

Abstract
Diaryl disulfides and diaryl thiosulfonates were synthesized with the two phenyl rings of all compounds bearing identical halide substituents. Because of structural similarity to the potent antimitotic natural product combretastatin A-4 (CA-4), the compounds were examined for inhibition of tubulin polymerization, and the thiosulfonates were more active than the disulfides. The nine thiosulfonates had IC50 values ranging from 1.2 to 9.1 μM, as compared with 1.3 μM obtained with CA-4. The compounds thus ranged from equipotent with CA-4 to 7-fold less active. The nine disulfides had IC50 values ranging from 1.2 to 5.1 μM, as compared with 0.54 μM obtained with CA-4. The compounds thus ranged from less than half as active as CA-4 to over 9-fold less active. The most active members of each group, 2g and 3c, in the assembly assay were modeled into the colchicine site. Compound 3c had significant hydrophobic interactions with β-tubulin residues CYS 241 and ALA 250, and its thiosulfonate bridge made a hydrogen bond with β-tubulin residue ASN 258. Compound 2g had hydrophobic interactions with β-tubulin residues ALA 250, CYS 241 and ALA 254, but there was no significant interaction of the disulfide bridge with tubulin.
Keywords: Diaryl disulfides, Diaryl thiosulfonates, Combretastatin A-4, Tubulin, Cytotoxicity
Graphical Abstract
Introduction
The attachment of halogen atoms to the structures of the potent antitubulin agent combretastatin A-4 (CA-4) led to analogues with equivalent antitubulin activity (Figure 1) [1]. Aryl thiosulfonates and their direct precursors aryl disulfides are known for their importance in life science, pharmaceutical potential, and food chemistry [2,3]. However, their biological actions, particularly antiproliferative effects, are still unexplored. As a part of our work on the synthesis of CA-4 analogues, we planned and synthesized diaryl disulfides and thiosulfonates bearing halides (Figure 1) in order to investigate their cytotoxicity against the MCF-7 tumor cell line and their potential to inhibit tubulin polymerization.
Figure 1.

Structures of CA-4, halide analogues of CA-4 and diaryl disulfides and diaryl thiosulfonates
Methodologies aimed at direct conversion of halogenated thiols into the corresponding diaryl disulfides, followed by formation of diaryl thiosulfonates in reasonable yields, are limited to a few reports [4–7], making the search for new and effective procedures very desirable. Catalysts such as AgNO3/BF3·OEt2 [8] and Al(H2PO4)3, HNO3 [9] have been used for nitration of aromatic rings. Making use of these systems, our research group successfully achieved the synthesis of diaryl thiosulfonates from thiols.
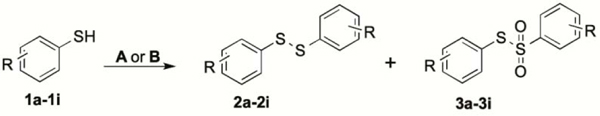
Here we report an extension of the previous protocol that enabled us to perform a one-pot novel synthesis of different symmetrical halide-substituted diaryl disulfides and diaryl thiosulfonates directly from thiols with good yields. Since these compounds can be regarded as analogues of CA-4, they were evaluated for cytotoxicity against the human breast cancer cell line MCF-7 and for activity against purified tubulin.
Results and Discussion
Chemistry
Aspects of the previously described synthetic process were revised to obtain significantly better yields of the target compounds. After establishing optimal conditions, the diverse halogenated thiols underwent ArS-SAr coupling. At room temperature, the reactions were slow, and diaryl disulfides were the predominant products using either system A or B (Table 1). Acetonitrile (ACN) efficiently increased the reaction rate, and heating reaction mixtures to ~ 85 °C resulted in oxidation of the disulfides to thiosulfonates. These compounds were obtained in high yields by the treatment of thiols with Al(H2PO4)3-HNO3 in ACN (System B, Table 1). Both protocols can also be applied to significantly larger reaction mixtures, and this scale-up also provided excellent yields.
Table 1.
| |||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Yield (%) | |||||||
|
|
|||||||
| Entry | Thiol (R) | System A (r.t.) | System B (r.t.) | System B (heat) | |||
| Diaryl disulfide | Diaryl thiosulfonate | Diaryl disulfide | Diaryl thiosulfonate | Diaryl disulfide | Diaryl thiosulfonate | ||
|
| |||||||
| 1 | 1a (o-F) | 2a (74.3) | 3a (10.2) | 2a (72.0) | 3a (12.4) | 2a (6.2) | 3a (87.0) |
| 2 | 1b (m-F) | 2b (54.0) | - | 2b (61.5) | 3bc (20.3) | 2b (16.5) | 3bc (63.0) |
| 3 | 1c (p-F) | 2c (83.0) | 3c (13.7) | 2c (78.3) | 3c (9.0) | 2c (0.0) | 3c (95.0) |
| 4 | 1d (o-Cl) | 2d (63.2) | 3d (15.5) | 2d (69.9) | 3d (11.5) | 2d (0.0) | 3d (83.8) |
| 5 | 1e (m-Cl) | - | - | - | - | 2e (18.5) | 3e (67.3) |
| 6 | 1f (p-Cl) | 2f (70.2) | 3f (12.0) | 2f (75.0) | 3f (13.8) | 2f (0.0) | 3f (92.0) |
| 7 | 1g (o-Br) | 2g (47.6) | 3g (9.2) | 2g (35.8) | 3g (0.0) | 2g (9.3) | 3gc (80.6) |
| 8 | 1h (m-Br) | 2h (59.3) | 3h (0.0) | 2h (44.1) | 3h (0.0) | 2h (10.1) | 3h (84.4) |
| 9 | 1i (p-Br) | 2i (76.6) | 3i (0.0) | 2i (62.7) | 3i (17.0) | 2i (0.0) | 3i (93.0) |
AgNO3/BF3·OEt2, ACN (dry)
Al(H2PO4)3-HNO3, ACN (dry)
reaction not performed
The compounds were unambiguously characterized by NMR and IR spectra and by low and high-resolution mass spectrometry. The fluorinated diaryl disulfides 2a-2c and diaryl thiosulfonates 3a-3c presented the majority of 13C NMR signals as doublets because of 1J, 2J and 3J fluorine (spin ½) coupling with the respective carbons in both aromatic rings. Due to symmetry, the disulfides 2a-2i showed a reduced number of chemical shift signals. When compared to starting materials, the compounds do not present SH group signals in the 1H NMR spectra. NMR data were compared with those from the literature [7, 10–19] and the structures of the synthesized compounds were confirmed. In the IR spectra of 3a-3i, the presence of the SO2 group was verified by strong absorption bands in the regions 1350–1300 cm−1 and 1160–1120 cm−1. The high-resolution mass spectra showed similar fragmentation patterns for the compounds bearing the same aromatic substituents, allowing ready assignment of mass units.
Biological evaluation
The compounds described here were evaluated for effects on the growth of MCF-7 human breast cancer cells and for inhibitory effects on tubulin polymerization and the binding of [3H]colchicine to tubulin, in comparison with the potent colchicine site agent CA-4 [20]. Overall, they had minimal cytotoxic activity and relatively weak effects on colchicine binding, but all the compounds had significant activity as inhibitors of tubulin assembly.
The disulfides are compared with CA-4 in the data presented in Table 2. No disulfide had activity in inhibiting the growth of MCF-7 cells even at 10 μM, as compared with an IC50 of 39 nM obtained with CA-4. All the compounds, however, had activity as inhibitors of tubulin assembly, sometimes approaching that of CA-4. This result resembles those previously described with halogenated (Br, Cl and F) analogues of CA-4 [1].
Table 2:
Bioassays with diaryl disulfides
|
IC50 μM ± SD |
Inhibition of colchicine bindingc % Inhibition ± SD | ||
|---|---|---|---|---|
| Inhibition of tubulin assemblya (relative to CSA4) | Inhibition of MCF-7 cell growthb | |||
|
|
||||
| R = ortho (o), meta (m), para (p) - F, Cl, Br | 5 μM inhibitor | 50 μM inhibitor | ||
|
| ||||
| CA-4 | 0.54 ± 0.06 | 0.039 ± 0.006 | 98 ± 1 | 100 ± 0.04 |
| (2a) o-F | 2.4 ± 0.3 (4.4) | > 10 | 0 ± 4 | 53 ± 5 |
| (2b) m-F | 4.8 ± 0.1 (8.9) | > 10 | 1.4 ± 4 | 12 ± 2 |
| (2c) p-F | 1.9 ± 0.06 (3.5) | > 10 | 2.6 ± 4 | 17 ± 4 |
| (2d) o-Cl | 1.4 ± 0.2 (2.6) | > 10 | 15 ± 5 | 36 ± 2 |
| (2e) m-Cl | 3.1 ± 0.4 (5.7) | > 10 | 12 ± 2 | 18 ± 5 |
| (2f) p-Cl | 4.6 ± 0.7 (8.5) | > 10 | 9.8 ± 2 | 0 ± 4 |
| (2g) o-Br | 1.2 ± 0.1 (2.2) | > 10 | 27 ± 3 | 34 ± 4 |
| (2h) m-Br | 4.0 ±0.5 (7.4) | > 10 | 6.6 ± 5 | 17 ± 4 |
| (2i) p-Br | 5.1 ± 0.5 (9.4) | > 10 | 11 ± 0.3 | 0 ± 2 |
Averages of three independent experiments.
Single determination, except for CA-4.
Averages of two independent experiments.
The most active assembly inhibitors among the diaryl disulfides were 2a, 2c, 2d and 2g, with IC50 values ranging from 1.2 (2.2-fold that of CA-4) to 2.4 μM (4.4-fold that of CA-4). Despite the strong inhibition of assembly observed with these diaryl disulfides, none of them was a strong inhibitor of [3H]colchicine binding at 5 μM. Therefore, they were also examined as inhibitors at a 50 μM concentration, and > 30% inhibition was observed with three of these compounds (Table 2). Compound 2a was the best inhibitor of colchicine binding (53%), while the best inhibitor of tubulin assembly, compound 2g, inhibited colchicine binding by 34%.
Considering only the four most active inhibitors of assembly, three had the halide substituent in the ortho position, with the o-bromo (2g) the most active and the o-fluoro (2a) the least active. The third of the best assembly inhibitors was the p-fluoro compound 2c.
It is important to note that the sulfides are symmetrical compounds, whereas the diaryl thiosulfonates are not, although in the latter the halide substituents on the two phenyl rings are also identical. The data obtained with the thiosulfonates in presented in Table 2. Again, most of the compounds did not inhibit MCF-7 growth at 10 μM, but 3c yielded an IC50 of 7.5 μM and 3f of 10 μM. It is important to point out that the tubulin assembly experiments with the thiosulfonates were performed with a different tubulin preparation than that used with the thioethers, and the values obtained should be compared with those obtained for CA-4 in each experimental series. Using the same criterion with the diaryl thiosulfonates as with the thioethers for activity as inhibitors of assembly, two diaryl thiosulfonates (3c and 3g) were as active as CA-4, while 4 other compounds (3b, 3e, 3h and 3i) were one-half to one-fourth as active as CA-4. The remaining three compounds were 15–20% as active as CA-4 as inhibitors of tubulin assembly. What immediately stands out is that three of the least active compounds (3d, 3e and 3f) all bear chloro substituents on the phenyl rings (Table 3). Compound 3c, with p-fluoro substituents, was the best of the diaryl thiosulfonate compounds for all three biological parameters examined, with activity slightly better than CA-4 as an inhibitor of tubulin assembly. Compound 3g was as active as CA-4 as an inhibitor of tubulin assembly, and it was reasonably active as an inhibitor of colchicine binding.
Table 3:
Bioassays with diaryl thiosulfonates
|
IC50 μM ± SD |
Inhibition of colchicine bindingc % Inhibition ± SD | ||
|---|---|---|---|---|
| Inhibition of tubulin assemblya (relative to CSA4) | Inhibition of MCF-7 cell growthb | |||
|
|
||||
| R = ortho (o), meta (m), para (p) - F, Cl, Br | 5 μinhibitor | 50 μM inhibitor | ||
|
| ||||
| CA-4 | 1.3 ± 0.1 | 0.021 ± 0.004 | 98 ± 1 | 100 ± 0.04 |
| (3a) o-F | 5.5 ± 0.1 (4.2) | > 10 | 0 | 75 ± 5 |
| (3b) m-F | 3.1 ± 0.2 (2.4) | > 10 | 8.9 ± 0.9 | 97 ± 0.1 |
| (3c) p-F | 1.2 ± 0.06 (0.92) | 7.5 ± 0d | 52 ± 5 | 100 ± 0.3 |
| (3d) o-Cl | 6.3 ± 0.3 (4.8) | > 10 | 12 ± 2 | 31 ± 4 |
| (3e) m-Cl | 4.7 ± 0.04 (3.6) | > 10 | 93 ± 1 | |
| (3f) p-Cl | 9.1 ± 0.9 (7.0) | 10 ± 0d | 0 | 75 ± 2 |
| (3g) o-Br | 1.3 ± 0.06 (1.0) | > 10 | 18 ± 5 | 90 ± 0.3 |
| (3h) m-Br | 3.3 ± 0.04 (2.5) | > 10 | 20 ± 1 | 99 ± 1 |
| (3i) p-Br | 4.2 ± 0.2 (3.2) | > 10 | 6.6 ± 4 | 80 ± 2 |
Averages of three independent experiments.
Single determination, except for CA-4, 3c and 3f.
Averages of two independent experiments.
The same value was obtained twice.
Compound 3c is of note because, among the diaryl thiosulfonates, it had the best activity in all three biological parameters studied, and fluorinated stilbenes showed broad-spectrum anticancer activity in different cell lines [1, 21], especially against the MCF-7 cell line [1]. Compound 3f was the least active of the diaryl thiosulfonates in the assembly assay and weakly cytotoxic, with an IC50 of 10 μM against the MCF-7 cells. Aryl chlorine compounds in a different chemotype showed potent cytotoxic activity in the MCF-7 cell line [22]. Other disulfides [23–25] and thiosulfonates [25] have shown cytotoxic activity, but a comparison between these compounds reveals that thiosulfonates are the more active [25].
Perhaps the most striking feature in our results is that the diaryl thiosulfonates were much better inhibitors than the thioethers of [3H]colchicine binding to tubulin, granted that this activity was still very weak compared with that of CA-4. The colchicine inhibition effect was most prominent with the compounds at 50 μM, where all except 3d inhibited binding of the radiolabeled ligand by over 50%.
Molecular modeling
A bridge containing at least one sulfur atom seems to play an important role in binding to the colchicine site of tubulin, probably due to its atomic volume and electronegativity, aided by both steric and hydrophobic factors [23]. However, the differences between the results of the biological assays of diaryl disulfide and diaryl thiosulfonate derivatives indicate that the chemical features of the bridge have a direct influence on the biological potential of the compounds
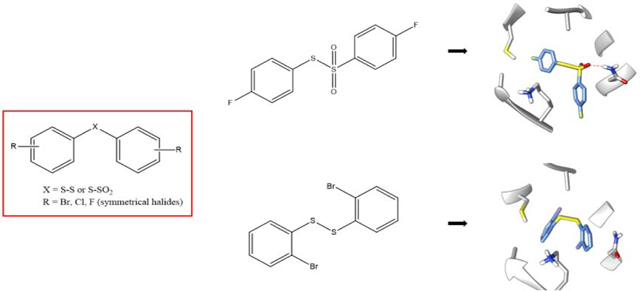
Our modeling studies did show that the diaryl disulfides and diaryl thiosulfonates had noticeable differences in their binding mode positions and in the intermolecular interactions made with the amino acid residues of the active site. The interaction of compound 3c, the most active of the diaryl thiosulfonates, bound in the colchicine site on β-tubulin, is shown in Figure 2A. The thiosulfonate moiety made hydrophobic interactions with CYS 241 and ALA 250 and a hydrogen bond with ASN 258 (Figure 2A). These observed intermolecular interactions are consistent with those already reported for this type of compound at the colchicine site [26]. In contrast, considering the same amino acid residues, the most active disulfide compound 2g only made hydrophobic interactions with ALA 354, CYS 241 and ALA 250 (Figure 2B).
Figure 2.

Binding modes of the diaryl thiosulfonate 3c (A) and the diaryl disulfide 2g (B) at the colchicine site in β-tubulin (PDB ID: 5LYJ).
The absence of the hydrogen bond with ASN 258 made the disulfide molecule more flexible at the active site, and the bridge had a different final conformation in comparison with CA-4. Despite the similarity of the three compounds, the overlay after energy minimization demonstrated that CA-4 and the diaryl thiosulfonate derivatives have a more promising 3D similarity in their final conformations (Figure 3). As a result, the binding position of these two compounds is very similar and, consequently, so are most of the intermolecular interactions. It correlates well with the better results observed in the bioassays with the thiosulfonates.
Figure 3.

Overlay of CA-4 (pink), diaryl thiosulfonate 3c (blue) and diaryl disulfide 2g (orange)
Considering that these compounds were conceived as analogues of CA-4, we should note that typically analogues of CA-4 with the greatest activity have two phenyl rings with asymmetric substituents, and this asymmetry was an important feature for potent antitubulin activity [27, 28]. In addition, a number of biphenyls, modeled on colchicine, were evaluated for antitubulin activity, and these were generally inactive, although the substituents on the two phenyl rings were asymmetric [29]. Overall, these results point to the importance of a rigid bridge between the two phenyl rings, as found in CA-4 or with polar groups capable of making hydrogen bonds, as is the case with the thiosulfonates.
Conclusion
In summary, both synthetic protocols are versatile systems for one-pot synthesis of diaryl disulfides and diaryl thiosulfonates from thiols. The fundamental advantages include efficiency and selectivity, determined by the specific reaction conditions. The thiosulfonates seem more promising than the disulfides as leads for compounds that will bind in the colchicine site of tubulin. Eight of nine thiosulfonates, but only four of nine disulfides, were at least 20% as active as CA-4 as inhibitors of tubulin assembly, and eight of nine thiosulfonates were more potent than all the disulfides as inhibitors of colchicine binding to tubulin. Our molecular models of 2g and 3c bound in the colchicine site were consistent with these findings, in that the thiosulfonate 3c had demonstrated a higher number of intermolecular interactions in comparison with disulfide 2g. This could be rationalized in that the bridge interaction of 3c through a hydrogen bond with tubulin imparted rigidity to the bound ligand, while such an interaction could not occur with 2g. Our next step will be to extend our synthetic work to prepare diaryl sulfonates with asymmetrically substituted phenyl rings to better mimic the asymmetric substituents on the phenyl rings of CA-4.
Experimental
Chemistry
All reagents were analytical grade and used without further purification. TLC was performed on Merck 60 F254 precoated silica plates, and spots were detected by UV light and a solution of sulfur vanillin [0.5 g vanillin in 100 mL sulfuric acid/methanol (40:10)]. Purification of products was carried out by column chromatography using silica gel 60 (0.035–0.075 mm) and preparative thin-layer chromatography on silica gel 60 F254 (0.063–0.2 mm). The solvents employed in the reactions and silica gel column chromatography were purified and dried according to procedures found in the literature [30]. All melting points were determined using a Uniscience of Brazil model 498 instrument.
IR spectra were recorded on KBr pellets or thin-film using Perkin-Elmer 683 FTIR and FTIR MB100 Boomen spectrometers and reported as wave lengths (cm−1). All samples submitted for 1H and 13C NMR spectroscopy were dissolved in CDCl3, which was also the internal reference. 1H NMR (300 MHz) and 13C NMR (75 MHz) spectra were measured with a Bruker Avance DPX-300 spectrometer with CDCl3 as the solvent. Chemical shifts are reported in delta (δ) units, parts per million (ppm) relative to TMS (δ = 0.0) or the residual CHCl3 (δ = 7.24) used as an internal reference standard. The 1H NMR spectra are reported as follows: ppm (multiplicity, coupling constant J/Hz, number of protons). Multiplicity is abbreviated as follows: s (singlet), d (doublet), dd (doublet of doublets), dq (doublet of quartets), dt (doublet of triplets), t (triplet), q (quartet), m (multiplet), td (triplet of doublets). Coupling constants (J) are quoted in Hertz and recorded to the nearest 0.1 Hz. Low-resolution mass spectra were run on a Shimadzu GCMS-QP2010 Plus, working in ionization mode by electronic impact (EI) at 70 eV, where some samples were introduced through a direct exposition probe. High-resolution mass spectrometry (HRMS) was acquired on a Micromass Autospec working in ionization mode by EI at 70 eV and on a MicroTOF Bruker Daltonics instrument and analyzed by electrospray ionization (ESI). The structures assigned to the compounds were thus confirmed by the spectral data and fit with the MS data.
General Procedure using AgNO3 / BF3 · OEt2/ ACN (dry) (System A)
7 mmol of halogenated thiol was dissolved in anhydrous ACN (23 mL). Then, 7.7 mmol of AgNO3 and 0.8 mL of BF3O(Et)2 were added dropwise to the reaction mixture. The mixture was stirred and monitored at intervals by TLC chromatography (hexane / EtOAc 9:1). After 24 h, the reaction was stopped and treated with ice. After extraction with ethyl acetate, the organic layer was washed with distilled water and brine and dried over MgSO4, followed by filtration under reduced pressure. The products were purified by silica gel (GF DE 500 μm, UNIPLAT) chromatography using hexane/EtOAc (9:1) as the eluent.
General Procedure using Al(H2PO4)3 / HNO3 /ACN (dry) (System B)
32 mmol of halogenated thiol was dissolved in anhydrous ACN. 0.16 mmol of aluminium dihydrogen phosphate (freshly prepared) and concentrated HNO3 (3 mL) were added to this solution, which was stirred at room temperature (r.t.) and monitored by TLC for 24 h, when it was observed that the consumption of the starting material was complete. The method was optimized by increasing the temperature to 85 °C, leading to faster formation of the thiosulfonate products with high yields. The workup was performed by neutralizing the mixture by careful addition of a sodium bicarbonate solution. The mixture was extracted using ethyl acetate. The organic layer was washed with distilled water and brine and dried over MgSO4, followed by filtration under reduced pressure. The products were purified by silica gel (GF DE 500 μm, UNIPLAT) chromatography using hexane/EtOAc (9:1) as the eluent.
Disulfides
1-Fluoro-2-[(2-fluorophenyl)disulfanyl]benzene (2a)
Light yellow oil, (A (r.t.) 74.3%, B (r.t.) 72.0%, B (heat) 6.2% yield). FT-IR (KBr, cm−1) νmax: 3071, 1593, 1574, 1470, 1447, 1260, 1122, 821, 752. 1H NMR (300 MHz, CDCl3) δ: 7.02–7.13 (m, 4H), 7.22–7.29 (m, 2H), 7.61 (td, J = 7.7 Hz, J = 1.5 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ: 115.5–115.8 (d, 2JC-F = 21.5 Hz (C-H)), 123.3–123.5 (d, 2JC-F = 17.1 Hz (C-S)), 124.6–124.7 (d, 3JC-F = 3.6 Hz (C-H)), 129.6–129.7 (d, 3JC-F = 7.6 Hz (C-H)), 131.1 (s, (C-H)), 158.8–162.0 (d, 1JC-F = 245.0 Hz (C-F)); MS (EI) m/z (%): 254.05 [M+] (71.79), 190.05 (15.67), 127.05 (100.00), 83 (68.64), 57.00 (16.60).
1-Fluoro-3-[(3-fluorophenyl)disulfanyl]benzene (2b)
Light yellow oil, (A (r.t.) 54.0%, B (r.t.) 61.5%, B (heat) 16.5% yield). FT-IR (KBr, cm−1) νmax: 3062, 1596, 1579, 1472, 1429, 1264, 1215, 876, 774, 675. 1H NMR (300 MHz, CDCl3) δ: 6.93–6.98 (m, 2H), 7.27–7.3 (m, 6H); 13C NMR (75 MHz, CDCl3) δ: 113.7–114.0 (d, 2JC-F = 24.6 Hz (C-H)), 114.0–114.3 (d, 2JC-F = 21.7 Hz (C-H)), 122.5–122.5 (d, 4JC-F = 2.8 Hz (C-H)), 130.3–130.4 (d, 3JC-F = 8.6 Hz (C-H)), 138.7–138.8 (d, 3JC-F = 6.8 Hz (C-S)), 161.3–164.6 (d, 1JC-F = 247.9 Hz (C-F)); MS (EI) m/z (%): 254.00 [M+] (76.46), 221.00 (21.56), 190.05 (23.15), 127.00 (100), 83.00 (88.73).
1-Fluoro-4-[(4-fluorophenyl)disulfanyl]benzene (2c)
White powder, (A (r.t.) 83.0%, B (r.t.) 78.3% yield); m.p. 67–69 °C. FT-IR (KBr, cm−1) νmax: 3069, 1589, 1488, 1229, 1155, 824, 621. 1H NMR (300 MHz, CDCl3) δ: 6.99 (t, J = 8.4, 4H), 7.43 (q, J = 5.1 Hz, J = 3.5 Hz, 4H); 13C NMR (75 MHz, CDCl3) δ: 116.1–116.4 (d, 2JC-F = 22.2 Hz (C-H)), 131.2–131.3 (d, 3JC-F = 7.9 Hz (C-H)), 132.1–132.2 (d, 4JC-F = 3.8 Hz (C-S)), 160.9–164.2 (d, 1JC-F = 246.7 Hz (C-F)); MS (EI) m/z (%): 254.00 [M+] (88.30), 190.00 (17.67), 127.00 (100), 83.00 (62.11).
1-Chloro-2-[(2-chlorophenyl)disulfanyl]benzene (2d)
White powder, (A (r.t.) 63.0%, B (r.t.) 69.9% yield); m.p. 73–75 ºC. FT-IR (KBr, cm−1) νmax: 3056, 1574, 1449, 1432, 1029, 747, 722, 660. 1H NMR (300 MHz, CDCl3) δ: 7.11–7.23 (m, 4H), 7.35 (dd, J = 7.6 Hz, J = 1.4 Hz, 2H), 7.54 (dd, J = 7.9 Hz, J = 1.5 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ: 127.2 (C-H), 127.6 (C-H), 127.8 (C-H), 129.7 (C-H), 131.9 (C-S), 134.4 (C-Cl); MS (EI) m/z (%): 285.90 [M+] (60.24), 287.90 [M+2] (43.12), 289.90 [M+4] (10.16), 215.95 (21.77), 217.90 (21.65), 221.90 (13.47), 142.95 (82.24), 144.90 (30.59), 108.00 (100.00).
1-Chloro-3-[(3-chlorophenyl)disulfanyl]benzene (2e)
Light yellow oil, (B (heat) 18.5% yield). FT-IR (KBr, cm−1) νmax: 3053, 1574, 1460, 1406, 1116, 1072, 1072, 867, 772, 675. 1H NMR (300 MHz, CDCl3) δ: 7.19–7.21 (m, 4H), 7.32–7.36 (m, 2H), 7.48 (s, 2H); 13C NMR (75 MHz, CDCl3) δ: 125.2 (C-H), 126.8 (C-H), 127.4 (C-H), 130.1 (C-H), 135.0 (C-Cl), 138.3 (C-S); MS (EI) m/z (%): 285.95 [M+] (80.55), 287.95 [M+2] (58.93), 289.95 [M+4] (13.85), 218.00 (33.88), 222.00 (27.00), 224.00 (17.37), 143.00 (84.11), 144.95 (31.30), 108.00 (100.00).
1-Chloro-4-[(4-chlorophenyl)disulfanyl]benzene (2f)
White powder, (A (r.t.) 70.2%, B (r.t.) 75.0% yield); m.p. 69–70 °C. FT-IR (KBr, cm−1) νmax: 3078, 1896, 1473, 1396, 1094, 1010, 816, 741. 1H NMR (300 MHz, CDCl3) δ: 7.25 (d, J = 8,6 Hz, 4H), 7.38 (d, J = 8.5 Hz, 4H); 13C NMR (75 MHz, CDCl3) δ: 129.2 (C-H), 133.5 (C-H), 135.1 (C-Cl), 137.6 (C-S); MS (EI) m/z (%): 285.90 [M+] (39.49), 287.90 [M+2] (28.23), 289.90 [M+4] (6.62), 142.95 (100.00), 144.95 (37.24), 108.00 (64.00), 109,00 (11.90).
1-Bromo-2-[(2-bromophenyl)disulfanyl]benzene (2g)
White powder, (A (r.t.) 47.6%, B (r.t.) 35.8%, B (heat) 9.3% yield); m.p. 86–89 °C. FT-IR (KBr, cm−1) νmax: 3052, 1444, 1427, 1016, 742, 704, 650. 1H NMR (300 MHz, CDCl3) δ: 7.06 (td, J = 7.9 Hz, J = 1.3 Hz, 2H), 7.24 (td, J = 7.7 Hz, J = 1.8 Hz, 2H), 7.51 (d, J = 8.0 Hz, 4H); 13C NMR (75 MHz, CDCl3) δ: 121.0 (C-Br), 126.9 (C-H), 127.9 (C-H), 128.2 (C-H), 132.9 (C-H), 136.1 (C-S); MS (EI) m/z (%): 373.75 [M+] (14.57), 375.75 [M+2] (29.97), 377.75 [M+4] (16.38), 215.90 (52.20), 186.85 (10.57), 188.85 (11.03), 108.00 (100.00).
1-Bromo-3-[(3-bromophenyl)disulfanyl]benzene (2h)
Dark yellow oil, (A (r.t.) 59.3%, B (r.t.) 44.1%, B (heat) 10.1% yield). FT-IR (KBr, cm−1) νmax: 3051, 1572, 1561, 1456, 1399, 1066, 771, 743, 674. 1H NMR (300 MHz, CDCl3) δ: 7.14 (t, J = 7.9 Hz, 2H), 7.33–7.40 (m, 4H), 7.62 (d, J = 1.6 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ: 123.1 (C-Br), 125.7 (C-H), 129.8 (C-H), 130.4 (2C-H), 138.5 (C-S); MS (EI) m/z (%): 373.75 [M+] (21.00), 375.75 [M+2] (41.19), 377.75 [M+4] (23.11), 215.95 (16.29), 186.85 (14.30), 188.85 (14.81), 108.00 (100.00).
1-Bromo-4-[(4-bromophenyl)disulfanyl]benzene (2i)
White powder, (A (r.t.) 76.6%, B (r.t.) 62.7% yield); m.p. 84–85 ºC. FT-IR (KBr, cm−1) νmax: 3073, 1468, 1384, 1080, 1006, 826, 812, 723.1H NMR (300 MHz, CDCl3) δ: 7.31 (d, J = 8.59 Hz, 4H), 7.41 (d, J = 8.3 Hz, 4H); 13C NMR (75 MHz, CDCl3) δ: 121.5 (C-Br), 129.4 (C-H), 132.2 (C-H), 135.7 (C-S); MS (EI) m/z (%): 373.80 [M+] (23.72), 375.80 [M+2] (48.89), 377.80 [M+4] (26.09), 186.90 (40.59), 188.90 (41.60), 108.00 (100.00).
Thiosulfonates
1‐Fluoro‐2‐[(2‐fluorobenzenesulfonyl)sulfanyl]benzene (3a)
Light yellow oil, (A (r.t.) 10.2%, B (r.t.) 12.4%, B (heat) 87.0% yield). FT-IR (KBr, cm−1) νmax: 3071, 1574, 1470. 1447, 1260, 1222, 1156, 821, 752, 671, 549. 1H NMR (300 MHz, CDCl3) δ: 7.02 (td, J = 8.5 Hz, J = 1.0 Hz, 1H), 7.12 (td, J = 7.5 Hz, J = 1.1 Hz, 1H), 7.15 (td, J = 6.4 Hz, J = 1.1 Hz, 1H), 7.23 (td, J = 8.5 Hz, J = 1.0 Hz, 1H), 7.42–7.52 (m, 3H), 7.57–7.64 (m, 1H); 13C NMR (75 MHz, CDCl3) δ: 114.6–114.9 (d, 2JC-F = 18.9 Hz (C-S)), 116.2–116.5 (d, 2JC-F = 22.8 Hz (C-H)), 117.5–117.8 (d, 2JC-F = 21.1 Hz (C-H)), 123.8–123.9 (d, 3JC-F = 3.4 Hz (C-H)), 125.0–125.1 (d, 3JC-F = 4.0 Hz (C-H)), 130.2 (s, (C-H)), 131.1–131.3 (d, 2JC-F = 12.0 Hz (C-S)), 134.5–134.6 (d, 3JC-F = 8.0 Hz (C-H)), 136.3–136.4 (d, 3JC-F = 8.5 Hz (C-H)), 139.1 (s, (C-H)), 157.2–160.6 (d, 1JC-F = 258.8 (C-F)), 161.2–164.6 (d, 1JC-F = 251.8 Hz (C-F)); HRMS (ESI) C12H8F2O2S2 [M+] m/z (%) calc. 285.9933, found 285.9933.
1-Fluoro-3-[(3-fluorobenzenesulfonyl)sulfanyl]benzene (3b)
Yellow oil, (B (r.t.) 20.3%, B (heat) 63.0% yield). FT-IR (KBr, cm−1) νmax: 3062, 1596, 1582, 1472, 1429, 1264, 1215, 876, 774, 675, 520. 1H NMR (300 MHz, CDCl3) δ: 7.04–7.16 (m, 3H), 7.21–7.31 (m, 4H), 7.35–7.41 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 114.6–114.9 (d, 2JC-F = 24.6 Hz (C-H)), 118.8–119.1 (d, 2JC-F = 21.1 Hz (C-H)), 121.0–121.3 (d, 2JC-F = 21.1 Hz (C-H)), 123.0–123.3 (d, 2JC-F = 22.3 Hz (C-H)), 123.3 (s, (C-H)), 128.9–129.0 (d, 3JC-F = 8.3 Hz (C-S)), 130.6–130.7 (d, 3JC-F = 8.5 Hz (C-H)), 130.8 (s, (C-H)),132.1–132.2 (d, 3JC-F = 3.0 Hz (C-H)), 144.3–144.4 (d, 3JC-F = 6.4 Hz (C-S)), 160.3–163.7 (d, 1JC-F = 251.4 Hz (C-F)), 160.6–163.9 (d, 1JC-F = 250.1 Hz (C-F)); HRMS (ESI) C12H8F2O2S2 [M+] m/z (%), calc. 285.9933, found 285.9934.
1-Fluoro-4-[(4-fluorobenzenesulfonyl)sulfanyl]benzene (3c)
White powder, (A (r.t.) 13.7%, B (r.t.) 9.0%, B (heat) 95.0% yield); m.p. 67–69 °C. FT-IR (KBr, cm−1) νmax: 3101, 3070, 1585, 1489, 1335, 1238, 1146, 1072, 833, 660, 586, 517. 1H NMR (300 MHz, CDCl3) δ: 7.00–7.12 (m, 4H), 7.34 (q, J = 8.6 Hz, J = 1.7 Hz, 2H), 7.56 (q, J = 8.8 Hz, J = 1.9 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ: 116.0–116.3 (d, 2JC-F = 22.3 Hz, (C-H)), 116.8–117.1 (d, 2JC-F = 22,0 Hz, (C-H)), 123.2 (d, 4JC-F = 2.9 Hz (C-S)), 130.4–130.5 (d, 3JC-F = 9.2 Hz, (C-H)), 138.8–138.9 (d, 3JC-F = 9.2 Hz, (C-H)), 163.2–166.5 (d, 1JC-F = 254.4 Hz, (C-F)), 163.9–167.3 (d, 1JC-F = 255.9 Hz, (C-F)); HRMS (ESI) C12H8F2O2S2 [M+] m/z (%), calc. 285.9933, found 285.9927.
1-Chloro-2-[(2-chlorobenzenesulfonyl)sulfanyl]benzene (3d)
White powder, (A (r.t.) 15.5%, B (r.t.) 11.5, B (heat) 83.8% yield); m.p. 81–83 °C. FT-IR (KBr, cm−1) νmax: 3055, 1821, 1783, 1597, 1573, 1564, 1477, 1431, 1029, 747, 723, 660. 1H NMR (300 MHz, CDCl3) δ: 7.19–7.40 (m, 4H), 7.48–7.58 (m, 3H), 7.63 (dd, J = 7.7 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ: 126.6 (C-H), 126.8 (C-Cl), 127.6 (C-H), 130.2 (C-H), 131.0 (C-H), 132.4 (C-H), 133.0 (C-Cl), 133.1 (C-H), 134.7 (C-H), 139.9 (C-H), 140.3(C-S), 140.4 (C-S); MS (EI) m/z (%): 317.85 [M+] (21.93), 319.85 [M+2] (16.20), 321.85 [M+4] (3.69), 174.95 (34.49), 176.90 (12.33), 158.95 (100.0), 160.90 (36.48), 143,95 (30.87), 107.95 (55.79), 110.95 (41.25), 112.95 (13.66).
1-Chloro-3-[(3-chlorobenzenesulfonyl)sulfanyl]benzene (3e)
Yellow oil, (B (heat) 67.3% yield). FT-IR (KBr, cm−1) νmax: 2926, 1573, 1460, 1338, 1151, 1074, 783, 676, 604, 535. 1H NMR (300 MHz, CDCl3) δ: 7.15–7.29 (m, 3H), 7.31–7.44 (m, 3H), 7.47–7.53 (m, 2H); 13C NMR (75 MHz, CDCl3) δ: 125.6 (C-H), 127.6 (C-H), 129.0 (C-Cl), 130.2 (C-H), 130.6 (C-H), 131.9 (C-H), 134.0 (C-H), 134.6 (C-H), 135.1 (C-Cl), 135.3 (C-S), 136.1 (C-H), 144.0 (C-S); MS (EI) m/z (%): 317.95 [M+] (18.11), 319.95[M+2] (13.14), 321.90 [M+4] (3.84), 281.05 (9.02), 207.00 (14.50), 175.95 (41.23), 177.00 (16.82), 159.00 (74.25), 160.95 (27.28), 143.0 (34.77), 143.95 (71.66), 146.00 (28.74), 111.00 (100.00), 108.00 (94.99), 75.00 (71.82).
1-Chloro-4-[(4-chlorobenzenesulfonyl)sulfanyl]benzene (3f)
White powder, (A (r.t.) 12.0%, B (r.t.) 13.8%, B (heat) 92.0% yield); m.p. 134–135 °C. FT-IR (KBr, cm−1) νmax: 2923, 1573, 1450, 1330, 1149, 1033, 756, 597. 1H NMR (300 MHz, CDCl3) δ: 7.27–7.31 (m, 4H), 7.41 (dt, J = 8.9 Hz, J = 2.1 Hz, 2H), 7.50 (dt, J = 9.1 Hz, J = 1.7 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ: 125.9 (C-Cl), 128.9 (C-H), 129.2 (C-H), 129.9 (C-H), 137.6 (C-H), 138.5 (C-S), 140.5 (C-Cl), 141.2 (C-S); HRMS (ESI) C12H8Cl2O2S2 [M+] m/z (%), calc. 317.9342, found 317.9342.
1-Bromo-2-[(2-bromobenzenesulfonyl)sulfanyl]benzene (3g)
White powder, (A (r.t.) 9.2%, B (heat) 80.6% yield); m.p. 128–131 ºC. FT-IR (KBr, cm−1) νmax: 3082, 1569, 1446, 1326, 1149, 1018, 752, 594, 540. 1H NMR (300 MHz, CDCl3) δ: 7.25–7.29 (m, 2H), 7.33 (td, J = 4.6 Hz, J = 0.9 Hz, 1H), 7.41 (td, J = 4.6 Hz, J = 1.0 Hz, 1H), 7.52 (dd, J = 4.7 Hz, J = 0.8 Hz, 1H), 7.56 (dd, J = 4.8 Hz, J = 1.0 Hz, 1H), 7.68 (dd, J = 4.7 Hz, J = 1.1 Hz, 1H), 7.78 (dd, J = 4.7 Hz, J = 0.7 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ: 121.3 (C-Br), 127.2 (C-H), 128.3 (C-H), 129.1 (C-Br), 131.2 (C-S), 131.3 (C-H), 133.0 (C-H), 133.6 (C-H), 134.6 (C-H), 136.0 (C-H), 140.1 (C-H), 142.0 (C-S); HRMS (ESI) C12H8Br2O2S2 [M+] m/z (%), calc. 405.8332, found 405.8334.
1-Bromo-3-[(3-bromobenzenesulfonyl)sulfanyl]benzene (3h)
White powder, (B (heat) 84.4% yield); m.p. 91–93 ºC. FT-IR (KBr, cm−1) νmax: 3086, 3070, 1570, 1558, 1458, 1327, 1146, 787, 756, 671, 602, 528. 1H NMR (300 MHz, CDCl3) δ: 7.26 (t, J = 7.8 Hz, 1H), 7.33 (td, J = 7.8 Hz, J = 1.8 Hz, 2H), 7.44 (t, J = 1.8 Hz, 1H), 7.49 (dq, J = 7.9 Hz, J = 0.9 Hz, 1H), 7.63 (dq, J = 7.9 Hz, J+ = 1.2 Hz, 2H), 7.72 (dq, J = 78.0 Hz, J = 1.0 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ: 122.9 (2C-Br), 126.0 (C-H), 129.3 (C-S), 130.4 (C-H), 130.5 (C-H), 130.9 (C-H), 134.8 (C-H), 135.1 (C-H), 136.9 (C-H), 138.9 (C-H), 144.1 (C-S); HRMS (ESI) C12H8Br2O2S2 [M+] m/z (%), calc. 405.8332, found 405.8326.
1-Bromo-4-[(4-bromobenzenesulfonyl)sulfanyl]benzene (3i)
White powder, (A (r.t.) 17.0%, B (heat) 93.0% yield); m.p. 153–154 °C. FT-IR (KBr, cm−1) νmax: 3086, 2924, 1566, 1331, 1146, 1068, 1007, 822, 741, 598, 552. 1H NMR (300 MHz, CDCl3) δ: 7.22 (dt, J = 9.2 Hz, J = 2.2 Hz, 2H), 7.42 (dt, J = 9.1 Hz, J = 2.6 Hz, 2H), 7.50 (dt, J = 9.6 Hz, J = 2.4 Hz, 2H), 7.58 (dt, J = 9.1 Hz, J = 2.1 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ: 126.6 (C-Br), 127.0 (C-Br), 129.0 (C-H), 129.2 (C-S), 132.3 (C-H), 132.9 (C-H), 137.8 (C-H), 141.9 (C-S); HRMS (ESI) C12H8Br2O2S2 [M+] m/z (%), calc. 405.8332, found 405.8314.
Bioassays
The tubulin assembly [31] and inhibition of colchicine binding to tubulin [32] assays were performed as described before. In the assembly assay, the tubulin concentration was 10 μM, and the parameter measured was the extent of assembly after 20 min at 30 °C. In the colchicine binding assay, the tubulin concentration was 1.0 μM, the [3H]colchicine concentration was 5.0 μM, and the inhibitor concentration was 5 or 50 μM, as indicated. Incubation was for 10 min at 37 °C, a time point chosen because the control reaction is about 40–60% complete. The MCF-7 cytotoxicity assay was performed as described by Monks et al., and protein, stained by sulforhodamine B, was the cell parameter measured [33].
Molecular modeling
The 3D structures of the most active disulfide and thiosulfonate compounds were drawn using the program MarvinSketch 16.9.5 (ChemAxon Ltd.). The structural optimization was made through the PM7 semi-empirical method incorporated in the software MOPAC2016 [34].
To determine the potential binding model assumed by both compounds (2g and 3c), molecular docking simulations were carried out using AutoDockVINA [35]. For the simulations, the protein from PDB ID 5LYJ with CA-4 as the bound ligand (ligand PDB ID: 7BA) was used and the grid box determined was large enough to contain the binding site of the target. Before each simulation, the crystallographic ligand was deleted. The best result obtained by the molecular docking simulation for both compounds was energy minimized with the program GROMAC 5 package [36] and CHARMM force field [37]. The properties of the solvent were determined based on the TIP3P water model, and a cubic box that guarantees a space of 1.2 nm between the protein and box walls was used. All system charges were neutralized with the addition of ions at physiological concentration (0.15 μM). The algorithm steepest descent minimization and conjugated gradients were used with a maximum force of 10 KJ/mol.
Supplementary Material
Development high yield and versatile one-pot synthesis for diaryl disulfides and diaryl thiosulfonates;
The thiosulfonates have demonstrated to be promising as a lead compound for the development of new inhibitors of colchicine site of tubulin.
Molecular modeling studies were consistent with the in vitro results
Acknowledgments
This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Brazil (CAPES) - Finance Code 001. The authors are also indebted to Conselho Nacional de Desenvolvimento Científico e Tecnológico - Brazil (CNPq), PROPP-UFMS and Fundação de Apoio ao Desenvolvimento do Ensino, Ciência e Tecnologia – Brazil (FUNDECT – MS). This research was supported in part by the Developmental Therapeutics Program in the Division of Cancer Treatment and Diagnosis of the National Cancer Institute, which includes federal funds under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
Conflicts of interest
The authors declare that the research was conducted in the absence of any conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Pettit GR, Minardi MD, Rosenberg HJ, Hamel E, Bibby MC, Martin SW, Jung MK, Pettit RK, Cuthbertson TJ, Chapuis JC, Antineoplastic agents. 509. Synthesis of fluorcombstatin phosphate and related 3-halostilbenes, J. Nat. Prod 68 (2005) 1450–1458. [ 10.1021/np058038i]. [DOI] [PubMed] [Google Scholar]
- [2].Mampuys P, McElroy CR, Clark JH, Orru RVA, Maes BUW, Thiosulfonates as emerging reactants: synthesis and applications, Adv. Synth. Catal 362 (2020) 3–64. [ 10.1002/adsc.201900864]. [DOI] [Google Scholar]
- [3].Xiao X, Xue J, Jiang X, Polysulfurating reagent design for unsymmetrical polysulfide construction, Nat. Commun 9 (2018) 2191. [ 10.1038/s41467-018-04306-55]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Mahieu J-P, Gosselet M, Sebille B, Beuzard Y, Synthesis of new thiosulfonates and disulfides from sulfonyl chlorides and thiols, Synt. Commun 16 (1986) 1709–1722. [ 10.1080/00397918608056430]. [DOI] [Google Scholar]
- [5].Keshari T, Kapoorr R, Yadav LDS, Iron(III)-catalyzed radical cross-coupling of thiols with sodium sulfinates: a facile access to thiosulfonates, Synlett. 27 (2016) 1878–1882. [ 10.1055/s-0035-1562101]. [DOI] [Google Scholar]
- [6].Yang Z, Shi Y, Zhan Z, Zhang H, Xing H, Lu R, Zhang Y, Guan M, Wu Y, Sustainable electrocatalytic oxidant-free syntheses of thiosulfonates from thiols, ChemElectroChem. 5 (2018) 3619–3623. [ 10.1002/celc.201801058]. [DOI] [Google Scholar]
- [7].Sobhani S, Aryanejad S, Maleki MF, Zinc dichromate trihydrate (ZnCr2O7×3H2O) as an efficient reagent for the one-pot synthesis of thiosulfonates from thiols, Synlett 3 (2011) 319–322. [ 10.1055/s-0030-1259506]. [DOI] [Google Scholar]
- [8].Olah GA, Fung AP, Narang SC, Olah JA, Aromatic substitution. 48. Boron trifluoride catalyzed nitration of aromatics with silver nitrate in acetonitrile solution, J. Org. Chem 46 (1981) 3533–3537. [ 10.1021/jo00330a032]. [DOI] [Google Scholar]
- [9].Bharadwaj SK, Hussain S, Kar M, Chaudhuri MK, Al(H2PO4)3: An efficient catalyst for nitration of organic compounds with nitric acid, Catal. Commun 9 (2008) 919–923. [ 10.1016/j.catcom.2007.09.020]. [DOI] [Google Scholar]
- [10].Talla A, Driessen B, Straathof NJW, Milroy LG, Brunsveld L, Hessel V, Noël T, Metal-free photocatalytic aerobic oxidation of thiols to disulfides in batch and continuous-flow, Adv. Synth. Catal 357 (2015) 2180–2186. [ 10.1002/adsc.201401010]. [DOI] [Google Scholar]
- [11].Howard JL, Schotten C, Alston ST, Browne DL, Preparation of difluoromethylthioethers through difluoromethylation of disulfides using TMS-CF2H, Chem. Commun 52 (2016) 8448–8451. [ 10.1039/c6cc02693a]. [DOI] [PubMed] [Google Scholar]
- [12].Paul S, Islam SM, Oxidative dehydrogenation of thiols to disulfides at room temperature using silica supported iron oxide as an efficient solid catalyst, RSC Adv. 6 (2016) 95753–95759. [ 10.1039/C6RA19832E]. [DOI] [Google Scholar]
- [13].Zheng Y, Qing FL, Huang Y, Xu XH, Tunable and practical synthesis of thiosulfonates and disulfides from sulfonyl chlorides in the presence of tetrabutylammonium iodide, Adv. Synth. Catal 358 (2016) 3477–3481. [ 10.1002/adsc.201600633]. [DOI] [Google Scholar]
- [14].Leng J, Wang SM, Qin HL, Chemoselective synthesis of diaryl disulfides via a visible light-mediated coupling of arenediazonium tetrafluoroborates and CS2, Beilstein J. Org. Chem 13 (2017) 903–909. [ 10.3762/bjoc.13.91]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Chatterjee T, Ranu BC, Aerobic oxidation of thiols to disulfides under ball-milling in the absence of any catalyst, solvent, or base, RSC Adv. 3 (2013) 10680. [ 10.1039/c3ra41996g]. [DOI] [Google Scholar]
- [16].Zhao X, Liu TX, Zhang G, Synthesis of thiosulfonates via CuI-catalyzed reductive coupling of arenesulfonyl chlorides using Na2SO3 or NaHSO3 as reductants, Asian J. Org. Chem 6 (2017) 677–681. [ 10.1002/ajoc.201700090]. [DOI] [Google Scholar]
- [17].Iwata S, Senoo M, Hata T, Urabe H, Synthesis of S-aryl arenethiosulfonates from N,N-di(arenesulfonyl) hydrazines: reduction of sulfonyl chlorides with an organic reagent, Heteroat. Chem 24 (2013) 336–344. [ 10.1002/hc.21088]. [DOI] [Google Scholar]
- [18].Peng Z, Zheng X, Zhang Y, An D, Dong W, H2O2-mediated metal-free protocol towards unsymmetrical thiosulfonates from sulfonyl hydrazides and disulfides in PEG-400, Green Chem. 20 (2018) 1760–1764. [ 10.1039/c8gc00381e]. [DOI] [Google Scholar]
- [19].Xia M, Cheng J, Catalyst- and oxidant-free coupling of disulfides with H-phosphine oxide: construction of P–S bond leading to thiophosphinates, Tetrahedron Lett. 57 (2016) 4702–4704. [ 10.1016/j.tetlet.2016.09.021]. [DOI] [Google Scholar]
- [20].Lin CM, Ho HH, Pettit GR, Hamel E, Antimitotic natural products combretastatin A-4 and combretastatin A-2: studies on the mechanism of their inhibition of the binding of colchicine to tubulin, Biochemistry 28 (1989) 6984–6991. [ 10.1021/bi00443a031]. [DOI] [PubMed] [Google Scholar]
- [21].De Filippis B, Ammazzalorso A, Fantacuzzi M, Giampietro L, Maccallini C, Amoroso R, Anticancer activity of stilbene-based derivatives, ChemMedChem 12 (2017) 558–570. [ 10.1002/cmdc.201700045]. [DOI] [PubMed] [Google Scholar]
- [22].De Martino G, La Regina G, Coluccia A, Edler MC, Barbera MC, Brancale A, Wilcox E, Hamel E, Artico M, Silvestri R, Arylthioindoles, potent inhibitors of tubulin polymerization, J. Med. Chem 47 (2004) 6120–6123. [ 10.1021/jm049360d]. [DOI] [PubMed] [Google Scholar]
- [23].dos A. dos Santos E, Hamel E, Bai R, Burnett JC, Tozatti CSS, Bogo D, Perdomo RT, Antunes AMM, Marques MM, de Matos MFC, de Lima DP, Synthesis and evaluation of diaryl sulfides and diaryl selenide compounds for antitubulin and cytotoxic activity, Bioorg. Med. Chem. Lett 23 (2013) 4669–4673. [ 10.1016/j.bmcl.2013.06.009]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Siyo V, Schäfer G, Hunter R, Grafov A, Grafona I, Nieger M, Katz AA, Parker MI, Kaschula CH, The cytotoxicity of the ajoene analogue BisPMB in WHCO1 oesophageal cancer cells is mediated by CHOP/GADD153, Molecules 26 (2017) 892. [ 10.3390/molecules22060892]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Smith M, Hunter R, Stellenboom N, Kusza DA, Parker MI, Hammouda ANH, Jackson G, Kaschula CH, The cytotoxicity of garlic-related disulphides and thiosulfonates in WHCO1 oesophageal cancer cells is dependent on S-thiolation and not production of ROS, Biochim. Biophys. Acta - Gen. Subj 1860 (2016) 1439–1449. [ 10.1016/j.bbagen.2016.03.032]. [DOI] [PubMed] [Google Scholar]
- [26].Nantes CI, Pereira ID, Bai R, Hamel E, Burnett JC, De Oliveira RJO, de Matos MFC, Beatriz A, Yonekawa MKA, Perdomo RT, de Lima DP, Bogo D, dos Santos E. dos A., S-(4-methoxyphenyl)-4-methoxybenzenesulfonothioate as a promising lead compound for the development of a renal carcinoma agent, ChemMedChem 15 (2020) 449–458. [ 10.1002/cmdc.201900566]. [DOI] [PubMed] [Google Scholar]
- [27].Cushman M, Nagarathnam D, Gopal D, Chakraborti AK, Lin CM, Hamel E, Synthesis and evaluation of stilbene and dihydrostilbene derivatives as potential anticancer agents that inhibit tubulin polymerization, J. Med. Chem 34 (1991) 2579–2588. [ 10.1021/jm00112a036]. [DOI] [PubMed] [Google Scholar]
- [28].Cushman M, He HM, Lin CM, Hamel E, Synthesis and evaluation of a series of benzylaniline hydrochlorides as potential cytotoxic and antimitotic agents acting by inhibition of tubulin polymerization, J. Med. Chem 36 (1993) 2817–2821. [ 10.1021/jm00071a012]. [DOI] [PubMed] [Google Scholar]
- [29].Itoh Y, Brossi A, Hamel E, George JLFC, Colchicine models: synthesis and antitubulin activity of 2′‐monosubstituted and 2′,5‐disubstituted 2,3,4,4′‐tetramethoxy‐1,1′‐biphenyls. Synthesis of 4,4′,5′,6′‐tetramethoxy‐1,1′‐biphenyl‐2,3′‐dicarboxylic acid, Helv. Chim. Acta 72 (1989) 196–204. [ 10.1002/hlca.19890720203]. [DOI] [Google Scholar]
- [30].Perrin DD, Armarego WLF, Purification of Laboratory Chemicals, third ed., Pergamon Press, Oxford, 1988. [Google Scholar]
- [31].Hamel E, Evaluation of antimitotic agents by quantitative comparisons of their effects on the polymerization of purified tubulin, Cell Biochem. Biophys 38 (2003) 1–22. [ 10.1385/CBB:38:1:1]. [DOI] [PubMed] [Google Scholar]
- [32].Verdier-Pinard P, Lai JY, Yoo HD, Yu J, Marquez B, Nagle DG, Nambu M, White JD, Falck JR, Gerwick WH, Day BW, Hamel E, Structure-activity analysis of the interaction of curacin A, the potent colchicine site antimitotic agent, with tubulin and effects of analogs on the growth of MCF-7 breast cancer cells, Mol. Pharmacol 53 (1998) 62–76. [ 10.1124/mol.53.1.62]. [DOI] [PubMed] [Google Scholar]
- [33].Monks A, Scudiero D, Skehan P, Shoemaker R, Paull K, Vistica D, Hose C, Langley J, Cronise P, Vaigro-Wolff A, Gray-Goodrich M, Campbell H, Mayo J, Boyd M, Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines, J. Natl. Cancer Inst 83 (1991) 757–766. [ 10.1093/jnci/83.11.757]. [DOI] [PubMed] [Google Scholar]
- [34].James J U, Stewart P, Computational Chemistry, Colorado Springs, CO, MOPAC2016. [Google Scholar]
- [35].Olson OT, Arthur J, AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading, NIH Public Access 31 (2010) 455–461. [ 10.1002/jcc.21334.AutoDock]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJC, GROMACS: fast, flexible, and free, J. Comput. Chem 26 (2005) 1701–1718. [ 10.1002/jcc.20291]. [DOI] [PubMed] [Google Scholar]
- [37].Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, Mackerell AD, CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields, J. Comput. Chem 31 (2010) 671–690. [ 10.1002/jcc.21367]. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.