

Abstract
We previously identified a novel molecular subtype of idiopathic pulmonary fibrosis (IPF) defined by increased expression of cilium-associated genes, airway mucin gene MUC5B, and KRT5 marker of basal cell airway progenitors. Here we show the association of MUC5B and cilia gene expression in human IPF airway epithelial cells, providing further rationale for examining the role of cilium genes in the pathogenesis of IPF. We demonstrate increased multiciliogenesis and changes in motile cilia structure of multiciliated cells both in IPF and bleomycin lung fibrosis models. Importantly, conditional deletion of a cilium gene, Ift88 (intraflagellar transport 88), in Krt5 basal cells reduces Krt5 pod formation and lung fibrosis, whereas no changes are observed in Ift88 conditional deletion in club cell progenitors. Our findings indicate that aberrant injury-activated primary ciliogenesis and Hedgehog signaling may play a causative role in Krt5 pod formation, which leads to aberrant multiciliogenesis and lung fibrosis. This implies that modulating cilium gene expression in Krt5 cell progenitors is a potential therapeutic target for IPF.
Keywords: injury-induced multiciliogenesis, abnormal structure of multiciliated cells, Ift88, Krt5 cell
Idiopathic pulmonary fibrosis (IPF) is a largely untreatable and heterogeneous disease that has a strong genetic basis but is also associated with environmental factors such as cigarette smoke and occupational exposures. Genetic studies to date point to at least two subtypes of disease, one caused by a common variant in the airway mucin MUC5B gene and the other by common and rare variants in telomerase pathway genes (1). Our transcriptome profiling study of whole lung tissue also identified two molecular subtypes of IPF, one associated with increased expression of airway genes (cilia-associated structural components and transcription factors, mucins, and keratins) and the other with increased expression of alveolar genes (surfactant and others) (2). We have proposed that the airway subtype of IPF is a mucociliary disease caused by recurrent injury/inflammation/repair at the bronchoalveolar junction, which is initiated and exacerbated by overexpression of MUC5B, reduced mucociliary function, retention of particles, and enhanced lung injury (3). On the basis of the relationship between the MUC5B promoter variant rs35705950 and excess production of MUC5B, specifically at the bronchoalveolar junction, it has been suggested that too much MUC5B impairs mucociliary function (4, 5), causes excessive retention of inhaled substances, and, over time, the foci of lung injury lead to scar tissue and persistent fibroproliferation that expands and displaces normal lung tissue. Given the importance of environmental exposures, it is logical to speculate that common inhaled particles might cause exaggerated interstitial injury in individuals who have defects in mucociliary clearance (MCC) and mucosal host defense.
The expression of cilium-associated genes defines a novel molecular subtype of IPF that is associated with the presence of microscopic honeycombing but not fibroblastic foci. Moreover, higher expression of cilium genes is associated with expression of the airway mucin gene MUC5B and KRT5 marker of basal cell airway progenitors (2). Ectopic presence of KRT5+ progenitor populations in the fibrotic areas has been observed in IPF and mouse models of injury, mostly in the form of Krt5/KRT5 pods. In mice, it has been shown that Krt5 pods form cysts that persist and generate cyst-like structures, similar to microscopic honeycombing observed in humans (6–10). This suggests the potential importance of cilium gene expression in the injured lung. Moreover, primary cilia are transiently present on Krt5 basal cells in adult mouse airways during normal airway cell differentiation and repair after respiratory virus injury (11), and indeed multiciliated cells originate from primary ciliated Krt5 cells through multiciliogenesis (12, 13), although club cells are also progenitors for multiciliated cells (14). No study to date has examined the potential role of cilium gene expression in airway regeneration after injury and the development of lung fibrosis.
Here we show the association of MUC5B and cilia gene expression in IPF airway epithelial cells, experimentally confirming our initial observations from whole lung tissue. In the bleomycin-induced injury murine model, we demonstrate dynamic changes in bleomycin-induced multiciliogenesis for both the number of multiciliated cells and changes in the motile cilia structure of multiciliated cells. In addition, conditional deletion of a cilium-associated gene Ift88 in Krt5 basal progenitor cells reduces Krt5 pod formation and lung fibrosis and delays multiciliogenesis after bleomycin injury. These findings have broad implications for the functional role of cilium genes in epithelial biology and epithelial–stromal crosstalk in the lung.
Methods
Lung Tissue Gene Expression
Our published lung tissue gene expression dataset on Affymetrix Gene ST 1.0 microarrays was used (2).
Airway Cell Gene Expression
RNA was extracted using AllPep DNA/RNA kit (Qiagen). qRT-PCR for MUC5B was performed using Taqman reagents (Thermo Fisher, Hs00861595_m1) and normalized to GAPDH. Another aliquot of total RNA was labeled and hybridized to Agilent SurePrint G3 Human Gene Expression Microarray. Array data were log-transformed and quantile normalized. Principal components analysis was used to exclude outliers (more than 3 standard deviations from the mean in any given PC). Data were then analyzed using a mixed-effects model controlling for age, sex, smoking status, and batch effects (random effect). Analyses were done in R Studio.
Air–Liquid Interface Culture of Human Bronchial Epithelial Cells
Primary airway epithelial cells were obtained and cultured as previously described (15).
Mouse Husbandry
Mice were housed and maintained as previously described (16).
Mouse Strains
C57BL/6J male mice were purchased from Jackson Laboratory. Krt5-CreERT2 (17) and were crossed to Ift88f/+ (18) to produce Krt5-CreERT2-Ift88f/+ and Krt5-CreERT2/+-Ift88+/+. Heterozygous males and females were bred to obtain Krt5-CreERT2-Ift88f/f, Krt5-CreERT2-Ift88f/+, and Krt5-CreERT2-Ift88f/+.
Bleomycin Exposure
All mice began bleomycin treatment between 8 and 12 weeks of age, as previously described (16). Studies were conducted after a single bleomycin challenge with 2.5 U/kg bleomycin. Mice were injected with EdU (5-ethynyl-2′-deoxyuridine) 24 hours before harvesting.
Hydroxyproline
The entire right lung was removed and used to measure lung fibrosis as previously described (16).
Second-Harmonic Generation (SHG)
SHG signals detection has been described previously (16).
Histochemistry and Immunohistochemistry
Lung fixation and staining are on the basis of established and published protocols (16).
Single-Cell Isolation and Flow Cytometry Analysis
Epithelial cells were isolated from adult mouse lungs with antibody-conjugated magnetic beads as previously described (19).
Dot Blot Analysis
Muc5b concentrations from mouse lung lavage were measured via dot blot analysis performed using a Minifold I dot blot filtration manifold (Schleicher and Schuell).
Dissection of Conducting Airways and Epithelial Cell Enrichment
Mice treated with bleomycin or saline at 12 weeks of age were used. Mouse lungs were cleared of blood and inflated and incubated with dispase for 30 minutes at room temperature. Parenchymal cells were gently removed from the lung lobe, and the remaining airway tree was gently washed with PBS. After separation, the airways were minced and mixed with RNA lysis buffer for RNA extraction.
Quantitative RT-PCR
mRNA expression amounts were determined by established and published protocols (16).
Electron Microscopy
Eighty nm ultrathin sections were prepared using a Leica UC7 μltramicrotome, and the sections were poststained with uranyl acetate and lead citrate. Imaging was performed with JEOL JEM1400plus transition electron microscopy (TEM) operated at 120kV and using a Gatan Orius 832 camera.
Statistical Analysis
Immunofluorescence, flow cytometry, SHG, and hydroxyproline data were analyzed for statistical significance using a one-way ANOVA, and significantly different means were identified using the Dunnett’s post hoc test (GraphPad). The significant comparisons were obtained on the basis of the adjusted P values < 0.05.
More detailed methods are available in the online supplement.
Results
Multiciliogenesis Is Induced in IPF-Conducting Airways
Previously, we found that the expression of cilium-associated genes, as well as MUC5B and other airway genes, defines a novel molecular subtype of IPF using whole lung tissue (2). To understand the relationship of MUC5B with the cilium-associated genes further, we examined gene expression changes associated with MUC5B in airway epithelial cells (new dataset) and whole lung tissue (2). All analyses were adjusted for age, sex, and smoking status. In IPF, 2,401 transcripts were associated with MUC5B gene expression in airway epithelial cells and 5,123 in lung tissue (false discovery rate [FDR] < 0.05) (Figure 1A). In normal airway epithelial cells, 204 transcripts were associated with MUC5B gene expression (FDR < 0.05), and only two transcripts were associated with MUC5B gene expression in normal lung tissue (SLC25A17 and BPIFB1; FDR < 0.05). To explore the function of transcripts associated with MUC5B gene expression in IPF, we examined for enrichment in gene ontology cellular component categories using 500 genes with the strongest correlations to MUC5B in IPF airway epithelial cells and IPF lung tissue. In IPF airway epithelial cells, four of the top five cellular component categories enriched were cilia-related (cilium [FDR, 1.57 × 10−11], ciliary part [FDR, 2.38 × 10−10], motile cilia [FDR, 5.12 × 10−7], and dynein complex [FDR, 9.2 × 10−6]). Similarly, in IPF lung tissue, the top five cellular component categories enriched were also related to cilia (cilium [FDR, 1.47 × 10−46], ciliary plasm [FDR, 1.79 × 10−32], axoneme [FDR, 2.00 × 10−32], motile cilia [FDR, 3.36 × 10−30], and ciliary part [FDR, 7.76 × 10−29]) (Figure 1B). In IPF, airway epithelial cells and lung tissue had 356 overlapping genes (Figure 1A) significantly associated with MUC5B expression, and the top two enriched categories for these overlapping genes were also cilia-related (cilium [FDR, 1.14 × 10−19]; ciliary part [FDR, 3.56 × 10−18]). Importantly, two transcription factors that are key markers of multiciliogenesis, MYB and FOXJ1 (12), were present in the overlapping gene list (Figure E1A in the online supplement). MYB is a transcription factor acting downstream of Notch signaling that has a conserved role in early multiciliogenesis and activates transcription factor FOXJ1, which can drive the complete multiciliogenesis (Figure E1B) (12).
Figure 1.
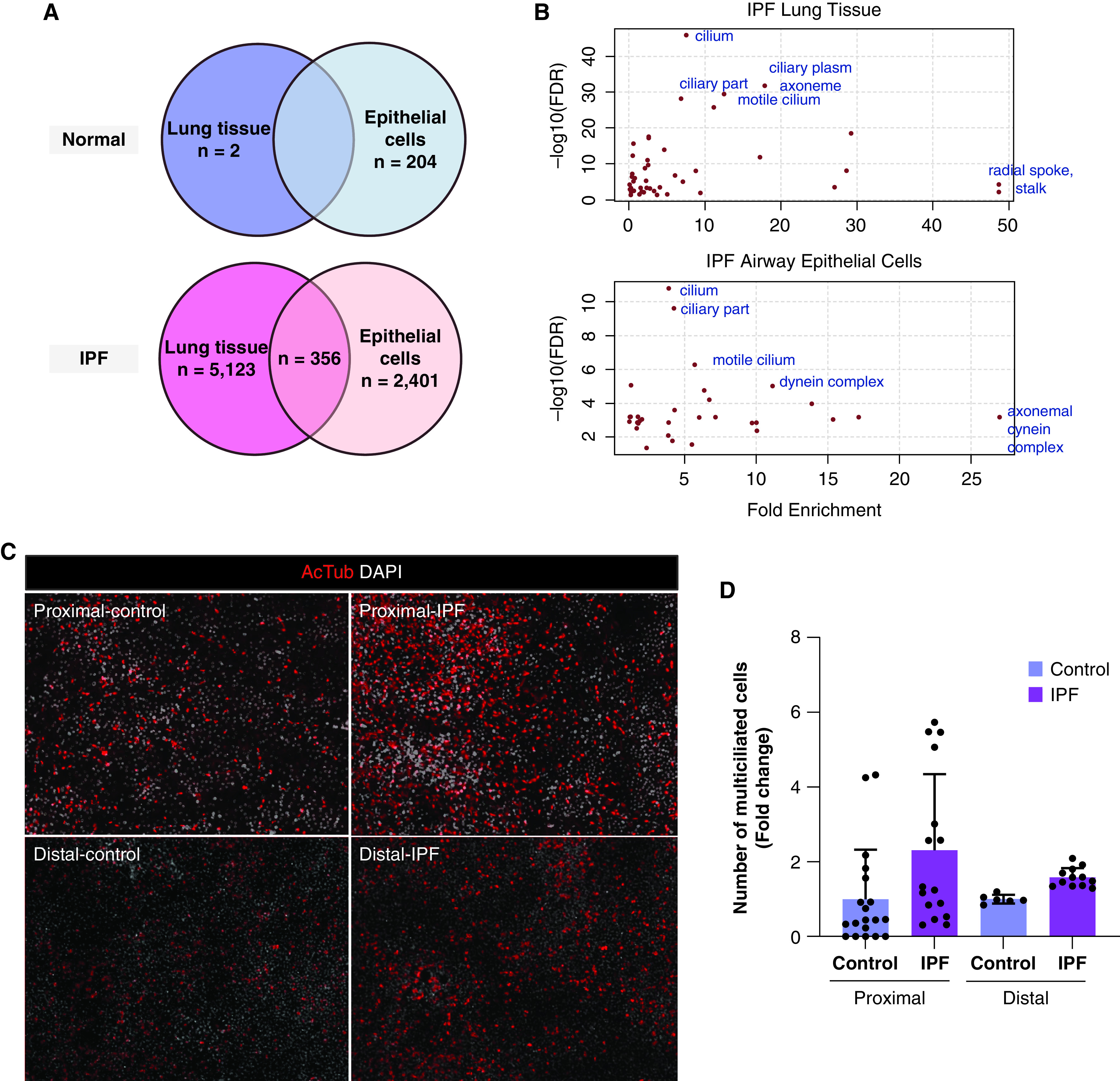
Multiciliated cells are increased in idiopathic pulmonary fibrosis (IPF). (A) Summary of genes whose expression is significantly associated (false discovery rate < 0.05) with MUC5B gene expression in proximal airway epithelia and whole lung tissue of subjects with IPF and nondiseased control subjects. (B) Pathway enrichment using gene ontology categories for cellular components. (C) Acetylated tubulin (red) and DAPI (white) expression at 28 days after establishing ALI culture using control and IPF airway epithelia. (D) Fold changes in the number of multiciliated cells from ALI cultures in panel (C). Data are presented as dot plots with means ± SD (n = 3 donors per group). Statistical significance was determined using a one-way ANOVA. ALI = air–liquid interface; DAPI = 4′,6-diamidino-2-phenylindole; FDR = false discovery rate.
Next, we evaluated whether the number of multiciliated cells increased in IPF. We stained airway epithelial cells with acetylated tubulin to stain motile cilia and quantified the number of multiciliated cells in the air–liquid interface (ALI) cultures from donors grown for 28 days at ALI (Figure 1C). The proportion of multiciliated cells as a percentage of total cell number was 2.3- and 1.6-fold increased both in the proximal and distal airway, respectively, in IPF compared with control subjects (Figure 1D). We next assessed the extent of multiciliogenesis by quantifying the percentage of MYB (actively ciliating cells) and FOXJ1 (fully ciliated cells) by immunofluorescence (IF) in lung tissue (Figure E2A). We observed more than a 20% increase in multiciliogenesis markers both in proximal and distal airways in IPF compared with control lung tissue (Figure E2B), implying that multiciliogenesis may be activated in IPF airways compared with healthy donors. This led us to postulate that multiciliogenesis may be aberrant in IPF and to examine cilia structure in IPF. To this end, we assessed motile cilium structure by TEM (Figure 2). In contrast to IPF proximal airway tissue, which shows the complete classical 9 + 2 microtubule organization of motile cilia similar to healthy tissue (Figure 2A), IPF distal airway tissue displayed defects such as incomplete or absent microtubule central pairs (Figure 2B). These types of defects in cilia structure are detrimental to cilia function (20, 21). Our results raise the possibility that the defective repair/regeneration process may play a role in mucociliary dysfunction and the development of the airway molecular subtype of IPF.
Figure 2.
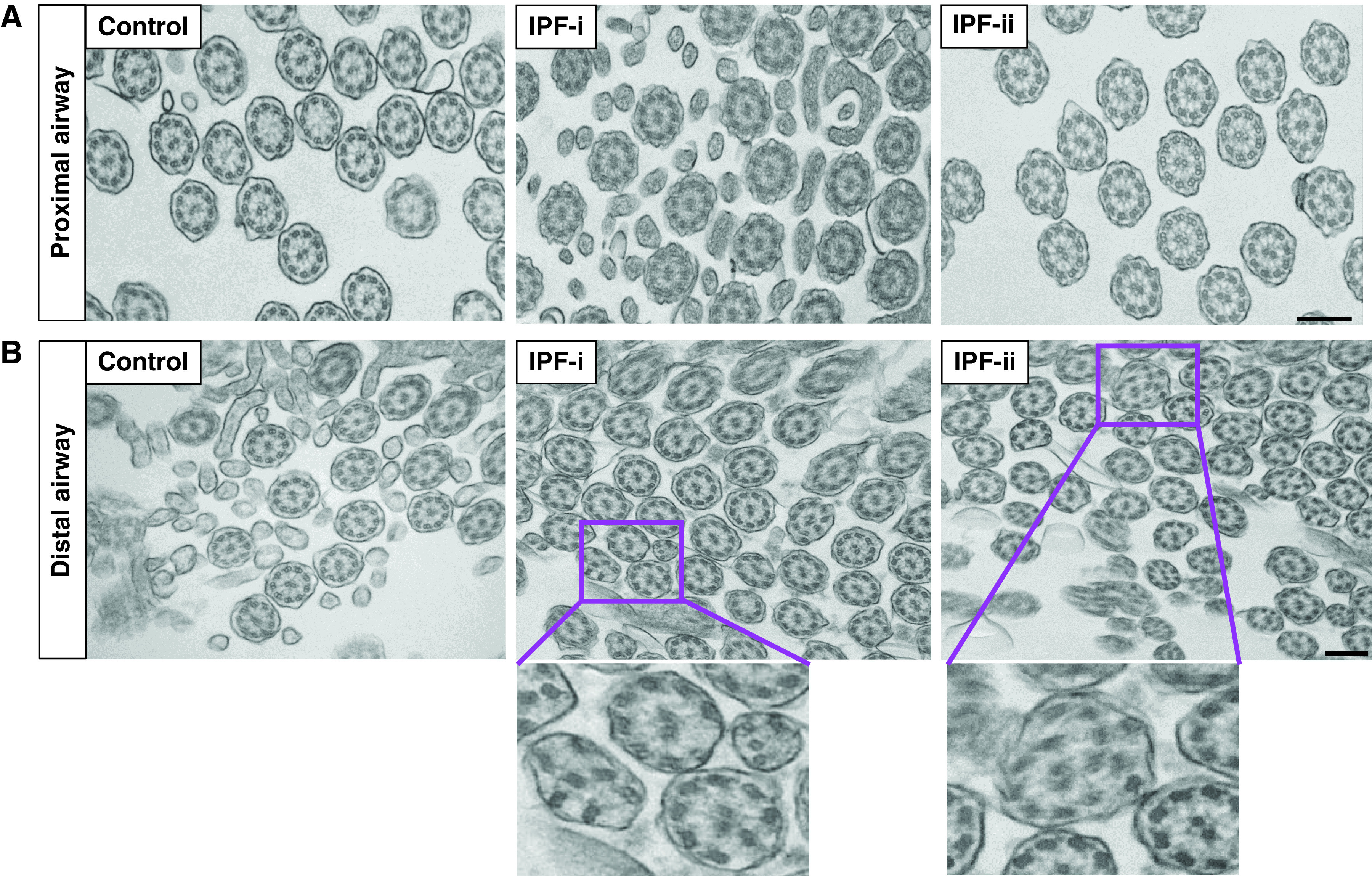
Motile cilia structure is disrupted in the IPF distal airway. Transition electron microscopy of axoneme of motile cilia of (A) proximal airway and (B) distal airway in healthy control subjects and patients with IPF. IPF-i and IPF-ii are high-magnification images of the boxed region in IPF-i and IPF-ii. Data are generated from n = 3 individuals per disease group. Scale bar, 200 μm. TEM = transition electron microscopy.
Multiciliogenesis Is Induced in Mouse Lungs upon Injury
On the basis of our findings of the elevated number of multiciliated cells and emergence of incomplete 9 + 2 structure in patients with IPF, we sought to study multiciliogenesis in animal models of lung fibrosis (22). To characterize multiciliogenesis in the airways, C57/B6J mice were challenged with single intratracheal (IT) bleomycin (2.5 U/kg), and bleomycin-induced multiciliogenesis was assessed on Weeks 1, 2, 3, 5, 7, 10, 13, and 16 after IT bleomycin, and saline at Weeks 0 and 16 were used as control subjects (Figure 3A). First, to study the dynamics of multiciliated cells in response to IT bleomycin, we stained motile cilia with acetylated tubulin. Remarkably, there was a decrease in staining of motile cilia in the airways, and large areas of deciliated airway epithelium were evident at 1–3 weeks after bleomycin compared with saline control subjects (Figure 3B). Fully recovered multiciliated cells are first observed at 5 weeks after injury, and, surprisingly, the number of multiciliated cells was significantly increased compared with baseline; this increase persists up to 16 weeks compared with control subjects.
Figure 3.

Intratracheal bleomycin induces multiciliogenesis upon lung injury. (A) Schematic representation of intratracheal (IT) single-dose bleomycin-induced lung injury and subsequent tissue collection from wild-type (WT) mice. (B) Immunostaining for acTub (magenta) and DAPI (blue) in proximal and distal airways at each time point. Scale bar, 20 μm. (C) Immunostaining for cMyb (red), Foxj1 (white), and DAPI (blue) in proximal and distal airways at each time point. Scale bar, 50 μm. (D) Percentage of cMyb+ and Foxj1+ cells per 100 DAPI+ cells over time by counting immunostained sections in proximal (left) and distal (right) airways. Data are means ± SD (n = 6–8 mice per group). Statistical significance was assessed by ANOVA using the Dunnett test for multiple comparisons to S0 control subjects. B1 = bleomycin 1 week; B16 = bleomycin 16 weeks; Bleo = bleomycin; S0 = saline 0 weeks; S16 = saline 16 weeks. *P < 0.05.
Next, we quantified the percentage of cMyb+ and Foxj1+ cells by using both immunofluorescence and flow cytometry. The number of cMyb+ cells increases rapidly as early as 1 week and reaches over 60% of airway cells, with significant differences compared with saline control subjects in both proximal and distal airways (Figures 3C and 3D). At 5 weeks, a significant recovery in Foxj1+ cell numbers was evident. Interestingly, Foxj1+ cells also persisted at high concentrations compared with saline control subjects for up to 16 weeks. Importantly, a similar trend was also found in H1N1 influenza virus-exposed mice (Figure E3). Generally consistent with immunofluorescence staining of the airways, flow cytometry analysis of Epcam+ cells isolated from whole lung digests (Figure E4A) demonstrated slight but significant changes in the number of Foxj1+ and cMyb+ cells at 3, 5, and 16 weeks after IT bleomycin treatment. The attenuation of the signal in flow cytometry compared with immunofluorescence data is likely because the effect of airway cells is diluted by alveolar type II cells present in the majority of the Epcam+ cell fraction that we analyzed. It is notable that the percentage of cMyb+ and Foxj1+ cells showed the highest point at 16 weeks. These changes observed in flow cytometry are probably because of multiciliated cells in a cyst-like structure formed in the parenchymal region (Figure E4B) in addition to multiciliogenesis in the airways. Correlating generally with the number of multiciliated cells, IT bleomycin-induced lung fibrosis measured both by hydroxyproline content in the lung and SHG imaging of fibrillar collagen in the parenchyma also persists at 5–10 weeks (Dobrinskikh and colleagues, under review) (23).
We next performed TEM imaging to examine cilia structure in the bleomycin model. TEM images demonstrated the presence of some cells with (1) irregular length of cilia or (2) swollen cilia and weak 9 + 2 structure in bleomycin groups, which is not found in saline control groups (Figure 4A). This suggests that aberrant repair of damaged motile cilia results in ciliary abnormalities, indicating that altered cilia structure could be one of the early markers of lung fibrosis and may affect mucociliary function in the airways.
Figure 4.

Motile cilia structure is altered in the bleomycin model. (A) TEM of a thin section cut through the bronchiolar epithelium of the mouse lung. (B) Quantitative RT-PCR analysis of the changes in expression of cilia structural genes together with Foxj1 in airway-enriched cells (top) and whole lung tissue (bottom). Data are the mean ± SD (n = 3–4 mice per group). Statistical significance was assessed by ANOVA using the Dunnett test for multiple comparisons to Saline control subjects. *P < 0.05.
To test whether motile cilia functional genes were affected in airway epithelial cells by bleomycin treatment, the expression of three different categories of the ciliated cell-related genes was examined: 1) transcription factor for multiciliogenesis Foxj1; 2) cilia intraflagellar transport gene Ift88; and 3) cilia motility and structural integrity genes Dnah5 (dynein axonemal heavy chain 5), Dnah9, Dnah10, Dnah11, and Spag6 (sperm-associated antigen 6). Airway trees were isolated from the whole lung, and airway cell-enriched RNA samples were used for quantitative RT-PCR. The expression of the cilia-related genes was significantly downregulated in the airway at 1 week after bleomycin treatment, back to saline amounts at 5 weeks, and was higher than saline at 16 weeks, which is consistent with dynamic changes we observed in the motile cilia in response to bleomycin treatment (Figure 4B). Our results from the bleomycin mouse model are consistent with human IPF data, raising the possibility that the defective repair/regeneration process in multiciliated cells may play a role in mucociliary dysfunction and the development of lung fibrosis.
Stem/Progenitor Cells Involved in Damage-induced Multiciliogenesis in the Mouse Lung
Krt5 basal cells are believed to arise from immature stem/progenitor cells that are named distal airway stem cells or lineage-negative epithelial progenitors, and these cells proliferate to form clusters of cells in the damaged alveolar regions named keratin 5 pods and are dominant in the bronchiolized area (6–10) (Figure E4C) and act as progenitors for both club and multiciliated cells. Krt5-traced cells are known to express cilia-associated genes after injury in mouse lungs (7). Club cells are also known to give rise to ciliated cells (14), as well as alveolar type I and II cells after injury (24). Thus, we examined how progenitors of multiciliated cells respond to IT bleomycin. We confirmed that both Krt5 basal cells and club cells are increased when multiciliogenesis in the lung is induced (Figure 5A), suggesting compensatory changes involved in epithelial repair because of deciliation in the airway and loss of alveolar type II cells in the distal lung on injury (Figure E4D). Muc5b expression was increased with the number of club cells in a parallel manner (Figure 5B). Next, we costained cMyb with Krt5 and Scgb1a1 (club cell marker) to determine the progenitor cell type responsible for generating multiciliated cells. In accordance with previous publications, only a few Krt5 cells were detected in the airways in control mice; however, the number of Krt5 cells was increased by IT bleomycin both in proximal airways and parenchymal region (Krt5 pods). These Krt5 cells, both in airway and Krt5 pods, expressed cMyb in the nucleus but not Foxj1, indicating initiation of multiciliogenesis (Figures 5C and 5D). Club cells residing in both proximal and distal airways also express cMyb in the nucleus, indicating that both Krt5 cells and club cells give rise to multiciliated cells after IT bleomycin but in different lung regions. These data suggest that both Krt5 cells and club cells contribute to an increase in multiciliated cell numbers after the injury with bleomycin. However, lineage-tracing experiments will be needed to confirm these results.
Figure 5.

Intratracheal bleomycin induces the proliferation of basal stem cells and progenitor cells. (A) Quantification of the percentage of Krt5+ (basal stem cells) and CC10 (club cells) cells per total number of EpCam+ cells over time. Data are means ± SD (n = 5–6 mice per group). (B) Muc5b protein secretion in the lung lavage of bleomycin-treated mice over time measured by dot blot. Data are presented as dot plots with means ± SD (n = 6–8 mice per group). Statistical significance was assessed by ANOVA using the Dunnett test for multiple comparisons to S0 control subjects. (C) Immunostaining for cMyb (red), Scgb1a1 (white), and DAPI (blue) in proximal airways at 5 weeks after IT bleomycin (left). Immunostaining for cMyb (red), Foxj1 (white), Krt5 (green), and DAPI (blue) in proximal airways at 5 weeks after IT bleomycin (right). Scale bar, 50 μm. (D) Immunostaining for cMyb (red), Foxj1 (white), Krt5 (green), and DAPI (blue) in Krt5 pods in distal airways at 5 (left) and 16 (right) weeks after IT bleomycin (left). Scale bar, 50 μm. Statistical significance was assessed by ANOVA using the Dunnett test for multiple comparisons to S0 control subjects. *P < 0.05.
To confirm that ciliated cells were differentiated from Krt5 and club cells without undergoing cell division, we labeled proliferating cells using the EdU assay. The percentage of proliferating cells was at its peak 3 weeks after bleomycin but only in alveolar regions, whereas proliferating cells in the airways were barely detectable (Figure E4E).
Ift88 Conditional Deletion in Krt5 Cells Results in Reduced Krt5 Pods and Lung Fibrosis
Previously, we reported that expression of the KRT5 gene is highly correlated with cilium genes in IPF (2). KRT5+ cells are only present in the normal airway epithelium; however, this cell population is greatly expanded in IPF interstitium, likely attempting to regenerate the epithelium (10). Primary cilia on basal airway progenitors precede multiciliogenesis during normal airway cell differentiation and repair after respiratory virus injury (11). The IPF cilia gene expression signature contains both motile and primary cilia genes. One of the key cilia-associated genes is IFT88 because it is required for cilium assembly (25) as well as suggested to contribute to fibrosis in IPF (26) by activating Sonic Hedgehog (SHH) signaling (26). However, the role of Ift88 in primary ciliated Krt5 cells in the development of fibrosis and multiciliogenesis has not been explored.
Thus, we tested whether we could reduce bleomycin-induced fibrosis by conditional deletion of the Ift88 gene in Krt5 cells by treating Krt5-CreERT2-Ift88flox/flox mice with tamoxifen. Somatic deletion of the floxed Ift88 allele in lung tissue was confirmed using PCR with allele-specific primers and immunofluorescence staining for Ift88 (Figure E5). In a steady-state, Ift88 conditional deletion in Krt5 cells (Figure 6A) generates no motile cilia; however, multiciliated cells are still generated by club cells (Figures 6B and E6A), and the number of multiciliated cells and club cells were not changed (Figure E6B). Lung fibrosis in Krt5-CreERT2-Ift88flox/flox was assessed at 1, 2, and 3 weeks after a single IT bleomycin dose by measuring hydroxyproline content. Compared with Ift88 wild-type mice, mice with the Ift88 conditional deletion in Krt5 cells had a significant reduction in fibrosis and injury at 2 and 3 weeks after bleomycin (Figures 6C and E7A). This difference in collagen was also quantified by confocal/multiphoton–excitation fluorescence microscopy with SHG at 3 weeks after bleomycin, and the results were consistent with hydroxyproline assay (Figure E7B). We have shown that Krt5 pods are positively associated with the extent of fibrosis (27). To test whether Krt5 pods and fibrosis are affected by Ift88 deletion in Krt5 cells, we quantified the area and number of Krt5 pods in mouse lungs. Compared with Ift88 wild-type mice, mice with the Ift88 conditional deletion in Krt5 cells had a significant reduction in the area and number of Krt5 pods (Figure 6D). On the other hand, we could not detect any protective effect of Ift88 conditional deletion in club cell progenitors (CCSP-CreERT2-Ift88F/F mice) on fibrosis and the area of Krt5 pods on injury (Figures E7C and E7D).
Figure 6.

Impact of Ift88 deletion in Krt5 basal cells upon lung fibrosis. (A) Schematic representation of single-dose IT bleomycin-induced lung injury and subsequent tissue collection from Krt5-CreERT2-Ift88F/F mice. (B) Immunostaining for Ift88 (red), Krt5-derived cell (yellow), and DAPI (blue) in Krt5-CreERT2-Ift88+/+-ROSA-mT/mG and Krt5-CreERT2-Ift88F/F-ROSA-mT/mG mice treated with saline. (C) Hydroxyproline concentrations were measured by colorimetric assay in the entire left lung at 1, 2, and 3 weeks after IT bleomycin. Data are presented as dot plots with means ± SD (n = 6–7 mice per group). Statistical significance was assessed by ANOVA using the Dunnett test for multiple comparisons to saline control subjects. (D) Area of Krt5 pods per mouse (left) and the number of Krt5 pods per mouse (right) in Krt5-CreERT2-Ift88+/+ and Krt5-CreERT2-Ift88F/F at 3 weeks after IT bleomycin. Data are presented as dot plots with means ± SD (n = 6–7 mice per group). Statistical significance for the area of the Krt5 pod was assessed by ANOVA using the Dunnett test for multiple comparisons to WT Bleo. Statistical significance for the number of Krt5 pods was assessed by Fisher’s exact test. (E) Representative images of airway epithelia of Krt5-CreERT2-Ift88+/+ and Krt5-CreERT2-Ift88F/F mice upon injury stained for Krt5 (white) and DAPI (blue). (F) Immunostaining for cMyb (red), acTub (white), and DAPI (blue) in proximal airways of Krt5-CreERT2-Ift88+/+ and Krt5-CreERT2-Ift88F/F mice at 3 weeks after IT bleomycin. Krt5-CreERT2-Ift88+/+ with saline treatment was used for control subjects. (G) Immunostaining for Gli1 and Gli2 (red) and DAPI (blue) in proximal airways of Krt5-CreERT2-Ift88+/+ and Krt5-CreERT2-Ift88F/F mice at 3 weeks after IT bleomycin. Krt5-CreERT2-Ift88+/+ with saline treatment was used for control subjects. KO = knockout. *P < 0.05.
Krt5-CreERT2-Ift88+/+ mice showed induced cMyb in the airways on bleomycin treatment as expected (Figure 6F); however, the expression of cMyb-positive cells was almost similar to the basal concentration in Krt5-CreERT2-Ift88F/F mice, suggesting multiciliogenesis is delayed or reduced by Ift88 deletion in Krt5 cells. Further studies will be needed to delineate signaling pathways that link defects in primary cilia on progenitor cells to defects in multiciliogenesis and the development of lung fibrosis. However, these data suggest that modulating cilium gene expression in Krt5 cell progenitors is a potential therapeutic target for IPF modulating abnormal multiciliogenesis and fibrosis. Next, we stained for Gli2 (activator Gli) and Gli3 (inhibitory Gli) (28) to confirm whether Ift88 deletion in Krt5 cells causes dampened expression of SHH signaling target genes. Gli2 was localized in the cytoplasm and nucleus both in Krt5-CreERT2-Ift88+/+ mice in steady status and translocated to the nucleus upon bleomycin treatment; however, nuclear staining of Gli2 in Krt5-CreERT2-Ift88flox/flox mice is barely detectable. However, Gli3 was localized in the nucleus only in Krt5-CreERT2-Ift88flox/flox mice on injury, implying strong inhibition of SHH signaling because of Ift88 deletion in Krt5 cells (Figure 6G). These results indicate that Ift88 deletion in Krt5 cells inhibits SHH signaling, contributing to the reduction in Krt5 pod formation as well as multiciliogenesis, which both contribute to fibrosis (Figure E8).
Discussion
This study builds on our recent discovery that the expression of cilium-associated genes defines a novel airway-based molecular subtype of IPF in lung tissue (2) and extends these findings specifically to the airway epithelium of patients with IPF and animal models of lung fibrosis. Work presented herein demonstrates, for the first time, that the number of multiciliated cells with disrupted motile cilia structure is significantly increased both in IPF and in the bleomycin mouse model at later time points after the airway cells have regenerated after injury. These findings are in line with recent work of our group that demonstrated dysfunction of the IPF distal epithelia (15) and further support the idea of disrupted mucociliary clearance in the distal lung as an important event in the pathogenesis of IPF. We also demonstrate that the development of lung fibrosis can be reduced in part by modulating a cilium gene in Krt5 progenitor cells, which provides evidence for the role of ciliogenesis in the epithelial–stromal crosstalk in the bleomycin model.
In human IPF lungs, we observed an increased number of ciliated cells consistent with published single-cell RNA sequencing data (29). Using the bleomycin model of lung injury and fibrosis, we show, for the first time, that multiciliated cells shed their ciliary axonemes 1–3 weeks after IT bleomycin, which is consistent with previously documented acute damage response by cigarette smoke in the human airway epithelium (30), severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)-infected cells (31), and oxygen toxicity in the newborn mice (32). After this initial deciliation, lung epithelial cells induce multiciliogenesis to regenerate damaged cilia in the airways; this occurs after 5 weeks after bleomycin. Analogous to IPF lungs, we observe an increase in the number of ciliated (Foxj1+) and actively ciliating (cMyb+) cells in the bleomycin model at later time points. The number of cMyb+ cells persisted even after multiciliated cells were regenerated. In patients with severe chronic obstructive pulmonary disease (COPD), the number of Myb+ cells is also increased and abundant in regions of the lung that contain both high numbers of ciliated cells and secretory cells (13), similar to the persistence of cMyb+ cells we observed (Figure 3). It has been demonstrated that a subpopulation of club cells expresses cMyb in the nucleus after naphthalene injury, permitting multilineage airway epithelial cell differentiation (13). This observation suggested that cMyb function is not restricted to early events in ciliated cell differentiation.
In addition, we found ciliary structural defects in motile cilia in IPF distal airways that were not observed in proximal airways. Analogous to IPF lungs, we observe ciliary structural abnormalities of the recovered motile cilia on multiciliated cells after bleomycin injury. In addition to ciliary structural defects observed by TEM, we also demonstrate that Dnah5, Dnah9, and Dnah11 are initially downregulated by bleomycin-induced injury, followed by enhanced expression compared with control subjects after 5 weeks. This is important because it has been previously shown that the loss of function of Dnah5 can cause immotile cilia (33), the loss of function of Dnah9 causes defects in distal movement of the cilia (34, 35), and the loss of function of Dnah11 results in defective proximal ciliary movement impairment (36). The data we show here are reminiscent of multiciliated cells infected by SARS-CoV-2, which is characterized by downregulation of the master regulator of ciliogenesis FOXJ1 before extensive cilia loss and misoriented basal body and compromised motile cilia function accompanied by an increase in KRT5 cells and airway mucins (31). Taken together, our data in the bleomycin model recapitulate and extend the findings in IPF and provide additional evidence for MCC defects, building on our previous observations that overexpression of Muc5b in the distal lung leads to impaired mucociliary clearance in response to bleomycin (16).
Our work provides evidence that IPF lungs share features of ciliary dysfunction observed in other chronic airway disorders. Diseased airway tissues show a range of epithelial structural and functional abnormalities (37). Multiciliated cell loss or damage in chronic airway diseases such as cystic fibrosis is thought to be caused by infections that target multiciliated cells or indirectly by inflammation (38). In cystic fibrosis, the damage is localized sporadically throughout the airways and may be reversible, although chronic injury to the epithelial surface eventually gives rise to epithelial remodeling with irreversible structural changes (37). In addition, cilia are poorly organized even in more ciliated areas, with many misaligned cilia (39), indicating that MCC over that area would be disrupted. Abnormalities of MCC, and consequently reduced host defenses of the lung, are a common theme in many acquired lung disorders (40). In many of these disorders, the airway cilia demonstrate acquired structural and/or functional abnormalities with associated abnormalities in mucociliary clearance (40). Recently, bronchial epithelium in COPD has been shown to have mucociliary defects, specifically lower ciliary beat frequency and higher dyskinesia index measured by TEM (41), in addition to earlier observations of persistent increase in the number of Myb+ cells in COPD lung tissue (19). This suggests commonalities across COPD and IPF that are likely the result of recurrent injury by environmental exposures such as cigarette smoke.
Our results add to the body of evidence that IPF is likely a disease characterized by defective MCC (3). Multiciliated cells with disrupted motile cilia in IPF lungs possibly impede MCC, and mucus accumulates in the airways, contributing to increased inflammation and infection and, thus, to progressive pulmonary degeneration (42, 43). Irregularly arranged cilia structure has been associated with cytoskeletal disruption and actin dynamics, either because of genetic mutations (44) or in response to mechanical stress (45). Cilia damage and reduced cilia beat frequency have been implicated as causes of reduced MCC (20, 46). Airway clearance is also known to be disrupted by cigarette smoking and environmental pollutants and in airway diseases such as COPD, asthma, acute and chronic infection, CF, and others (40). Our research requires more studies on whether the mucociliary function is compromised by disrupted cilia structure in IPF and how it impacts disease pathogenesis when combined with MUC5B overexpression.
A robust expression of regenerative Krt5 cells in the lung parenchyma after injury has been observed in mice (7), which we confirmed. Krt5 cells in parenchyma persist at 5–16 weeks on bleomycin treatment in our study; however, we have not yet studied how these Krt5 cells that reside in the parenchyma and crosstalk with fibroblasts lead to the development of fibrosis as a result of injury to the distal airway. The potential pathway is the promotion of fibrosis through signaling crosstalk with resident lung fibroblasts by unjamming in airway epithelia (47), as we have recently demonstrated (15). This implies that the IPF distal airway epithelium, especially KRT5 cells, plays an active role in driving fibrosis, which is consistent with our findings. SHH pathway is known to be activated in IPF lungs and contribute to IPF pathogenesis by increasing proliferation, migration, extracellular matrix production, and survival of fibroblasts (48–50). It has been suggested that the IFT88 gene may contribute to the development of fibrosis in IPF (26) by activating SHH signaling (26). We demonstrate that deletion of Ift88 in Krt5 cells results in reduced Krt5 pod formation, reduced or delayed multiciliogenesis, and reduced lung fibrosis. We also show that this is at least in part mediated by reducing SHH signaling, as it has been previously shown that conditional inactivation of Ift88 results in the inactivation of SHH signaling (26). Krt5-traced cells expressed cilia-associated genes (7), implying that primary cilia on Krt5 progenitor cells are critical for the development of lung fibrosis via epithelial–stromal crosstalk in the bleomycin model. Our group recently showed that airway epithelia from IPF can induce a profibrotic response in healthy human lung fibroblasts (15), providing strong evidence that airway epithelia can modulate lung fibroblasts. In this study, we showed that deletion of Ift88 in Krt5 progenitor cells results in reduced or delayed multiciliogenesis; however, future work should also examine how ablation of Ift88 in multiciliated cells affects lung fibrosis.
Conclusions
We provide evidence from IPF tissue and the bleomycin model that aberrant primary and multiciliogenesis as well as defective cilia structure may contribute to the pathogenesis of IPF. We also show that conditional deletion of a cilium gene in basal progenitor cells reduces lung fibrosis. This has broad implications for the functional role of multiciliated cells as well as the understanding of the epithelial regeneration response to injury. These findings highlight key cellular mechanisms of how the respiratory airway epithelial cells respond to injury and contribute to the development of disease.
Footnotes
Supported by the National Institutes of Health (NIH) (R24AA019661), NIH/National Heart, Lung, and Blood Institute (F32HL154666), and the Department of Defence (DoD W81XWH-16-PRMRP-FPA).
Author Contributions: E.K. performed data collection and writing. S.K.M. and I.T.S. performed part of the human data collection. X.M., A.H.-G., J.N.B., and E.M.-T. performed part of the mouse data collection. C.E.H., K.H., E.P.W., A.E., J.P.H., and J.H.C. assisted in sample collection. E.L.B., Y.Z., C.M.E., E.K.V., D.A.S., E.D., and I.V.Y. provided comments.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2021-0554OC on May 24, 2022
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Mathai SK, Newton CA, Schwartz DA, Garcia CK. Pulmonary fibrosis in the era of stratified medicine. Thorax . 2016;71:1154–1160. doi: 10.1136/thoraxjnl-2016-209172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yang IV, Coldren CD, Leach SM, Seibold MA, Murphy E, Lin J, et al. Expression of cilium-associated genes defines novel molecular subtypes of idiopathic pulmonary fibrosis. Thorax . 2013;68:1114–1121. doi: 10.1136/thoraxjnl-2012-202943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Evans CM, Fingerlin TE, Schwarz MI, Lynch D, Kurche J, Warg L, et al. Idiopathic pulmonary fibrosis: a genetic disease that involves mucociliary dysfunction of the peripheral airways. Physiol Rev . 2016;96:1567–1591. doi: 10.1152/physrev.00004.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Boucher RC. Idiopathic pulmonary fibrosis--a sticky business. N Engl J Med . 2011;364:1560–1561. doi: 10.1056/NEJMe1014191. [DOI] [PubMed] [Google Scholar]
- 5. Button B, Cai LH, Ehre C, Kesimer M, Hill DB, Sheehan JK, et al. A periciliary brush promotes the lung health by separating the mucus layer from airway epithelia. Science . 2012;337:937–941. doi: 10.1126/science.1223012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kurche JS, Dobrinskikh E, Hennessy CE, Huber J, Estrella A, Hancock LA, et al. Muc5b enhances murine honeycomb-like cyst formation. Am J Respir Cell Mol Biol . 2019;61:544–546. doi: 10.1165/rcmb.2019-0138LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vaughan AE, Brumwell AN, Xi Y, Gotts JE, Brownfield DG, Treutlein B, et al. Lineage-negative progenitors mobilize to regenerate lung epithelium after major injury. Nature . 2015;517:621–625. doi: 10.1038/nature14112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Seibold MA, Smith RW, Urbanek C, Groshong SD, Cosgrove GP, Brown KK, et al. The idiopathic pulmonary fibrosis honeycomb cyst contains a mucocilary pseudostratified epithelium. PLoS One . 2013;8:e58658. doi: 10.1371/journal.pone.0058658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tata PR, Rajagopal J. Plasticity in the lung: making and breaking cell identity. Development . 2017;144:755–766. doi: 10.1242/dev.143784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Smirnova NF, Schamberger AC, Nayakanti S, Hatz R, Behr J, Eickelberg O. Detection and quantification of epithelial progenitor cell populations in human healthy and IPF lungs. Respir Res . 2016;17:83. doi: 10.1186/s12931-016-0404-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jain R, Pan J, Driscoll JA, Wisner JW, Huang T, Gunsten SP, et al. Temporal relationship between primary and motile ciliogenesis in airway epithelial cells. Am J Respir Cell Mol Biol . 2010;43:731–739. doi: 10.1165/rcmb.2009-0328OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tan FE, Vladar EK, Ma L, Fuentealba LC, Hoh R, Espinoza FH, et al. Myb promotes centriole amplification and later steps of the multiciliogenesis program. Development . 2013;140:4277–4286. doi: 10.1242/dev.094102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pan JH, Adair-Kirk TL, Patel AC, Huang T, Yozamp NS, Xu J, et al. Myb permits multilineage airway epithelial cell differentiation. Stem Cells . 2014;32:3245–3256. doi: 10.1002/stem.1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rawlins EL, Okubo T, Xue Y, Brass DM, Auten RL, Hasegawa H, et al. The role of Scgb1a1+ Clara cells in the long-term maintenance and repair of lung airway, but not alveolar, epithelium. Cell Stem Cell . 2009;4:525–534. doi: 10.1016/j.stem.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Stancil IT, Michalski JE, Davis-Hall D, Chu HW, Park JA, Magin CM, et al. Pulmonary fibrosis distal airway epithelia are dynamically and structurally dysfunctional. Nat Commun . 2021;12:4566. doi: 10.1038/s41467-021-24853-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hancock LA, Hennessy CE, Solomon GM, Dobrinskikh E, Estrella A, Hara N, et al. Muc5b overexpression causes mucociliary dysfunction and enhances lung fibrosis in mice. Nat Commun . 2018;9:5363. doi: 10.1038/s41467-018-07768-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rock JR, Onaitis MW, Rawlins EL, Lu Y, Clark CP, Xue Y, et al. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc Natl Acad Sci USA . 2009;106:12771–12775. doi: 10.1073/pnas.0906850106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Haycraft CJ, Zhang Q, Song B, Jackson WS, Detloff PJ, Serra R, et al. Intraflagellar transport is essential for endochondral bone formation. Development . 2007;134:307–316. doi: 10.1242/dev.02732. [DOI] [PubMed] [Google Scholar]
- 19. Yamada M, Kubo H, Ota C, Takahashi T, Tando Y, Suzuki T, et al. The increase of microRNA-21 during lung fibrosis and its contribution to epithelial-mesenchymal transition in pulmonary epithelial cells. Respir Res . 2013;14:95. doi: 10.1186/1465-9921-14-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Leopold PL, O’Mahony MJ, Lian XJ, Tilley AE, Harvey BG, Crystal RG. Smoking is associated with shortened airway cilia. PLoS One . 2009;4:e8157. doi: 10.1371/journal.pone.0008157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kunimoto K, Yamazaki Y, Nishida T, Shinohara K, Ishikawa H, Hasegawa T, et al. Coordinated ciliary beating requires Odf2-mediated polarization of basal bodies via basal feet. Cell . 2012;148:189–200. doi: 10.1016/j.cell.2011.10.052. [DOI] [PubMed] [Google Scholar]
- 22. Liu T, De Los Santos FG, Phan SH. The bleomycin model of pulmonary fibrosis. Methods Mol Biol . 2017;1627:27–42. doi: 10.1007/978-1-4939-7113-8_2. [DOI] [PubMed] [Google Scholar]
- 23. Scotton CJ, Hayes B, Alexander R, Datta A, Forty EJ, Mercer PF, et al. Ex vivo micro-computed tomography analysis of bleomycin-induced lung fibrosis for preclinical drug evaluation. Eur Respir J . 2013;42:1633–1645. doi: 10.1183/09031936.00182412. [DOI] [PubMed] [Google Scholar]
- 24. Zheng D, Limmon GV, Yin L, Leung NH, Yu H, Chow VT, et al. Regeneration of alveolar type I and II cells from Scgb1a1-expressing cells following severe pulmonary damage induced by bleomycin and influenza. PLoS One . 2012;7:e48451. doi: 10.1371/journal.pone.0048451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rosenbaum JL, Witman GB. Intraflagellar transport. Nat Rev Mol Cell Biol . 2002;3:813–825. doi: 10.1038/nrm952. [DOI] [PubMed] [Google Scholar]
- 26. Lee J, Oh DH, Park KC, Choi JE, Kwon JB, Lee J, et al. Increased primary cilia in idiopathic pulmonary fibrosis. Mol Cells . 2018;41:224–233. doi: 10.14348/molcells.2018.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dobrinskikh E, Estrella AM, Hennessy CE, Hara N, Schwarz MI, Kurche JS, et al. Genes, other than Muc5b, play a role in bleomycin-induced lung fibrosis. Am J Physiol Lung Cell Mol Physiol . 2021;321:L440–L450. doi: 10.1152/ajplung.00615.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Moshai EF, Wémeau-Stervinou L, Cigna N, Brayer S, Sommé JM, Crestani B, et al. Targeting the hedgehog-glioma-associated oncogene homolog pathway inhibits bleomycin-induced lung fibrosis in mice. Am J Respir Cell Mol Biol . 2014;51:11–25. doi: 10.1165/rcmb.2013-0154OC. [DOI] [PubMed] [Google Scholar]
- 29. Adams TS, Schupp JC, Poli S, Ayaub EA, Neumark N, Ahangari F, et al. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci Adv . 2020;6:eaba1983. doi: 10.1126/sciadv.aba1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Brekman A, Walters MS, Tilley AE, Crystal RG. FOXJ1 prevents cilia growth inhibition by cigarette smoke in human airway epithelium in vitro. Am J Respir Cell Mol Biol . 2014;51:688–700. doi: 10.1165/rcmb.2013-0363OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Robinot R, Hubert M, de Melo GD, Lazarini F, Bruel T, Smith N, et al. SARS-CoV-2 infection induces the dedifferentiation of multiciliated cells and impairs mucociliary clearance. Nat Commun . 2021;12:4354. doi: 10.1038/s41467-021-24521-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bonikos DS, Bensch KG, Northway WH., Jr Oxygen toxicity in the newborn. The effect of chronic continuous 100 percent oxygen exposure on the lungs of newborn mice. Am J Pathol . 1976;85:623–650. [PMC free article] [PubMed] [Google Scholar]
- 33. Olbrich H, Häffner K, Kispert A, Völkel A, Volz A, Sasmaz G, et al. Mutations in DNAH5 cause primary ciliary dyskinesia and randomization of left-right asymmetry. Nat Genet . 2002;30:143–144. doi: 10.1038/ng817. [DOI] [PubMed] [Google Scholar]
- 34. Loges NT, Antony D, Maver A, Deardorff MA, Güleç EY, Gezdirici A, et al. Recessive DNAH9 loss-of-function mutations cause laterality defects and subtle respiratory ciliary-beating defects. Am J Hum Genet . 2018;103:995–1008. doi: 10.1016/j.ajhg.2018.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fliegauf M, Olbrich H, Horvath J, Wildhaber JH, Zariwala MA, Kennedy M, et al. Mislocalization of DNAH5 and DNAH9 in respiratory cells from patients with primary ciliary dyskinesia. Am J Respir Crit Care Med . 2005;171:1343–1349. doi: 10.1164/rccm.200411-1583OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dougherty GW, Loges NT, Klinkenbusch JA, Olbrich H, Pennekamp P, Menchen T, et al. DNAH11 localization in the proximal region of respiratory cilia defines distinct outer dynein arm complexes. Am J Respir Cell Mol Biol . 2016;55:213–224. doi: 10.1165/rcmb.2015-0353OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Adam D, Roux-Delrieu J, Luczka E, Bonnomet A, Lesage J, Mérol JC, et al. Cystic fibrosis airway epithelium remodeling: involvement of inflammation. J Pathol . 2015;235:408–419. doi: 10.1002/path.4471. [DOI] [PubMed] [Google Scholar]
- 38. Heijerman H. Infection and inflammation in cystic fibrosis: a short review. J Cyst Fibros . 2005;4:3–5. doi: 10.1016/j.jcf.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 39. Vladar EK, Nayak JV, Milla CE, Axelrod JD. Airway epithelial homeostasis and planar cell polarity signaling depend on multiciliated cell differentiation. JCI Insight . 2016;1:e88027. doi: 10.1172/jci.insight.88027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tilley AE, Walters MS, Shaykhiev R, Crystal RG. Cilia dysfunction in lung disease. Annu Rev Physiol . 2015;77:379–406. doi: 10.1146/annurev-physiol-021014-071931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Thomas B, Koh MS, O’Callaghan C, Allen JC, Jr, Rutman A, Hirst RA, et al. Dysfunctional bronchial cilia are a feature of chronic obstructive pulmonary disease (COPD) COPD . 2021;18:657–663. doi: 10.1080/15412555.2021.1963695. [DOI] [PubMed] [Google Scholar]
- 42. Hylkema MN, Sterk PJ, de Boer WI, Postma DS. Tobacco use in relation to COPD and asthma. Eur Respir J . 2007;29:438–445. doi: 10.1183/09031936.00124506. [DOI] [PubMed] [Google Scholar]
- 43. Brody JS, Spira A. State of the art. Chronic obstructive pulmonary disease, inflammation, and lung cancer. Proc Am Thorac Soc . 2006;3:535–537. doi: 10.1513/pats.200603-089MS. [DOI] [PubMed] [Google Scholar]
- 44. Kim J, Lee JE, Heynen-Genel S, Suyama E, Ono K, Lee K, et al. Functional genomic screen for modulators of ciliogenesis and cilium length. Nature . 2010;464:1048–1051. doi: 10.1038/nature08895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. McMurray RJ, Wann AK, Thompson CL, Connelly JT, Knight MM. Surface topography regulates wnt signaling through control of primary cilia structure in mesenchymal stem cells. Sci Rep . 2013;3:3545. doi: 10.1038/srep03545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wanner A, Salathé M, O’Riordan TG. Mucociliary clearance in the airways. Am J Respir Crit Care Med . 1996;154:1868–1902. doi: 10.1164/ajrccm.154.6.8970383. [DOI] [PubMed] [Google Scholar]
- 47. Mitchel JA, Das A, O’Sullivan MJ, Stancil IT, DeCamp SJ, Koehler S, et al. In primary airway epithelial cells, the unjamming transition is distinct from the epithelial-to-mesenchymal transition. Nat Commun . 2020;11:5053. doi: 10.1038/s41467-020-18841-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bolaños AL, Milla CM, Lira JC, Ramírez R, Checa M, Barrera L, et al. Role of Sonic Hedgehog in idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol . 2012;303:L978–L990. doi: 10.1152/ajplung.00184.2012. [DOI] [PubMed] [Google Scholar]
- 49. Cigna N, Farrokhi Moshai E, Brayer S, Marchal-Somme J, Wémeau-Stervinou L, Fabre A, et al. The hedgehog system machinery controls transforming growth factor-β-dependent myofibroblastic differentiation in humans: involvement in idiopathic pulmonary fibrosis. Am J Pathol . 2012;181:2126–2137. doi: 10.1016/j.ajpath.2012.08.019. [DOI] [PubMed] [Google Scholar]
- 50. Fitch PM, Howie SE, Wallace WA. Oxidative damage and TGF-β differentially induce lung epithelial cell sonic hedgehog and tenascin-C expression: implications for the regulation of lung remodeling in idiopathic interstitial lung disease. Int J Exp Pathol . 2011;92:8–17. doi: 10.1111/j.1365-2613.2010.00743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]