

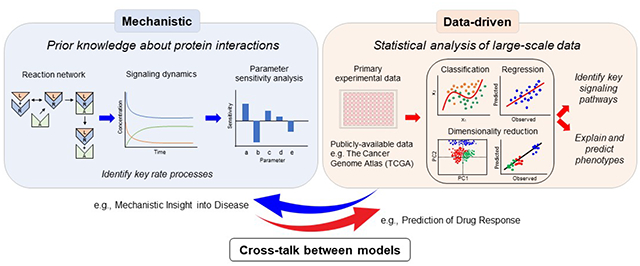
Abstract
A full understanding of cell signaling processes requires knowledge of protein structure/function relationships, protein-protein interactions, and the abilities of pathways to control phenotypes. Computational models offer a valuable framework for integrating that knowledge to predict the effects of system perturbations and interventions in health and disease. Whereas mechanistic models are well suited for understanding the biophysical basis for signal transduction and principles of therapeutic design, data-driven models are particularly suited to distill complex signaling relationships among samples and between multivariate signaling changes and phenotypes. Both approaches have limitations and provide incomplete representations of signaling biology, but their careful implementation and integration can provide new understanding for how manipulating system variables impacts cellular decisions.
Keywords: systems biology, uncertainty, sensitivity, parameter sampling, parameter estimation, regression, clustering, classification, cancer, immunology
Graphical Abstract
INTRODUCTION
Computational systems biology models have become virtually indispensable tools for integrating our knowledge of complex cell signaling processes. As biochemists characterized elegant protein structure/function relationships and post-translational modifications, the need to synthesize that understanding quantitatively to identify rate-limiting steps in signal transduction became apparent. Mechanistic models are well suited for this because of their ability to capture relevant biophysical processes through kinetic, constitutive, and conservation equations. As omics techniques eventually became common for measuring signaling processes at the network level, a need grew to cluster samples and predict phenotypes based on high-dimensional features. Data-driven models are ideal for these applications because of their ability to reduce system dimensionality while identifying key features and because of a general lack of mechanistic equations connecting phenotypes to multivariate signaling.
The distinction between mechanistic and data-driven modeling approaches can also be illustrated in terms of the useful “cue-signal-response” paradigm [1]. In that conceptual model, extracellular cues (e.g., growth factors) activate receptors, which initiate intracellular signaling processes, and thus elicit cellular responses (Fig. 1). In our view, mechanistic and data-driven models work best at the cue-signal and signal-response levels, respectively.
Figure 1. Modeling intracellular signaling and disease in context of “cue-signal-response” paradigm.
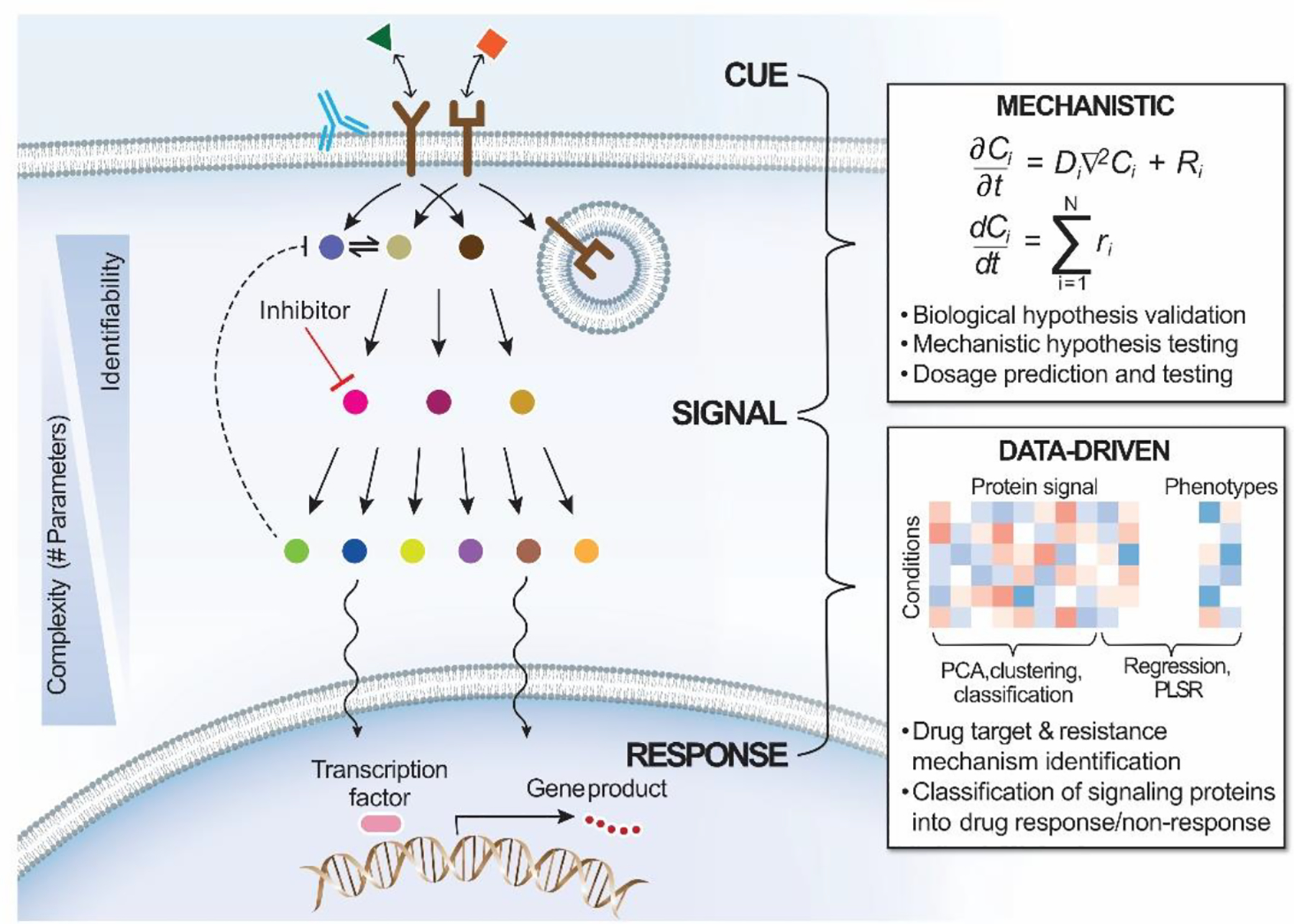
Intracellular signaling generally proceeds with a cue from the extracellular environment in form of ligands. Ligands bind to cognate receptors at the plasma membrane to activate receptors and downstream signaling pathways. Downstream effectors often undergo nucleocytoplasmic shuttling and regulate cell response via regulation of gene transcription. These processes are regulated by a variety of mechanisms, including receptor endocytic trafficking and feedbacks. In mechanistic models of signal transduction, biochemical reactions are typically represented by systems of differential equations, which include numerous parameters reflecting the rates of various binding and catalytic steps. These parameters determine model complexity and identifiability. Signaling dynamics and parameter relations deduced from mechanistic models help to form and validate biological hypotheses and can be applied to predict drug dosing regimens. Insight into signal-to-response relationships is more readily gained by data-driven models, which can reveal relevant structures in large signaling-relevant data sets and predict the importance of groups of signaling pathways in determining complex cell phenotypes. Common applications for data-driven models of signaling include identifying drug targets and resistance mechanisms.
This review highlights recent examples of mechanistic and data-driven signaling models that demonstrate the types of applications described above. Specific methods, model limitations, and implementation issues investigators should consider are described. Use of publicly available data and signaling modeling efforts by the biopharma industry are also discussed.
MECHANISTIC MODELS
Mechanistic signaling models synthesize biophysical understanding of network interactions to predict protein concentrations, post-translational modifications, or localization in response to system perturbations. Within the conceptual framework of Fig. 1, mechanistic models are most appropriate for cue-signal processes because they are governed by conservation and reaction rate laws that can be used to build differential equations. Well-mixed compartmentalized models are common, but mass transfer limitations can exist in signaling and have been incorporated, for example, in modeling signaling at the membrane-cytosol interface and in morphogenic gradients [2,3]. Deterministic solvers are used for abundant species, and stochastic methods can be employed for dilute species (e.g., < 100 molecules per compartment) [4].
While mechanistic models have a powerful ability to adapt to different physical scenarios, they tend to be populated by numerous parameters that are difficult to measure directly and are trained on experimental data with associated error. Thus, even though they are based on physical laws, mechanistic models do not produce output that is free from uncertainty. Moreover, signaling network structures often create scenarios where only a few combinations of rate processes exert meaningful control over system behavior. Dealing with these issues is often the most challenging aspect of mechanistic model design. The tools and methods described below help navigate these issues and should be considered in mechanistic model development.
Parameter Estimation and Inference
In many mechanistic models, protein-protein interactions are encoded based on experimental knowledge, and model tuning focuses primarily on parameters. Often, parameters believed to be well known are fixed, and those that are unknown or whose perturbations produce large changes in model output are estimated through fitting. In other situations, all parameters may be fit simultaneously. In either case, investigators should use caution that fitting results are consistent with magnitudes of established signaling process rates. Investigators should also keep in mind that poor fit may indicate a need to revise model structure and that model selection based on information criteria (e.g., Bayesian) or Bayesian evidence can be used for optimization.
Parameter estimation requires an objective function for minimization. In many cases, that function is defined as the sum of squared errors between model predictions and data [5], a choice used in fitting tools available in MATLAB and COPASI [6]. If errors are normally distributed, maximum likelihood estimation (MLE) provides parameter estimates equivalent to least squares while also providing the joint probability that the estimates explain model fit [7]. When errors are not normally distributed, profile likelihood provides more accurate estimates by finding parameters that minimize the chi-square distribution of the log-likelihood ratio [8]. For signaling models with up to thousands of parameters, the computational cost of estimating the gradient of the objective function can be prohibitive. Adjoint sensitivity analysis can substantially reduce computational costs by efficiently estimating the objective function gradient using backpropagation of the solution in a manner independent of the number of parameters [9].
When parameter distributions rather than point estimates are of interest, inference methods can be used. In Bayesian inference, each parameter is assigned a prior probability distribution representing uncertainty, and this is updated based on available data and optimization to generate a final posterior distribution [7,10–12]. In one recent example, an adaptive metropolis algorithm was used to calculate posterior parameter distributions describing the phosphorylation kinetics of different receptor tyrosine kinases [10]. When both the likelihood function and prior probability distribution are difficult to obtain, as in stochastic models, optimization can proceed via likelihood-free nested sampling, which iteratively simulates the model with parameter combinations sampled from posterior distributions to compute probabilities [13].
The efficiency and accuracy of the methods discussed here and others (e.g., Monte Carlo, Markov Chain Monte Carlo, and iterative filtering inference) are still being explored. Efforts have also recently been made to devise systematic platforms for obtaining probability density functions that work well for differential equation-based models when standard approaches fail [14].
Sensitivity Analysis
Local sensitivity analysis is the simplest technique to identify parameters and rate processes that most impact model output. It involves calculating a change in model output for a perturbation in an individual parameter. Local sensitivity analysis has been used, for example, to identify factors determining the combined effects of two RAF inhibitors on ERK activity [15], signaling protein complex persistence in response to EGFR activation [16], and efficacy of different classes of EGFR-targeted therapies [17]. Global sensitivity analysis is a more sophisticated and computationally demanding method in which all parameters are perturbed simultaneously. This approach more realistically depicts the biological fact that multiple parameters are perturbed at once in signaling systems (e.g., abundances of multiple proteins) [18]. Global sensitivity analysis may identify parameters that do not emerge through local analysis and may suppress the apparent importance of parameters that only exert control near the base model parameters. The discrepancy between local and global analyses tends to grow with more parameters [19], and it is useful to compare the methods to generate biological hypotheses.
One widely used global sensitivity metric is the partial rank correlation coefficient (PRCC), which quantifies the effects of parameter variations on model output using values chosen by random or pseudorandom methods such as Latin Hypercube Sampling (LHS) [20], as in recent immune signaling models [21,22]. PRCC magnitude and significance are assessed to identify important parameters. A more computationally intensive, yet popular, method is extended Fourier Amplitude Sensitivity Test (eFAST), a hybrid local/global sensitivity analysis in which variance in model output, represented by a Fourier series, is decomposed into variance from each parameter sampled from sinusoidal functions. eFAST was recently employed to identify parameters that exert strong influence on ERK activation individually or in groups [23].
Because global sensitivity analysis involves evaluating the model with multiple parameter sets sampled from a defined space, computation time depends on sampling method, with options including random, LHS, and Sobol sequences [20,24]. Although there is no established guideline for choice of sampling method to our knowledge, specific methods have typically been paired with specific sensitivity analysis approaches (i.e., LHS-PRCC or Sobol sequence-Sobol). Pseudorandom sampling such as LHS is generally preferred to random because the former samples parameter space homogenously and efficiently. Kolmogorov-Smirnov statistics is a simpler global method that measures separation between two cumulative trajectory distributions caused by solving the system with nominal parameter and perturbed parameter values [19,25].
Sloppiness, Identifiability, and Uncertainty
Due to lack of prior knowledge of some parameters and the locations of specific parameters within model equations (i.e., model structure), most mechanistic models of cell signaling are “sloppy” [26–28]. In such models, only a small number of parameter combinations control model output. This is characterized mathematically by log-linearly spaced eigenvalues of the Hessian matrix (second derivatives of the chi-squared quality of fit metric with respect to parameters) [12,29]. Uncertainty in model output can be formally quantified and attributed to combinations of “sloppy” or “stiff” parameters. Sloppiness analysis provides a range of fluctuations around the best fit parameters in each eigendirection of the Hessian that produces nearly equivalent model output. Thus, unlike sensitivity analysis, sloppiness directly accounts for error in the data used to fit and identify important parameters.
Many mechanistic models are also structurally non-identifiable or poorly conditioned, meaning that a unique set of parameters that provides a good fit to data cannot be determined, or is practically non-identifiable because parameter confidence intervals cannot be determined due to insufficient amount or quality of data [30]. While it may be tempting to consider model sloppiness and non-identifiability as synonymous, a sloppy model may be structurally identifiable and have unique values of parameter estimates for experimental data [31].
Modeling Cellular Responses using Mechanistic Models
While mechanistic models are typically best-suited to cue-signal problems, their use to understand transcriptional processes can provide a connection to phenotype determination. In one recent example, an ensemble ODE-based model was used to predict prolactin-mediated STAT5 transcriptional regulatory activity and Bcl-XL gene expression to identify strategies to enhance beta cell survival [32]. In another example, delay differential equations were trained on RNA-seq data to demonstrate that the transcription factors EGR1 and FRA-1 filter ERK activity to regulate DNA damage response [33]. In these examples and others (e.g., modeling macrophage phenotypes [34,35] or epithelial-mesenchymal transition [36]), a key element of a successful approach is that phenotypes are controlled by a known and circumscribed set of transcription factors such that a meaningful mechanistic model can be constructed.
DATA-DRIVEN MODELS
Data-driven models use computational algorithms to analyze signaling data without mechanistic knowledge. Datasets must include enough independent observations of the system (e.g., responses to different signaling agonists or drugs) and have sufficiently broad feature measurement (e.g., phosphoprotein measurements) to capture the biochemical processes that distinguish samples and explain how signaling leads to phenotypic changes. A variety of experimental methods can be used to gather these data, depending on the application and sample availability [37]. Within the conceptual framework of Fig. 1, data-driven models are most appropriate for signal-response relationships because of their ability to distill the complexity of high-dimensional features and quantitatively map those features to outcomes despite the lack of physically-based governing equations.
Depending on the approach, data-driven methods are sometimes referred to as machine learning models or statistical models and can be categorized as unsupervised or supervised. Unsupervised approaches analyze unlabeled data (i.e., outcomes are unknown) and focus primarily on sample clustering and dimensionality reduction. Common examples include principal components analysis (PCA) and t-stochastic neighbor embedding (t-SNE). Supervised approaches (i.e., regression and classification) predict outcomes and identify predictive signaling features by building models on labeled training data. Response data can include signals themselves, phenotypes, or group identity (e.g., tumor subtype). Successful data-driven modeling depends on pairing datasets with appropriate computational methods and on model refinement through cross validation.
Clustering and Dimensionality Reduction
For large data sets, dimensionality reduction and clustering has become increasingly common for understanding data structures. These methods identify latent dimensions in the data that best explain sample variance or minimize stress. PCA is a widely known technique based on maximizing variance among samples as they are projected into orthogonal, rank-ordered principal component vectors, defined by linear combinations of all measured features. PCA has become commonplace in signaling research, with recent examples including phosphoprotein analysis to stratify drug-responsive melanoma cell lines [38] and identification of pathways that differentiate primary Sjögren’s syndrome patients from healthy subjects [39].
In recent years, non-linear dimensionality reduction techniques including t-SNE and uniform manifold approximation and projection (UMAP) have also become common. These methods work through nonlinear projections of data into lower dimensional spaces in a way that minimizes the differences between inter-sample distance metrics in the high- and low-dimensional representations of the data. They generally perform better than PCA for separating single-cell measurements (e.g., mass cytometry, single-cell RNA-seq) [40,41] and produce low-dimensional embeddings that are useful as inputs to clustering algorithms (e.g., k-means) or for visualization. One drawback is that quantitative information and interpretations are lost in the non-linear projections. One recent study used t-SNE and decision tree-based classification to identify several clinically relevant epithelial-mesenchymal transition states in lung cancer from mass cytometry data [42]. A similar study used t-SNE and UMAP projections of flow cytometry data as inputs to a self-organizing maps algorithm to identify tumor cell groups with distinct signaling profiles that stratified patients for glioblastoma progression [43]. While the authors concluded that t-SNE and UMAP provided comparable performance, UMAP is generally faster and more accurately represents latent local and global structures within high-dimensional data sets [40]. Dimensionality reduction and cluster analysis can also identify signaling pathways warranting further investigation in mechanistic models [33,44] and supervised regression models [45–47].
Predicting Phenotypic Responses from Multivariate Signaling Data
Data-driven models can also be used to build quantitative relationships between phenotypes and high-dimensional feature data that provide direct or indirect information about signaling. A pervasive approach in the literature is to rely on next-generation sequencing for feature data and some form of pathway analysis for the coarse identification of relevant signaling pathways for different known sample classes (e.g., tumor cell subtypes). While this approach can be useful for a coarse initial survey, it should not be used as the only approach for identifying key signaling pathways for phenotype determination because transcriptomic measurements typically lack temporal resolution and provide only an indirect measure of signaling pathway activity. Phosphoproteomics provide a more direct indication of signaling pathway activity (at least as regulated by kinases) and can be obtained at the network level using various approaches [37]. Phosphoproteomic data can be subjected to differential phosphorylation analyses analogous to gene set enrichment [48–51], but these approaches still lack a connection between signaling measurements and outcomes.
To connect signaling measurements and outcomes, some studies have used partial least squares regression (PLSR), a linear approach that projects signaling and phenotype measurements into a reduced dimensional space by optimizing the alignment of scores vectors for paired matrices of signaling and phenotype measurements. Recent examples include predictions of cell responses to different classes of drugs [47,52,53] or the human immunodeficiency virus [54]. Nonlinear partial least squares regression can also be performed, and the optimal choice of model order can be informed by considering information criteria (e.g., Akaike). Other studies have used elastic net regression [46,55,56], a regularized version of linear regression that allows for correlated predictors without needing to project data onto a separate regression space (as in PLSR). When discrete phenotype or response data are provided (e.g., patient disease or treatment status), classification methods such as logistic regression, partial least squares discriminant analysis (PLS-DA), and support vector machines can be used to identify signaling processes that are predictive of sample class [57–60]. In one recent example, PLS-DA was used to identify novel immune cell-specific phosphoprotein signatures from mass cytometry data that most discriminated between healthy and treatment-naïve juvenile dermatomyositis patients [60].
PUBLICLY AVAILABLE DATASETS
A variety of sources of publicly available data can assist investigators in developing signaling hypotheses. These datasets generally lack dynamic resolution and are typically useful only for data-driven models. For oncology, the Cancer Dependency Map (DepMap) and The Cancer Genome Atlas (TCGA) provide cell line and patient tumor data that have been widely used for bioinformatics approaches. For example, TCGA data sets have been used in approaches similar to pathway analysis to identify activated kinase and oncoprotein networks in KRAS mutant contexts [51,61]. While publicly available snapshot transcriptomic data and cellular drug sensitivity data have been used in both data science and mechanistic models to predict cellular responses to drug perturbations [52,56,62–64], the general lack of dynamic measurements and emphasis on transcript data limits the insight that can be gained. Dynamic data provides richer information for mechanistic model building, but the same is true of data-driven models [56]. Incorporation of dynamic data, especially of phosphoprotein measurements and for complex perturbations (e.g., with combination therapies), in publicly available datasets will facilitate more powerful signaling models.
APPLICATIONS OF SIGNALING MODELS IN INDUSTRY
The biopharma industry has increasingly recognized systems signaling models as useful tools for developing and deploying therapeutics. Recent examples have leveraged mechanistic models to predict the utility of combining DNA damage response inhibitors with doxorubicin or gemcitabine to treat cancer [65], identify cell settings where a bispecific antibody would outperform a monovalent antibody targeting the MET receptor [66], develop design principles for drugs targeting signaling proteins for degradation [67], and design clinical trials for an ErbB3-targeting antibody in ovarian, breast, and lung cancer [68]. Industry practitioners have also used non-mechanistic models such as a decision trees to predict cancer cell line response to targeted inhibitors based on ligand addiction [69].
Industry colleagues have also advocated for advancing computational models, including those for signaling. For example, they have promoted data science as a core discipline in drug discovery and development [70] and urged thinking beyond drug-target binding energetics to build models incorporating the nonidealities of cellular environments from which toxicodynamic effects emerge [71]. They are also leading efforts to develop and disseminate prototype models for refinement by the research community, as in a recent mechanistic model of SARS-CoV-2 based on viral dynamics, immune and inflammatory signaling, and tissue damage [72].
CONCLUDING REMARKS
By necessity, our coverage of signaling modeling approaches was targeted and brief. A lengthier discussion would be needed to survey other relevant topics including multiscale [73,74], logic-based [75,76], Boolean [77], pharmacokinetic/pharmacodynamic [78] and evolutionary models, as well as genetic algorithms and constraint-based approaches [79]. It is also worth noting that distinctions between modeling approaches drawn here are not totally general and that crossover applications are increasingly occurring. For example, data-driven approaches can be used in the development of mechanistic models, as in a recent example using PLSR to interpret global sensitivity analysis for a model of EGF-mediated RAF membrane localization [80]. As another example, some machine learning approaches have modeled signaling dynamics and phenotypes using differential equations, with weighted protein interaction parameters used to fit model outputs to measured phosphoprotein and cell response dynamics [64].
We anticipate that integrated mechanistic and data-driven models will eventually enable quantitative predictions for how changes to the biophysical properties of cellular cues propagate through multivariate signaling processes to drive changes in cell phenotype. The key and challenge to accomplishing that goal is at the signaling effector level, where the output of mechanistic models will become the input of data-driven models. That is, enough pathway nodes will need to be mechanistically modeled to generate meaningful data-driven models of phenotype. Accomplishing that goal will create new and useful modeling frameworks for the development of basic science hypotheses and for the design of targeted therapy approaches for disease.
HIGHLIGHTS.
Mechanistic models describe the physical basis for signaling network function.
Parameter sensitivity and uncertainty analyses aid mechanistic model interpretation.
Data-driven models distill high-dimensional omics into cell phenotype models.
Data-driven and mechanistic models inform and constrain one another.
ACKNOWLEDGEMENTS
This was supported by NSF MCB 1716537, NIH T32 LM012416, and NCI U01 CA243007. The authors gratefully acknowledge these colleagues for helpful discussions or manuscript reviews: Birgit Schoerbl (Novartis), Douglas Lauffenburger (MIT), Kevin Janes (University of Virginia), Bill Hlavacek (Los Alamos National Laboratory), and Kevin Brown (Oregon State University). The authors also acknowledge the contributions of Ms. Anita Imagliazzo for her compensated assistance with figure preparation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Lazzara MJ, Lauffenburger DA: Quantitative modeling perspectives on the ErbB system of cell regulatory processes. Exp Cell Res 2009, 315:717–725. [DOI] [PubMed] [Google Scholar]
- 2.Giese W, Milicic G, Schroder A, Klipp E: Spatial modeling of the membrane-cytosolic interface in protein kinase signal transduction. PLoS Comput Biol 2018, 14:e1006075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Osterfield M, Berg CA, Shvartsman SY: Epithelial Patterning, Morphogenesis, and Evolution: Drosophila Eggshell as a Model. Dev Cell 2017, 41:337–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cowan AE, Moraru II, Schaff JC, Slepchenko BM, Loew LM: Spatial modeling of cell signaling networks. Methods Cell Biol 2012, 110:195–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Degasperi A, Fey D, Kholodenko BN: Performance of objective functions and optimisation procedures for parameter estimation in system biology models. NPJ Syst Biol Appl 2017, 3:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoops S, Sahle S, Gauges R, Lee C, Pahle J, Simus N, Singhal M, Xu L, Mendes P, Kummer U: COPASI--a COmplex PAthway SImulator. Bioinformatics 2006, 22:3067–3074. [DOI] [PubMed] [Google Scholar]
- 7.Blum Y, Mikelson J, Dobrzynski M, Ryu H, Jacques MA, Jeon NL, Khammash M, Pertz O: Temporal perturbation of ERK dynamics reveals network architecture of FGF2/MAPK signaling. Mol Syst Biol 2019, 15:e8947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kok F, Rosenblatt M, Teusel M, Nizharadze T, Goncalves Magalhaes V, Dachert C, Maiwald T, Vlasov A, Wasch M, Tyufekchieva S, et al. : Disentangling molecular mechanisms regulating sensitization of interferon alpha signal transduction. Mol Syst Biol 2020, 16:e8955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frohlich F, Kessler T, Weindl D, Shadrin A, Schmiester L, Hache H, Muradyan A, Schutte M, Lim JH, Heinig M, et al. : Efficient Parameter Estimation Enables the Prediction of Drug Response Using a Mechanistic Pan-Cancer Pathway Model. Cell Syst 2018, 7:567–579 e566. [DOI] [PubMed] [Google Scholar]
- 10.••.Claas AM, Atta L, Gordonov S, Meyer AS, Lauffenburger DA: Systems Modeling Identifies Divergent Receptor Tyrosine Kinase Reprogramming to MAPK Pathway Inhibition. Cell Mol Bioeng 2018, 11:451–469. [DOI] [PMC free article] [PubMed] [Google Scholar]; Using an ODE-based mechanistic model fitted against steady state and time-course measurements of receptor tyrosine kinase levels in MDAMB231 and SUM159 cells, the authors identify the main drivers of resistance to MEK and ERK inhibitors. Parameter probability distributions obtained via Bayesian inference were compared with literature values to identify sources of uncertainty in data and used to predict response to receptor-specific perturbations. In addition, PCA was used to identify parameters that explain the most variation across cell lines and inhibitors in response to the perturbations.
- 11.Yeung E, McFann S, Marsh L, Dufresne E, Filippi S, Harrington HA, Shvartsman SY, Wuhr M: Inference of Multisite Phosphorylation Rate Constants and Their Modulation by Pathogenic Mutations. Curr Biol 2020, 30:877–882 e876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown KS, Sethna JP: Statistical mechanical approaches to models with many poorly known parameters. Phys Rev E Stat Nonlin Soft Matter Phys 2003, 68:021904. [DOI] [PubMed] [Google Scholar]
- 13.Mikelson J, Khammash M: Likelihood-free nested sampling for parameter inference of biochemical reaction networks. PLoS Comput Biol 2020, 16:e1008264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.•.Joslyn LR, Kirschner DE, Linderman JJ: CaliPro: A Calibration Protocol That Utilizes Parameter Density Estimation to Explore Parameter Space and Calibrate Complex Biological Models. Cell Mol Bioeng 2021, 14:31–47. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors introduce an efficient, metaheuristic parameter inference protocol for detecting a parameter space where distinct and biologically reasonable dynamics are produced in a variety of ODE-based and stochastic mechanistic models. This overcomes the typical need for distribution of values within experimental data or initial guesses of model parameters in Bayesian inference.
- 15.Rukhlenko OS, Khorsand F, Krstic A, Rozanc J, Alexopoulos LG, Rauch N, Erickson KE, Hlavacek WS, Posner RG, Gomez-Coca S, et al. : Dissecting RAF Inhibitor Resistance by Structure-based Modeling Reveals Ways to Overcome Oncogenic RAS Signaling. Cell Syst 2018, 7:161–179 e114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Furcht CM, Buonato JM, Lazzara MJ: EGFR-activated Src family kinases maintain GAB1-SHP2 complexes distal from EGFR. Sci Signal 2015, 8:ra46. [DOI] [PubMed] [Google Scholar]
- 17.Monast CS, Lazzara MJ: Identifying Determinants of EGFR-Targeted Therapeutic Biochemical Efficacy Using Computational Modeling. CPT: Pharmacometrics & Systems Pharmacology 2014, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lebedeva G, Sorokin A, Faratian D, Mullen P, Goltsov A, Langdon SP, Harrison DJ, Goryanin I: Model-based global sensitivity analysis as applied to identification of anti-cancer drug targets and biomarkers of drug resistance in the ErbB2/3 network. Eur J Pharm Sci 2012, 46:244–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee D, Beste MT, Anderson NR, Koretzky GA, Hammer DA: Identifying Key Pathways and Components in Chemokine-Triggered T Lymphocyte Arrest Dynamics Using a Multi-Parametric Global Sensitivity Analysis. Cell Mol Bioeng 2019, 12:193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marino S, Hogue IB, Ray CJ, Kirschner DE: A methodology for performing global uncertainty and sensitivity analysis in systems biology. J Theor Biol 2008, 254:178–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao C, Mirando AC, Sove RJ, Medeiros TX, Annex BH, Popel AS: A mechanistic integrative computational model of macrophage polarization: Implications in human pathophysiology. PLoS Comput Biol 2019, 15:e1007468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marino S, Cilfone NA, Mattila JT, Linderman JJ, Flynn JL, Kirschner DE: Macrophage polarization drives granuloma outcome during Mycobacterium tuberculosis infection. Infect Immun 2015, 83:324–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.••.Rohrs JA, Siegler EL, Wang P, Finley SD: ERK Activation in CAR T Cells Is Amplified by CD28-Mediated Increase in CD3zeta Phosphorylation. iScience 2020, 23:101023. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors demonstrate that the ERK response time via activation of chimeric antigen receptor (CAR)-engineered T cells in MAPK pathway is most strongly controlled by catalytic rates of LCK, ZAP-70, and the phosphatase CD45, parameters that were previously not well defined. This identification is possible by performing global sensitivity analysis on the mechanistic model that incorporates four other co-stimulatory pathways identified in previous studies. This study exmplifies the use of sensitivity analysis results in generating testable, mechanistic hypotheses.
- 24.Nguyen Quang M, Rogers T, Hofman J, Lanham AB: Global Sensitivity Analysis of Metabolic Models for Phosphorus Accumulating Organisms in Enhanced Biological Phosphorus Removal. Front Bioeng Biotechnol 2019, 7:234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arulraj T, Barik D: Mathematical modeling identifies Lck as a potential mediator for PD-1 induced inhibition of early TCR signaling. PLoS One 2018, 13:e0206232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Transtrum MK, Machta BB, Brown KS, Daniels BC, Myers CR, Sethna JP: Perspective: Sloppiness and emergent theories in physics, biology, and beyond. J Chem Phys 2015, 143:010901. [DOI] [PubMed] [Google Scholar]
- 27.Gutenkunst RN, Waterfall JJ, Casey FP, Brown KS, Myers CR, Sethna JP: Universally sloppy parameter sensitivities in systems biology models. PLoS Comput Biol 2007, 3:1871–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Waterfall JJ, Casey FP, Gutenkunst RN, Brown KS, Myers CR, Brouwer PW, Elser V, Sethna JP: Sloppy-model universality class and the Vandermonde matrix. Phys Rev Lett 2006, 97 :150601. [DOI] [PubMed] [Google Scholar]
- 29.Apgar JF, Witmer DK, White FM, Tidor B: Sloppy models, parameter uncertainty, and the role of experimental design. Mol Biosyst 2010, 6:1890–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Raue A, Kreutz C, Maiwald T, Bachmann J, Schilling M, Klingmuller U, Timmer J: Structural and practical identifiability analysis of partially observed dynamical models by exploiting the profile likelihood. Bioinformatics 2009, 25:1923–1929. [DOI] [PubMed] [Google Scholar]
- 31.Chis OT, Villaverde AF, Banga JR, Balsa-Canto E: On the relationship between sloppiness and identifiability. Math Biosci 2016, 282:147–161. [DOI] [PubMed] [Google Scholar]
- 32.Mortlock RD, Georgia SK, Finley SD: Dynamic Regulation of JAK-STAT Signaling Through the Prolactin Receptor Predicted by Computational Modeling. Cell Mol Bioeng 2021, 14:15–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davies AE, Pargett M, Siebert S, Gillies TE, Choi Y, Tobin SJ, Ram AR, Murthy V, Juliano C, Quon G, et al. : Systems-Level Properties of EGFR-RAS-ERK Signaling Amplify Local Signals to Generate Dynamic Gene Expression Heterogeneity. Cell Syst 2020, 11:161–175 e165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu X, Zhang J, Zeigler AC, Nelson AR, Lindsey ML, Saucerman JJ: Network Analysis Reveals a Distinct Axis of Macrophage Activation in Response to Conflicting Inflammatory Cues. J Immunol 2021, 206:883–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao C, Medeiros TX, Sove RJ, Annex BH, Popel AS: A data-driven computational model enables integrative and mechanistic characterization of dynamic macrophage polarization. iScience 2021, 24:102112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wade JD, Lun XK, Zivanovic N, Voit EO, Bodenmiller B: Mechanistic Model of Signaling Dynamics Across an Epithelial Mesenchymal Transition. Front Physiol 2020, 11:579117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Day EK, Sosale NG, Lazzara MJ: Cell signaling regulation by protein phosphorylation: a multivariate, heterogeneous, and context-dependent process. Curr Opin Biotechnol 2016, 40:185–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.•.Rozanc J, Sakellaropoulos T, Antoranz A, Gutta C, Podder B, Vetma V, Rufo N, Agostinis P, Pliaka V, Sauter T, et al. : Phosphoprotein patterns predict trametinib responsiveness and optimal trametinib sensitisation strategies in melanoma. Cell Death Differ 2019, 26:1365–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]; Using a diverse set of melanoma cell lines with clinically relevant mutations, the authors use principal components analysis and linear discriminant analysis to show that cell line sensitivity to the MEK inhibitor trametinib can be reliably predicted from phosphoprotein profiles independent of the mutational status of the cell lines. Additionally, the combination of supervised and unsupervised modeling approaches identifies specific, targetable proteins whose inhibition sensitizes melanoma cells to trametinib.
- 39.Davies R, Sarkar I, Hammenfors D, Bergum B, Vogelsang P, Solberg SM, Gavasso S, Brun JG, Jonsson R, Appel S: Single Cell Based Phosphorylation Profiling Identifies Alterations in Toll-Like Receptor 7 and 9 Signaling in Patients With Primary Sjogren’s Syndrome. Front Immunol 2019, 10:281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Becht E, McInnes L, Healy J, Dutertre C-A, Kwok IWH, Ng LG, Ginhoux F, Newell EW: Dimensionality reduction for visualizing single-cell data using UMAP. Nature Biotechnology 2019, 37:38–44. [DOI] [PubMed] [Google Scholar]
- 41.Luecken MD, Theis FJ: Current best practices in single‐cell RNA‐seq analysis: a tutorial. Molecular Systems Biology 2019, 15:e8746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Karacosta LG, Anchang B, Ignatiadis N, Kimmey SC, Benson JA, Shrager JB, Tibshirani R, Bendall SC, Plevritis SK: Mapping lung cancer epithelial-mesenchymal transition states and trajectories with single-cell resolution. Nat Commun 2019, 10:5587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.•.Leelatian N, Sinnaeve J, Mistry AM, Barone SM, Brockman AA, Diggins KE, Greenplate AR, Weaver KD, Thompson RC, Chambless LB, et al. : Unsupervised machine learning reveals risk stratifying glioblastoma tumor cells. Elife 2020, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]; Coupling single-cell proteomics with a novel unsupervised clustering method, the authors demonstrate the ability of distinct cell populations within a tumor to predict patient outcomes in glioblastoma. The unsupervised model provides quantitative information about the signaling characteristics of these cell populations and insight for designing treatment strategies and for understanding the mechanisms of disease progression.
- 44.Pereira EJ, Burns JS, Lee CY, Marohl T, Calderon D, Wang L, Atkins KA, Wang CC, Janes KA: Sporadic activation of an oxidative stress-dependent NRF2-p53 signaling network in breast epithelial spheroids and premalignancies. Sci Signal 2020, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tong M, Yu C, Shi J, Huang W, Ge S, Liu M, Song L, Zhan D, Xia X, Liu W, et al. : Phosphoproteomics Enables Molecular Subtyping and Nomination of Kinase Candidates for Individual Patients of Diffuse-Type Gastric Cancer. iScience 2019, 22:44–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Good Z, Sarno J, Jager A, Samusik N, Aghaeepour N, Simonds EF, White L, Lacayo NJ, Fantl WJ, Fazio G, et al. : Single-cell developmental classification of B cell precursor acute lymphoblastic leukemia at diagnosis reveals predictors of relapse. Nature Medicine 2018, 24:474–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Khaliq M, Manikkam M, Martinez ED, Fallahi-Sichani M: Epigenetic modulation reveals differentiation state specificity of oncogene addiction. Nat Commun 2021, 12:1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Krug K: A Curated Resource for Phosphosite-specific Signature Analysis. Molecular & cellular proteomics 2019, 18:576–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Katsogiannou M, Boyer J-B, Valdeolivas A, Remy E, Calzone L, Audebert S, Rocchi P, Camoin L, Baudot A: Integrative proteomic and phosphoproteomic profiling of prostate cell lines. PLOS ONE 2019, 14:e0224148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Poulin EJ, Bera AK, Lu J, Lin YJ, Strasser SD, Paulo JA, Huang TQ, Morales C, Yan W, Cook J, et al. : Tissue-Specific Oncogenic Activity of KRAS(A146T). Cancer Discov 2019, 9:738–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brubaker DK, Paulo JA, Sheth S, Poulin EJ, Popow O, Joughin BA, Strasser SD, Starchenko A, Gygi SP, Lauffenburger DA, et al. : Proteogenomic Network Analysis of Context-Specific KRAS Signaling in Mouse-to-Human Cross-Species Translation. Cell Systems 2019, 9:258–270.e256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rydenfelt M, Wongchenko M, Klinger B, Yan Y, Bluthgen N: The cancer cell proteome and transcriptome predicts sensitivity to targeted and cytotoxic drugs. Life Sci Alliance 2019, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.•.Day EK, Zhong Q, Purow B, Lazzara MJ: Data-driven computational modeling identifies determinants of glioblastoma response to SHP2 inhibition. Cancer Res 2021, 81:2056–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors develop a partial least squares regression model of the effects of antagonizing the oncogenic protein tyrosine phosphatase SHP2 in glioblastoma. The study demonstrates the utility of data-driven models for predicting the effects of targeting phosphatases, which have multiple substrates. For example, the model led to experimentally validated conclusion that SHP2 inhibition antagonizes the effects of temozolomide chemotherapy in a PTEN-dependent manner.
- 54.Fong LE, Sulistijo ES, Miller-Jensen K: Systems analysis of latent HIV reversal reveals altered stress kinase signaling and increased cell death in infected T cells. Sci Rep 2017, 7:16179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang JH, Wright SN, Hamblin M, McCloskey D, Alcantar MA, Schrubbers L, Lopatkin AJ, Satish S, Nili A, Palsson BO, et al. : A White-Box Machine Learning Approach for Revealing Antibiotic Mechanisms of Action. Cell 2019, 177:1649–1661 e1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.•.Zhao W, Li J, Chen MM, Luo Y, Ju Z, Nesser NK, Johnson-Camacho K, Boniface CT, Lawrence Y, Pande NT, et al. : Large-Scale Characterization of Drug Responses of Clinically Relevant Proteins in Cancer Cell Lines. Cancer Cell 2020, 38:829–843 e824. [DOI] [PMC free article] [PubMed] [Google Scholar]; Using reverse-phase protein arrays and publicly available data, the authors describe one of the first and largest collections of publicly available phosphoprotein measurements in drug-perturbed cancer cell lines. The authors show that these dynamic measurements in perturbed cells greatly increase the ability of data-driven models to predict cellular responses to drug treatment.
- 57.Zhou W, Sailani MR, Contrepois K, Zhou Y, Ahadi S, Leopold SR, Zhang MJ, Rao V, Avina M, Mishra T, et al. : Longitudinal multi-omics of host-microbe dynamics in prediabetes. Nature 2019, 569:663–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lin YT, Way GP, Barwick BG, Mariano MC, Marcoulis M, Ferguson ID, Driessen C, Boise LH, Greene CS, Wiita AP: Integrated phosphoproteomics and transcriptional classifiers reveal hidden RAS signaling dynamics in multiple myeloma. Blood Adv 2019, 3:3214–3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Goutman SA, Boss J, Guo K, Alakwaa FM, Patterson A, Kim S, Savelieff MG, Hur J, Feldman EL: Untargeted metabolomics yields insight into ALS disease mechanisms. J Neurol Neurosurg Psychiatry 2020, 91:1329–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Throm AA, Alinger JB, Pingel JT, Daugherty AL, Pachman LM, French AR: Dysregulated NK cell PLCgamma2 signaling and activity in juvenile dermatomyositis. JCI Insight 2018, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Broyde J, Simpson DR, Murray D, Paull EO, Chu BW, Tagore S, Jones SJ, Griffin AT, Giorgi FM, Lachmann A, et al. : Oncoprotein-specific molecular interaction maps (SigMaps) for cancer network analyses. Nat Biotechnol 2021, 39:215–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ma W, Chen LS, Ozbek U, Han SW, Lin C, Paulovich AG, Zhong H, Wang P: Integrative Proteo-genomic Analysis to Construct CNA-protein Regulatory Map in Breast and Ovarian Tumors. Mol Cell Proteomics 2019, 18:S66–S81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fröhlich F, Kessler T, Weindl D, Shadrin A, Schmiester L, Hache H, Muradyan A, Schütte M, Lim J-H, Heinig M, et al. : Efficient Parameter Estimation Enables the Prediction of Drug Response Using a Mechanistic Pan-Cancer Pathway Model. Cell Systems 2018, 7:567–579.e566. [DOI] [PubMed] [Google Scholar]
- 64.••.Yuan B, Shen C, Luna A, Korkut A, Marks DS, Ingraham J, Sander C: CellBox: Interpretable Machine Learning for Perturbation Biology with Application to the Design of Cancer Combination Therapy. Cell Syst 2021, 12:128–140 e124. [DOI] [PubMed] [Google Scholar]; The authors describe a unique data-driven, ODE-based machine-learning model that is predictive of both cell phenotypes and phosphoprotein dynamics in response to drug perturbations. The model’s predictive power enables it to serve as a hypothesis generator for combination therapies, and its ODE framework allows it to provide quantitative information about the protein-protein interactions that are predictive of cell responses without any prior biological knowledge.
- 65.Alkan O, Schoeberl B, Shah M, Koshkaryev A, Heinemann T, Drummond DC, Yaffe MB, Raue A: Modeling chemotherapy-induced stress to identify rational combination therapies in the DNA damage response pathway. Sci Signal 2018, 11. [DOI] [PubMed] [Google Scholar]
- 66.Casaletto JB, Geddie ML, Abu-Yousif AO, Masson K, Fulgham A, Boudot A, Maiwald T, Kearns JD, Kohli N, Su S, et al. : MM-131, a bispecific anti-Met/EpCAM mAb, inhibits HGF-dependent and HGF-independent Met signaling through concurrent binding to EpCAM. Proc Natl Acad Sci U S A 2019, 116:7533–7542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Han B: A suite of mathematical solutions to describe ternary complex formation and their application to targeted protein degradation by heterobifunctional ligands. J Biol Chem 2020, 295:15280–15291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schoeberl B, Kudla A, Masson K, Kalra A, Curley M, Finn G, Pace E, Harms B, Kim J, Kearns J, et al. : Systems biology driving drug development: from design to the clinical testing of the anti-ErbB3 antibody seribantumab (MM-121). NPJ Syst Biol Appl 2017, 3:16034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hass H, Masson K, Wohlgemuth S, Paragas V, Allen JE, Sevecka M, Pace E, Timmer J, Stelling J, MacBeath G, et al. : Predicting ligand-dependent tumors from multidimensional signaling features. NPJ Syst Biol Appl 2017, 3:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ferrero E, Brachat S, Jenkins JL, Marc P, Skewes-Cox P, Altshuler RC, Gubser Keller C, Kauffmann A, Sassaman EK, Laramie JM, et al. : Ten simple rules to power drug discovery with data science. PLoS Comput Biol 2020, 16:e1008126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pearlstein RA, Wan H, Aravamuthan V: Toward in vivo relevant drug design. Drug Discov Today 2021, 26:637–650. [DOI] [PubMed] [Google Scholar]
- 72.Dai W, Rao R, Sher A, Tania N, Musante CJ, Allen R: A Prototype QSP Model of the Immune Response to SARS-CoV-2 for Community Development. CPT Pharmacometrics Syst Pharmacol 2021, 10:18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Warner HV, Sivakumar N, Peirce SM, Lazzara MJ: Multiscale computational models of cancer. Current Opinion in Biomedical Engineering 2019, 11:137–144. [Google Scholar]
- 74.Rikard SM, Athey TL, Nelson AR, Christiansen SLM, Lee JJ, Holmes JW, Peirce SM, Saucerman JJ: Multiscale Coupling of an Agent-Based Model of Tissue Fibrosis and a Logic-Based Model of Intracellular Signaling. Front Physiol 2019, 10:1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Eduati F, Doldan-Martelli V, Klinger B, Cokelaer T, Sieber A, Kogera F, Dorel M, Garnett MJ, Bluthgen N, Saez-Rodriguez J: Drug Resistance Mechanisms in Colorectal Cancer Dissected with Cell Type-Specific Dynamic Logic Models. Cancer Res 2017, 77:3364–3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Thobe K, Kuznia C, Sers C, Siebert H: Evaluating Uncertainty in Signaling Networks Using Logical Modeling. Front Physiol 2018, 9:1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.•.Silverbush D, Grosskurth S, Wang D, Powell F, Gottgens B, Dry J, Fisher J: Cell-Specific Computational Modeling of the PIM Pathway in Acute Myeloid Leukemia. Cancer Res 2017, 77:827–838. [DOI] [PubMed] [Google Scholar]; Using a Qualitative Networks model, an extension of a Boolean model, the authors identify the source of PIM inhibitor resistance in acute myeloid leukemia. The model was calibrated with reverse phase protein array data and trained against perturbation data from literature for multiple cell lines. This study offers an example of successful integration of mechanistic and data-driven modeling.
- 78.Betts A, Haddish-Berhane N, Shah DK, van der Graaf PH, Barletta F, King L, Clark T, Kamperschroer C, Root A, Hooper A, et al. : A Translational Quantitative Systems Pharmacology Model for CD3 Bispecific Molecules: Application to Quantify T Cell-Mediated Tumor Cell Killing by P-Cadherin LP DART((R)). AAPS J 2019, 21:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yu H, Blair RH: Integration of probabilistic regulatory networks into constraint-based models of metabolism with applications to Alzheimer’s disease. BMC Bioinformatics 2019, 20:386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Surve SV, Myers PJ, Clayton SA, Watkins SC, Lazzara MJ, Sorkin A: Localization dynamics of endogenous fluorescently labeled RAF1 in EGF-stimulated cells. Mol Biol Cell 2019, 30:506–523. [DOI] [PMC free article] [PubMed] [Google Scholar]