Qiagen’s RLT cell lysis buffer (Qiagen), and total RNA was isolated using a total RNA isolation kit (Qiagen). Total RNA obtained was sent to the UK genomics core laboratory (Lexington, KY), where total RNA was mixed with nCounter mouse inflammation panel (NanoString Technologies, Seattle, WA), which can measure over 547 genes involved in immune response. Binding of mRNA was detected with the NanoString nCounter analysis system, and raw count data were normalized and statistically analyzed using the SAS program by the UK genomics core laboratory.
IFN and TLR reporter cell assays
For detecting IFN, the supernatants from type I IFN–producing cells were added to B16-Blue IFN-α/β cells in a 96-well plate. After a 24-h incubation, the supernatants were assayed for secreted embryonic alkaline phosphatase (SEAP) activity using QUANTI-Blue reagent (InvivoGen) according to the manufacturer’s instructions. Briefly, 50 μl of cell supernatant containing SEAP was mixed with 150 μl of QUANTI-Blue reagent and incubated for 3–4 h at 37°C, and absorbance was measured at 650 nm using a spectrometer. The concentrations of type I IFN in test samples were quantified using a standard curve generated from recombinant type I IFN-β–treated B16-Blue IFN-μ/β cells in the same assay. For assessing TLR7 or TLR9 activation by CLs, HEK-Blue null, HEK-Blue human TLR7, and HEK-Blue human TLR9 reporter cells (InvivoGen) were stimulated with indicated concentrations of R-DOTAP, PMA (Sigma), R848 (InvivoGen), ODN2006 (InvivoGen), or ODN2395 (InvivoGen) for 24 h. After stimulation, cell supernatants were assayed for TLR and NF-κB–induced SEAP activity using QUANTI-Blue reagent according to the manufacturer’s instruction.
Assessment of Ag-specific T cell responses
Mice received two 100-μl doses of formulations containing antigenic peptides delivered s.c. on day 0 and day 7 on the shaved hind limb flanks. Seven days after the second injection, spleen cells from the euthanized mice were used to detect Ag-specific T cell responses using an IFN-g ELISPOT assay. Processed spleen cells (2.5 × 105) were stimulated for 18–24 h at 37°C with CD8 T cell epitope peptides of interest (stimulatory peptides) or no peptides (control) in a 96-well plate precoated with the mouse IFN-γ capture Ab (Mabtech, Cincinnati, OH). After stimulation, wells were washed with PBS and incubated with biotin-conjugated anti–IFN-γ Ab followed by streptavidin–HRP Ab. To visualize the Ag-specific IFN-gγ–producing cells, wells were incubated for 6 min with TMB substrate, washed with water, and air dried. Spots were scanned and counted using a CTL ImmunoSpot Analyzer and ImmunoSpot Version 4 software (Cellular Technology, Cleveland, OH). Spot counts were summarized as median values from triplicate samples. Each sample had unstimulated and PMA/ionomycin control wells to detect background and a positive control. Wells were considered positive if the spot count exceeded five spots, and Ag-specific response was considered positive if the spot count was 3-fold greater when compared with nonpeptide controls. For intracellular protein analysis, single-cell suspensions of spleen cells were stimulated with the indicated stimulatory antigenic peptide for 6 h at 37°C in cRPMI media supplemented with 1× GolgiStop (BD Biosciences) protein transport inhibitor containing monensin. Following stimulation, cells were washed with FACS buffer and stained with fluorochrome-conjugated anti-mouse CD3, CD4, and CD8 Abs. The cells were then washed, fixed and permeabilized using a fixation/permeabilization kit (BD Biosciences), and stained with flurochrome-conjugated anti-mouse IFN-γ, TNF-α, and IL-2. Cells were washed with FACS buffer after intracellular staining and analyzed immediately using a flow cytometer.
Equipment, software, and statistical analysis
Flow cytometry was performed using a BD LSRII flow cytometer equipped with BD FACSDiva software or BD FACSCaliber or Beckman Coulter’s Cytoflex LX cytometer. All flow data were analyzed using FlowJo version 10.0 software. NanoString data analysis was performed by the UK genomic core laboratory using the SAS program. Statistical analysis for all other studies was performed using GraphPad Prism 7.0 software and comparing means by a simple Student t test or by ANOVA with Tukey multiple comparison correction. The Mantel–Cox test was used for survival curves. A p value <0.05 was considered statistically significant.
Results
R-DOTAP formulations containing multiple tumor-associated peptide Ags induced robust CD8 T cell responses
One of the crucial steps in successful immunotherapy is the induction of robust CTL responses against epitopes derived from tumor-associated Ags. Previous studies demonstrated that enantiomerically specific R-DOTAP CL nanoparticles encapsulated with a single tumor-associated HPV peptide Ag resulted in strong Ag-specific CTL responses and tumor regression (24, 33). In an effort to determine whether this platform would be useful for the formulation with other tumor-associated Ags, we created a formulation containing modified peptides derived from human MUC1. MUC1 is a tumor-associated Ag that is overexpressed in the majority of human carcinomas. The C-terminal portion of MUC1 is also an oncoprotein, and its expression correlates with poor prognosis in a variety of tumor types. Recently, a number of agonist CD8 epitope peptides have been produced that show enhanced recognition by T cells from cancer patients and can induce lysis of human tumor targets in an HLA-restricted manner (38).
To evaluate the therapeutic potential of R-DOTAP formulations for the MUC1 Ags, we prepared a MUC1 peptide mixture containing multiple modified agonist peptides, including the HLA-A2–restricted epitopes V1A, V2A, C1A, and C2A (Table I). To detect HLA-A2–restricted CTL responses, we used HLA-A2 transgenic mice (AAD). Groups of AAD mice were s.c. injected with two doses of R-DOTAP–MUC1 peptide formulation on day 0 and day 7, and the Ag-induced CD8 T cell responses were measured on day 14 using an ELISPOT assay with human HLA-A2–restricted epitopes as stimulatory peptides. As shown in Fig. 1A, R-DOTAP–based formulations induced robust Ag-specific CD8 T cell responses to multiple HLA-A2–restricted epitopes, as indicated by a significant increase in the IFN-γ–producing T cells compared with groups receiving peptide Ags mixed with sucrose buffer.
FIGURE 1.
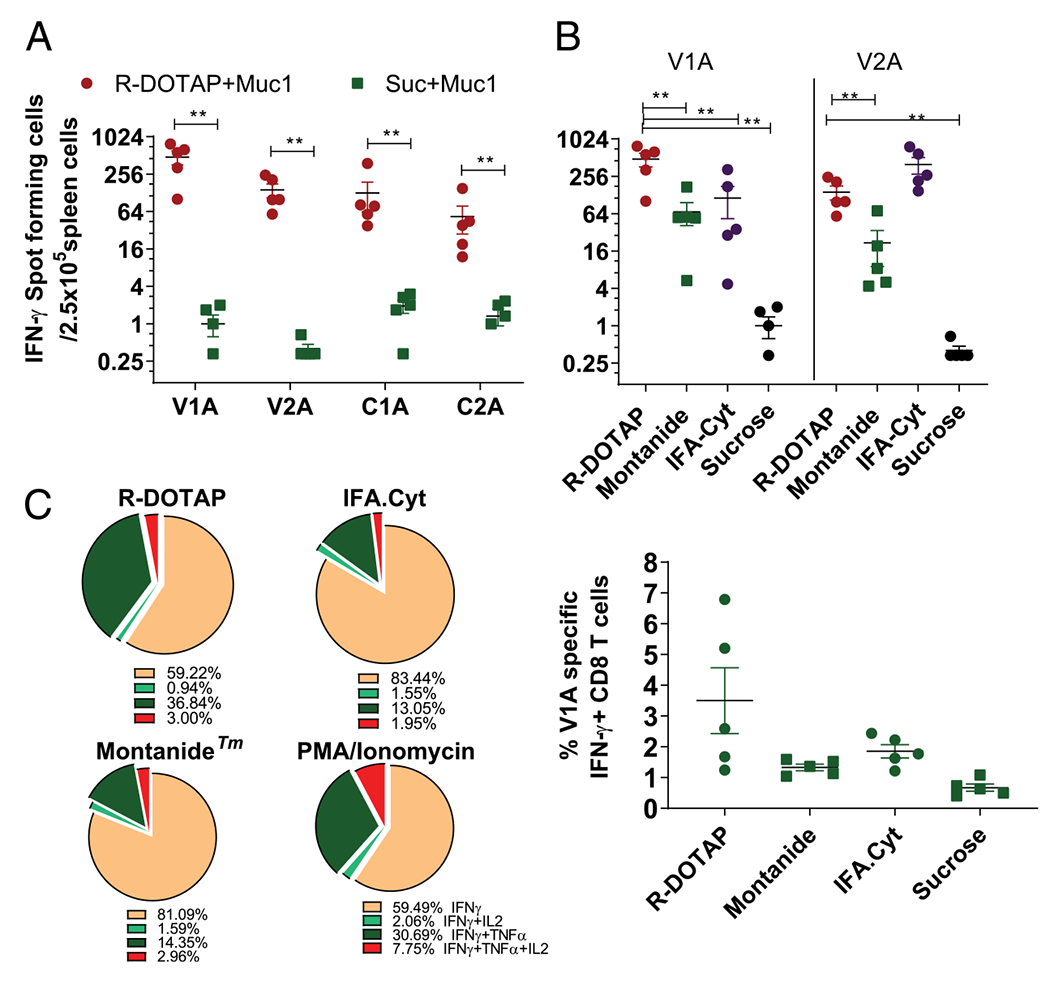
R-DOTAP formulations containing multiple tumor-associated Ags induce quantitatively and qualitatively superior CD8 T cell responses. Groups of AAD mice (n = 6) were s.c. injected with a human MUC1 multipeptide formulation containing the indicated MUC1 CD8 T cell epitopes formulated with R-DOTAP or other adjuvants. Mice were injected on day 0 and day 7, and Ag-specific CD8 T cell responses in spleen were assessed 7 d after the second injection. (A) ELISPOT analysis measuring the number of V1A, V2A, C1A, and C2A specific IFN-γ–producing cells in spleens from mice injected with an R-DOTAP–MUC1 formulation. (B) ELISPOT analysis measuring the number of V1A and V2A specific IFN-γ–producing cells in spleen from mice injected with R-DOTAP, IFA-Cyt, Montanide, or sucrose formulations containing MUC1 peptides. (C) Intracellular cytokine staining of VIA- or PMA/ionomycin–stimulated spleen cells following immunization with the indicated formulations. Graph represents the percentage of IFN-γ–producing cells among total CD8 T cells. Pie charts represent the percentage of cells producing additional cytokines IL-2 and TNF-± among total IFN-γ–producing cells. Data represent the mean ± SEM of n = 5–6 mice in each group. Experiments were repeated at least three times with similar results. **p < 0.05.
To better understand the strength of the immune responses and clinical relevance, head-to-head comparisons were made with adjuvant-based therapeutic vaccine formulations, which have shown promise in preclinical and clinical studies. We first compared the R-DOTAP–MUC1 formulation to two commonly used emulsion-based adjuvants. Montanide is a proprietary emulsion adjuvant frequently used for peptide-based cancer vaccines (39). Another emulsion-based combination adjuvant formulation specifically designed to induce strong in vivo CD8 T cell responses is IFA-Cyt (35). Mice receiving R-DOTAP–MUC1 showed strong responses to both V1A and V2A stimulatory peptides (Fig. 1B). In contrast, IFA-Cyt generated an equivalent strong response only to V2A, and Montanide-induced responses were significantly lower for both V1A and V2A peptides. We further tested the polyfunctionality (ability to produce multiple cytokines) of Ag-specific CD8 T cells by measuring their ability to produce IFN-γ, TNF-α, or IL-2 by intracellular cytokine staining. R-DOTAP–based formulations stimulated significantly higher percentages of polyfunctional Ag-specific CD8 T cells compared with the other two tested emulsion-based lipid formulations (Fig. 1C), indicating that R-DOTAP induced not only a higher number of Ag-specific CD8 T cells in vivo but also qualitatively superior T cells compared with other typical immunotherapy approaches. In separate experiments discussed later, administration of R-DOTAP–based formulations induced stronger CD8 T cell responses as compared with the potent adjuvants CFA or GM-CSF containing the same peptide Ag. Thus, R-DOTAP provides a versatile T cell immunotherapeutic platform capable of formulation with multiple protein and peptide Ags and can induce stronger in vivo CD8 T cell responses than most current immunotherapeutic approaches.
R-DOTAP–based HPV immunotherapy stimulated potent tumor regression and a high level of Ag-specific CD8 T cell infiltration into the tumor
Earlier studies showed that a single injection of DOTAP nanoparticles that contained a short HPV-16 E7 peptide, RF9, given 1 wk after TC-1 tumor implantation, induced complete regression of implanted TC-1 tumors (24). However, a clinically useful Ag-specific immunotherapy would need to contain proteins or multiple long HPV peptides to cover multiple HLA-restricted epitopes. We have prepared such an HPV mix also contains KF18, a long peptide from the HPV-16 E7 protein modified to interact optimally with R-DOTAP as previously described (32). KF18 contains the H-2Db–restricted CD8 epitope RF9 (Table I). In addition, we sought to determine whether the addition of clinically proven cytokines would synergize with R-DOTAP and augment an already strong antitumor response. For this, we chose to examine GM-CSF as a cytokine that has shown promise in multiple preclinical and clinical studies (7). GM-CSF has proven to be very effective at inducing recruitment, maturation, and function of DC (40), properties which might synergize nicely with R-DOTAP.
Groups of mice were injected s.c. with R-DOTAP plus KF18 with or without GM-CSF, and the T cell response to RF9 was evaluated by ELISPOT analysis. Mice treated with the R-DOTAP–KF18 formulation induced an extremely strong IFN-γ ELISPOT response to the RF9 CD8 T cell epitope. This response was not further elevated when GM-CSF was added to the R-DOTAP–based immunotherapy. Mice treated with GM-CSF and KF18 stimulated a modest Ag-specific T cell ELISPOT response, whereas no response was observed with KF18 alone (Fig. 2A). Thus, R-DOTAP was clearly superior to GM-CSF at stimulating a CD8 T cell response to a long HPV peptide, and GM-CSF did not augment the already strong response mediated by R-DOTAP alone.
FIGURE 2.
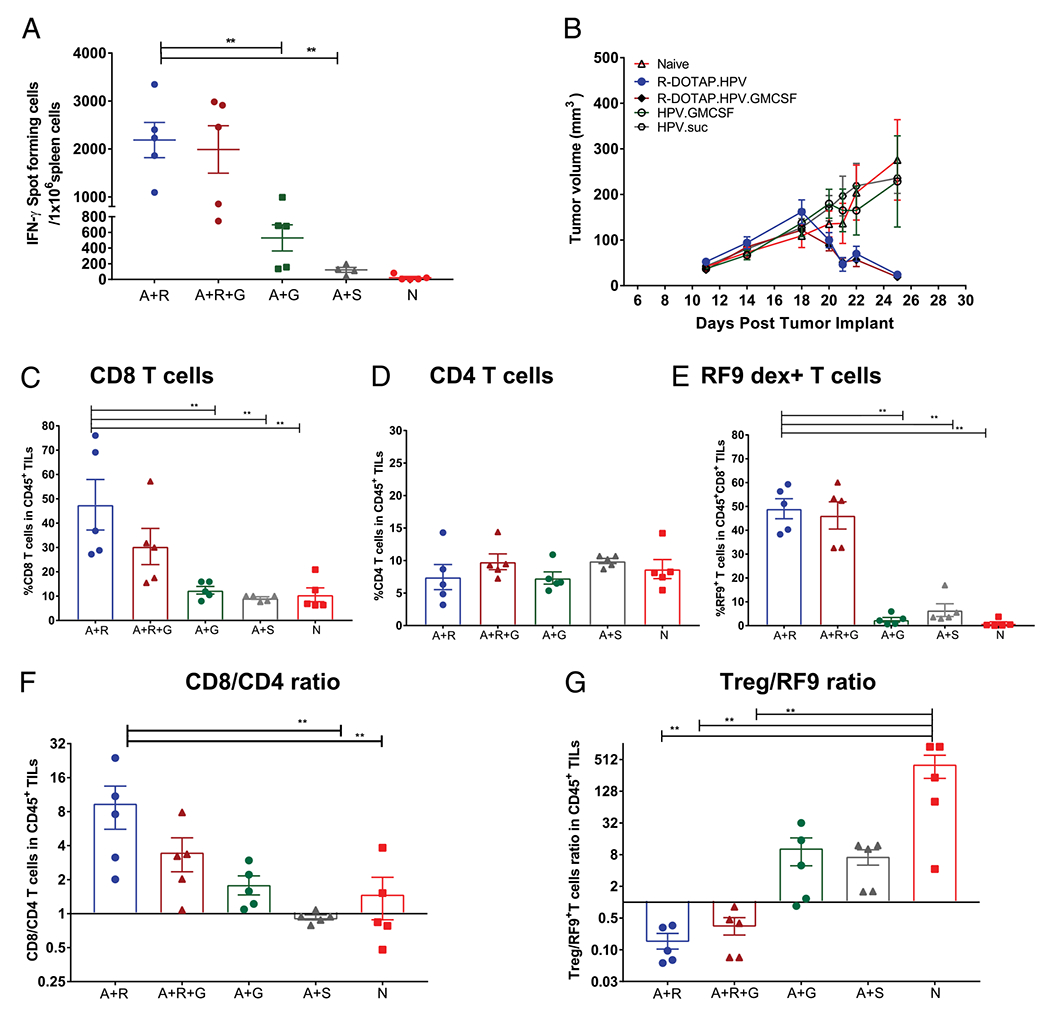
R-DOTAP efficiently alters effector to suppressor T cell ratio, promoting tumor regression. (A) Groups of C57BL/6J mice (n = 5) were s.c. injected with Ag KF18 plus R-DOTAP (A+R), KF18 plus R-DOTAP plus GM-CSF (A+R+G), KF18 plus GM-CSF (A+G), or KF18 plus sucrose (A+S) and compared with naive (N) mice. Mice were boosted on day 7 with the same formulations and analyzed by IFN-γ ELISPOT on day 14 using RF9 as a stimulatory peptide. (B) Mice (n = 10) were implanted s.c. with 1 × 105 TC-1 tumor cells and, when the tumors reached an average diameter of 4–5 mm (day 11), were administered a single dose of formulation containing R-DOTAP mixed with HPV mix (R-DOTAP.HPV), R-DOTAP plus HPV mix plus GM-CSF (R-DOTAP.HPV.GM-CSF), HPV mix plus GM-CSF (HPV.GM-CSF), or HPV mix plus sucrose (HPV.suc), and tumor growth was monitored. (C-G) Parallel groups of tumor-bearing mice (n = 5), treated as in (B), were euthanized 20 d after tumor implant when some groups showed initial signs of regression, and tumors were processed to assess tumor-infiltrating lymphocytes (TILs). Live CD45+CD3+ cells were gated, and the percentages of CD8+ (C), CD4+ (D), and RF9-Db dextramer+ cells among CD8+ TILs (E) were determined. (F) The CD8/CD4 ratio and (G) the T regulatory cell (Treg) (CD3+CD4+Foxp3+CD25+) to RF9-Db dextramer binding (Treg/RF9 ratio) were calculated. Data represent the mean ± SEM from each group (n = 5), and experiments were repeated at least three times with similar results. **p < 0.05.
To better understand the R-DOTAP–induced changes within the tumor microenvironment and to evaluate the potential synergistic effect of GM-CSF in tumor regression, we s.c. injected TC-1 tumor-bearing B6 mice a single time when the tumors measured 4–5 mm. Mice were injected with R-DOTAP plus HPV mix containing KF18, with or without GM-CSF, and the effector and regulatory T cell recruitment to the tumor microenvironment was assessed on day 26. We observed complete tumor regression in the groups injected with R-DOTAP plus HPV mix. Addition of GM-CSF to the R-DOTAP plus HPV mix also resulted in tumor regression with the same kinetics. Thus, there was no obvious effect of GM-CSF addition on tumor regression mediated by R-DOTAP plus HPV mix. In contrast, injection with GM-CSF plus HPV mix alone did not result in tumor regression, indicating that tumor regression was only obtained with formulations containing R-DOTAP. As expected, no tumor regression was observed in naive groups or groups receiving HPV mix alone (Fig. 2B).
To assess the tumor-infiltrating lymphocytes present within the tumor after various treatments, parallel groups of mice were treated identically to those in Fig. 2B, and tumors were removed on day 9 postimmunization, a time at which tumor regression was just apparent. Tumors were enzymatically digested, and cell populations were analyzed by flow cytometry. R-DOTAP plus HPV mix–treated mice showed the highest percentage of CD8 T cells within the tumor, and ~50% of these cells were RF9-specific, whether or not GM-CSF was combined with R-DOTAP (Fig. 2C, 2E). CD4 T cell infiltration into tumors was not statistically different among the different groups, but the CD8/CD4 ratio was significantly greater in the R-DOTAP plus HPV mix–injected group (Fig. 2D, 2F). GM-CSF and Ag or Ag alone did not induce significant CD8 or CD8–RF9–specific T cell infiltration into the tumors (Fig. 2C, 2E). Finally, the T regulatory cell (Treg)/RF9-specific T cell ratio within the tumors was dramatically lower in the R-DOTAP plus HPV mix groups (Fig. 2G). These data collectively indicate that R-DOTAP–based formulations induced a fundamental change in the tumor microenvironment, which correlated with tumor regression, characterized by a high level of CD8 Ag-specific T cell infiltration and a dramatic lowering of Treg/T effector cell ratio. GM-CSF did not enhance the effector cell infiltration into tumors, and we therefore conclude that GM-CSF does not provide any synergistic benefit to R-DOTAP plus HPV under these conditions.
HPV Ags formulated with R-DOTAP cause regression of large, established tumors
To test the ability of R-DOTAP to induce regression of larger tumors, we implanted B6 mice with TC-1 tumor cells s.c. and allowed the tumor to grow to larger sizes (4–9 mm) prior to treatment. We then gave one dose of R-DOTAP plus KF18. A single s.c. injection with R-DOTAP plus KF18 caused complete regression in seven out of eight mice bearing tumors measuring 4–5 mm, six out of eight mice bearing tumors of 5–7 mm, and three out of eight mice bearing tumors of 7–9 mm (Fig. 3A–C). Mice that received R-DOTAP alone or naive mice were unable to effect tumor regression (Fig. 3D, 3E). To assess the memory T cell responses, we rechallenged the mice that had completely regressed tumors with a second dose of TC-1 tumor cells implanted 50 d after the first tumor implant. All mice that had regressed primary tumors also completely prevented the establishment and growth of reintroduced tumors, demonstrating the induction of robust memory T cell responses. The untreated control mice that received tumor cell implants on day 50 showed establishment and progression of tumor (Fig. 3F). These results clearly indicated that R-DOTAP–based formulations were efficient in inducing processing and cross-presentation of a long peptide Ag and inducing the required phenotype of T cells, resulting in robust antitumor responses capable of eradicating large, established tumors.
FIGURE 3.
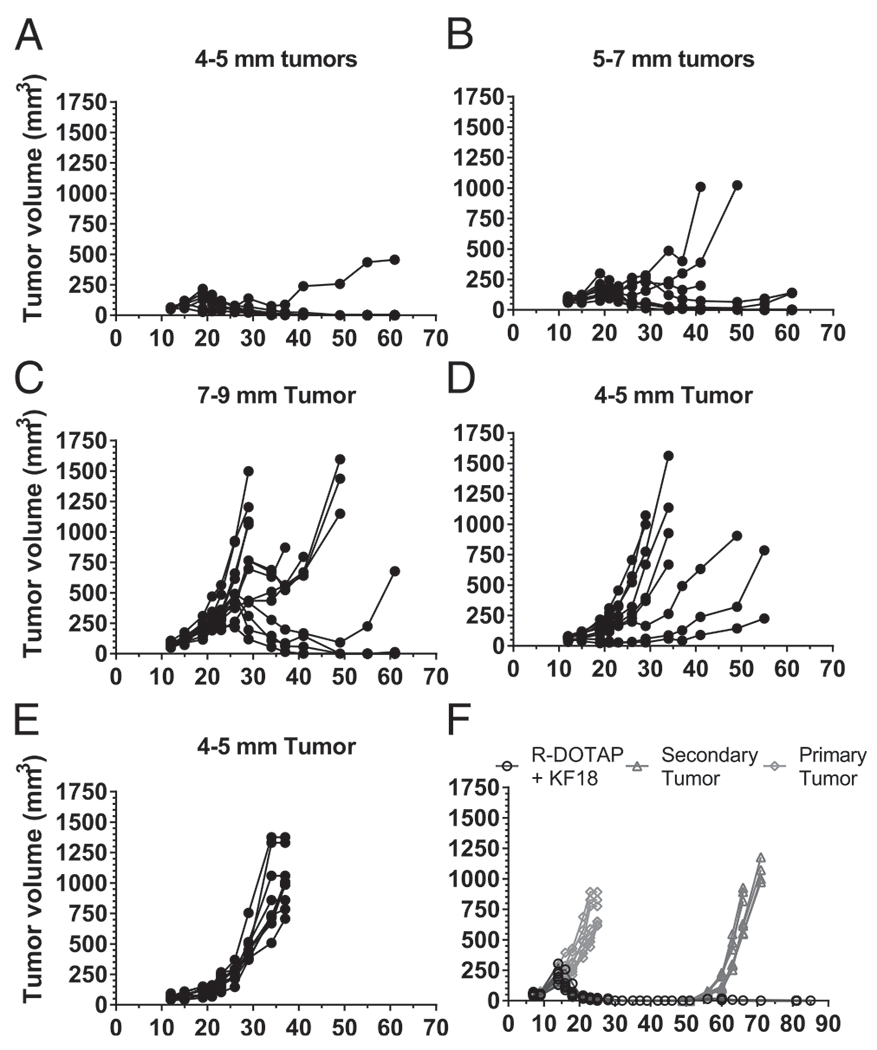
A single peptide and R-DOTAP immunotherapy can regress large TC-1 tumors and induce durable immunity. Mice implanted s.c. with 1 × 105 TC-1 tumor cells were injected with one dose of KF18 and R-DOTAP when the tumors reached the average diameters shown, ranging from 4 to 9 mm (A-C). (D) Mice were injected with R-DOTAP without peptide. (E) Naive mice received no treatment. (F) Mice bearing primary tumors (4–5 mm) were injected with one dose of an R-DOTAP formulation containing KF18 peptide on day 7 (open black circles), and after complete tumor regression, the mice were rechallenged with a second dose of 1 × 105 TC-1 tumor cells. Naive mice receiving a tumor implant at the time of primary injection are shown as gray diamonds, and naive mice receiving a tumor implant at the time of rechallenge are shown as gray triangles. Data represent tumor measurements of each mouse (n = 8–12 mice) over a period of 85 d.
R-DOTAP administration with the melanoma-associated peptide Trp2 in combination with checkpoint inhibition resulted in significant inhibition of melanoma growth
Checkpoint inhibition has been remarkably effective in tumor immunotherapy, sometimes resulting in complete regression of tumors (41). However, checkpoint inhibition relies on the existence of sufficient tumor-specific T cells in the patient capable of responding once the checkpoint inhibition is relieved (4). Thus, a significant number of patients do not respond to this therapy. To overcome this limitation, there is a considerable effort to combine checkpoint inhibitors with treatments designed to increase the levels of tumor-specific CD8 T cells, such as simultaneous vaccination against tumor-specific Ags. To determine if checkpoint inhibitors enhanced the antitumor response of R-DOTAP, we employed the B16F10 melanoma model. B16F10 is a notoriously difficult tumor to successfully treat with Ag-specific immunotherapy. One reason is that many of the Ags targeted are selfantigens to which there is some degree of tolerance (42). A previous study demonstrated that Trp2 Ag dose was important in the ability of R-DOTAP to break the tumor’s immune tolerance, and a 75-μmol dose was demonstrated to inhibit tumor growth but did not induce regression (43). We first tested whether an R-DOTAP plus Trp2 formulation would induce a strong CD8 T cell response. Trp2 is a 9-aa tyrosinase related peptide presented by the H-2Kb molecule (Table I). s.c. injection of mice with R-DOTAP plus Trp2 resulted in strong CD8 ELISPOT responses, whereas Trp2 alone did not elicit a response (Fig. 4A). To determine whether anti-PD1 treatment synergized with R-DOTAP plus Trp2 treatment in slowing the growth of B16 melanoma, mice were implanted with B16F10 melanoma and injected with R-DOTAP plus Trp2 when tumors reached a size of 3 mm. In addition, some groups received five injections of anti-PD1 Ab. Treatment with R-DOTAP plus Trp2 resulted in some slowing of tumor growth compared with naive or anti-PD1–only groups. When the R-DOTAP plus Trp2 formulation was combined with anti-PD1 treatment, a synergistic effect was apparent, resulting in a dramatic inhibition of tumor growth and an extension of survival (Fig. 4B–D). These results strongly suggest an effective immunotherapeutic synergy between the R-DOTAP T cell–activating platform and anti-PD1 therapy. Thus, R-DOTAP may be successfully combined with a checkpoint inhibitor in combination immunotherapy strategies.
FIGURE 4.
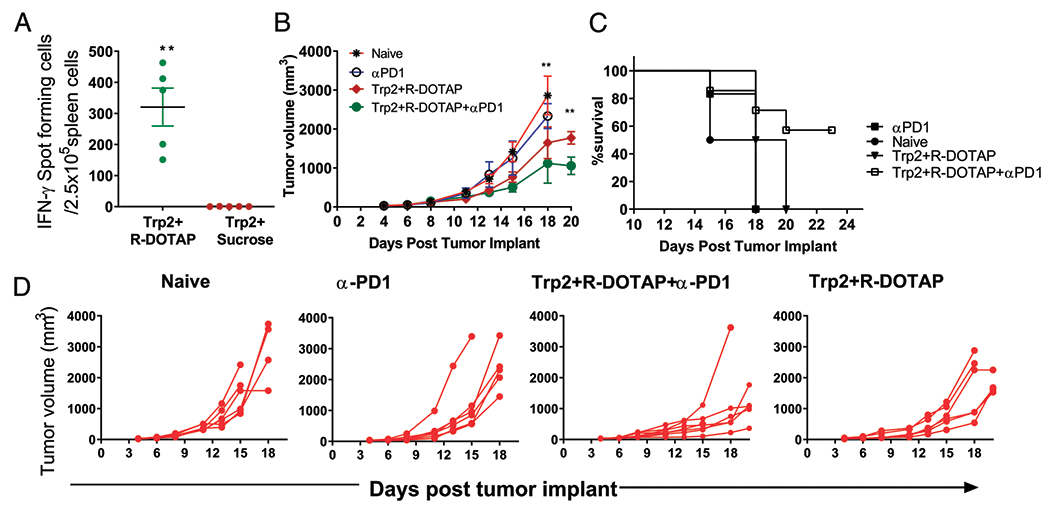
R-DOTAP synergizes with anti-mouse PD1 treatment to significantly alter B16 melanoma tumor growth in vivo. (A) Groups of C57BL/6J mice (n = 5) were s.c. injected with Trp2-encapsulated R-DOTAP (Trp2+R-DOTAP) nanoparticles or Trp2 mixed in sucrose buffer (Trp2+Sucrose) on day 0 and boosted on day 7. Trp2-specific CD8 T cell responses in spleen were assessed 7 d after the second injection by ELISPOT assay. (B-D) Mice were implanted s.c. with 1 × 105 B16F10 melanoma cells and received two doses (days 5 and 12 after tumor implant) of Trp2 plus R-DOTAP, Trp2 plus R-DOTAP plus anti-PD1, or anti-PD1 alone. For anti-PD1 treatment, each mouse received five doses of 200 μg of anti-mouse PD1 Ab delivered i.p. at 3-d intervals starting on day 5 after tumor implant. (B) Mean tumor volume ± SEM (n = 5) in treated or naive mice. (C) Survival over the course of the study. (D) Tumor growth kinetics in each individual mouse. Experiments were repeated three times with similar results. **p < 0.05.
R-DOTAP promoted DC uptake and Ag cross-presentation in vivo and in vitro
Numerous studies have shown that CLs are efficient at transporting nucleic acid and protein/peptides across cell membranes, suggesting enhanced cross-presentation of Ags as a mechanism of action of CL nanoparticles (8, 9, 16, 18, 19). To further examine the ability of R-DOTAP to induce Ag uptake and processing, we first visualized the uptake of OVA into BMDCs using Alexa 647–OVA. BMDCs were incubated for various times with R-DOTAP and Alexa 647–OVA or Alexa 647–OVA alone, followed by measurement of Alexa 647–OVA fluorescence by flow cytometry. Although some Alexa 647–OVA uptake was observed in BMDCs incubated with Alexa 647–OVA alone, uptake was dramatically enhanced in the presence of R-DOTAP. Notably, R-DOTAP facilitated significant uptake within the first 10 min and continued throughout the hour (Fig. 5A). Presumably, OVA uptake mediated by R-DOTAP would deliver OVA into acidic endosomes, where OVA processing would be expected to occur. To evaluate processing, we used DQ-OVA, which is a heavily fluorescenated OVA that self-quenches in the intact molecule but fluoresces when degraded (44). Incubation of BMDCs with DQ-OVA alone resulted in measurable uptake and some processing, as indicated by an increase in green fluorescence. However, incubation of BMDCs with DQ-OVA and R-DOTAP resulted in a significant shift to red fluorescence indicative of extensive processing and endosomal accumulation. Incubation of BMDCs with DQ-OVA and LPS did not result in enhanced processing (Fig. 5B). Thus, R-DOTAP facilitated rapid protein uptake and processing in BMDCs, presumably in endosomal compartments.
FIGURE 5.
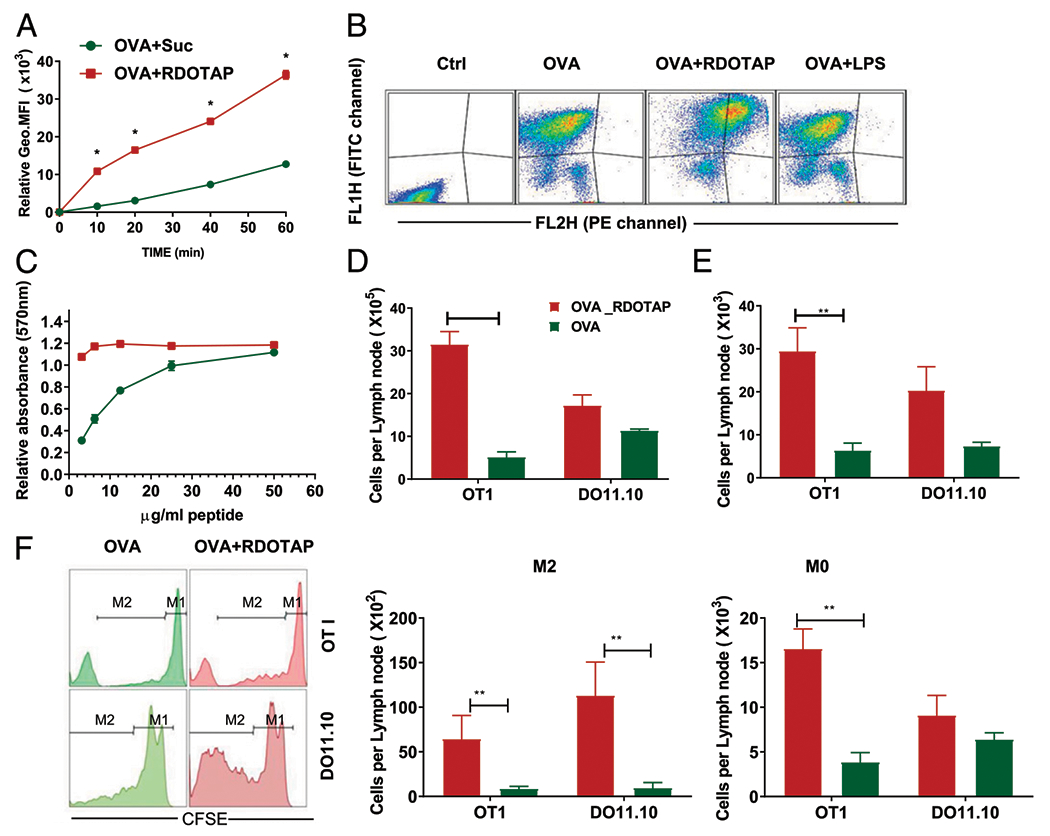
R-DOTAP enhances peptide cross-presentation in vitro and in vivo. (A) BMDCs were incubated with Alexa 647–conjugated OVA admixed with sucrose or R-DOTAP nanoparticles for the indicated times, and the association of OVA with BMDCs was determined by flow cytometry and represented as the geometric mean fluorescence intensity. (B) BMDCs were incubated with DQ-OVA, DQ-OVA plus R-DOTAP (OVA+RDOTAP), or DQ-OVA plus LPS (OVA+LPS) for 60 min, washed, and analyzed by flow cytometry. DQ-OVA processing was measured by assessing the fluorescence in the FITC channel (FL1H) and the fluorescence in the PE-channel (FL2H) representing OVA processing. (C) BMDCs were pulsed for 10 min with the indicated concentrations of OVA241–270 peptide admixed with sucrose (green circles) or R-DOTAP (red squares) and cocultured with B3Z cells overnight. Production of lacZ by OVA peptide-stimulated B3Z was measured using a lacZ colorimetric assay and displayed as relative absorbance compared with untreated B3Z cells. (D-F) C57BL/6 or BALB/c mice were adoptively transferred with CFSE-labeled OT1 or DO11.10 spleen cells and, after 24 h, injected s.c. with 1 μg OVA admixed with 4 mm R-DOTAP (OVA+R-DOTAP) or sucrose buffer (OVA). (D) The total number of cells in the draining popliteal LNs in each treated mouse were enumerated. (E) Total CFSE-labeled cells per LN. (F) Total Ag-specific CD8 T cell expansion was measured by CFSE dilution assay and quantitating cells that have undergone at least one division (M2) or no divisions (M0) using a flow cytometer. n = 4 mice per group. Results represent the mean ± SD. All experiments were repeated at least three times with similar results. *p < 0.05, **p < 0.05.
To further examine the ability of R-DOTAP to influence cross-presentation by DC, we used the B3Z T cell hybridoma, which expresses a TCR specific for the SL9 peptide of OVA presented by H-2Kb. B3Z cells express a reporter lacZ gene under the control of the NFAT promoter, providing a rapid and sensitive assay for the processing and presentation of SL9 Ag by DC (36). We incubated BMDCs with a long OVA peptide (OVA241–270) containing the SL9 epitope formulated with R-DOTAP nanoparticles or sucrose buffer for 1 h at 37°C to load the peptide on to BMDCs. Excess peptide was removed by washing, and BMDCs were then cocultured with B3Z cells overnight. The efficiency of SL9 peptide cross-presentation by BMDCs was measured using a colorimetric lacZ detection assay. Whereas incubation of BMDCs with peptide alone resulted in some cross-presentation to B3Z cells, the addition of R-DOTAP nanoparticles resulted in maximal stimulation with ~10-fold less peptide (Fig. 5C). These results suggested that R-DOTAP potently enhanced cross-presentation of peptide Ags.
To directly examine cross-presentation in vivo following R-DOTAP administration with Ag, we used an adoptive transfer model in which transgenic T cells specific for the CD8 epitope (OT1) or the CD4 epitope (DO11.10) of OVA were labeled with CFSE and transferred into normal mice. Activation and proliferation of OT1 cells in the adoptive transfer mice requires in vivo processing of whole OVA through endosomal uptake and processing into the SL9 epitope and presentation on the H-2Kb class I molecule (i.e., cross-presentation). Likewise, activation and proliferation of DO11.10 T cells would require in vivo processing of whole OVA through endosomal uptake and processing into the OVA323–339 epitope and presentation on the I-Ad class II molecule. Adoptive transfer mice were injected in the footpad with 1 μg of whole OVA admixed with either sucrose or R-DOTAP. After 3 d, proliferation of the transgenic T cells in the draining popliteal LN was assessed by flow cytometry measuring CFSE dilution in the transgenic T cells. The LN draining the R-DOTAP and OVA–injected footpads were noticeably enlarged, and this was reflected in an increased total cell number isolated per LN. There was also a significant increase in total OT1 cells, both divided and undivided, in the R-DOTAP plus OVAȓtreated mice compared with OVA alone. Similar results were obtained when identical studies were performed using DO11.10 transgenic T cells specific for the CD4 epitope of OVA (Fig. 5D–F). Thus, R-DOTAP enhances MHC class I cross-presentation and MHC class II presentation of whole protein to CD8+ or CD4+ T cells in the DLN when administered s.c.
R-DOTAP alone stimulated a strong type I IFN response, essential for its immunotherapeutic activity in vivo
To examine the immunostimulatory effect of R-DOTAP in the DLN, mice were injected with R-DOTAP nanoparticles or LPS s.c. in the nape of the neck, and CD11c+ DC were purified from pooled DLN after 4 or 24 h. Inflammatory gene expression was measured in purified RNA by NanoString multiplex analysis. Among the inflammatory genes examined, the strongest genes upregulated were those involved in the type I IFN pathway. These included IFN-α, IFN-β, CXCL10, and Stat 1. No induction of classical NF-κB–dependent cytokines was observed (Fig. 6A). This result suggested that R-DOTAP alone was capable of inducing type I IFN in DC. To directly examine type I IFN production by DC, BMDCs were incubated with R-DOTAP or LPS as a positive control for 18 h, and type I IFN was measured in the B16-Blue bioassay. There was a significant dose-dependent induction of type I IFN in BMDCs, peaking at a dose of 50–100 μM. Although the type I IFN production was ~10-fold less than that induced by LPS, it was nonetheless highly significant and consistent over multiple experiments (Fig. 6B).
FIGURE 6.
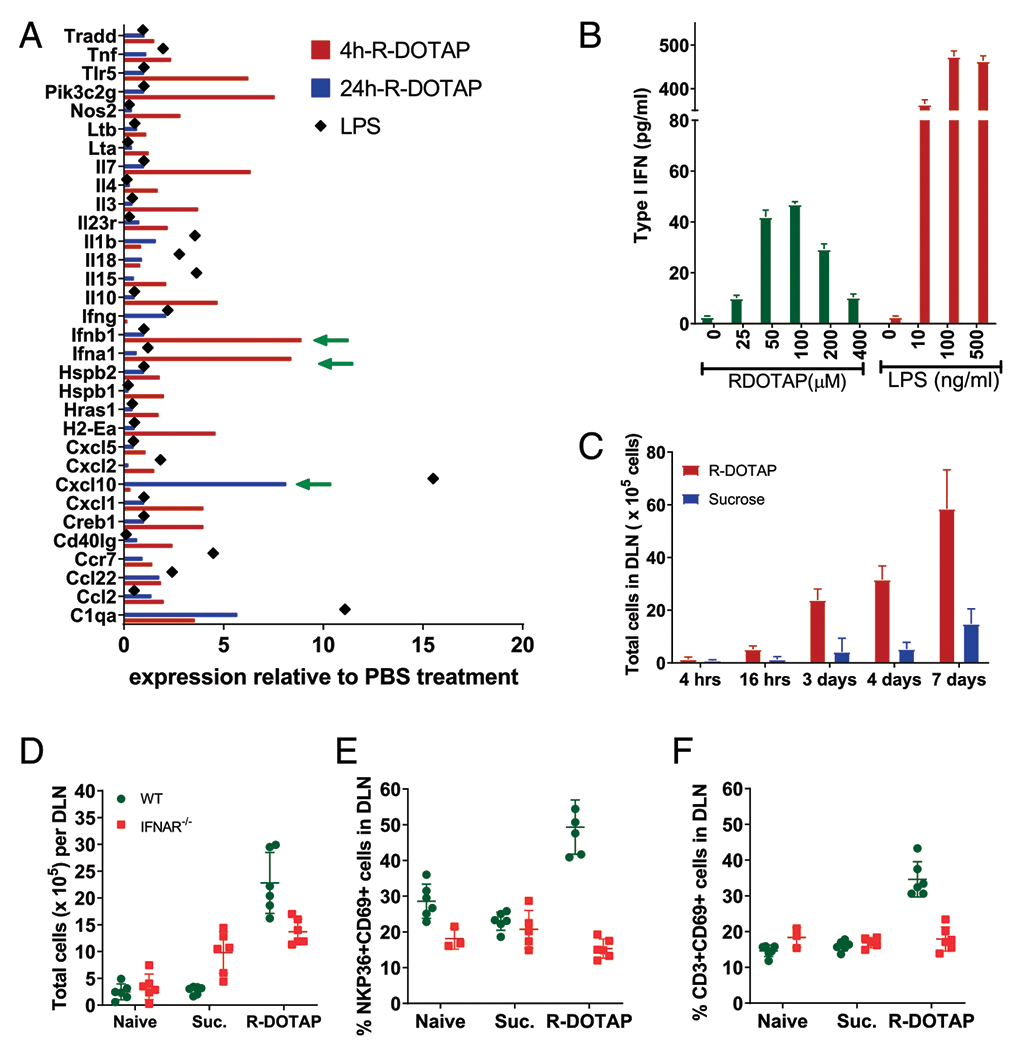
R-DOTAP administration induces production of type I IFN in the DLN in vivo. (A) Groups of four C57BL/6 mice were injected with R-DOTAP, PBS, or LPS at the nape of the neck, and draining axillary and brachial LNs were harvested from each mouse after 4 or 24 h. CD11c+ cells from pooled LNs of each individual mouse were sort-purified, and relative gene expression was analyzed using Nanostring technology. Shown are mean RNA expression levels for genes from the R-DOTAP or LPS groups that showed >0.5-fold difference relative to PBS treatment. (B) BMDCs from IFNAR−/− mice were stimulated with the indicated concentrations of R-DOTAP nanoparticles or LPS for 24 h, and type I IFN production was measured using an IFN-α/β reporter assay. (C) Mice were injected with R-DOTAP or sucrose in the footpad, DLN were harvested at the indicated times and enzymatically digested, and total cell number was enumerated. (D) Wild-type (WT) or IFNAR−/− mice were administered R-DOTAP or sucrose (Suc.) in the footpad, and total cells per DLN were quantitated after 24 h. (E) DLN from mice described in (D) were analyzed for the percentage of CD69+ NKP36+ NK cells or (F) the percent of CD69+ CD3+ T cells. Experiments were repeated at least three times with similar results.
Because we consistently observed significant enlargement of the DLN following R-DOTAP and OVA injection, we sought to determine whether R-DOTAP alone was responsible and the role type I IFN played in this process. Therefore, we injected wild-type or IFN-αR knockout (KO) mice in the footpads with either R-DOTAP alone (no Ag) or sucrose diluent and characterized cell populations in the draining popliteal LNs. R-DOTAP alone resulted in a visible increase in DLN size of wild-type mice, and this was due to a steady increase in total cell number over a 7-d period (Fig. 6C). This increase in total cell number was dependent on type I IFN signaling, as it was greatly reduced in the IFN-αR KO mouse (Fig. 6D). Type I IFN is known to inhibit lymphocyte egress from lymphoid organs through the upregulation of CD69, which, in turn, inhibits the sphingosine 1 phosphate receptor required for lymphocyte egress (45). We therefore postulated that R-DOTAP injection would result in the upregulation of CD69 in the DLN. Indeed, R-DOTAP injection of wild-type mice resulted in the upregulation of CD69 on both NK cells (Fig. 6E) and T cells (Fig. 6F). In contrast, no CD69 upregulation was seen after R-DOTAP injection of IFN-αR KO mice, demonstrating that the R-DOTAP–induced upregulation of CD69 was type I IFN–dependent. Therefore, R-DOTAP in the absence of Ag induces type I IFN in the DLN, which in turn upregulates CD69 in T cells and NK cells, resulting in their accumulation in the DLN.
Based on the documented importance of the type I IFN pathway in CD8 T cell immunity (46), we postulated that the potent CD8 T cell-inducing capacity of R-DOTAP and Ag was due to its intrinsic ability to induce type I IFN in the DLN. To test this, we injected wild-type or IFN-αR KO mice with OVA257–264 (SL9) mixed with R-DOTAP or SL9 peptide emulsified in CFA on days 0 and 7 and analyzed the T cell response by ELISPOT assay on day 14. Administration of SL9 plus R-DOTAP stimulated a very strong ELISPOT response in wild-type mice but almost no response in IFN-αR KO mice. In contrast, administration of SL9 plus CFA induced a weaker but significant ELISPOT response in both wild-type and IFN-αR KO mice, indicating that IFN-αR KO mice are fully capable of mounting a CD8 T cell response to SL9 (Fig. 7A). Thus, the CD8 T cell–stimulatory activity of R-DOTAP appears to be highly specific and entirely dependent on type I IFN signaling, whereas adjuvants such as CFA can stimulate CD8 T cells through other, non–type I IFN–dependent pathways.
FIGURE 7.
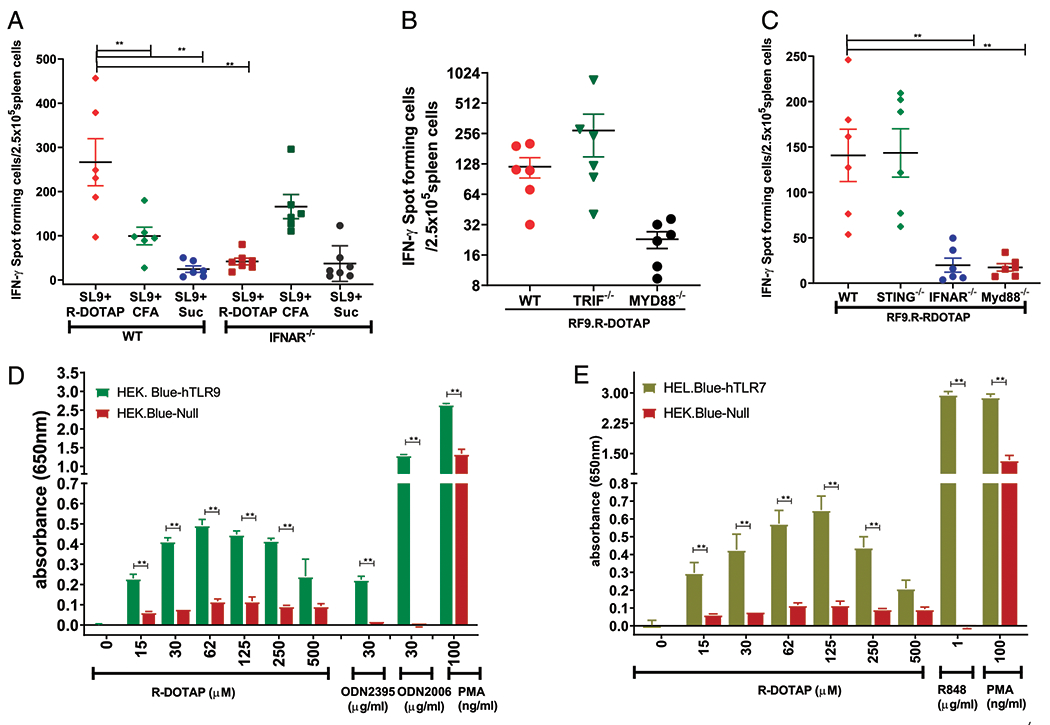
R-DOTAP mediates its CTL-inducing effects by activating type I IFN in a Myd88-dependent manner. (A) Wild-type (WT) or IFNAR−/− mice were injected on days 0 and 7 with the OVA SL9 peptide admixed with R-DOTAP (SL9+R-DOTAP), SL9 peptide emulsified with CFA (SL9+CFA), or SL9 in sucrose (SL9+Suc), and ELISPOTs were performed against the SL9 peptide on day 14. (B) WT, TRIF−/−, or MYD88−/− mice were injected on days 0 and 7 with the RF9 peptide encapsulated in R-DOTAP nanoparticles (RF9.R-DOTAP), and ELISPOTs were performed against the RF9 peptide on day 14. (C) WT, STING−/−, IFNAR−/−, or Myd88−/− mice were injected on days 0 and 7 with RF9 peptide encapsulated in R-DOTAP nanoparticles (RF9.R-DOTAP), and ELISPOTs were performed against the RF9 peptide on day 14. Data represent the mean ± SD of peptide-specific IFN-γ–producing cells in spleens from treated mice (n = 5). (D) HEK-Blue null, HEK-Blue human TLR9, and (E) HEK-Blue human TLR7 reporter cells were stimulated for 24 h with the indicated concentration of R-DOTAP nanoparticles, ODN2006 or ODN2395 (D), R848 (E), or PMA; the cell supernatants were assayed for SEAP activity using QUANTI-Blue reagent; and the background subtracted relative absorbance in triplicate wells were plotted. Data represent mean absorbance ± SEM of triplicate cultures, and all experiments were repeated at least two times with similar results. **p < 0.05.
Either TLR-dependent pathways or intracellular sensors like STING can induce type I IFN in cells (47). To determine the relative contributions of these pathways to the immunostimulatory activity of R-DOTAP, we injected wild-type, STING KO, IFN-αR KO, TRIF KO, or Myd88 KO mice with R-DOTAP and RF9 peptide. As expected, injection of RF9 plus R-DOTAP into wild-type mice stimulated a strong CD8 response that was completely abrogated in IFN-αR KO mice. Injection of R-DOTAP/peptide into STING KO or TRIF KO mice resulted in a strong CD8 response identical to that in wild-type mice. In contrast, injection of R-DOTAP/peptide into Myd88 KO mice resulted in no CD8 response (Fig. 7B, 7C). Thus, these results clearly show that the CD8 T cell immunostimulatory activity of R-DOTAP requires the type I IFN pathway and uses the adaptor molecule Myd88 but not TRIF or STING.
The TLR9 family of receptors (TLR7, TLR8, and TLR9) expressed in endosomes use Myd88 and IRF-7 for the induction of type I IFN (48). To test if R-DOTAP can mediate its effects through engaging the TLR9 family receptors, we used HEK-Blue reporter cells expressing individual human TLR7 or 9 and a SEAP reporter under the control of an NF-κB promoter. Incubation of either HEK-Blue TLR9 or TLR7 cells with R-DOTAP resulted in a dose-dependent induction of NF-κB–SEAP, peaking at ~100 μM R-DOTAP. No SEAP expression was observed with HEK-Blue null control cells in response to R-DOTAP, whereas strong SEAP expression was observed with the TLR-independent stimulus, PMA (Fig. 7D, 7E). The magnitude of the R-DOTAP–stimulated TLR9 response was comparable to that of the class C CpG (ODN2395). The number of R-DOTAP–stimulated HEK-Blue TLR7 cells was ~5-fold less than that of the TLR7 agonist R848. Therefore, we conclude that R-DOTAP activates both TLR7 and TLR9, resulting in moderate NF-κB activation.
Discussion
The mechanistic studies in this report primarily focused on combinations of two components: the prototype member of this group of immunostimulatory CLs, R-DOTAP nanoparticles, and cancer-derived peptide Ags that function effectively as immunotherapeutics when combined with the immunostimulatory nanoparticles. The immunotherapeutic formulations induced robust CD8 T cell responses in mice to a variety of multiepitopic and single-epitope peptide Ags. Our data confirm and extend earlier pivotal studies with R-DOTAP in in vitro and in vivo model systems of HPV-related cancer. The Huang group first demonstrated that a single s.c. injection of TC-1 tumor-bearing mice with HPV E6 and E7 peptide Ags or whole protein complexed with R-DOTAP nanoparticles effected complete regression of implanted tumor. They also demonstrated that lipidation of the N terminus of the peptide with a palmitoyl group followed by a K-S-S linker greatly enhanced the peptide interaction with R-DOTAP nanoparticles and amplified the resulting immunogenicity (24). In this study, lipidated long multiepitopic peptides were delivered and processed effectively by APC and stimulated strong CD8 T cell responses. Treatment with HPV E7 peptide containing the nominal 9-aa CD8 epitope codelivered with R-DOTAP nanoparticles was capable of eliciting robust and durable antitumor responses capable of regressing large 5–7-mm tumors. Although the Huang group reported that R-DOTAP is immunostimulatory and induced the maturation of DC, chemokine expression, and activation of PI3 kinase and Erk but not NF-κB (29–31), the complete immunostimulatory mechanism of action in vivo has remained undefined. In this study, we show that R-DOTAP functions in vivo primarily through the induction of type I IFN. Considering the known actions of type I IFN, it is likely that type I IFN induction explains many of the previously described actions of R-DOTAP, including promotion of cross-presentation, induction of chemokines, and promotion of CD8 T cell differentiation.
Cancer vaccine or Ag-specific immunotherapeutic approaches have primarily focused on optimizing Ag-specific CD8 T cell induction. These approaches include designs to enhance Ag delivery, uptake, and presentation of Ag, including DNA (49), viral (50) or intracellular bacterial vectors (51), nanoparticles (52), and targeting of the Ag to DC through conjugation (53) or pulsing DC in vitro with Ag (54). Most approaches also include immunostimulatory compounds, typically TLR agonists designed to induce the desired cytokine production (7). Still others include recombinant cytokines like IL-2, IL-12, or GM-CSF (35). Finally, many cancer vaccines are designed to create an Ag depot through the creation of emulsions (e.g., CFA, IFA, or Montanide). Many of these approaches show CD8 T cell stimulation and tumor regression to various degrees. CLs have been appreciated for some time to facilitate delivery, uptake, and cross-presentation of Ag (9, 16–18). However, the nature of the immunostimulatory activity of certain CLs, and R-DOTAP in particular, has not been completely understood until now. Our demonstration that the structurally specific CL R-DOTAP functions as an activator of the type I IFN pathway explains the ability of R-DOTAP formulations to induce potent responses without inclusion of extraneous cytokines or TLR agonists. One of the most effective cancer vaccines previously reported consists of antitumor Ab, IL-2, and lipid modified peptide plus CpG and anti-PD1. This multicomponent vaccine plus immune modifiers induced strong CD8 responses and tumor regression with multiple doses. However, in the absence of anti PD1 checkpoint blockade, this vaccine did not result in complete regression of a TC-1 tumor using the same RF9 peptide Ag as in our studies (55). In contrast, we show that R-DOTAP nanoparticles formulated with HPV peptide Ag KF18 effected complete regression of large TC-1 tumors with a single s.c. injection. Previous studies and the data presented in this study now support the notion that R-DOTAP nanoparticles, combined with protein or peptide Ags, possess all the critical properties for a powerful CD8 T cell immunotherapy. These include excellent Ag delivery, Ag uptake, and cross-presentation; intrinsic and specific immunostimulatory properties through activation of type I IFN; and the formation of an Ag depot without the severe injection site reactions observed with emulsions and other approaches (56). Thus, R-DOTAP nanoparticles, combined with various tumor Ags, because of their unique multifunctional immunological properties, produce a powerful platform for the development of potentially safe and effective cancer immunotherapies.
Certain CLs are excellent at promoting cross-presentation of associated proteins or peptides on MHC class I (18, 19, 57). This is largely related to the cationic surface charge of the liposomal nanoparticles and optimal size in the 100–200-nm range. This facilitates rapid binding to cell membranes, rapid internalization into endosomes, and destabilization of the endosomal membranes releasing contents into the cytoplasm (10, 15). Whereas most studies of cross-presentation involved in vitro systems, our results in this study extend this property to in vivo systems. ELISPOT assays in which animals were injected with a long peptide but stimulated in the ELISPOT plate using the corresponding short specific 9-aa epitope demonstrate that cross-presentation must have occurred during T cell priming. We also demonstrate this formally by adoptive transfer of OT1 TCR transgenic T cells, which recognize OVA257–264 in the context of H-2Kb, thus providing a sensitive assay for the presentation of OVA257–264-Kb complexes in the DLN. There are two non–mutually exclusive mechanisms to explain the ability of R-DOTAP nanoparticles to promote cross-presentation in vivo. The first mechanism involves the same principles as have been described in vitro. In this scenario, R-DOTAP nanoparticles containing Ag transit to the DLN and bind to DC, resulting in endosomal uptake and cross-presentation through mechanisms already described. In this case, R-DOTAP nanoparticles serve as a delivery vehicle. The second possibility, based on our findings that R-DOTAP induces type I IFN production, is that type I IFN licenses DC within the DLN to greatly enhance cross-presentation pathways (46), such that even proteins or peptides not directly associated with R-DOTAP nanoparticles are taken up and cross-presented efficiently. In this case, the delivery vehicle capacity of R-DOTAP nanoparticles would be less important. Both mechanisms would also promote cross-presentation of multiple peptides or proteins at once, resulting in effective CTL priming to multiple epitopes. It is likely that both mechanisms occur in vivo, resulting in strongly enhanced cross-presentation.
We have presented several pieces of evidence pointing to the conclusion that R-DOTAP activates the type I IFN pathway and that this is necessary for the immunostimulatory activity of R-DOTAP. By 4 h after R-DOTAP injection into the footpad, NanoString RNA analysis of purified CD11c DC from DLN shows that the most strongly upregulated genes are IFN-α and IFN-β. The most strongly upregulated gene 24 h after either R-DOTAP injection was CXCL10, a cytokine gene strongly inducible by type I IFN. In vitro, BMDCs secrete type I IFN following incubation with R-DOTAP, indicating that the IFN-inducing effect of R-DOTAP is direct. IFN-αR KO mice were unable to generate Ag-specific CD8 T cell responses following R-DOTAP and peptide administration, indicating that type I IFN was necessary for the biologic activity of R-DOTAP. Interestingly, injection of IFN-αR KO mice with the same peptide emulsified in CFA resulted in a strong CD8 T cell response equivalent to, if not greater than, that in wild-type mice. Thus, although Ag-specific CD8 T cells can be induced in the absence of type I IFN with an adjuvant such as CFA, CD8 T cell induction by R-DOTAP primarily uses the type I IFN pathway. Consistent with type I IFN induction by R-DOTAP, CD69 was strongly upregulated on T cells and NK cells, and lymphocyte cell number steadily increased in the DLN over a 7-d period. Consistent with previous studies (45), both CD69 upregulation and the increase in DLN cell number were abrogated in IFN-αR KO mice, confirming the dependence on type I IFN. Presumably, CD69 upregulation is the primary cause of the increase in cell number in the DLN, as it has been demonstrated that CD69 blocks the S1P receptor required for lymphocyte egress from lymphoid tissue (45). Therefore, we conclude that R-DOTAP alone induces type I IFN in the DLN following s.c. injection. One measurable consequence of this is the upregulation of CD69 on T cells, the subsequent inhibition of egress from the LNs resulting in greatly increased cell number over an extended period of time and the development of T cell memory (58). Although it has been previously demonstrated that DOTAP racemic mixture complexed with either RNA or DNA can induce type I IFN production (59–61), this is the first study, to our knowledge, showing that a CL independently induces type I IFN. Considering our data on in vivo cross-presentation, we postulate that exposure of DC with R-DOTAP nanoparticles and associated Ag at the site of administration and/or in the DLN subsequently creates the ideal environment in the DLN for priming and differentiation of Ag-specific CD8 CTL.
To ascertain the upstream events leading to type I IFN induction by R-DOTAP, we used Myd88, TRIF, and STING KO mice. Myd88 KO mice were unable to mount a CD8 T cell response following R-DOTAP–peptide administration, whereas TRIF or STING KO mice responded normally. Likewise, Myd88 KO mice did not show an upregulation of CD69, nor did they show an increase in DLN cell number following R-DOTAP administration. Thus, we conclude that R-DOTAP induces type I IFN through a pathway using the Myd88, but not TRIF or STING, pathway.
Type I IFN can be produced through multiple innate stimuli. The cell type producing the highest level of type I IFN is the plasmacytoid DC, capable of producing 1000 times more type I IFN than other DC subtypes (61). Plasmacytoid DC, as well as other types of DC, and macrophages express TLR9 subfamily members (TLR7, 8, and 9) in endosomal compartments. These TLRs all use Myd88 as the adaptor molecule and stimulate type I IFN production, making them the likely targets of R-DOTAP. TLR7 recognizes ssRNA typically found in viruses but can also recognize mammalian RNA (62). TLR7 can also recognize non–nucleic acid imidizole–quinoline compounds like R848 and imiquimod, which is currently approved as an immunotherapeutic cream for genital warts and skin cancer (63). TLR8 is functional in humans but not mice and also recognizes ssRNA, resulting in type I IFN expression through Myd88. TLR9 recognizes unmethylated CpG DNA, which typically occurs more frequently in bacterial DNA, but vertebrate DNA can also stimulate through TLR9 (64). TLR7 and 9 both stimulate type I IFN production through an IRF-7– and Myd88-dependent pathway (48). Our findings that R-DOTAP directly stimulates both TLR7 and TLR9 suggest a model for the immunostimulatory properties of R-DOTAP. It has already been well-documented that R-DOTAP nanoparticles are rapidly internalized into endosomes, resulting in endosomal destabilization and alteration of normal endosomal trafficking (14, 61). We postulate that this endosomal destabilization promotes TLR dimerization and signal activation, resulting in type I IFN induction. This could be through direct interaction of R-DOTAP with the TLR or through indirect endosomal perturbation. An alternative possibility is that endogenous cellular RNA and DNA released from neighboring cells are transported into endosomes by R-DOTAP nanoparticles, resulting in agonistic TLR7 and 9 stimulation. Careful cellular and biochemical approaches will, however, be required to confirm the detailed mechanism by which TLR7 and 9 are activated.
In summary, these studies describe the elucidation of the mechanism of action of an Ag-specific immunotherapeutic platform based upon the use of a novel enantiomeric CL, R-DOTAP. Treatment with this immunotherapeutic has been shown to initiate a cascade of responses in dendritic and T cells required for effective immunotherapy. R-DOTAP is the first CL to be shown to mediate both effective Ag cross-presentation and stimulation of type I IFN, leading to the robust Ag-specific and antitumor immune responses (33). As expected from an immunotherapeutic capable of generating high levels of polyfunctional Ag-specific effector CD8 T cells, combination therapy with checkpoint inhibitors resulted in a significant synergy in a rigorous B16 melanoma model. A first-generation Versamune immunotherapeutic containing multiple HPV E6 and E7 Ags has now completed phase 1/2a clinical trials in humans. Data analysis not only demonstrated excellent safety but confirmed preclinical observations of high-level generation of Ag-specific CD4 and CD8 T cell responses in human subjects (G. Conn, F. Bedu-Addo, and J. Woodward, manuscript in preparation).
Acknowledgments
We thank Dr. Charles Snow and Dr. Mark Einstein for critical manuscript review, helpful advice, and scientific discussion.
This work was supported by a PDS Biotechnology Corporation research grant (to J.G.W.).
Disclosures
J.G.W. has received research funding from PDS Biotechnology. J.G.W., S.K.G., and M.W. have received stock options from PDS Biotechnology. F.B.-A. and G.C. are principal officers in PDS Biotechnology.
Abbreviations used in this article:
- BMDC
bone marrow–derived DC
- CGMP
current good manufacturing practice–grade
- CL
cationic lipid
- cRPMI
complete RPMI 1640
- DC
dendritic cell
- DLN
draining lymph node
- DOTAP
1,2-dioleoyl-3-trimethyl-ammonium-propane
- HPV
human papillomavirus
- IFA-Cyt
IFA, IL-12, GM-CSF, and HBV128–140 helper epitope
- KO
knockout
- LN
lymph node
- MUC1
mucin 1
- PD1
programmed cell death protein 1
- SEAP
secreted embryonic alkaline phosphatase
- Treg
T regulatory cell
- UK
University of Kentucky
References
- 1.Kamphorst AO, and Ahmed R. 2013. Manipulating the PD-1 pathway to improve immunity. Curr. Opin. Immunol 25: 381–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sharma P, and Allison JP 2015. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell 161: 205–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tang H, Wang Y, Chlewicki LK, Zhang Y, Guo J, Liang W, Wang J, Wang X, and Fu YX. 2016. Facilitating T cell infiltration in tumor microenvironment overcomes resistance to PD-L1 blockade. [Published erratum appears in 2016 Cancer Cell 30: 500.] Cancer Cell 29: 285–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. 2014. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515: 568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN, et al. 2014. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 515: 563–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. 2012. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med 366: 2443–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tagliamonte M, Petrizzo A, Tornesello ML, Buonaguro FM, and Buonaguro L. 2014. Antigen-specific vaccines for cancer treatment. Hum. Vaccin. Immunother 10: 3332–3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Felgner PL, Gadek TR, Holm M, Roman R, Chan HW, Wenz M, Northrop JP, Ringold GM, and Danielsen M. 1987. Lipofection: a highly efficient, lipid-mediated DNA-transfection procedure. Proc. Natl. Acad. Sci. USA 84: 7413–7417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zelphati O, Wang Y, Kitada S, Reed JC, Felgner PL, and Corbeil J. 2001. Intracellular delivery of proteins with a new lipid-mediated delivery system. J. Biol. Chem 276: 35103–35110. [DOI] [PubMed] [Google Scholar]
- 10.Wrobel I, and Collins D. 1995. Fusion of cationic liposomes with mammalian cells occurs after endocytosis. Biochim. Biophys. Acta 1235: 296–304. [DOI] [PubMed] [Google Scholar]
- 11.Rejman J, Bragonzi A, and Conese M. 2005. Role of clathrin- and caveolae-mediated endocytosis in gene transfer mediated by lipo- and polyplexes. Mol. Ther 12: 468–474. [DOI] [PubMed] [Google Scholar]
- 12.Rejman J, Conese M, and Hoekstra D. 2006. Gene transfer by means of lipo- and polyplexes: role of clathrin and caveolae-mediated endocytosis. J. Liposome Res 16: 237–247. [DOI] [PubMed] [Google Scholar]
- 13.Zuhorn IS, Kalicharan R, and Hoekstra D. 2002. Lipoplex-mediated transfection of mammalian cells occurs through the cholesterol-dependent clathrin-mediated pathway of endocytosis. J. Biol. Chem 277: 18021–18028. [DOI] [PubMed] [Google Scholar]
- 14.Xu Y, and Szoka FC Jr. 1996. Mechanism of DNA release from cationic liposome/DNA complexes used in cell transfection. Biochemistry 35: 5616–5623. [DOI] [PubMed] [Google Scholar]
- 15.Zabner J, Fasbender AJ, Moninger T, Poellinger KA, and Welsh MJ. 1995. Cellular and molecular barriers to gene transfer by a cationic lipid. J. Biol. Chem 270: 18997–19007. [DOI] [PubMed] [Google Scholar]
- 16.Walker C, Selby M, Erickson A, Cataldo D, Valensi JP, and Van Nest GV. 1992. Cationic lipids direct a viral glycoprotein into the class I major histocompatibility complex antigen-presentation pathway. Proc. Natl. Acad. Sci. USA 89: 7915–7918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nakanishi T, Kunisawa J, Hayashi A, Tsutsumi Y, Kubo K, Nakagawa S, Fujiwara H, Hamaoka T, and Mayumi T. 1997. Positively charged liposome functions as an efficient immunoadjuvant in inducing immune responses to soluble proteins. Biochem. Biophys. Res. Commun 240: 793–797. [DOI] [PubMed] [Google Scholar]
- 18.Chikh G, and Schutze-Redelmeier MP. 2002. Liposomal delivery of CTL epitopes to dendritic cells. Biosci. Rep 22: 339–353. [DOI] [PubMed] [Google Scholar]
- 19.Okada N, Saito T, Mori K, Masunaga Y, Fujii Y, Fujita J, Fujimoto K, Nakanishi T, Tanaka K, Nakagawa S, et al. 2001. Effects of lipofectin-antigen complexes on major histocompatibility complex class I-restricted antigen presentation pathway in murine dendritic cells and on dendritic cell maturation. Biochim. Biophys. Acta 1527: 97–101. [DOI] [PubMed] [Google Scholar]
- 20.Bei R, Guptill V, Masuelli L, Kashmiri SV, Muraro R, Frati L, Schlom J, and Kantor J. 1998. The use of a cationic liposome formulation (DOTAP) mixed with a recombinant tumor-associated antigen to induce immune responses and protective immunity in mice. J. Immunother 21: 159–169. [DOI] [PubMed] [Google Scholar]
- 21.Heravi Shargh V, Jaafari MR, Khamesipour A, Jalali SA, Firouzmand H, Abbasi A, and Badiee A. 2012. Cationic liposomes containing soluble Leishmania antigens (SLA) plus CpG ODNs induce protection against murine model of leishmaniasis. Parasitol. Res 111: 105–114. [DOI] [PubMed] [Google Scholar]
- 22.Lay M, Callejo B, Chang S, Hong DK, Lewis DB, Carroll TD, Matzinger S, Fritts L, Miller CJ, Warner JF, et al. 2009. Cationic lipid/DNA complexes (JVRS-100) combined with influenza vaccine (Fluzone) increases antibody response, cellular immunity, and antigenically drifted protection. Vaccine 27: 3811–3820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guy B, Pascal N, Françon A, Bonnin A, Gimenez S, Lafay-Vialon E, Trannoy D, and Haensler J. 2001. Design, characterization and preclinical efficacy of a cationic lipid adjuvant for influenza split vaccine. Vaccine 19: 1794–1805. [DOI] [PubMed] [Google Scholar]
- 24.Chen W, Yan W, and Huang L. 2008. A simple but effective cancer vaccine consisting of an antigen and a cationic lipid. Cancer Immunol. Immunother 57: 517–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pizzuto M, Gangloff M, Scherman D, Gay NJ, Escriou V, Ruysschaert JM, and Lonez C. 2017. Toll-like receptor 2 promiscuity is responsible for the immunostimulatory activity of nucleic acid nanocarriers. J. Control. Release 247: 182–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lonez C, Irvine KL, Pizzuto M, Schmidt BI, Gay NJ, Ruysschaert JM, Gangloff M, and Bryant CE. 2015. Critical residues involved in Toll-like receptor 4 activation by cationic lipid nanocarriers are not located at the lipopolysaccharide-binding interface. Cell. Mol. Life Sci 72: 3971–3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lonez C, Bessodes M, Scherman D, Vandenbranden M, Escriou V, and Ruysschaert JM. 2014. Cationic lipid nanocarriers activate Toll-like receptor 2 and NLRP3 inflammasome pathways. Nanomedicine (Lond.) 10: 775–782. [DOI] [PubMed] [Google Scholar]
- 28.Wilmar A, Lonez C, Vermeersch M, Andrianne M, Pérez-Morga D, Ruysschaert JM, Vandenbranden M, Leo O, and Temmerman ST. 2012. The cationic lipid, diC14 amidine, extends the adjuvant properties of aluminum salts through a TLR-4- and caspase-1-independent mechanism. Vaccine 30: 414–424. [DOI] [PubMed] [Google Scholar]
- 29.Yan W, Chen W, and Huang L. 2007. Mechanism of adjuvant activity of cationic liposome: phosphorylation of a MAP kinase, ERK and induction of chemokines. Mol. Immunol 44: 3672–3681. [DOI] [PubMed] [Google Scholar]
- 30.Vangasseri DP, Cui Z, Chen W, Hokey DA, Falo LD Jr., and Huang L. 2006. Immunostimulation of dendritic cells by cationic liposomes. Mol. Membr. Biol 23: 385–395. [DOI] [PubMed] [Google Scholar]
- 31.Yan W, Chen W, and Huang L. 2008. Reactive oxygen species play a central role in the activity of cationic liposome based cancer vaccine. J. Control. Release 130: 22–28. [DOI] [PubMed] [Google Scholar]
- 32.Chen W, and Huang L. 2008. Induction of cytotoxic T-lymphocytes and antitumor activity by a liposomal lipopeptide vaccine. Mol. Pharm 5: 464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vasievich EA, Chen W, and Huang L. 2011. Enantiospecific adjuvant activity of cationic lipid DOTAP in cancer vaccine. Cancer Immunol. Immunother 60: 629–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lin KY, Guarnieri FG, Staveley-O’Carroll KF, Levitsky HI, August JT, Pardoll DM, and Wu TC. 1996. Treatment of established tumors with a novel vaccine that enhances major histocompatibility class II presentation of tumor antigen. Cancer Res. 56: 21–26. [PubMed] [Google Scholar]
- 35.Epel M, Carmi I, Soueid-Baumgarten S, Oh S, Bera T, Pastan I, Berzofsky J, and Reiter Y. 2008. Targeting TARP, a novel breast and prostate tumor-associated antigen, with T cell receptor-like human recombinant antibodies. Eur. J. Immunol 38: 1706–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sanderson S, and Shastri N. 1994. LacZ inducible, antigen/MHC-specific T cell hybrids. Int. Immunol 6: 369–376. [DOI] [PubMed] [Google Scholar]
- 37.Egan RM, Yorkey C, Black R, Loh WK, Stevens JL, and Woodward JG. 1996. Peptide-specific T cell clonal expansion in vivo following immunization in the eye, an immune-privileged site. J. Immunol 157: 2262–2271. [PubMed] [Google Scholar]
- 38.Jochems C, Tucker JA, Vergati M, Boyerinas B, Gulley JL, Schlom J, and Tsang KY. 2014. Identification and characterization of agonist epitopes of the MUC1-C oncoprotein. Cancer Immunol. Immunother 63: 161–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wada H, Isobe M, Kakimi K, Mizote Y, Eikawa S, Sato E, Takigawa N, Kiura K, Tsuji K, Iwatsuki K, et al. 2014. Vaccination with NY-ESO-1 overlapping peptides mixed with Picibanil OK-432 and montanide ISA-51 in patients with cancers expressing the NY-ESO-1 antigen. J. Immunother 37: 84–92. [DOI] [PubMed] [Google Scholar]
- 40.Jinushi M, Hodi FS, and Dranoff G. 2008. Enhancing the clinical activity of granulocyte-macrophage colony-stimulating factor-secreting tumor cell vaccines. Immunol. Rev 222: 287–298. [DOI] [PubMed] [Google Scholar]
- 41.Sun C, Mezzadra R, and Schumacher TN. 2018. Regulation and function of the PD-L1 checkpoint. Immunity 48: 434–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Colella TA, Bullock TN, Russell LB, Mullins DW, Overwijk WW, Luckey CJ, Pierce RA, Restifo NP, and Engelhard VH. 2000. Self-tolerance to the murine homologue of a tyrosinase-derived melanoma antigen: implications for tumor immunotherapy. J. Exp. Med 191: 1221–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vasievich EA, Ramishetti S, Zhang Y, and Huang L. 2012. Trp2 peptide vaccine adjuvanted with (R)-DOTAP inhibits tumor growth in an advanced melanoma model. Mol. Pharm 9: 261–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Quinn KM, Yamamoto A, Costa A, Darrah PA, Lindsay RW, Hegde ST, Johnson TR, Flynn BJ, Loré K, and Seder RA. 2013. Coadministration of polyinosinic:polycytidylic acid and immunostimulatory complexes modifies antigen processing in dendritic cell subsets and enhances HIV gag-specific T cell immunity. J. Immunol 191: 5085–5096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shiow LR, Rosen DB, Brdicková N, Xu Y, An J, Lanier LL, Cyster JG, and Matloubian M. 2006. CD69 acts downstream of interferon-alpha/beta to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature 440: 540–544. [DOI] [PubMed] [Google Scholar]
- 46.Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, and Gajewski TF. 2011. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8alpha+ dendritic cells. J. Exp. Med 208: 2005–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ishikawa H, and Barber GN. 2011. The STING pathway and regulation of innate immune signaling in response to DNA pathogens. Cell. Mol. Life Sci 68: 1157–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, and Taniguchi T. 2005. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 434: 772–777. [DOI] [PubMed] [Google Scholar]
- 49.Rice J, Ottensmeier CH, and Stevenson FK. 2008. DNA vaccines: precision tools for activating effective immunity against cancer. Nat. Rev. Cancer 8: 108–120. [DOI] [PubMed] [Google Scholar]
- 50.Kallert SM, Darbre S, Bonilla WV, Kreutzfeldt M, Page N, Müller P, Kreuzaler M, Lu M, Favre S, Kreppel F, et al. 2017. Replicating viral vector platform exploits alarmin signals for potent CD8+ T cell-mediated tumour immunotherapy. Nat. Commun 8: 15327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bolhassani A, Naderi N, and Soleymani S. 2017. Prospects and progress of Listeria-based cancer vaccines. Expert Opin. Biol. Ther 17: 1389–1400. [DOI] [PubMed] [Google Scholar]
- 52.Alshamsan A 2017. Nanotechnology-based cancer vaccine. Methods Mol. Biol 1530: 257–270. [DOI] [PubMed] [Google Scholar]
- 53.Cohn L, Chatterjee B, Esselborn F, Smed-Sörensen A, Nakamura N, Chalouni C, Lee BC, Vandlen R, Keler T, Lauer P, et al. 2013. Antigen delivery to early endosomes eliminates the superiority of human blood BDCA3+ dendritic cells at cross presentation. J. Exp. Med 210: 1049–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, Redfern CH, Ferrari AC, Dreicer R, Sims RB, et al. ; IMPACT Study Investigators. 2010. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med 363: 411–422. [DOI] [PubMed] [Google Scholar]
- 55.Moynihan KD, Opel CF, Szeto GL, Tzeng A, Zhu EF, Engreitz JM, Williams RT, Rakhra K, Zhang MH, Rothschilds AM, et al. 2016. Eradication of large established tumors in mice by combination immunotherapy that engages innate and adaptive immune responses. Nat. Med 22: 1402–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Korsholm KS, Agger EM, Foged C, Christensen D, Dietrich J, Andersen CS, Geisler C, and Andersen P. 2007. The adjuvant mechanism of cationic dimethyldioctadecylammonium liposomes. Immunology 121: 216–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zheng L, Huang XL, Fan Z, Borowski L, Wilson CC, and Rinaldo CR Jr. 1999. Delivery of liposome-encapsulated HIV type 1 proteins to human dendritic cells for stimulation of HIV type 1-specific memory cytotoxic T lymphocyte responses. AIDS Res. Hum. Retroviruses 15: 1011–1020. [DOI] [PubMed] [Google Scholar]
- 58.Schoenberger SP 2012. CD69 guides CD4+ T cells to the seat of memory. Proc. Natl. Acad. Sci. USA 109: 8358–8359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ma Z, Li J, He F, Wilson A, Pitt B, and Li S. 2005. Cationic lipids enhance siRNA-mediated interferon response in mice. Biochem. Biophys. Res. Commun 330: 755–759. [DOI] [PubMed] [Google Scholar]
- 60.Wang C, Zhuang Y, Zhang Y, Luo Z, Gao N, Li P, Pan H, Cai L, and Ma Y. 2012. Toll-like receptor 3 agonist complexed with cationic liposome augments vaccine-elicited antitumor immunity by enhancing TLR3-IRF3 signaling and type I interferons in dendritic cells. Vaccine 30: 4790–4799. [DOI] [PubMed] [Google Scholar]
- 61.Honda K, Ohba Y, Yanai H, Negishi H, Mizutani T, Takaoka A, Taya C, and Taniguchi T. 2005. Spatiotemporal regulation of MyD88-IRF-7 signalling for robust type-I interferon induction. Nature 434: 1035–1040. [DOI] [PubMed] [Google Scholar]
- 62.Barrat FJ, Meeker T, Gregorio J, Chan JH, Uematsu S, Akira S, Chang B, Duramad O, and Coffman RL. 2005. Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus. J. Exp. Med 202: 1131–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Johnston D, and Bystryn JC. 2006. Topical imiquimod is a potent adjuvant to a weakly-immunogenic protein prototype vaccine. Vaccine 24: 1958–1965. [DOI] [PubMed] [Google Scholar]
- 64.Yasuda K, Yu P, Kirschning CJ, Schlatter B, Schmitz F, Heit A, Bauer S, Hochrein H, and Wagner H. 2005. Endosomal translocation of vertebrate DNA activates dendritic cells via TLR9-dependent and-independent pathways. J. Immunol 174: 6129–6136. [DOI] [PubMed] [Google Scholar]