

Abstract
The dynamics describing the vicious cycle characteristic of cystic fibrosis (CF) lung disease, initiated by stagnant mucus and perpetuated by infection and inflammation, remain unclear. Here we determine the effect of the CF airway milieu, with persistent mucoobstruction, resident pathogens, and inflammation, on the mucin quantity and quality that govern lung disease pathogenesis and progression. The concentrations of MUC5AC and MUC5B were measured and characterized in sputum samples from subjects with CF (N = 44) and healthy subjects (N = 29) with respect to their macromolecular properties, degree of proteolysis, and glycomics diversity. These parameters were related to quantitative microbiome and clinical data. MUC5AC and MUC5B concentrations were elevated, 30- and 8-fold, respectively, in CF as compared with control sputum. Mucin parameters did not correlate with hypertonic saline, inhaled corticosteroids, or antibiotics use. No differences in mucin parameters were detected at baseline versus during exacerbations. Mucin concentrations significantly correlated with the age and sputum human neutrophil elastase activity. Although significantly more proteolytic cleavages were detected in CF mucins, their macromolecular properties (e.g., size and molecular weight) were not significantly different than control mucins, likely reflecting the role of S-S bonds in maintaining multimeric structures. No evidence of giant mucin macromolecule reflecting oxidative stress–induced cross-linking was found. Mucin glycomic analysis revealed significantly more sialylated glycans in CF, and the total abundance of nonsulfated O-glycans correlated with the relative abundance of pathogens. Collectively, the interaction of mucins, pathogens, epithelium, and inflammatory cells promotes proteomic and glycomic changes that reflect a persistent mucoobstructive, infectious, and inflammatory state.
Keywords: mucins, cystic fibrosis, mucus obstruction, microbiome, inflammation
Cystic fibrosis (CF) is an autosomal recessive genetic disorder caused by defects in the CFTR (cystic fibrosis transmembrane conductance regulator) (1). Although systemic in nature, most CF-associated morbidity and mortality reflects a respiratory disease that arises from ineffective mucus clearance and, ultimately, mucus stasis that is associated with chronic inflammation and infection (2). The large, highly glycosylated polymeric mucins are the major macromolecules in the airway mucus layer (3). The two most abundant secreted airway mucins are MUC5B, secreted from both submucosal glands and superficial airway epithelium, and MUC5AC secreted predominantly from the superficial airway epithelium (4–6). The function of these two mucins differs, and their relative concentrations differ in mucoobstructive diseases (e.g., chronic obstructive pulmonary disease [COPD]) versus asthma (7, 8). The mucus stasis characteristic of CF reflects largely raised mucin concentrations, increased mucus layer osmotic pressure, ciliary compression, and increased adhesive interactions between the mucus layer and epithelial surfaces. Mucus adhesion to epithelial surfaces leads to defective mucus clearance by both cilial and cough-dependent mechanisms (9–13).
Bacteria interact with the static mucus that serves as the nidus for bacterial infection in CF. The complexity and diversity of microbial communities in the CF lung have been elucidated by molecular characterization of microbes, which have revealed a succession from obligate and facultative anaerobic oral flora species to classic CF pathogens (e.g., Pseudomonas aeruginosa, Staphyloccocus aureus, and Haemophilus influenzae) in the progression of CF lung disease (14–16). Recent in vivo studies have shown that mucins can be degraded by bacterial glycosylhydrolases and used as energy sources by anaerobic bacteria aspirated into the lung from the oral cavity (17, 18). Proteases secreted by bacteria and host inflammatory cells also create a highly proteolytic CF airway environment, which overwhelms the lung’s antiprotease systems (19–21). It was proposed that this proteolytic activity can degrade mucin protein backbones and alter mucus rheological properties (22, 23).
The aim of this study was to characterize the interaction of the vicious cycle of CF lung disease (i.e., mucus obstruction, infection, and inflammation) on the properties of the mucins that provide the biophysical properties to mucus that mediates cilial and cough-dependent clearance. Total mucin, and the individual gel-forming mucin, MUC5AC and MUC5B, concentrations were measured and characterized with respect to modification of their macromolecular properties by proteolysis and glycosidases. These findings were related to clinical and quantitative microbiologic data to elucidate bacterial–mucin interactions in the CF lung environment. In parallel, a controlled ex vivo model system in which primary cell cultures were challenged with Pseudomonas aeruginosa was used to investigate specific roles for this CF pathogen in the modification of secreted mucins.
Methods
Study Population
Spontaneously expectorated sputum samples were collected from subjects with CF when clinically stable (N = 18) and at the onset of antibiotic treatment for an acute exacerbation (N = 25) (14). Induced sputum samples were collected from 29 healthy control subjects without CF. Sputum samples were frozen immediately after collection. Demographics and clinical data, including spirometry measurements, were obtained at each sample collection. Frozen sputum samples were thawed on ice and solubilized in 4M guanidine hydrochloride (GuHCl). Demographics and clinical data from all subjects are provided in Table 1.
Table 1.
Study Cohort Characteristics for Nondisease Controls and Cystic Fibrosis Patient Populations
| Nondisease Controls | Cystic Fibrosis | |
|---|---|---|
| Subjects | 29 | 44 |
| Age, mean (SD) | 49.4 (16.7) | 27 (10) |
| Gender, n (%) | ||
| Male | 10 (34.5) | 23 (52.3) |
| Female | 19 (65.5) | 21 (47.7) |
| Body mass index, mean (SD) | 27.1 (5.2) | 21.4 (3.0) |
| FEV1 % predicted, mean (SD) | 96.3 (18.4) | 52.2 (19.9) |
| Disease status at time of collection, n (%) | ||
| Stable (%) | N/A | 18 (42) |
| Exacerbation (%) | N/A | 25 (58) |
| Δ508 status, n (%) | ||
| Homozygous | N/A | 25 (57) |
| Heterozygous | N/A | 14 (32) |
| Other | N/A | 5 (11) |
Definition of abbreviations: FEV1 = forced expiratory volume in 1 second; N/A = not applicable.
Mucin Quantitation
Total mucin concentrations
Total sputum mucin concentrations were measured using size exclusion chromatography coupled to laser photometry (DAWN HELEOS II; Wyatt Technology) and refractometery (Optilab T-rEX; Wyatt Technology) after treatment with DNAse (Ambion; Life Technologies) as previously described (11). Data were analyzed with the Wyatt Technology ASTRA software, version 7.1.2.
MUC5AC and MUC5B concentrations
Samples were prepared for label-free and internal-labeled mass spectrometry analysis using a modified filter-aided sample preparation method (7, 24). After sample digestion, the tryptic peptides were analyzed by liquid chromatography–tandem mass spectrometry (a hybrid quadrupole Orbitrap mass spectrometer with a nanospray source, Q Exactive; Thermo Fisher Scientific) using data-dependent analysis.
Purification and Characterization of Gel-Forming Mucins
Mucin isolation and macromolecular measurements
A two-step isopycnic centrifugation method was used to separate the gel-forming mucins from globular proteins and DNA (4). Molecular weight and radius of gyration of the purified gel-forming mucins were measured using size exclusion chromatography coupled with laser photometry (DAWN HELEOS II) and refractometery (Optilab T-rEX) as described previously (11).
Glycomics analysis
The purified mucins were dialyzed into PBS, precipitated from acetone, and subjected to glycan analysis as previously described (25, 26). O-linked glycans were released from the precipitated mucins by reductive β-elimination, yielding intact, reduced structures. The released O-glycans were permethylated and analyzed by nanospray ionization multidimensional mass spectrometry using instruments with linear and orbital ion trap capabilities (LTQ-Orbi and Fusion Lumos; ThermoFisher) (27). Graphical representations of monosaccharide residues are presented in accordance with the broadly accepted Symbolic Nomenclature for Glycans, and O-glycan analysis was performed in keeping with the MIRAGE (Minimum Information Required for a Glycomics Experiment) guidelines for glycomic studies (28, 29).
Microbiota
Aliquots of sputum from each patient with CF were used for microbiota analysis using both clinical culture and bacterial 16S rRNA gene-sequencing methods as detailed previously (16).
Statistics
Paired statistical or repeated measure analyses were used for the in vitro experiments. Data were log2 transformed, and mixed models were used to analyze the sputum samples that were collected from the same individual at both stable and exacerbation timepoints. In addition, sex and age were accounted for as covariates. Scaled Bray-Curtis distance nonmetric multidimensional scaling analyses were used to test whether significant segregation occurred between the CF and control sputum on the basis of a range parameters.
Study Approval
Informed consent was obtained from all study participants, and the protocol was submitted to and approved by the University of North Carolina at Chapel Hill Biomedical Institutional Review Board.
Additional methods for more details can be found in the data supplement.
Results
Total Mucin Concentrations in CF versus Normal Subjects
Total mucin concentrations in sputum were raised in subjects with CF at baseline and during exacerbations as compared with control subjects (Figure 1A). Specifically, stable subjects with CF (n = 19) exhibited an approximately fourfold increase in mean total mucin concentrations (±SE) as compared with control subjects without disease (n = 10) (5477 ± 687.9 μg/ml vs. 1468 ± 400.4 μg/ml sputum) (Figure 1A). Subjects with CF exacerbating at the time of sputum collection (n = 24) also exhibited elevated total mucin concentration as compared with control subjects (5817 ± 576.4 μg/m), but this value was not significantly different than the stable subjects with CF. There was a significant correlation between the age of the patient within the CF cohort and the total mucin concentration (Pearson r = 0.427; P = 0.017) (Figure 1B) and also between sputum HNE (human neutrophil elastase) activity and total mucin concentration (Pearson r = 0.643; P = 0.007) (Figure 1C). Although there was a trend, no significant correlation was observed between total mucin and forced expiratory volume in 1 second percent predicted (Pearson r = 0.124; P = 0.47) (see Figure E1 in the data supplement) likely owing to smaller size of the cohort. No differences in total mucin concentrations were found in the CF cohort with reference to medication use, including hypertonic saline, inhaled corticosteroids, and antibiotics (e.g., tobramycin and colistin) (Figure E1).
Figure 1.
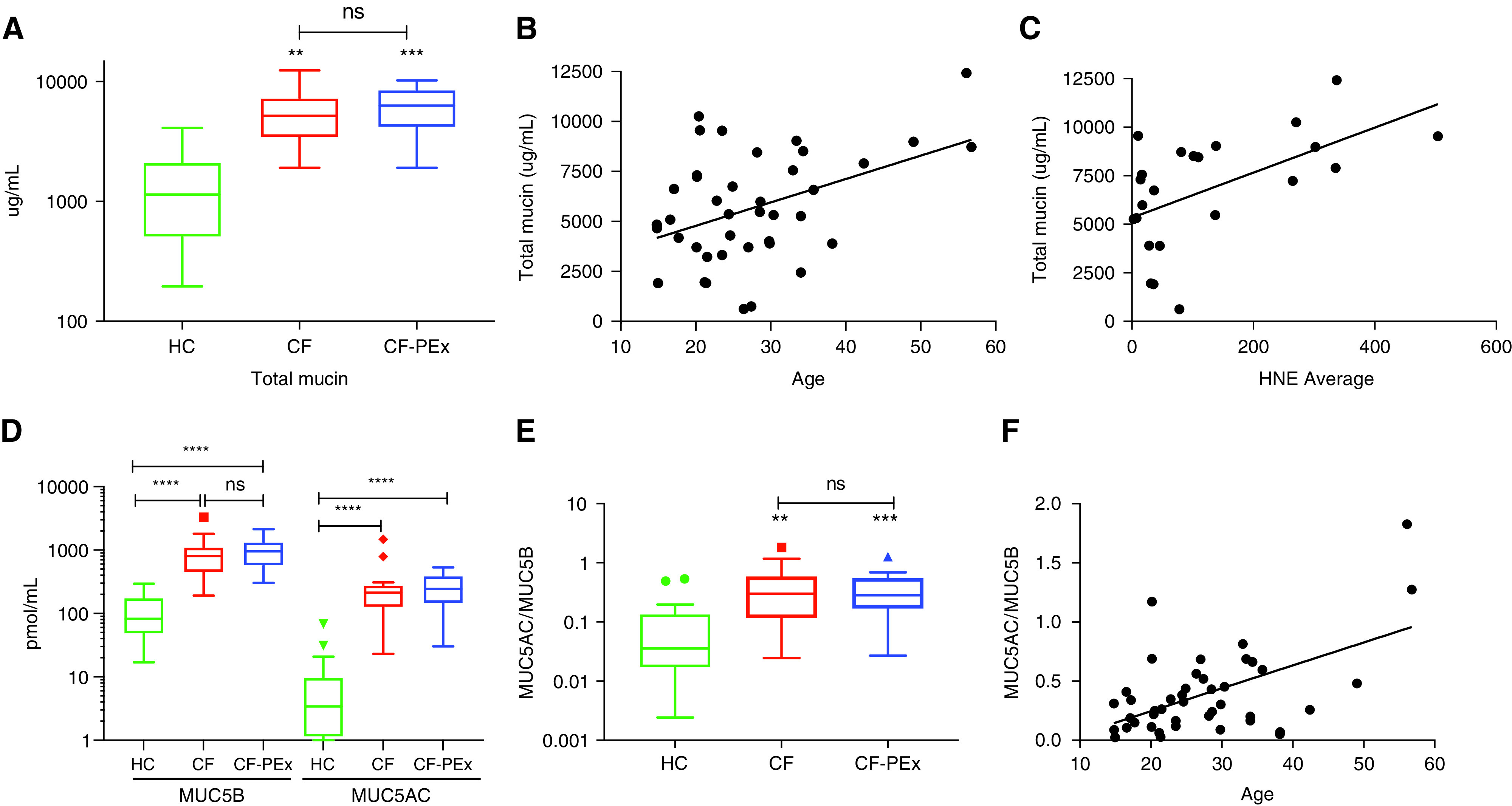
Mucin concentration increases significantly in cystic fibrosis (CF). (A) Total mucin concentrations of CF sputum is significantly higher than healthy control (HC) but does not differ between stable CF and CF with pulmonary exacerbation (CF-PEx) disease status. (B and C) Correlation between total mucin concentration and age (B) (n = 40) (Pearson r = 0.430; P = 0.006) and HNE (C) (n = 23) (Pearson r = 0.552; P = 0.006). (D and E) Absolute MUC5B and MUC5AC concentrations and the MUC5AC/MUC5B ratio are significantly elevated in CF but do not differ between stable and exacerbation disease status (healthy, n = 19; CF stable, n = 17; and CF exacerbation, n = 24). (F) Correlation of the ratio of MUC5AC/MUC5B ratio and age (n = 43) (Pearson r = 0.546; P = 0.00015). **P < 0.01, ***P < 0.005, and ****P < 0.001. FEV1 = forced expiratory volume in 1 second; HNE = human neutrophil elastase; ns = not significant.
MUC5B and MUC5AC Concentrations in CF versus Normal Sputum
The absolute MUC5B and MUC5AC concentrations in sputum from subjects with CF (n = 17) were also significantly greater than those in control subjects (n = 19) (Figure 1D). The absolute MUC5B concentration in the stable CF cohort was increased by approximately eightfold to 927.4 ± 179.3 picomol/ml as compared with values from control subjects, 108.2 ± 20.38 picomol/ml. MUC5AC concentrations were relatively lower in control normal subjects (9.514 ± 3.74 picomol/ml) than MUC5B values (108.2 ± 20.38 picomol/ml) and were increased approximately 30-fold in subjects with CF (288.7 ± 84.4 picomol/ml). Reflecting the disproportionate increase in MUC5AC versus MUC5B in CF, the ratio of MUC5AC to MUC5B was significantly higher in stable subjects with CF compared than in control subjects (0.44 ± 0.114 vs. 0.10 ± 0.036) (Figure 1E). As with the total mucin concentrations, significant positive correlations (Pearson r = 0.597; P ± 0.00025) were observed between the MUC5AC-to-MUC5B ratio and the age of the subjects with CF (Figure 1F). Neither the absolute concentrations of MUC5B (976.4 ± 103.3 picomol/ml) and MUC5AC (267.8 ± 28.19 picomol/ml) (Figure 1D) nor the ratio of MUC5AC to MUC5B (0.37 ± 0.06) (Figure 1E) were different as a function of exacerbation status in patients with CF (Figures 1D and 1E).
The Macromolecular Properties of the Gel-Forming Mucins Purified from Healthy Controls and CF Sputum
The mean (±SE) molecular weights of isolated mucins in sputum from stable subjects with CF (19.7 × 106 ± 2.18 × 106 g/mol, n = 15) versus that from normal subjects (26.5 × 106 ± 2.88 × 106 g/mol, n = 10) were lower but not significantly different (n = 10; P = 0.074) (Figure 2A). The radius of gyration (Rg) of the mucins isolated from stable CF sputum (182.8 ± 7.07 nm) was also not significantly different form normal subject samples (195.0 ± 8.93 nm) (Figure 2B). As with the concentration measurements, molecular weight did not differentiate CF sputum on the basis of disease status (exacerbation, 19.7 × 106 ± 1.84 × 106 g/mol) (Figure 2C). These findings were consistent for the Rg measurements, which did not change during exacerbation periods for CF (190.0 ± 7.91 nm; N ± 24).
Figure 2.
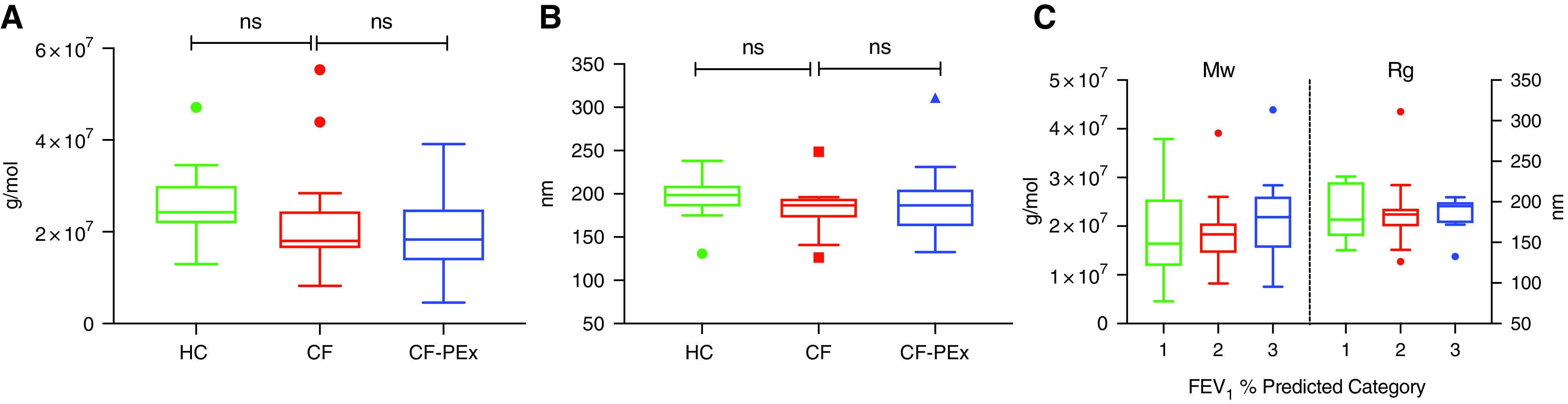
Macromolecular properties of the purified gel-forming mucins from sputum are not significantly different between healthy and CF. SEC-MALS measurement of (A) molecular weight (Mw) and (B) radius of gyration (Rg) of mucins purified by isopycnic centrifugation from sputum (healthy, n = 10; CF stable, n = 15; and CF exacerbation, n = 24). FEV1 percent predicted categories: 1: values ⩽36% (n = 10), 2: values >36% but ⩽65% (n = 17), and 3: values >65% (n = 10). (C) No significant differences in molecular weight and radius of gyration within the CF cohort based on FEV1 percent predicted category.
Quantity, Coverage, and Localization of MUC5B and MUC5AC Semitryptic Peptides in Mucins from Control and CF Sputum
To elucidate the effects of the highly proteolytic CF lung milieu on the mucin protein backbone, tryptic/semitryptic mucin mapping analyses were performed. A higher prevalence of semitryptic mucin peptides indicates in vivo proteolytic exposure preceding the in vitro trypsin digestion that cuts lysine or arginine residues. The ratio of semitryptic peptides to full tryptic peptides in sputum was significantly greater for both MUC5B (1.13 ± 0.06 vs. 0.51 ± 0.09) and MUC5AC (0.71 ± 0.07 vs. 0.07 ± 0.02) in CF than in healthy control sputum (Figure 3A). The increased ratio of semitryptic/full tryptic peptides corresponded to an increase in coverage of the MUC5B (11.04% ± 0.52% vs. 5.46% ± 0.74%) and MUC5AC (6.32% ± 0.57% vs. 0.62% ± 0.25%) protein backbones by semitryptic peptides in stable subjects with CF versus healthy control subjects (Figure 3B). There were no significant differences between CF sputum at baseline and during exacerbation based on the semitryptic/full tryptic peptide ratio or the semitryptic peptide coverage for MUC5B and MUC5AC (not shown).
Figure 3.
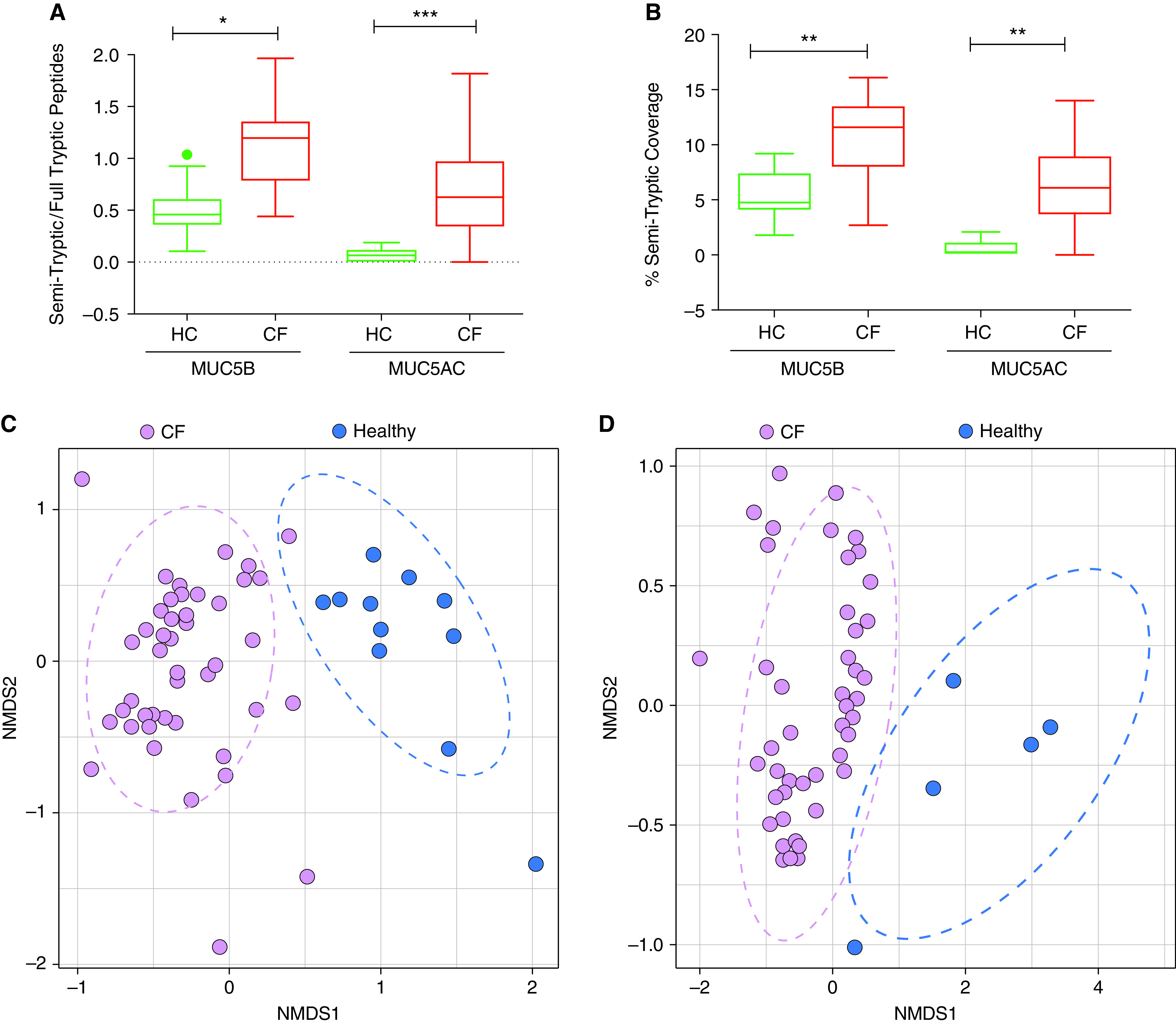
MUC5B and MUC5AC from CF sputum have a significantly higher ratio of semitryptic to fully tryptic peptides. Label-free LC-MS/MS analysis allowing (A) identification of unique semitryptic and fully tryptic peptides per sputum sample for MUC5B and MUC5AC and (B) the percentage of the mucin protein backbone covered by semitryptic peptides (control [HC], n = 10; CF, n = 41). (C and D) MUC5B and MUC5AC from CF (purple) and normal sputa (blue) show distinct signatures based on semitryptic peptides and the mucin domains they map to. Nonmetric multidimensional scaling (NMDS) analysis of unique MUC5B and MUC5AC semitryptic peptide sequences per sample from healthy and CF sputa and of their domain signature generated by mapping each semitryptic peptide to the MUC5B (C) or MUC5AC (D) backbone. There is significant separation between the CF and normal samples based on permutational multivariate analysis of variance (PERMANOVA) (P < 001) (MUC5B: normal, n = 11; CF, n = 44; MUC5AC: normal, n = 7; CF, n = 44.) Samples were excluded from analysis if no semitryptic peptides were identified. *P < 0.05, **P < 0.01, and ***P < 0.005.
To elucidate the differences in mucin backbone proteolysis between subjects with CF and healthy control subjects, a nonmetric multidimensional scaling analysis of the unique semitryptic peptides and the mucin protein domains onto which individual semitryptic peptides mapped was performed. This analysis revealed a significant segregation (permutational multivariate analysis of variance [PERMANOVA]; P < 0.001) of the healthy control and CF groups for both MUC5B (Figure 3C) and MUC5AC (Figure 3D).
A diagrammatic representation of the semitryptic peptide localization along the protein backbone for healthy control and CF MUC5B and MUC5AC mucins highlighted the differences in domain coverage between normal and CF (Figure 4). This analysis demonstrated significant differences in the percent of semitryptic peptides localized to specific mucin domains per sample for MUC5B and MUC5AC in the healthy control and CF groups. In general, there was a greater percentage of semitryptic peptides localized to the VWFD domains in the healthy control sputum than in CF. In contrast, CF showed a greater percentage of peptide coverage of the TIL, Cys-Rich, interdomain, and Ser/Thr rich regions. This finding is graphically demonstrated in the pie charts showing overall peptide localization based on a general domain designation for MUC5B and MUC5AC (Figure 4). Thus, the CF gel-forming mucins segregate from the normal on the basis of their absolute number of semitryptic peptides and their relative domain localization.
Figure 4.
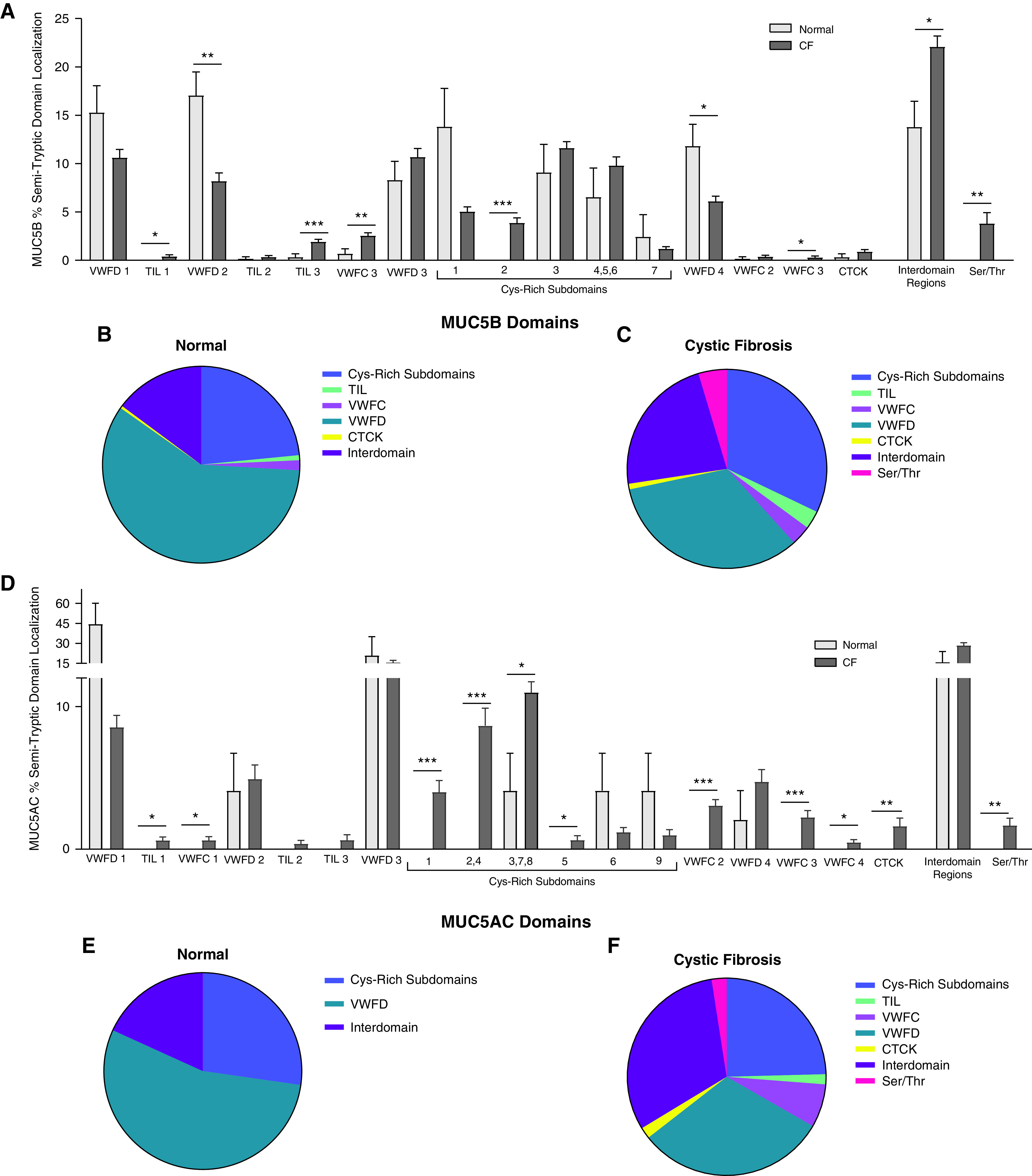
Specific domain localization of semitryptic peptides differs between CF and normal. Percentage of unique semitryptic peptides per sample mapped to individual Uniprot annotated protein domains for (A) MUC5B and (D) MUC5AC with pie charts showing localization based on region type for (B) normal and (C) CF for MUC5B and (E) normal and (F) CF for MUC5AC. *P < 0.05, **P < 0.01, and ***P < 0.005.
Sputum Mucin Concentrations and the CF Sputum Microbiota
The relative abundance of bacterial taxa within each CF sample was used to classify patients with CF on the basis of the proportions of oral flora, pathogens, and other flora (N = 18) (Figure 5A) and the microbiome data with respect to mucin analyses. Interesting trends were found between pathogen abundance and mucin measurements. Specifically, there was a positive trend between pathogen abundance and total mucin concentrations (Pearson r = 0.42; P = 0.053) (Figure 5B). There was a similar trend for the MUC5AC/MUC5B ratio (Pearson r = 0.48; P ± 0.062), which reflected a negative correlation between pathogen abundance and MUC5B concentrations (Pearson r ± −0.41; P = 0.11) and a positive correlation between pathogen abundance and MUC5AC concentrations (Pearson r = 0.32; P = 0.22) (Figure 5C and 5D). To test for effects of salivary contamination, a comparison between MUC5B concentration and salivary amylase (Figure E2) was performed, and no significant relationship was found between salivary amylase and oral flora (Pearson r = 0.008; P = 0.52), indicating that the oral flora detected in sputum were unlikely to be present owing to salivary contamination during sample collection.
Figure 5.
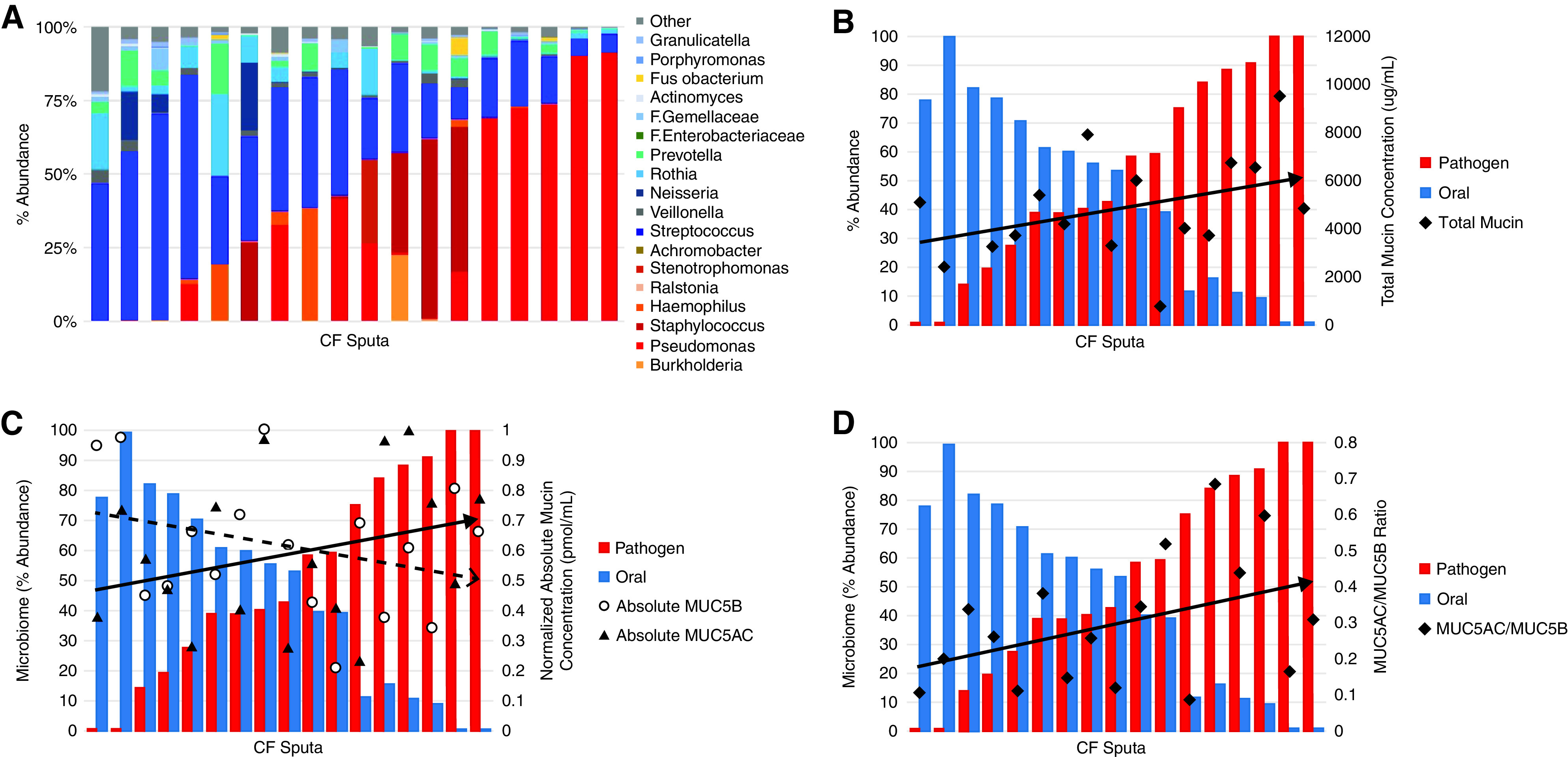
Relationship between microbiome and mucin concentrations. (A) Abundance of bacterial genera based on sputum microbiome sequence analysis shows stratification on the basis of oral flora or classic pathogen classification. 16s rRNA sequencing identified microbiome composition of a subset of CF sputum samples (n = 20). Each column represents one sputum sample. Cool-toned bars (blue/green) indicate that the genera is commonly identified as part of the oral flora community and warm (red/orange) toned bars indicate microbe is typically identified as a CF lung pathogen. (B) Total mucin concentrations compared to abundance of genera classified as either oral flora or pathogen (r = 0.42; P = 0.053). Absolute quantitative LS-MS/MS using labeled peptides was used to determine MUC5AC and MUC5B concentrations and the ratio between them (C), which was compared with microbiome data. Each pair of blue and red bars represents one CF sputum sample (n = 17), which were arranged by increasing pathogen abundance from left to right. The ratio reflects the relationship between the absolute concentrations (C) of MUC5AC (black triangles, r = 0.32; P = 0.22) and MUC5B (open circles, r = −0.41; P = 0.11). (D) MUC5AC/MUC5B ratio versus pathogen abundance (r = 0.48; P = 0.062).
Glycan Profiles of Gel-Forming Mucins and Correlations to Microbiota
Purified mucins were subjected to glycomics analyses to analyze the glycan abundance and structural diversity of subjects with CF versus control subjects (Tables E1 and E2). The total abundance of sulfated glycans in gel-forming mucins was sevenfold lower in CF mucins than in normal mucins (Figure 6A). In contrast, gel-forming mucins isolated from CF sputum (n = 10) exhibited significantly more nonsulfated glycans (14.6 ± 4.0 pmol/μg mucin) than healthy control subjects (3.3 ± 0.6 pmol/μg mucin; P < 0.05). Furthermore, these glycan structures were significantly more sialylated in CF than healthy control mucins (7.9 ± 2.1 pmol/μg vs. 1.1 ± 0.3 pmol/ug mucin; P < 0.01).
Figure 6.
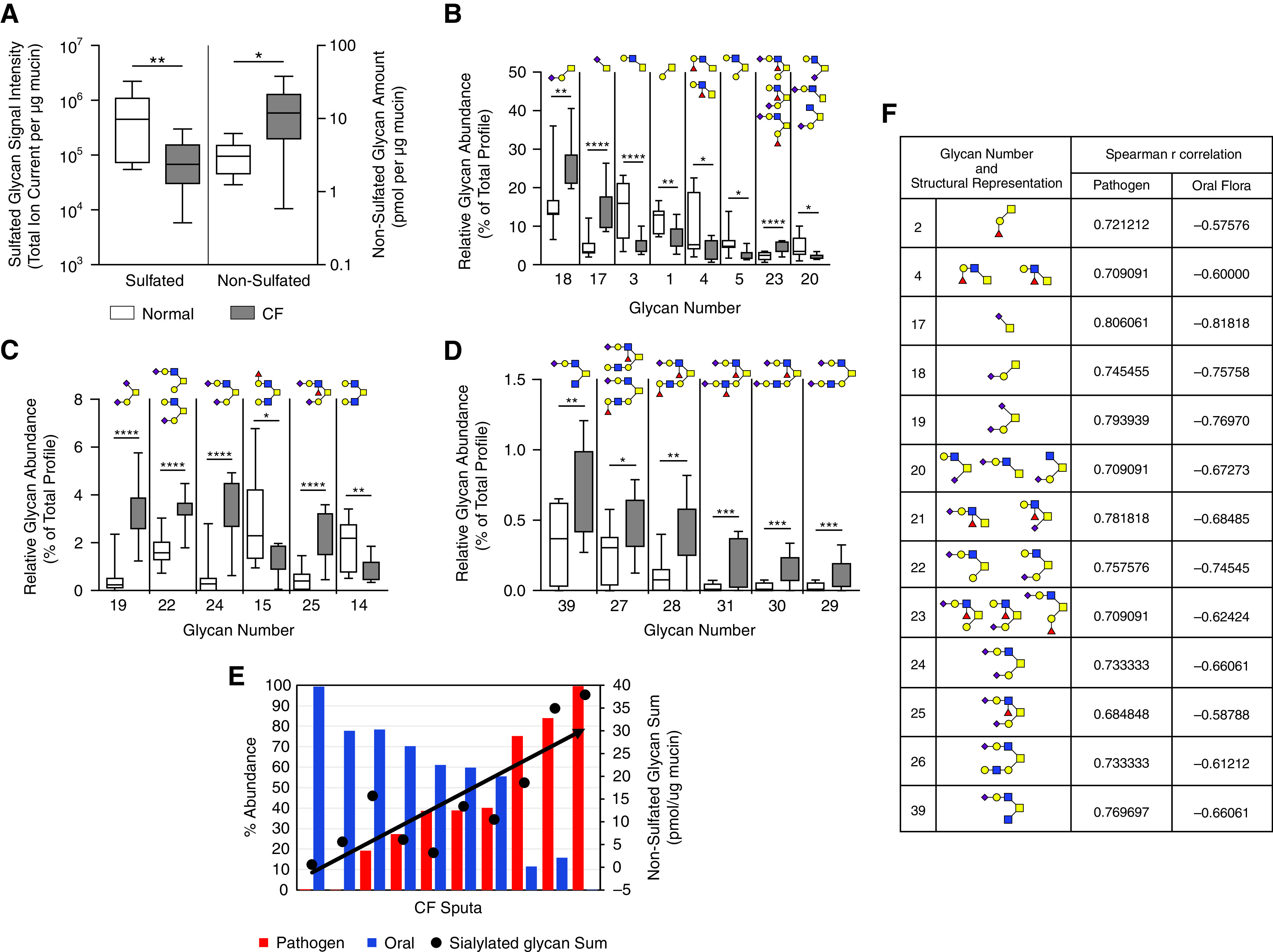
Purified gel forming mucins from control and CF sputum have significantly different glycan profiles. (A) Mass spectrometry analysis of sulfated and nonsulfated (sialylated) glycans of purified gel-forming mucins isolated via isopycnic centrifugation from healthy and CF sputa (healthy, n = 7; CF, n = 10). (B–D) Glycans of high, intermediate, and low abundance. (E) Significant positive correlation between pathogen abundance and cumulative nonsulfated glycan sum normalized per mg mucin (r = 0.82; P = 0.0001). Data are presented as box and whisker plots indicating the median, quartile, and data range. Glycan numbers refer to glycan structures in Table E1. *P < 0.05, **P < 0.01, ***P < 0.005, and ****P < 0.001. (F) Additional glycans that significantly associated with microbiome composition based on Spearman r correlation are listed in the inset table.
Sialylated core 1 and core 2 glycans were more highly abundant in CF than normal mucins (structures 17–19, 22–25, 27–29, and 31) (Figure 6), whereas nonsialylated core 1, core 2, core 3, and core 4 glycans (structures 1, 3–5, 14, and 15) (Figure 6) were less abundant in CF mucins. A sulfo-sialyl-Lewis type glycan, structure S10, was also significantly higher in CF mucins (Figure E3).
Within the CF cohort, glycomic data were compared to the microbiota on the basis of stratification by pathogen abundance. Among the CF samples, the total abundance of nonsulfated O-glycans (normalized to mucin concentration as measured by size exclusion chromatography-coupled to multi-angle laser light scattering [SEC-MALS]) significantly correlated with the percentage abundance of pathogens (Spearman r = 0.82; P = 0.001) (Figure 6E). Individual nonsulfated CF mucin glycans with significant correlations to the microbiota are listed in the inset table in Figure 4F, and, for the most part, these correlations mirrored the changes in relative glycan abundance detected in the CF mucin glycomics, namely sialylated core 1, 2, and 4 type glycans.
Relationship of Pseudomonas Exposure to Mucin Profiles In Vitro
To elucidate the effects of a specific pathogen (i.e., P. aeruginosa) infection on mucins in the absence other host factors, including HNE and other inflammatory cell–derived proteases, the apical surfaces of fully differentiated human tracheobrochial epithelial cell cultures derived from CF (CF-HTBE) were exposed to P. aeruginosa supernatants for 5 days. Apical lavages were analyzed by label-free liquid chromatography–tandem mass spectrometry (LC-MS/MS), SEC-MALS, and whole-mount immunohistochemistry (IHC) performed. Representative three-dimensional renderings of the whole-mount IHC show an increase in MUC5AC (red) and MUC5B (green) staining on the apical surface of Pseudomonas-exposed cultures as compared with the control culture after extensively washing with PBS (Figures 7A and 7B).
Figure 7.
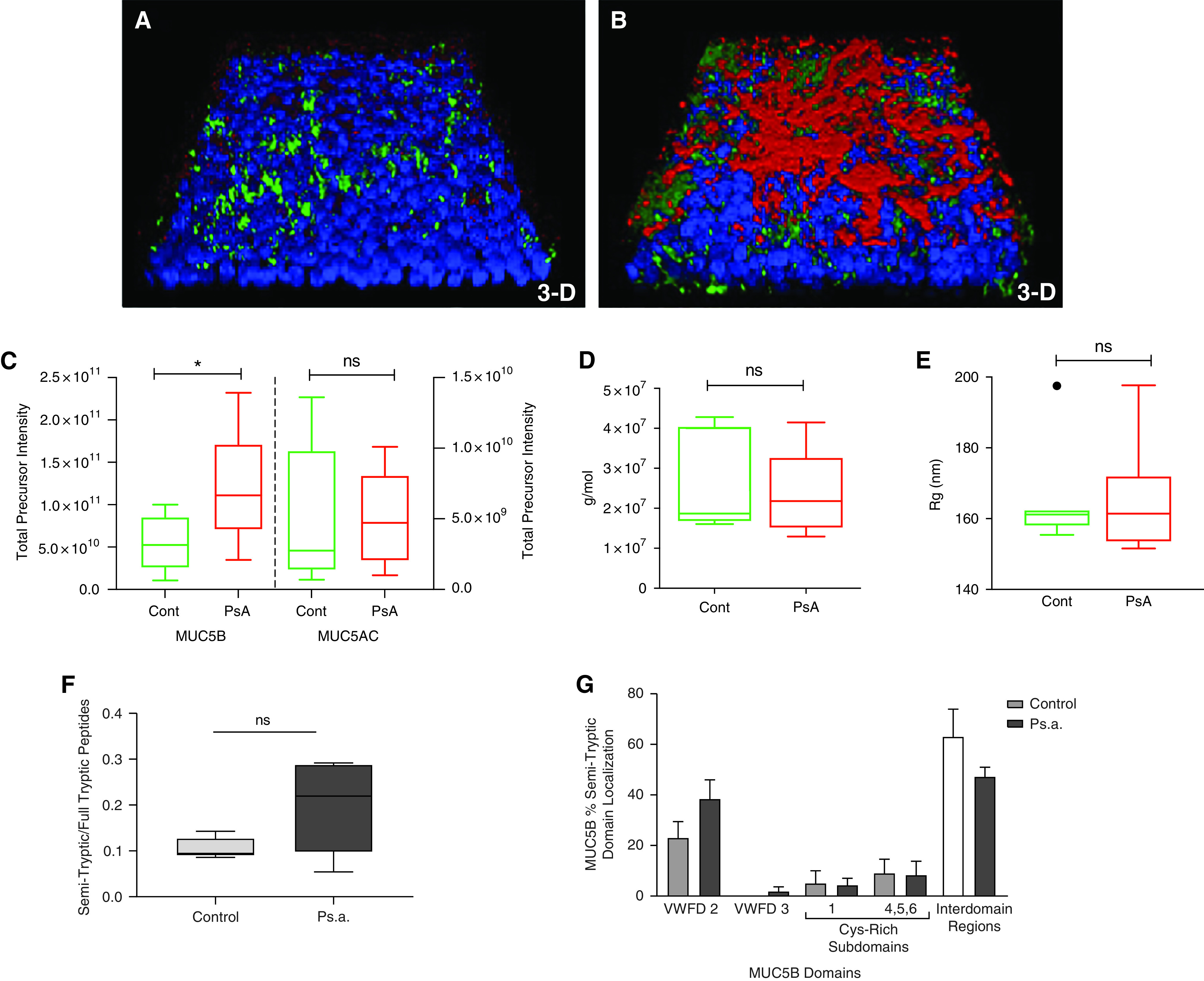
CF-HTBE cultures apically challenged with Pseudomonas aeruginosa exhibit mucin hypersecretion without alteration in macromolecular properties. (A and B) Representative images of whole-mount immunohistochemistry three-dimensional renderings of 120-hour tryptic soy broth (TSB) control (cont) (A) and P. aeruginosa (B) challenged CF-HTBE culture from the same donor. Blue: DAPI (nuclei). Red: MUC5AC. Green: MUC5B. (C) MUC5B and MUC5AC concentrations in HBE apical secretions after challenge as measured by label-free LC-MS/MS (n = 5). (D and E) Gel-forming mucins purified from apical secretions by isopycnic centrifugation show similar molecular weight (D) and radius of gyration (E) as measured by SEC-MALS (n = 7). (F and G) Ratio of unique MUC5B semitryptic peptides to fully tryptic peptides (F) and MUC5B semitryptic peptide localization is not significantly different between control and P. aeruginosa–challenged cultures (G). *P ⩽ 0.05.
Proteomic analyses of the apical washes from P. aeruginosa –exposed versus tryptic soy broth–exposed (control) cultures (mean total precursor intensity ± SE) revealed a significant twofold increase in MUC5B (11.9 × 1010 ± 3.17 × 1010, P. aeruginosa; 5.49 × 1010 ± 1.5 × 1010, control) intensity but not MUC5AC (5.03 × 109 ± 2.32 × 109, P. aeruginosa; 4.98 × 109 ± 1.53 × 109, control) (Figure 7C), suggesting that most of the MUC5AC on the epithelia surface is not washable with PBS. The gel-forming mucins purified from the apical secretions of Pseudomonas-treated cultures, analyzed by isopycnic centrifugation and SEC-MALS, were unchanged in molecular weight (25.1 × 106 ± 3.8 × 106 g/mol, P. aeruginosa, vs. 24.9 × 106 ± 4.4 × 106 g/mol, TSB (Figure 7D) or radius of gyration (165.5 ± 5.9 nm, P. aeruginosa, vs. 165.1 ± 5.5 nm, TSB) (Figure 7E) compared with the mucins from TSB-treated control cultures. Interestingly, the semitryptic analysis of the mucins after P. aeruginosa challenge showed fewer differences in both the domain localization and number of unique semitryptic peptides than the mucins isolated from TSB-treated control cultures (Figures 7F and 7G).
Timecourse Analysis of P. aeruginosa Supernatants and TSB with Purified Gel-Forming Mucins
To analyze effects of P. aeruginosa–derived proteases on mucin structure, purified MUC5AC or MUC5B was incubated with P. aeruginosa filtrate or TSB for 24 hours. Representative Western blots after agarose gel electrophoresis probed with polyclonal antibodies for MUC5B and MUC5AC revealed marked loss of antibody reactivity for both mucins exposed to P. aeruginosa during the 24-hour timecourse (quantitated below the Western blots) (Figures E4A and E4B). The mucin concentration, as measured by SEC-MALS, however, did not show a significant decrease after P. aeruginosa exposure to MUC5B or MUC5AC (Figures E4D and E4E). No loss of antibody reactivity was evident when mucin standards were incubated with the control, TSB (Figure E4C).
Discussion
Defective mucus clearance with mucus stasis and subsequent inflammation and infection are hallmarks of CF lung disease. Mucus layer hyperconcentration, via osmotic interactions with the periciliary layer (PCL), has been shown to be critical in initiating mucus stasis in CF (9, 11). However, recent data suggest MUC5AC and MUC5B have different properties and may differentially contribute to disease pathogenesis (30, 31). Neither the absolute concentrations of the individual MUC5AC and MUC5B gel-forming mucins to CF pathogenesis nor how their macromolecular properties are modified by the inflammatory and infectious milieu of the CF lung in the context of mucus transport versus stasis are known.
To study these processes, comprehensive sputum analyses were performed using samples from subjects with CF versus healthy, nonsmoking subjects. Because of the difficulty in quantifying and characterizing mucins antibody–based approaches in proteolytic environments (4, 11, 22), physical and mass spectrometry–based techniques were used to measure total mucin and individual mucin concentrations. Henderson and colleagues, using biophysical mucin measurements, were the first to show that total mucin concentrations were increased three- to fourfold in CF versus control sputum (10). Our current study, using a larger cohort, confirmed that observation by demonstrating that total mucin concentrations were increased approximately fourfold in CF versus control sputum.
Our studies extended these findings in several directions/dimensions. First, total mucin concentrations were positively correlated with age of the patients, but because of the limited subjects more than 40 years old (n = 4) in the cohort, this analysis was limited. Also, increased total mucin concentrations were correlated with sputum HNE activity. These relationships could reflect both HNE activities that increase mucin expression in the airways and the increase in HNE burden with age (32). Notably, HNE is one of the critical risk factors in developing bronchiectasis in children with CF, and HNE concentrations increase with age as more extensive and persistent neutrophilic inflammation evolves (33). The age- and HNE-related mucin associations are likely consequences of prolonged infection and inflammation seen in bronchiectasis.
Second, despite the heavy proteolytic and glycosidic activity and oxidative burden within the CF airways, the molecular integrity of mucins (i.e., Mw and Rg) isolated from CF sputum was not significantly different from that from healthy subjects. These findings suggest that mucins mostly remain intact in vivo likely via intramolecular disulfide bonds that maintain the structure of the mucin polymer despite sections of the backbone being cleaved by proteases. This “proteolytically cleaved, otherwise intact” structure may make CF mucins a favorable target for S-S bond–reducing agents as therapeutics. Consistent with this notion, previous studies have reported that CF mucins are more susceptible to reducing agents. In other words, they are reduced to many small species after administration of very small amounts of disulfide-reducing agents (34). Moreover, the similar macromolecular characteristics of mucins (Mw and Rg) isolated from CF and healthy sputum also suggest that “oxidation-induced cross-linking of mucin polymers”, as previously proposed (35) to explain increased viscoelasticity of “stiffened” airway mucus in CF, is unlikely. The CF mucin molecular mass/Rg data suggest that the increased viscoelasticity/stiffened nature of CF mucus is owing to increased mucin, including both MUC5AC and MUC5B, concentrations, and not increased mucin cross-linking. Similarly, these data suggest that the major target for S-S bond–reducing agents are intramucin, not polymeric intermucin, S-S bonds.
Third, important differences in the glycan composition of CF versus control gel-forming mucins were identified and were correlated to CF pathogens. CF gel-forming mucins were more sialylated and less sulfated than normal control mucins. The glycomic analyses demonstrated that CF mucins exhibited fewer sulfated glycans (approximately sevenfold), and more sialylated glycans (approximately fivefold), than healthy control mucins. Overall, the relative abundance of 19 out of 32 individual nonsulfated glycans were significantly altered (either increased or decreased) on mucins isolated from CF sputum compared with healthy subject sputum. In more detailed analyses, these alterations were shown to reflect direct microbiome effects and/or host responses in CF. Notably, the sputum with the highest abundance of oral flora/lowest abundance of pathogens had the lowest quantity of nonsulfated glycans decorating the mucin (0.58 pmol/μg mucin). In contrast, the sputum with the lowest abundance of oral flora, but the highest abundance of pathogens, had the highest quantity of nonsulfated glycans per μg of purified mucin (37.9 pmol/μg mucin). Furthermore, increases in lower anaerobe/higher pathogen abundance were correlated with increased quantities of sialylated glycans, including sialyl-Tn, sialyl-core 1, and sialyl-core2. Although glycan sulfation was decreased in total abundance among the CF mucins, a significant increase was detected for one of the most abundant sulfated O-glycans, a sulfo-sialyl-Lewis–type structure (Glycan S10). Sulfated, sialylated O-glycans have potent inflammomodulatory activities, and the increase in the relative abundance of this structure may provide a functional connection between mucins, the microbiota, and host immune modulation (36, 37).
Our mucin subtype quantitation data demonstrated that MUC5B and MUC5AC concentrations increased dramatically, approximately 8- and 30-fold, respectively, in CF versus control sputum, and the MUC5AC/MUC5B ratio rose to approximately 0.4 in CF from approximately 0.1 in healthy control sputum. The increase of MUC5B and MUC5AC concentrations are comparable to the values we reported for non-CF bronchiectasis, where their concentrations were also increased dramatically (6- and 17-fold, respectively) (38) (Figure E5). MUC5AC and MUC5B concentrations, like total mucins, correlated with age. When compared to other mucoobstructive diseases such as COPD, the concentrations of MUC5B and MUC5AC in CF are significantly more elevated (3.2- and 2.6-fold, CF vs. COPD, respectively), although interestingly the ratios of MUC5AC to MUC5B in CF and COPD sputum are similar (0.4 ± 0.08 vs. 0.5 ± 0.1) (7). These data suggest that the similar pathobiological processes with respect to regulation of MUC5AC and MUC5B secretion rates may be operating in a spectrum of mucoobstructive diseases but that mucus hyperconcentration, which may be more ion transport related, is more pronounced in CF than COPD.
We observed no differences in mucin parameters, including total and individual mucin concentrations at baseline patient versus during exacerbations. We can therefore surmise that the rheological properties of mucins per se will not change during exacerbation. However, studies have indicated that sputum viscoelasticity increases during exacerbation and returns to baseline after recovery (39). Studies have also shown that DNA content can increase owing to increased neutrophil extracellular trap formation (NETosis) during exacerbations (40, 41). Therefore, because DNA releases from neutrophils that affects the rheological measurements, it may explain the rheological changes in during exacerbations. Unfortunately, the samples we studied here were suitable neither for rheology studies nor DNA measurements to test this notion directly in this cohort. However, we compared the histone concentrations detected in the induced sputum as an indicator of DNA in the samples (Figure E6). The histone measurements indicated that subjects with CF have markedly elevated histone in the sputum, indicating the NETosis and likely DNA involvement in changes in rheological properties of CF sputum during exacerbations.
Analyses of proteolysis of mucins provided insights into interactions between mucin hydrolases and proteases in the CF environment. For example, the absolute quantities of cleaved mucin peptides were greater in CF than normal, and the localization of the semitryptic peptides identified a unique signature that discriminated CF from normal, healthy MUC5B and MUC5AC. Notably, control mucins yielded peptides that localized to the nonglycosylated mucin domains, whereas CF mucins exhibited a significantly greater percentage of the semitryptic peptides mapping to the unique interdomain regions and cyst-rich regions inside the highly glycosylated mucin domains. To test whether these effects were CF specific or reflected the chronic mucoinflammatory milieu associated with bronchiectasis, we compared the CF semitryptic/tryptic mucin peptide ratios to those from a previously characterized non-CF bronchiectasis (NCFB) cohort. The comparison indicates that both MUC5AC and MUC5B cleavage ratios are similar in CF and NCFB (Figure E5). These findings suggest that glycosylhydrolase enzymes are present in the CF bronchiectatic environments that removed protective glycans in the variable number of tandem repeat (VNTR) regions and allowed proteases to cleave the shielded mucin protein backbone. These inferences are consistent with our glycomic data and suggest that sulfated glycans may inhibit mucin proteolysis (see below).
To elucidate the effects of pseudomonal versus host proteases in the absence of host inflammatory cell proteases, primary airway epithelial cells were challenged with sterilized filtrates of P. aeruginosa. Analyses of the apical secretions after this challenge revealed MUC5B hypersecretion without a concurrent increase in MUC5AC (Figure 7C). However, whole-mount IHC examination of the cultures detected a larger increase in MUC5AC than MUC5B contained in adherent mucus layers overlying the cilia (PCL) (Figure 7B). These data suggest that secreted MUC5AC was more adherent to the cell surfaces and more resistant to removal by extensive apical washing with PBS. This adherent MUC5AC layer could only be removed by a reducing agent (DTT) added to the washing buffer (not shown). Notably, our in vivo (sputum) observations demonstrated a significant increase in the MUC5AC concentration (30-fold) in CF sputum, suggesting that although MUC5AC is associated an adherent/“tethered” mucus layer in vitro, MUC5AC is secreted into coughable mucus in vivo. The semitryptic peptide–mapping data after P. aeruginosa exposure revealed a very modest, insignificant increase in semitryptic peptides with a domain localization similar to TSB controls. These data are consistent with the P. aeruginosa time course incubation data, which showed minimal degradation of the gel-forming mucins but still loss of antibody epitopes. When compared with the semitryptic sputum data, the in vitro data highlight the combinatorial effect of the host inflammatory cell proteases (i.e., HNE) and bacterial glycosidase/proteases, both from oral flora and pathogenic bacteria, that are present and active in the in vivo environment.
The changes in glycan patterns that arise in the CF lung in vivo reflect a complex biology. One pathway reflects the bacterial glycosidases, including sulfatases and sialidases, which remove and use specific glycans. A pathway in series is likely the epithelium, which responds to the presence of specific bacteria by altering host mucin glycosylation. This complexity may explain in part why there have been numerous glycan studies of CF airway secretions using various methodologies that have produced conflicting results (36, 42–45).
The correlations between mucins glycan composition with the microbiota composition of the microbiome reveal unknown connections between mucins and the different bacterial communities colonizing the CF airways. The paucity of nonsulfated, but sialylated, glycans in the oral flora-dominated CF samples suggests that oral flora contain the enzymes necessary to degrade mucin to 1) provide an energy source for oral bacterial metabolism and 2) provide metabolic products that can be used as energy sources for other genera less adept at metabolizing mucins (17). Thus, the reduced abundance of nonsulfated, sialylated glycans decorating the gel-forming mucins in sputum with a predominance of oral flora may provide in vivo evidence that these microbes do in fact cleave and use the sugars decorating the mucin backbone. Robinson and colleagues (46) suggested that clinical (but not nonclinical) Pseudomonas strains were able to cleave the sulfur from mucins, although likely for non–growth-related purposes (e.g., to uncover putative binding sites or to facilitate further carbohydrate release from mucins and/or proteolysis of mucins). This notion is consistent with our data showing that the greatest amount of nonsulfated O-glycans was found in the sputum samples with the highest abundance of classic CF pathogens, including Pseudomonas. Lastly, previously reported alterations in mucin glycosylation have been hypothesized to reflect defective acidification of intracellular organelles secondary to CFTR mutations (47, 48). The trending changes in glycan diversity profiles within our CF cohort, which strongly correlate with the microbiota, suggest that extracellular environmental factors may drive the reported glycomic findings.
Numerous therapies and treatments are used clinically to facilitate the clearance of mucus from the CF lung by directly targeting the mucus hyperconcentration itself (e.g., hypertonic saline) or more indirectly by targeting the inflammatory and infectious stimuli worsening mucus hypersecretion (e.g., corticosteroids and antibiotics, respectively). Despite their frequent use in this CF population, no differences in total mucin concentration as a function of treatment were observed. It is possible that the results of these therapies may be more evident at an earlier timepoint in disease progression or the small sample sizes were not adequately powered to detect these associations. Thus, future studies should be extended to include samples obtained from a larger cohort of individuals with different degrees of disease severity to investigate how the concentrations and ratios of the gel-forming mucins and glycosylation pattern change with disease progression and in response to treatments, especially with highly effective CFTR modulators (49).
In summary, our data showed that both MUC5B and MUC5AC mucin concentrations were higher in CF than normal sputum. MUC5AC disproportionately increased in CF airways (30-fold) as compared with MUC5B (8-fold). Considering the distinct biophysical properties of MUC5AC (e.g., it forms stiffer and more viscoelastic mucus than MUC5B [30]) and its close association with the initiation, progression and exacerbation in mucoobstructive lung diseases (31), we can surmise that MUC5AC may play a significant role in the stagnant mucus formation in CF airways and may be resistant to hydration therapies and will require mucolytic approaches to restore/promote mucus clearance. Both MUC5AC and MUC5B in CF are subject to enzymatic attack by a combination of glycosidases, including sulfatases from pathogens, and proteases (dominated likely by host inflammatory cells). These enzymatic modifications, and increased oxidative stress, did not perturb mucin macromolecular properties (i.e., molecular weight or size [Rg]) that have been predicted to reflect mucin cross-linking. Rather, our data suggest that mucin concentrations and composition (MUC5AC/MUC5B ratio) are the main drivers of abnormal mucus with increases in osmotic pressure (11) and viscoelasticity (30) dominating the failure of mucus clearance. These enzymatic activities, however, do provide both sugar and amino acid substrates for bacterial growth. In addition, a shift of O-linked glycan diversity generally toward greater sialylation and decreased sulfation was observed in CF mucins that may reflect host responses to inflammation and infection. The mix of bacterial and host influences on mucins generated specific glycans of importance in inflammation signaling (e.g., sulfo-sialyl-Lewis–type structures) were enriched in CF versus normal control sputum. Collectively, the interaction of mucins, pathogens, airway mucosal epithelium/gland, and inflammatory cells promotes proteomic and glycomic changes in secreted mucins and produces a complex milieu with many features tailored to maintaining, in the absence of clearance of retained mucus, a persistent inflammatory/infectious environment.
Acknowledgments
Acknowledgment
Parts of this text were included in Bethany Batson’s dissertation (https://cdr.lib.unc.edu/downloads/j3860c53q?locale=en).
Footnotes
Supported by the Cystic Fibrosis Foundation (grant KESIME17G0) and National Heart, Lung, and Blood Institute (grants R01HL103940, R01HL110906, and HL092964).
Author Contributions: Conception and design: M.K., M. Tiemeyer, M. Tunney, J.S.E., N.G.M., R.C.B., M.S.M., and M.C.W. Data production, analysis, and interpretation: B.D.B., B.T.Z., G.R., S.S.L., T.K., H.D., A.C., and P.W.C. Writing the manuscript: B.D.B., M.K., R.C.B., and M.C.W. All authors reviewed the manuscript.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2021-0359OC on April 29, 2022
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science . 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- 2. Boucher RC. Airway surface dehydration in cystic fibrosis: pathogenesis and therapy. Annu Rev Med . 2007;58:157–170. doi: 10.1146/annurev.med.58.071905.105316. [DOI] [PubMed] [Google Scholar]
- 3. Thornton DJ, Rousseau K, McGuckin MA. Structure and function of the polymeric mucins in airways mucus. Annu Rev Physiol . 2008;70:459–486. doi: 10.1146/annurev.physiol.70.113006.100702. [DOI] [PubMed] [Google Scholar]
- 4. Kirkham S, Sheehan JK, Knight D, Richardson PS, Thornton DJ. Heterogeneity of airways mucus: variations in the amounts and glycoforms of the major oligomeric mucins MUC5AC and MUC5B. Biochem J . 2002;361:537–546. doi: 10.1042/0264-6021:3610537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kesimer M, Ehre C, Burns KA, Davis CW, Sheehan JK, Pickles RJ. Molecular organization of the mucins and glycocalyx underlying mucus transport over mucosal surfaces of the airways. Mucosal Immunol . 2013;6:379–392. doi: 10.1038/mi.2012.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Okuda K, Chen G, Subramani DB, Wolf M, Gilmore RC, Kato T, et al. Localization of secretory mucins muc5ac and muc5b in normal/healthy human airways. Am J Respir Crit Care Med . 2019;199:715–727. doi: 10.1164/rccm.201804-0734OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kesimer M, Ford AA, Ceppe A, Radicioni G, Cao R, Davis CW, et al. Airway mucin concentration as a marker of chronic bronchitis. N Engl J Med . 2017;377:911–922. doi: 10.1056/NEJMoa1701632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Welsh KG, Rousseau K, Fisher G, Bonser LR, Bradding P, Brightling CE, et al. Muc5ac and a glycosylated variant of muc5b alter mucin composition in children with acute asthma. Chest . 2017;152:771–779. doi: 10.1016/j.chest.2017.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Button B, Cai L-H, Ehre C, Kesimer M, Hill DB, Sheehan JK, et al. A periciliary brush promotes the lung health by separating the mucus layer from airway epithelia. Science . 2012;337:937–941. doi: 10.1126/science.1223012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Button B, Goodell HP, Atieh E, Chen Y-C, Williams R, Shenoy S, et al. Roles of mucus adhesion and cohesion in cough clearance. Proc Natl Acad Sci USA . 2018;115:12501–12506. doi: 10.1073/pnas.1811787115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Henderson AG, Ehre C, Button B, Abdullah LH, Cai L-H, Leigh MW, et al. Cystic fibrosis airway secretions exhibit mucin hyperconcentration and increased osmotic pressure. J Clin Invest . 2014;124:3047–3060. doi: 10.1172/JCI73469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hill DB, Long RF, Kissner WJ, Atieh E, Garbarine IC, Markovetz MR, et al. Pathological mucus and impaired mucus clearance in cystic fibrosis patients result from increased concentration, not altered pH. Eur Respir J . 2018;52:1801297. doi: 10.1183/13993003.01297-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hill DB, Vasquez PA, Mellnik J, McKinley SA, Vose A, Mu F, et al. A biophysical basis for mucus solids concentration as a candidate biomarker for airways disease. PLoS One . 2014;9:e87681. doi: 10.1371/journal.pone.0087681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Muhlebach MS, Hatch JE, Einarsson GG, McGrath SJ, Gilipin DF, Lavelle G, et al. Anaerobic bacteria cultured from cystic fibrosis airways correlate to milder disease: a multisite study. Eur Respir J . 2018;52:1800242. doi: 10.1183/13993003.00242-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tunney MM, Field TR, Moriarty TF, Patrick S, Doering G, Muhlebach MS, et al. Detection of anaerobic bacteria in high numbers in sputum from patients with cystic fibrosis. Am J Respir Crit Care Med . 2008;177:995–1001. doi: 10.1164/rccm.200708-1151OC. [DOI] [PubMed] [Google Scholar]
- 16. Muhlebach MS, Zorn BT, Esther CR, Hatch JE, Murray CP, Turkovic L, et al. Initial acquisition and succession of the cystic fibrosis lung microbiome is associated with disease progression in infants and preschool children. PLoS Pathog . 2018;14:e1006798. doi: 10.1371/journal.ppat.1006798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Flynn JM, Niccum D, Dunitz JM, Hunter RC. Evidence and role for bacterial mucin degradation in cystic fibrosis airway disease. PLoS Pathog . 2016;12:e1005846. doi: 10.1371/journal.ppat.1005846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Henke MO, John G, Rheineck C, Chillappagari S, Naehrlich L, Rubin BK. Serine proteases degrade airway mucins in cystic fibrosis. Infect Immun . 2011;79:3438–3444. doi: 10.1128/IAI.01252-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hentschel J, Fischer N, Janhsen WK, Markert UR, Lehmann T, Sonnemann J, et al. Protease-antiprotease imbalances differ between cystic fibrosis patients' upper and lower airway secretions. J Cyst Fibros . 2015;14:324–333. doi: 10.1016/j.jcf.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 20. Taylor SL, Rogers GB, Chen AC, Burr LD, McGuckin MA, Serisier DJ. Matrix metalloproteinases vary with airway microbiota composition and lung function in non-cystic fibrosis bronchiectasis. Ann Am Thorac Soc . 2015;12:701–707. doi: 10.1513/AnnalsATS.201411-513OC. [DOI] [PubMed] [Google Scholar]
- 21. Twigg MS, Brockbank S, Lowry P, FitzGerald SP, Taggart C, Weldon S. The role of serine proteases and antiproteases in the cystic fibrosis lung. Mediators Inflamm . 2015;2015:293053. doi: 10.1155/2015/293053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Horsley A, Rousseau K, Ridley C, Flight W, Jones A, Waigh TA, Thornton DJ. Reassessment of the importance of mucins in determining sputum properties in cystic fibrosis. J Cyst Fibros . 2014;13:260–266. doi: 10.1016/j.jcf.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Serisier DJ, Carroll MP, Shute JK, Young SA. Macrorheology of cystic fibrosis, chronic obstructive pulmonary disease & normal sputum. Respir Res . 2009;10:63. doi: 10.1186/1465-9921-10-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wiśniewski JR. In: Microbial proteomics: methods and protocols. Becher D, editor. New York: Springer; 2018. Filter-aided sample preparation for proteome analysis; pp. 3–10. [DOI] [PubMed] [Google Scholar]
- 25. Aoki K, Perlman M, Lim JM, Cantu R, Wells L, Tiemeyer M. Dynamic developmental elaboration of N-linked glycan complexity in the Drosophila melanogaster embryo. J Biol Chem . 2007;282:9127–9142. doi: 10.1074/jbc.M606711200. [DOI] [PubMed] [Google Scholar]
- 26. Aoki K, Porterfield M, Lee SS, Dong B, Nguyen K, McGlamry KH, et al. The diversity of O-linked glycans expressed during Drosophila melanogaster development reflects stage- and tissue-specific requirements for cell signaling. J Biol Chem . 2008;283:30385–30400. doi: 10.1074/jbc.M804925200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mehta N, Porterfield M, Struwe WB, Heiss C, Azadi P, Rudd PM, et al. Mass spectrometric quantification of n-linked glycans by reference to exogenous standards. J Proteome Res . 2016;15:2969–2980. doi: 10.1021/acs.jproteome.6b00132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Varki A, Cummings RD, Aebi M, Packer NH, Seeberger PH, Esko JD, et al. Symbol nomenclature for graphical representations of glycans. Glycobiology . 2015;25:1323–1324. doi: 10.1093/glycob/cwv091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. York WS, Agravat S, Aoki-Kinoshita KF, McBride R, Campbell MP, Costello CE, et al. MIRAGE: the minimum information required for a glycomics experiment. Glycobiology . 2014;24:402–406. doi: 10.1093/glycob/cwu018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Carpenter J, Wang Y, Gupta R, Li Y, Haridass P, Subramani DB, et al. Assembly and organization of the N-terminal region of mucin MUC5AC: indications for structural and functional distinction from MUC5B. Proc Natl Acad Sci USA . 2021;118:e2104490118. doi: 10.1073/pnas.2104490118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Radicioni G, Ceppe A, Ford AA, Alexis NE, Barr RG, Bleecker ER, et al. Airway mucin MUC5AC and MUC5B concentrations and the initiation and progression of chronic obstructive pulmonary disease: an analysis of the SPIROMICS cohort. Lancet Respir Med . 2021;9:1241–1254. doi: 10.1016/S2213-2600(21)00079-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Voynow JA, Young LR, Wang Y, Horger T, Rose MC, Fischer BM. Neutrophil elastase increases MUC5AC mRNA and protein expression in respiratory epithelial cells. Am J Physiol . 1999;276:L835–L843. doi: 10.1152/ajplung.1999.276.5.L835. [DOI] [PubMed] [Google Scholar]
- 33. Sly PD, Gangell CL, Chen L, Ware RS, Ranganathan S, Mott LS, et al. AREST CF Investigators Risk factors for bronchiectasis in children with cystic fibrosis. N Engl J Med . 2013;368:1963–1970. doi: 10.1056/NEJMoa1301725. [DOI] [PubMed] [Google Scholar]
- 34. Ehre C, Rushton ZL, Wang B, Hothem LN, Morrison CB, Fontana NC, et al. An improved inhaled mucolytic to treat airway muco-obstructive diseases. Am J Respir Crit Care Med . 2019;199:171–180. doi: 10.1164/rccm.201802-0245OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yuan S, Hollinger M, Lachowicz-Scroggins ME, Kerr SC, Dunican EM, Daniel BM, et al. Oxidation increases mucin polymer cross-links to stiffen airway mucus gels. Sci Transl Med . 2015;7:276ra27. doi: 10.1126/scitranslmed.3010525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shori DK, Kariyawasam HH, Knight RA, Hodson ME, Genter T, Hansen J, et al. Sulphation of the salivary mucin MG1 (MUC-5B) is not correlated to the degree of its sialylation and is unaffected by cystic fibrosis. Pflugers Arch . 2001;443:S50–S54. doi: 10.1007/s004240100644. [DOI] [PubMed] [Google Scholar]
- 37. Jeffries JL, Jia J, Choi W, Choe S, Miao J, Xu Y, et al. Pseudomonas aeruginosa pyocyanin modulates mucin glycosylation with sialyl-Lewis(x) to increase binding to airway epithelial cells. Mucosal Immunol . 2016;9:1039–1050. doi: 10.1038/mi.2015.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ramsey KA, Chen ACH, Radicioni G, Lourie R, Martin M, Broomfield A, et al. Airway mucus hyperconcentration in non-cystic fibrosis bronchiectasis. Am J Respir Crit Care Med . 2020;201:661–670. doi: 10.1164/rccm.201906-1219OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ma JT, Tang C, Kang L, Voynow JA, Rubin BK. Cystic fibrosis sputum rheology correlates with both acute and longitudinal changes in lung function. Chest . 2018;154:370–377. doi: 10.1016/j.chest.2018.03.005. [DOI] [PubMed] [Google Scholar]
- 40. Dicker AJ, Crichton ML, Pumphrey EG, Cassidy AJ, Suarez-Cuartin G, Sibila O, et al. Neutrophil extracellular traps are associated with disease severity and microbiota diversity in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol . 2018;141:117–127. doi: 10.1016/j.jaci.2017.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Toussaint M, Jackson DJ, Swieboda D, Guedán A, Tsourouktsoglou TD, Ching YM, et al. Host DNA released by NETosis promotes rhinovirus-induced type-2 allergic asthma exacerbation. Nat Med . 2017;23:681–691. doi: 10.1038/nm.4332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schulz BL, Sloane AJ, Robinson LJ, Prasad SS, Lindner RA, Robinson M, et al. Glycosylation of sputum mucins is altered in cystic fibrosis patients. Glycobiology . 2007;17:698–712. doi: 10.1093/glycob/cwm036. [DOI] [PubMed] [Google Scholar]
- 43. Holmén JM, Karlsson NG, Abdullah LH, Randell SH, Sheehan JK, Hansson GC, et al. Mucins and their O-Glycans from human bronchial epithelial cell cultures. Am J Physiol Lung Cell Mol Physiol . 2004;287:L824–L834. doi: 10.1152/ajplung.00108.2004. [DOI] [PubMed] [Google Scholar]
- 44. Schulz BL, Sloane AJ, Robinson LJ, Sebastian LT, Glanville AR, Song Y, et al. Mucin glycosylation changes in cystic fibrosis lung disease are not manifest in submucosal gland secretions. Biochem J . 2005;387:911–919. doi: 10.1042/BJ20041641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Boat TF, Cheng PW, Iyer RN, Carlson DM, Polony I. Human respiratory tract secretion. Mucous glycoproteins of nonpurulent tracheobronchial secretions, and sputum of patients with bronchitis and cystic fibrosis. Arch Biochem Biophys . 1976;177:95–104. doi: 10.1016/0003-9861(76)90419-7. [DOI] [PubMed] [Google Scholar]
- 46. Robinson CV, Elkins MR, Bialkowski KM, Thornton DJ, Kertesz MA. Desulfurization of mucin by Pseudomonas aeruginosa: influence of sulfate in the lungs of cystic fibrosis patients. J Med Microbiol . 2012;61:1644–1653. doi: 10.1099/jmm.0.047167-0. [DOI] [PubMed] [Google Scholar]
- 47. al-Awqati Q, Barasch J, Landry D. Chloride channels of intracellular organelles and their potential role in cystic fibrosis. J Exp Biol . 1992;172:245–266. doi: 10.1242/jeb.172.1.245. [DOI] [PubMed] [Google Scholar]
- 48. Barasch J, Kiss B, Prince A, Saiman L, Gruenert D, al-Awqati Q. Defective acidification of intracellular organelles in cystic fibrosis. Nature . 1991;352:70–73. doi: 10.1038/352070a0. [DOI] [PubMed] [Google Scholar]
- 49. Davies JC, Moskowitz SM, Brown C, Horsley A, Mall MA, McKone EF, et al. VX16-659-101 Study Group Vx-659-tezacaftor-ivacaftor in patients with cystic fibrosis and one or two phe508del alleles. N Engl J Med . 2018;379:1599–1611. doi: 10.1056/NEJMoa1807119. [DOI] [PMC free article] [PubMed] [Google Scholar]