

Abstract
Objective
To assess the efficacy and safety of upadacitinib (UPA), an oral Janus kinase inhibitor, as monotherapy or in combination with non-biologic DMARDs (nbDMARDs) in patients with PsA.
Methods
Pooled data were analysed from patients with prior inadequate response or intolerance to one or more nbDMARD (SELECT-PsA 1) or one or more biologic DMARD (SELECT-PsA 2) who received placebo, UPA 15 mg once daily (QD) or UPA 30 mg QD as monotherapy or in combination with two or fewer nbDMARDs for 24 weeks. Efficacy outcomes included achievement of ACR responses, Psoriasis Area and Severity Index responses, minimal disease activity and change from baseline and clinically meaningful improvement in the HAQ Disability Index. Adverse events (AEs) were summarized.
Results
A total of 1916 patients were included; 574 (30%) received monotherapy and 1342 (70%) received combination therapy. Placebo-subtracted treatment effects for a 20% improvement in ACR criteria at week 12 were 33.7% (95% CI 24.4, 43.1) and 34.0% (95% CI 27.9, 40.1) for UPA 15 mg QD monotherapy and combination therapy, respectively, and 45.7% (95% CI 36.9, 54.5) and 39.6% (95% CI 33.7, 45.5) for UPA 30 mg QD monotherapy and combination therapy, respectively. Treatment effects for other outcomes were consistent between monotherapy and combination therapy. AE frequency was generally similar for UPA monotherapy and combination therapy, although hepatic disorders and creatine phosphokinase elevation were more common with combination therapy vs monotherapy.
Conclusion
The efficacy and safety of UPA were generally consistent when administered as monotherapy or in combination with nbDMARDs through 24 weeks, supporting the use of UPA with or without nbDMARDs in PsA.
Trial registration
ClinicalTrials.gov (https://clinicaltrials.gov): SELECT-PsA 1 (NCT03104400), SELECT-PsA 2 (NCT03104374)
Keywords: psoriatic arthritis, Janus kinase inhibitor, monotherapy, upadacitinib
Rheumatology key messages.
Upadacitinib showed comparable efficacy as monotherapy and in combination with non-biologic DMARDs in PsA.
The safety profile of upadacitinib was generally similar when used as monotherapy and combination therapy.
Hepatic disorder events and creatine phosphokinase elevation were less common with monotherapy vs combination therapy.
Introduction
Research advances have translated into diverse treatment options for PsA, including conventional synthetic DMARDs (csDMARDs), biologic DMARDs (bDMARDs) and targeted synthetic DMARDs (tsDMARDs), with the potential to achieve low disease activity across various clinical domains [1–3]. However, questions remain regarding optimal treatment algorithms and treatment patterns and one key question for clinicians is whether comedication with csDMARDs is useful for patients with PsA [1–3].
The efficacy of csDMARDs such as MTX as concomitant therapy in PsA is not established, and several studies have demonstrated that MTX provides little additional benefit when combined with biologics or tsDMARDs [4–7]. For example, an analysis of two etanercept clinical trials found that etanercept was equally effective with or without MTX in patients with PsA [4]. Treatment guidelines for PsA differ on whether csDMARDs should be used as concomitant therapy; the EULAR guidelines [2] recommend combining biologics with csDMARDs (while acknowledging there is little evidence to support this), whereas the ACR guidelines [8] favour biologic monotherapy. In addition to a lack of clarity regarding the efficacy of combination therapy, many patients have contraindications to MTX or are unable to tolerate higher doses [5, 9]. Agents with novel mechanisms of action that are effective as monotherapy would therefore be a useful treatment option for PsA.
Upadacitinib (UPA) is an oral Janus kinase (JAK) inhibitor designed to selectively target JAK1 over the other JAK family enzymes: JAK2, JAK3 or tyrosine kinase 2 [10]. UPA has been assessed for the treatment of PsA in two global phase 3 trials, SELECT-PsA 1 and SELECT-PsA 2 [11, 12]. In both of these trials, UPA 15 mg and 30 mg once daily (QD) were significantly more effective than placebo in improving key clinical manifestations of PsA.
Here we report data from a pooled subgroup analysis of the two SELECT-PsA studies assessing efficacy and safety outcomes in patients who were treated with UPA as monotherapy or in combination with non-biologic DMARDs (nbDMARDs).
Methods
Patients
In SELECT-PsA 1 (NCT03104400) [11] and SELECT-PsA 2 (NCT03104374) [12], patients with active PsA (three or more swollen and three or more tender joints) and active or historical psoriasis were blindly randomized to UPA 15 mg QD, UPA 30 mg QD, placebo or adalimumab 40 mg every other week (SELECT-PsA 1 only) for 24 weeks. Patients in SELECT-PsA 1 had prior inadequate response (IR) or intolerance to one or more nbDMARD [11] and patients in SELECT-PsA 2 had prior IR or intolerance to one or more bDMARD [12]. Starting from week 16, patients who did not achieve ≥20% improvement in tender and swollen joint counts compared with baseline at both week 12 and week 16 were offered rescue therapy, which allowed patients to add or modify existing nbDMARDs, NSAIDs, acetaminophen, low-potency opioid medications or corticosteroids in accordance with the protocol.
The two trials were conducted according to the International Conference on Harmonization Guidelines, the Declaration of Helsinki and applicable local country regulations. All study-related documents were approved by independent ethics committees and institutional review boards of the participating centres (Supplementary Table S1, available at Rheumatology online). All patients provided written informed consent.
Comedications of interest
Patients were classed as receiving monotherapy if they received UPA alone or combination therapy if they received background treatment with one or two nbDMARDs [MTX (≤25 mg/week), SSZ (≤3000 mg/day), LEF (≤20 mg/day), apremilast (≤60 mg/day), HCQ (≤400 mg/day) and, less commonly, bucillamine (≤300 mg/day) and iguratimod (≤50 mg/day)]. Concomitant use of bDMARDs was not permitted.
Outcomes
Efficacy endpoints included the proportion of patients achieving 20%, 50% and 70% improvement in ACR criteria (ACR20, ACR50 and ACR70, respectively) at weeks 12 and 24; a Static Investigator Global Assessment of Psoriasis score of 0 or 1 (sIGA 0/1) and at least a 2 point improvement from baseline at week 16; 75%, 90% and 100% improvement in Psoriasis Area Severity Index (PASI 75, PASI 90 and PASI 100, respectively) responses at week 16; resolution of enthesitis at week 24; resolution of dactylitis at week 24; minimal disease activity (MDA) at week 24 and clinically meaningful improvement in the HAQ Disability Index (HAQ-DI; improvement of ≥0.35 vs baseline [13]) at week 12. Changes from baseline in pain and HAQ-DI at week 12 were also assessed. Safety outcomes were summarized by the frequency of adverse events (AEs) and laboratory abnormalities over 24 weeks.
Statistical analysis
All patients who had received at least one dose of study drug were pooled and included in the efficacy analyses. Patients receiving adalimumab in SELECT-PsA 1 were excluded from this analysis. The clinical trials were designed a priori for this analysis and patients were stratified by current use of one or more nbDMARD at randomization.
Demographic and clinical characteristics are presented using descriptive statistics. For binary efficacy endpoints, frequencies and percentages are reported, with non-responder imputation used for missing data; point estimates and 95% CIs for placebo-subtracted differences were calculated based on Cochran–Mantel–Haenszel analysis adjusting for study. For continuous endpoints, within-group least squares (LS) means (95% CI) and between-group LS means (95% CI) are presented and were calculated based on the mixed-effects model repeated measures analysis with unstructured variance–covariance matrix. The model included treatment, visit, treatment–visit interaction and study as fixed factors and the continuous fixed covariate of baseline measurement. Safety data in patients who received at least one dose of study drug are presented descriptively. Laboratory abnormalities were graded according to the Common Toxicity Criteria developed by the National Cancer Institute (version 4.03).
Results
Patients
In total, 1916 patients were included in the analysis, of whom 574 (30.0%) received UPA monotherapy [SELECT-PsA 1, n = 229 (39.9%); SELECT-PsA 2, n = 345 (60.1%)] and 1342 (70.0%) received UPA in combination with any nbDMARD [SELECT-PsA 1, n = 1046 (77.9%); SELECT-PsA 2, n = 296 (22.1%)]. Of the 1342 patients receiving combination therapy with any nbDMARD (including MTX), a subset of 1036 (77.2%) patients received UPA with MTX alone; this subgroup was analysed separately.
Baseline demographic and disease characteristics were generally balanced across the treatment arms and between patients receiving monotherapy and combination therapy, either with MTX only or with any nbDMARD (Table 1). Across all the groups, slightly more than half of patients were female and the mean age was 51–52 years. The mean duration since PsA diagnosis was longer in the monotherapy group compared with the combination therapy groups. The mean PASI score in patients with a body surface area >3% at baseline ranged from 10.2 to 12.7 in the monotherapy subgroup and from 8.8 to 11.5 in the combination therapy subgroups. At baseline, around one-quarter of patients had dactylitis and more than half of patients had enthesitis.
Table 1.
Baseline demographics and disease characteristics
| Parameter | Monotherapy |
Combination therapy with MTX |
Combination therapy with any nbDMARD (including MTX) |
||||||
|---|---|---|---|---|---|---|---|---|---|
| PBO (n = 188) | UPA 15 mg QD (n = 189) | UPA 30 mg QD (n = 197) | PBO (n = 342) | UPA 15 mg QD (n = 353) | UPA 30 mg QD (n = 341) | PBO (n = 447) | UPA 15 mg QD (n = 451) | UPA 30 mg QD (n = 444) | |
| Female, n (%) | 102 (54.3) | 105 (55.6) | 101 (51.3) | 173 (50.6) | 195 (55.2) | 190 (55.7) | 229 (51.2) | 246 (54.5) | 250 (56.3) |
| Age, years | 52.8 (11.5) | 52.2 (12.8) | 50.7 (11.5) | 51.2 (12.3) | 51.4 (12.0) | 51.3 (12.6) | 51.1 (12.3) | 52.0 (11.9) | 51.1 (12.7) |
| BMI ≥25 kg/m2, n (%) | 145 (77.1) | 152 (80.4) | 160 (81.2) | 274 (80.1) | 279 (79.0) | 267 (78.3) | 356 (79.6) | 361 (80.0) | 338 (76.1) |
| Duration since PsA diagnosis, years | 9.0 (9.5) | 8.6 (8.4) | 8.4 (8.7) | 7.2 (8.3) | 6.5 (7.3) | 6.8 (6.9) | 7.3 ( 8.1) | 6.8 (7.6) | 6.6 (6.8) |
| PASI (for baseline BSA ≥3%) | 12.7 (12.1) | 11.8 (10.8) | 10.2 (10.4) | 11.5 (11.5) | 9.5 (9.5) | 8.8 (8.2) | 10.8 (11.0) | 9.1 (9.0) | 8.8 (8.1) |
| Presence of dactylitis (LDI >0), n (%) | 54 (28.7) | 53 (28.0) | 50 (25.4) | 95 (27.8) | 108 (30.6) | 93 (27.3) | 136 (30.4) | 138 (30.6) | 127 (28.6) |
| Presence of enthesitis (LEI >0), n (%) | 118 (62.8) | 114 (60.3) | 134 (68.0) | 202 (59.1) | 222 (62.9) | 222 (65.1) | 267 (59.7) | 289 (64.1) | 285 (64.2) |
| TJC68 | 22.7 (16.8) | 23.4 (17.0) | 22.8 (15.2) | 21.5 (15.5) | 21.0 (14.7) | 20.5 (13.9) | 21.4 (15.2) | 21.2 (15.2) | 20.3 (14.0) |
| SJC66 | 10.5 (7.2) | 11.7 (9.1) | 11.7 ( 9.0) | 12.1 (9.1) | 11.5 (9.0) | 11.4 (8.0) | 11.7 (8.9) | 11.4 (8.9) | 11.3 (7.6) |
| Corticosteroid use at BL, n (%) | 18.9 (9.6) | 27 (14.3) | 12 (6.1) | 59 (17.3) | 52 (14.7) | 54 (15.8) | 76 (17.0) | 68 (15.1) | 72 (16.2) |
| MTX dose at BL, n (%) | |||||||||
| ≤15 mg | – | – | – | 209 (61.1) | 227 (64.3) | 201 (58.9) | 224 (50.1) | 239 (53.0) | 221 (49.8) |
| >15 mg | – | – | – | 131 (38.3) | 124 (35.1) | 139 (40.8) | 149 (33.3) | 138 (30.6) | 151 (34.0) |
| Patient’s assessment of pain | 6.5 (2.0) | 6.4 (2.1) | 6.1 (2.1) | 6.2 (2.2) | 6.2 (2.1) | 6.1 (2.1) | 6.2 (2.2) | 6.2 (2.1) | 6.0 (2.1) |
| HAQ-DI | 1.1 (0.7) | 1.1 (0.6) | 1.2 (0.6) | 1.2 (0.7) | 1.2 (0.6) | 1.1 (0.6) | 1.2 (0.7) | 1.1 (0.6) | 1.1 (0.6) |
Values are presented as mean (s.d.) unless stated otherwise. Non-biologic DMARDs permitted: MTX, SSZ, LEF, apremilast, HCQ, bucillamine and iguratimod.
BL: baseline; BSA: body surface area; LDI: Leeds Dactylitis Index; LEI: Leeds Enthesitis Index; PBO: placebo; SJC66: swollen joint count in 66 joints; TJC68: tender joint count in 68 joints.
Efficacy outcomes
The proportion of patients achieving efficacy outcomes (Table 2) and the corresponding placebo-subtracted treatment effects (Fig. 1) were consistent between UPA as monotherapy, UPA in combination with MTX and UPA in combination with any nbDMARD, with associated 95% CIs overlapping between the subgroups for each dose (Fig. 1). In addition, comparable treatment effects were mostly observed between the UPA 15 mg and 30 mg doses (Table 2 and Fig. 1).
Table 2.
Summary of efficacy by UPA as monotherapy or combination therapy
| Parameter | Monotherapy |
Combination therapy with MTX |
Combination therapy with any nbDMARD (including MTX) |
||||||
|---|---|---|---|---|---|---|---|---|---|
| PBO | UPA 15 mg QD | UPA 30 mg QD | PBO | UPA 15 mg QD | UPA 30 mg QD | PBO | UPA 15 mg QD | UPA 30 mg QD | |
| ACR20 at week 12, n/N (%) | 47/188 (25.0) | 111/189 (58.7) | 139/197 (70.6) | 120/342 (35.1) | 251/353 (71.1) | 254/341 (74.5) | 157/447 (35.1) | 312/451 (69.2) | 332/444 (74.8) |
| ACR50 at week 12, n/N (%) | 9/188 (4.8) | 56/189 (29.6) | 84/197 (42.6) | 44/342 (12.9) | 139/353 (39.4) | 168/341 (49.3) | 57/447 (12.8) | 172/451 (38.1) | 217/444 (48.9) |
| ACR70 at week 12, n/N (%) | 0 | 22/189 (11.6) | 39/197 (19.8) | 8/342 (2.3) | 51/353 (14.4) | 84/341 (24.6) | 11/447 (2.5) | 63/451 (14.0) | 104/444 (23.4) |
| Resolution of enthesitis (LEI = 0) at week 24, n/N (%)a | 23/118 (19.5) | 48/114 (42.1) | 66/134 (49.3) | 61/202 (30.2) | 121/222 (54.5) | 122/222 (55.0) | 77/267 (28.8) | 154/289 (53.3) | 156/285 (54.7) |
| Resolution of dactylitis (LDI = 0) at week 24, n/N (%)b | 12/54 (22.2) | 31/53 (58.5) | 33/50 (66.0) | 40/95 (42.1) | 85/108 (78.7) | 75/93 (80.6) | 56/136 (41.2) | 105/138 (76.1) | 102/127 (80.3) |
| sIGA 0/1 and ≥2 point improvement from BL at week 16, n/N (%) | 11/150 (7.3) | 56/153 (36.6) | 80/162 (49.4) | 32/264 (12.1) | 114/273 (41.8) | 125/256 (48.8) | 38/326 (11.7) | 142/340 (41.8) | 161/326 (49.4) |
| PASI 75 at week 16, n/N (%)c | 9/109 (8.3) | 53/106 (50.0) | 71/108 (65.7) | 46/194 (23.7) | 123/193 (63.7) | 108/187 (57.8) | 57/233 (24.5) | 149/238 (62.6) | 134/233 (57.5) |
| PASI 90 at week 16, n/N (%)c | 6/109 (5.5) | 30/106 (28.3) | 55/108 (50.9) | 25/194 (12.9) | 80/193 (41.5) | 92/187 (49.2) | 31/233 (13.3) | 97/238 (40.8) | 108/233 (46.4) |
| PASI 100 at week 16, n/N (%)c | 3/109 (2.8) | 15/106 (14.2) | 39/108 (36.1) | 16/194 (8.2) | 57/193 (29.5) | 61/187 (32.6) | 20/233 (8.6) | 69/238 (29.0) | 74/233 (31.8) |
| MDA at week 24, n/N (%) | 5/188 (2.7) | 52/189 (27.5) | 74/197 (37.6) | 43/342 (12.6) | 122/353 (34.6) | 139/341 (40.8) | 53/447 (11.9) | 158/451 (35.0) | 181/444 (40.8) |
| Change from BL in pain at week 12, Δ (95% CI) | –0.63 (–0.96, –0.30) | –1.96 (–2.28, –1.64) | –2.69 (–3.01, –2.38) | –0.91 (–1.16, –0.67) | –2.29 (–2.53, –2.05) | –2.73 (–2.97, –2.48) | –0.84 (–1.05, –0.63) | –2.21 (–2.42, –2.00) | –2.63 (–2.85, –2.42) |
| Change from BL in HAQ-DI at week 12, Δ (95% CI) | –0.14 (–0.21, –0.07) | –0.31 (–0.38, –0.25) | –0.49 (–0.55, –0.43) | –0.10 (–0.15, –0.04) | –0.43 (–0.49, –0.38) | –0.43 (–0.49, –0.38) | –0.11 (–0.16, –0.06) | –0.40 (–0.45, –0.36) | –0.43 (–0.48, –0.38) |
For patients with baseline LEI >0.
For patients with baseline LDI >0.
For patients with ≥3% body surface area psoriasis at baseline. BL: baseline; LDI: Leeds Dactylitis Index; LEI: Leeds Enthesitis Index; PBO: placebo.
Fig. 1.
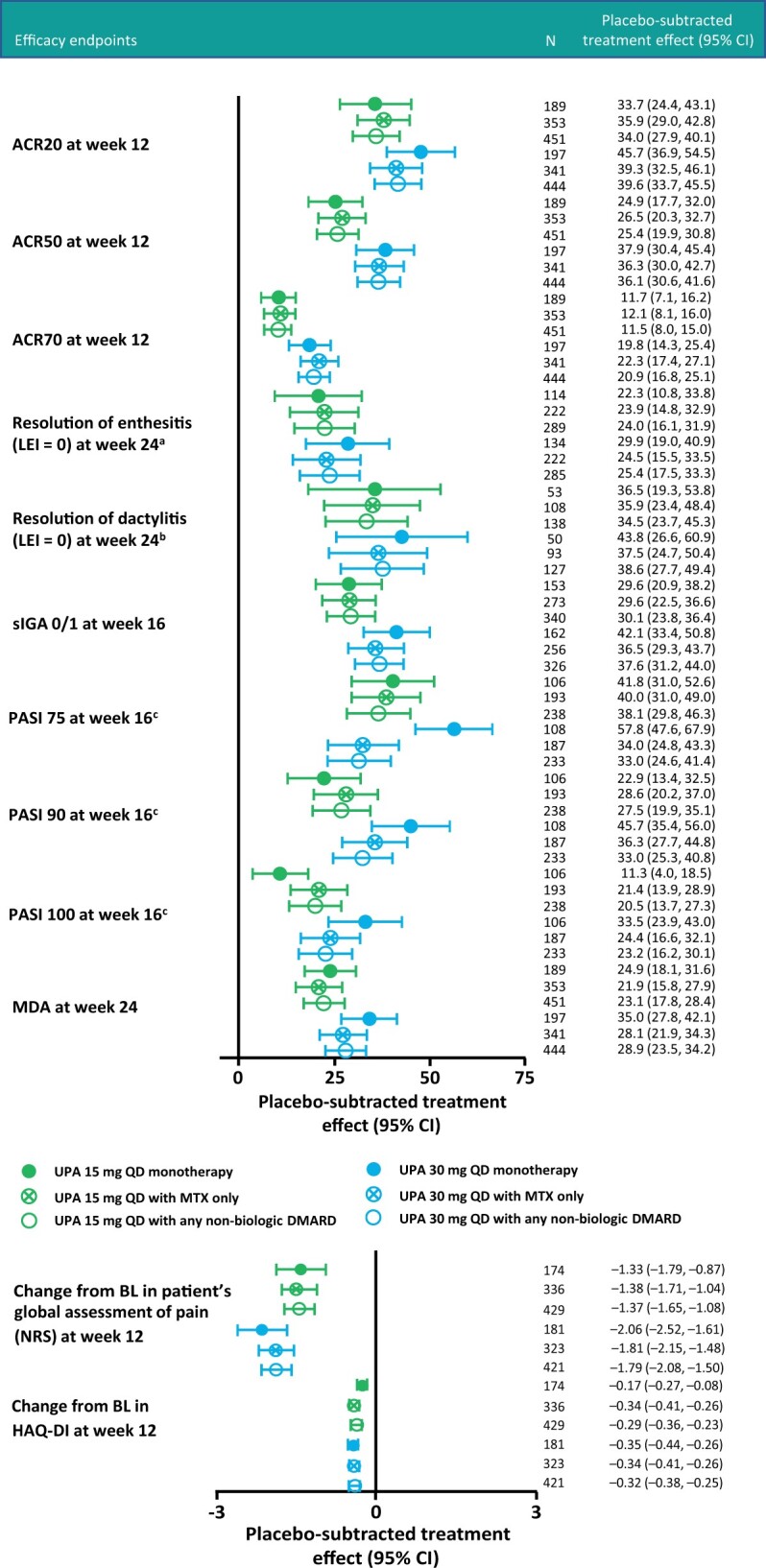
Integrated efficacy analysis of placebo-subtracted treatment effects
aFor patients with baseline LEI >0. bFor patients with baseline LDI >0. cFor patients with ≥3% body surface area psoriasis at baseline. LDI: Leeds Dactylitis Index; LEI: Leeds Enthesitis Index; NRS: numeric rating scale.
Placebo-subtracted treatment effects for achievement of ACR20 response at week 12 were 33.7% (95% CI 24.4, 43.1) and 34.0% (95% CI 27.9, 40.1) with UPA 15 mg QD monotherapy and combination therapy, respectively, and 45.7% (95% CI 36.9, 54.5) and 39.6% (95% CI 33.7, 45.5) with UPA 30 mg QD monotherapy and combination therapy, respectively (Fig. 1). Placebo-subtracted treatment effects for achievement of MDA at week 24 were 24.9% (95% CI 18.1, 31.6) and 23.1% (95% CI 17.8, 28.4) with UPA 15 mg QD monotherapy and combination therapy, respectively, and 35.0% (95% CI 27.8, 42.1) and 28.9% (95% CI 23.5, 34.2) with UPA 30 mg QD monotherapy and combination therapy, respectively. UPA also demonstrated consistency in placebo-subtracted treatment effects between the monotherapy and combination therapy groups for achievement of ACR50 and ACR70 responses at week 12 and resolution of dactylitis and enthesitis at week 24 (Fig. 1). For the skin endpoint PASI 75 response at week 16, UPA 15 mg demonstrated consistent placebo-subtracted treatment effects between monotherapy and combination therapy with overlapping CIs, while UPA 30 mg showed numerically greater placebo-subtracted values in monotherapy vs combination therapy (Fig. 1). Other skin endpoints such as achievement of PASI 90/100 and sIGA 0/1 with at least a 2 point improvement at week 16 demonstrated consistent placebo-subtracted treatment effects with overlapping CIs between monotherapy and combination therapy for both doses (Fig. 1). Change from baseline in pain and HAQ-DI scores at week 12 in the monotherapy and combination therapy groups also showed comparable results (Fig. 1 and Table 2). ACR20/50/70 responses at week 24 (Supplementary Table S2, available at Rheumatology online) were consistent with results at week 12.
Study-specific results for the SELECT-PsA 1 (nbDMARD-IR) and SELECT-PsA 2 (bDMARD-IR) studies reflect those of the integrated analysis, with generally comparable proportions of patients in the monotherapy and combination therapy subgroups of each study achieving ACR20/50/70 responses, MDA and sIGA 0/1 and at least a 2 point improvement across all treatment subgroups (Supplementary Table S3, available at Rheumatology online).
Safety outcomes
Generally, the frequency of AEs and serious AEs was comparable in patients receiving UPA 15 mg and 30 mg when administered as monotherapy and in combination with MTX or any nbDMARD through week 24 (Table 3). The frequency of discontinuation of study drug, patients lost to follow-up and discontinuation due to a lack of efficacy in patients receiving UPA 15 mg were higher in the monotherapy group compared with the combination therapy groups (Table 4). The higher frequency of discontinuation of study drug was attributed to a relatively smaller sample size in the monotherapy subgroup, and the occurrence of three cases of malignancy other than non-melanoma skin cancer with UPA 15 mg monotherapy (compared with no cases in the UPA 15 mg combination therapy group) for which discontinuation was required per the protocol.
Table 3.
Summary of AEs by UPA as monotherapy or combination therapy
| Parameter, n (%) | Monotherapy |
Combination therapy with MTX |
Combination therapy with any nbDMARD (including MTX) |
||||||
|---|---|---|---|---|---|---|---|---|---|
| PBO (n = 188) | UPA 15 mg QD (n = 189) | UPA 30 mg QD (n = 197) | PBO (n = 342) | UPA 15 mg QD (n = 353) | UPA 30 mg QD (n = 341) | PBO (n = 447) | UPA 15 mg QD (n = 451) | UPA 30 mg QD (n = 444) | |
| Any AE | 127 (67.6) | 124 (65.6) | 145 (73.6) | 191 (55.8) | 225 (63.7) | 248 (72.7) | 264 (59.1) | 298 (66.1) | 331 (74.5) |
| Serious AE | 8 (4.3) | 9 (4.8) | 9 (4.6) | 8 (2.3) | 14 (4.0) | 32 (9.4) | 9 (2.0) | 17 (3.8) | 35 (7.9) |
| AE leading to D/C of study drug | 13 (6.9) | 14 (7.4) | 14 (7.1) | 6 (1.8) | 10 (2.8) | 25 (7.3) | 11 (2.5) | 14 (3.1) | 27 (6.1) |
| Deaths | 1 (0.5) | 0 | 0 | 1 (0.3) | 0 | 0 | 1 (0.2) | 0 | 0 |
| Infection | 65 (34.6) | 67 (35.4) | 88 (44.7) | 97 (28.4) | 129 (36.5) | 155 (45.5) | 148 (33.1) | 173 (38.4) | 203 (45.7) |
| Serious infection | 2 (1.1) | 1 (0.5) | 2 (1.0) | 2 (0.6) | 3 (0.8) | 13 (3.8) | 3 (0.7) | 5 (1.1) | 15 (3.4) |
| Opportunistic infection excluding tuberculosis and herpes zoster | 0 | 0 | 2 (1.0) | 0 | 1 (0.3) | 2 (0.6) | 0 | 1 (0.2) | 2 (0.5) |
| Herpes zoster | 2 (1.1) | 2 (1.1) | 6 (3.0) | 2 (0.6) | 4 (1.1) | 6 (1.8) | 3 (0.7) | 5 (1.1) | 7 (1.6) |
| Active tuberculosis | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Malignancy other than NMSC | 0 | 3 (1.6) | 1 (0.5) | 0 | 0 | 2 (0.6) | 0 | 0 | 2 (0.5) |
| NMSC | 0 | 0 | 0 | 1 (0.3) | 0 | 2 (0.6) | 1 (0.2) | 1 (0.2) | 3 (0.7) |
| GI perforation (adjudicated) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| MACE (adjudicated) | 0 | 0 | 0 | 1 (0.3) | 1 (0.3) | 0 | 1 (0.2) | 1 (0.2) | 0 |
| VTE (adjudicated) | 1 (0.5) | 0 | 0 | 0 | 0 | 1 (0.3) | 0 | 1 (0.2) | 1 (0.2) |
| Hepatic disorder | 5 (2.7) | 8 (4.2) | 14 (7.1) | 12 (3.5) | 28 (7.9) | 45 (13.2) | 14 (3.1) | 35 (7.8) | 56 (12.6) |
| Anaemia | 3 (1.6) | 1 (0.5) | 11 (5.6) | 3 (0.9) | 5 (1.4) | 12 (3.5) | 3 (0.7) | 6 (1.3) | 23 (5.2) |
| Neutropenia | 1 (0.5) | 2 (1.1) | 6 (3.0) | 0 | 4 (1.1) | 12 (3.5) | 1 (0.2) | 4 (0.9) | 21 (4.7) |
| Lymphopenia | 0 | 2 (1.1) | 2 (1.0) | 4 (1.2) | 4 (1.1) | 12 (3.5) | 5 (1.1) | 6 (1.3) | 15 (3.4) |
| CPK elevation | 3 (1.6) | 10 (5.3) | 11 (5.6) | 5 (1.5) | 21 (5.9) | 34 (10.0) | 7 (1.6) | 32 (7.1) | 42 (9.5) |
D/C: discontinuation; GI: gastrointestinal; MACE: major adverse cardiovascular events; NMSC: non-melanoma skin cancer; PBO: placebo; VTE: venous thromboembolism.
Table 4.
Reasons for discontinuation through week 24 of monotherapy or combination therapy
| Parameter | Monotherapy |
Combination therapy with MTX |
Combination therapy with any nbDMARD (including MTX) |
||||||
|---|---|---|---|---|---|---|---|---|---|
| PBO (n = 188) | UPA 15 mg QD (n = 189) | UPA 30 mg QD (n = 197) | PBO (n = 342) | UPA 15 mg QD (n = 353) | UPA 30 mg QD (n = 341) | PBO (n = 447) | UPA 15 mg QD (n = 451) | UPA 30 mg QD (n = 444) | |
| Discontinuation prior to week 24, n (%) | 42 (22.3) | 26 (13.8) | 23 (11.7) | 33 (9.6) | 23 (6.5) | 32 (9.4) | 45 (10.1) | 30 (6.7) | 40 (9.0) |
| Adverse event | 13 (6.9) | 14 (7.4) | 12 (6.1) | 6 (1.8) | 9 (2.5) | 22 (6.5) | 11 (2.5) | 13 (2.9) | 24 (5.4) |
| Withdrawal by patient | 11 (5.9) | 1 (0.5) | 8 (4.1) | 17 (5.0) | 8 (2.3) | 6 (1.8) | 22 (4.9) | 9 (2.0) | 9 (2.0) |
| Lost to follow-up | 5 (2.7) | 6 (3.2) | 1 (0.5) | 4 (1.2) | 4 (1.1) | 1 (0.3) | 4 (0.9) | 4 (0.9) | 2 (0.5) |
| Lack of efficacy | 20 (10.6) | 5 (2.6) | 1 (0.5) | 6 (1.8) | 1 (0.3) | 1 (0.3) | 8 (1.8) | 1 (0.2) | 2 (0.5) |
| Other | 2 (1.1) | 2 (1.1) | 3 (1.5) | 2 (0.6) | 4 (1.1) | 3 (0.9) | 4 (0.9) | 6 (1.3) | 4 (0.9) |
Patients who discontinued study drug are counted under each reason given for discontinuation, therefore the sum of the counts given for the reasons may be greater than the overall number of discontinuations.
PBO: placebo.
The frequency of serious infections and herpes zoster was similar for placebo and UPA 15 mg QD as monotherapy or combination therapy but higher in the UPA 30 mg QD monotherapy and combination therapy subgroups (Table 3). All herpes zoster events were mild or moderate in severity except for one severe, non-serious event involving two dermatomes in a patient receiving UPA 30 mg QD with MTX. There were no major adverse cardiovascular events or venous thromboembolic events reported with UPA monotherapy. One non-fatal myocardial infarction was reported in a patient receiving UPA 15 mg QD with MTX, one pulmonary embolism was reported in a patient receiving UPA 15 mg QD with SSZ and one pulmonary embolism was reported in a patient receiving UPA 30 mg QD with MTX. In addition, one deep vein thrombosis was reported in a patient receiving placebo in the monotherapy group and one non-fatal myocardial infarction was reported in a patient receiving placebo in combination with MTX.
AEs of hepatic disorder (which were mostly non-serious transaminase elevation) and creatine phosphokinase (CPK) elevation were more common in the combination therapy groups than the monotherapy group, and more common with UPA 30 mg vs UPA 15 mg (Table 3). AEs of anaemia, neutropenia and lymphopenia were generally consistent across the monotherapy and combination therapy groups (Table 3). Grade 3 or 4 changes in laboratory values were infrequent (Table 5).
Table 5.
Percentage of patients with grade 3 or 4 laboratory abnormalitiesa
| Parameter, n (%) | Monotherapy |
Combination therapy with MTX |
Combination therapy with any nbDMARD (including MTX) |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| PBO (n = 183) | UPA 15 mg QD (n = 187) | UPA 30 mg QD (n = 195) | PBO (n = 339) | UPA 15 mg QD (n = 350) | UPA 30 mg QD (n = 340) | PBO (n = 444) | UPA 15 mg QD (n = 448) | UPA 30 mg QD (n = 443) | ||
| Alanine aminotransferase (U/l) | ||||||||||
| Grade 3 (>5.0–20.0× ULN) | 2 (1.1) | 1 (0.5) | 3 (1.5) | 3 (0.9)b | 4 (1.1) | 3 (0.9) | 6 (1.4)b | 5 (1.1) | 3 (0.7) | |
| Grade 4 (>20.0× ULN) | 0 | 0 | 0 | 0b | 0 | 0 | 0b | 0 | 0 | |
| Aspartate aminotransferase (U/l) | ||||||||||
| Grade 3 (>5.0–20.0× ULN) | 0 | 1 (0.5) | 0b | 2 (0.6)b | 1 (0.3) | 2 (0.6) | 3 (0.7)b | 1 (0.2) | 3 (0.7) | |
| Grade 4 (>20.0× ULN) | 0 | 0 | 0b | 0b | 0 | 1 (0.3) | 0b | 0 | 1 (0.2) | |
| Creatine kinase (U/l) | ||||||||||
| Grade 3 (>5.0–10.0× ULN) | 1 (0.5) | 2 (1.1) | 5 (2.6) | 1 (0.3) | 5 (1.4) | 6 (1.8) | 3 (0.7) | 5 (1.1) | 7 (1.6) | |
| Grade 4 (>10.0× ULN) | 1 (0.5) | 0 | 0 | 2 (0.6) | 2 (0.6) | 3 (0.9) | 2 (0.5) | 2 (0.4) | 4 (0.9) | |
| Haemoglobin (g/l) | ||||||||||
| Grade 3 (<80) | 0 | 0 | 1 (0.5) | 0 | 0 | 2 (0.6) | 0 | 0 | 2 (0.5) | |
| Lymphocytes (×109/l) | ||||||||||
| Grade 3 (0.2–<0.5) | 0 | 1 (0.5) | 2 (1.0) | 1 (0.3) | 3 (0.9) | 9 (2.6) | 1 (0.2) | 4 (0.9) | 9 (2.0) | |
| Grade 4 (<0.2) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Neutrophils (×109/l) | ||||||||||
| Grade 3 (0.5–<1.0) | 1 (0.5) | 2 (1.1) | 5 (2.6) | 1 (0.3) | 1 (0.3) | 4 (1.2) | 1 (0.2) | 2 (0.4) | 5 (1.1) | |
| Grade 4 (<0.5) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Platelets (×109/l) | ||||||||||
| Grade 3 (25–<50) | 0b | 0 | 0b | 0b | 0 | 1 (0.3) | 0b | 0 | 1 (0.2) | |
| Grade 4 (<25) | 0b | 0 | 0b | 0b | 0 | 0 | 0b | 0 | 0 | |
| Leucocytes (×109/l) | ||||||||||
| Grade 3 (1.0–<2.0) | 1 (0.5) | 0 | 1 (0.5) | 0 | 0 | 1 (0.3) | 0 | 0 | 1 (0.2) | |
| Grade 4 (<1.0) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
Abnormalities may reflect single, unconfirmed abnormalities.
Data missing for one patient. PBO: placebo; ULN: upper limit of normal.
Discussion
In this analysis, UPA used as a monotherapy or in combination with nbDMARDs (including MTX alone or any nbDMARD) was similarly well tolerated and effective in treating the major clinical manifestations of PsA, including musculoskeletal symptoms (peripheral arthritis, enthesitis and dactylitis), psoriasis, physical function and pain.
The finding that UPA combination therapy in PsA does not provide significant improvements in efficacy over monotherapy is consistent with observations investigating the efficacy of other PsA therapies used in combination with MTX or other nbDMARDs. A propensity score–matched analysis of a large registry of patients with PsA (n = 497) treated either with a combination of a TNF inhibitor (TNFi) and an nbDMARD or TNFi monotherapy demonstrated no difference between groups in time to remission, defined as achieving a 28-joint DAS using CRP (DAS28-CRP) <2.6 [6]. Similarly, a pooled analysis of two 24 week, placebo-controlled trials of subcutaneous etanercept (25 mg twice weekly or 50 mg once weekly) with (n = 322) or without MTX (n = 152) in patients with PsA showed a similar proportion of patients across the two groups achieving ACR20 [4]. Furthermore, in the Study of Etanercept and MTX in Subjects with PsA (SEAM‐PsA), both etanercept monotherapy and MTX combination therapy showed greater efficacy than MTX monotherapy in patients with PsA, according to ACR20 and MDA response rates and the extent of radiographic progression at follow-up [7]. Similarly, a post hoc analysis of 455 patients in the SPIRIT-P1 (NCT01695239) and SPIRIT-P2 (NCT02349295) trials found that treatment with once-monthly or once-fortnightly ixekizumab improved the signs and symptoms of PsA either alone or in combination with MTX [14]. More recently, a meta-analysis of randomized controlled trials found that the addition of MTX to biologics led to no clinical improvements vs biologic monotherapy in patients with PsA [15]. Within the same drug class, a study of tofacitinib found that withdrawal of MTX in patients receiving stable combination therapy did not result in clinically meaningful changes in disease activity or safety [16]. Interestingly, these data contrast with observations in RA, where combining bDMARDs with MTX results in increased efficacy [17, 18]. This is thought to be due to the reduction of anti-drug antibodies by MTX, resulting in increased drug survival [19]. However, this effect is not relevant to UPA since it does not induce immunogenicity in patients.
The data from our analysis also suggest that UPA was well tolerated when used as a monotherapy and when administered in combination with MTX alone or any nbDMARD, with the majority of AEs seen at comparable frequencies across the monotherapy and combination therapy groups. Hepatic disorders were more frequent with UPA as combination therapy compared with UPA as monotherapy, which is not surprising given the well-known effects of nbDMARDs such as MTX on liver function [20, 21]. CPK elevation also appeared to be higher in the combination therapy vs monotherapy groups, particularly in patients receiving UPA 30 mg. However, grade 3 or 4 changes in transaminases, CPK and other laboratory parameters were infrequent. Given that the efficacy of UPA monotherapy appeared to be comparable to that of UPA combination therapy, a reduction in the risk of mild laboratory abnormalities could be a benefit of treatment with UPA monotherapy, while also reducing the burden of medication use.
There appeared to be a higher rate of placebo response in the combination therapy groups compared with the monotherapy group. This may reflect the fact that the combination therapy groups had a higher proportion of patients from SELECT-PsA 1, which demonstrated higher placebo responses compared with SELECT-PsA 2 (Supplementary Table S3, available at Rheumatology online). In addition, patients in SELECT-PsA 1 and 2 were permitted to receive up to two concomitant nbDMARDs, which may have further increased the placebo response. The relatively high placebo response in SELECT-PsA 1 may be due to the fact that patients in this trial were less treatment refractory than those in SELECT-PsA 2 (nbDMARD-IR vs bDMARD-IR) [11, 12]. However, the placebo response in SELECT-PsA 1 was generally comparable to similar studies of JAK inhibitors in patients with PsA, such as the OPAL Broaden study (NCT01877668) of tofacitinib [22].
A primary strength of the current analysis is that it is based on a pooled analysis of data from two large, phase 3 clinical trials. Although the comparison of UPA as monotherapy vs combination therapy was not a primary objective of the studies, this analysis was planned prior to trial conduct and patients were stratified by current use of one or more nbDMARD at randomization. One limitation of the study is that the majority of patients taking a concomitant nbDMARD were receiving MTX, and thus it was not possible to individually assess UPA in combination with other nbDMARDs such as SSZ or LEF. In addition, although it was permitted, relatively few patients were receiving UPA in combination with two nbDMARDs, so the safety and efficacy of this treatment regimen could not be assessed. It should also be noted that all patients who were taking a nbDMARD at study entry met inclusion criteria related to active disease. Thus these data permit assessment of the safety and efficacy of treatment with UPA added to stable background therapy and are not able to inform the benefit or risk of starting both drugs simultaneously or adding a nbDMARD to existing UPA therapy. Finally, this analysis focussed on 24 week data; long-term efficacy and safety for UPA monotherapy and combination therapy, including any long-term benefits (such as exploring late-stage drug survival with or without combination therapy), will be assessed in the ongoing SELECT-PsA 1 and SELECT-PsA 2 studies.
In conclusion, the results of this analysis show that the efficacy and safety of UPA were generally consistent when administered as monotherapy or in combination with nbDMARDs. This supports the use of UPA with or without nbDMARDs in PsA and suggests that UPA monotherapy may be a useful treatment option in patients with contraindications to MTX or those who are unable to tolerate higher doses.
Supplementary Material
Acknowledgements
Medical writing support was provided by John Ewbank, PhD, of 2 the Nth (Cheshire, UK), and was funded by AbbVie.
Contributor Information
Peter Nash, School of Medicine, Griffith University, Brisbane, QLD, Australia.
Pascal Richette, Rheumatology Department, Lariboisière Hospital, AP-HP, Paris University; Bioscar Inserm U1132 and Université de Paris, Hôpital Lariboisière.
Laure Gossec, Sorbonne Université, INSERM, Institut Pierre Louis d'Epidémiologie et de Santé Publique; Rheumatology Department, Pitié-Salpêtrière Hospital, AP-HP, Sorbonne Université, Paris, France.
Antonio Marchesoni, Humanitas San Pio X, Milan, Italy.
Christopher Ritchlin, Allergy, Immunology and Rheumatology Division, Center for Musculoskeletal Medicine, University of Rochester Medical Center, Rochester, NY.
Koji Kato, AbbVie Inc, North Chicago, IL.
Erin L McDearmon-Blondell, AbbVie Inc, North Chicago, IL.
Elizabeth Lesser, AbbVie Inc, North Chicago, IL.
Reva McCaskill, AbbVie Inc, North Chicago, IL.
Dai Feng, AbbVie Inc, North Chicago, IL.
Jaclyn K Anderson, AbbVie Inc, North Chicago, IL.
Eric M Ruderman, Department of Medicine, Division of Rheumatology, Northwestern University Feinberg School of Medicine, Chicago, IL, USA.
Funding: This work was supported by AbbVie, who participated in the study design, research, analysis, data collection, interpretation of data, review and approval of the publication. All authors had access to relevant data and participated in the drafting, review and approval of this publication. No honoraria or payments were made for authorship.
Disclosure statement: P.N. has received funding for clinical trials, research grants and honoraria for lectures and advice from AbbVie, Amgen, Bristol Myers Squibb, Celgene, Eli Lilly, Janssen, Novartis, Pfizer, Roche, Sanofi-Aventis and UCB. P.R. has received honoraria from AbbVie, Amgen, Biogen, Bristol Myers Squibb, Celgene, Eli Lilly, Janssen, Novartis, Pfizer, Roche, Sanofi-Aventis and UCB. L.G. has received research grants from Amgen, Galapagos, Eli Lilly, Pfizer, Sandoz and Sanofi and consulting fees from AbbVie, Amgen, Bristol Myers Squibb, Biogen, Celgene, Galapagos, Gilead, Janssen, Eli Lilly, Novartis, Pfizer, Samsung Bioepis, Sanofi-Aventis and UCB. A.M. has received speaking fees and/or honoraria from AbbVie, Eli Lilly, Janssen, Novartis, Pfizer and UCB and consulting fees from AbbVie. C.R. has received consulting fees from AbbVie, Amgen, Bristol Myers Squibb, Janssen and Novartis; consulting for and as an independent contractor for Amgen and consulting for and research support from UCB. K.K., E.L.M.-B., E.L., R.M., D.F. and J.K.A. are AbbVie employees and may own stock or options. E.M.R. has received consulting fees from AbbVie, Amgen, Bristol Myers Squibb, Gilead, Janssen, Eli Lilly, Novartis and Pfizer.
Data availability statement
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual and trial-level data (analysis datasets), as well as other information (e.g. protocols and clinical study reports), provided the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. These clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research, and will be provided following review and approval of a research proposal and statistical analysis plan and execution of a Data Sharing Agreement.
Data requests can be submitted at any time and the data will be accessible for 12 months, with possible extensions considered. For more information on the process or to submit a request, visit https://www.abbvie.com/our-science/clinical-trials/clinical-trials-data-and-information-sharing/data-and-information-sharing-with-qualified-researchers.html.
Supplementary data
Supplementary data are available at Rheumatology online.
References
- 1. Coates LC, Gossec L, Ramiro S. et al. New GRAPPA and EULAR recommendations for the management of psoriatic arthritis. Rheumatology (Oxford) 2017;56:1251–3. [DOI] [PubMed] [Google Scholar]
- 2. Gossec L, Baraliakos X, Kerschbaumer A. et al. EULAR recommendations for the management of psoriatic arthritis with pharmacological therapies: 2019 update. Ann Rheum Dis 2020;79:700–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Singh JA, Guyatt G, Ogdie A. et al. 2018 American College of Rheumatology/National Psoriasis Foundation guideline for the treatment of psoriatic arthritis. Arthritis Care Res (Hoboken) 2019;71:2–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Combe B, Behrens F, McHugh N. et al. Comparison of etanercept monotherapy and combination therapy with methotrexate in psoriatic arthritis: results from 2 clinical trials. J Rheumatol 2016;43:1063–7. [DOI] [PubMed] [Google Scholar]
- 5. Ianculescu I, Weisman MH.. The role of methotrexate in psoriatic arthritis: what is the evidence? Clin Exp Rheumatol 2015;33(5 Suppl 93):S94–7. [PubMed] [Google Scholar]
- 6. Mease PJ, Collier DH, Saunders KC. et al. Comparative effectiveness of biologic monotherapy versus combination therapy for patients with psoriatic arthritis: results from the Corrona registry. RMD Open 2015;1:e000181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mease PJ, Gladman DD, Collier DH. et al. Etanercept and methotrexate as monotherapy or in combination for psoriatic arthritis: primary results from a randomized, controlled phase III trial. Arthritis Rheumatol 2019;71:1112–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Singh JA, Saag KG, Bridges SL Jr. et al. 2015 American College of Rheumatology guideline for the treatment of rheumatoid arthritis. Arthritis Rheumatol 2016;68:1–26. [DOI] [PubMed] [Google Scholar]
- 9. Ye W, Coates LC.. Should methotrexate have any place in the treatment of psoriatic arthritis? Rheum Dis Clin North Am 2019;45:325–39. [DOI] [PubMed] [Google Scholar]
- 10. Parmentier JM, Voss J, Graff C. et al. In vitro and in vivo characterization of the JAK1 selectivity of upadacitinib (ABT-494). BMC Rheumatol 2018;2:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. McInnes IB, Anderson JK, Magrey M. et al. Trial of upadacitinib and adalimumab for psoriatic arthritis. N Engl J Med 2021;384:1227–39. [DOI] [PubMed] [Google Scholar]
- 12. Mease PJ, Lertratanakul A, Anderson JK. et al. Upadacitinib for psoriatic arthritis refractory to biologics: SELECT-PsA 2. Ann Rheum Dis 2020;80:312–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mease PJ, Woolley JM, Bitman B. et al. Minimally important difference of Health Assessment Questionnaire in psoriatic arthritis: relating thresholds of improvement in functional ability to patient-rated importance and satisfaction. J Rheumatol 2011;38:2461–5. [DOI] [PubMed] [Google Scholar]
- 14. Combe B, Tsai TF, Huffstutter JE. et al. Ixekizumab, with or without concomitant methotrexate, improves signs and symptoms of PsA: week 52 results from Spirit-P1 and Spirit-P2 studies. Arthritis Res Ther 2021;23:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xie Y, Liu Y, Liu Y.. Are biologics combined with methotrexate better than biologics monotherapy in psoriasis and psoriatic arthritis: a meta-analysis of randomized controlled trials. Dermatol Ther 2021;34:e14926. [DOI] [PubMed] [Google Scholar]
- 16. Nash P, Mease PJ, Fleishaker D. et al. Tofacitinib as monotherapy following methotrexate withdrawal in patients with psoriatic arthritis previously treated with open-label tofacitinib plus methotrexate: a randomised, placebo-controlled substudy of OPAL balance. Lancet Rheumatol 2021;3:E28–E39. [DOI] [PubMed] [Google Scholar]
- 17. Breedveld FC, Weisman MH, Kavanaugh AF. et al. The PREMIER study: a multicenter, randomized, double-blind clinical trial of combination therapy with adalimumab plus methotrexate versus methotrexate alone or adalimumab alone in patients with early, aggressive rheumatoid arthritis who had not had previous methotrexate treatment. Arthritis Rheum 2006;54:26–37. [DOI] [PubMed] [Google Scholar]
- 18. Klareskog L, van der Heijde D, de Jager JP. et al. Therapeutic effect of the combination of etanercept and methotrexate compared with each treatment alone in patients with rheumatoid arthritis: double-blind randomised controlled trial. Lancet 2004;363:675–81. [DOI] [PubMed] [Google Scholar]
- 19. Friedman B, Cronstein B.. Methotrexate mechanism in treatment of rheumatoid arthritis. Joint Bone Spine 2019;86:301–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Conway R, Carey JJ.. Risk of liver disease in methotrexate treated patients. World J Hepatol 2017;9:1092–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Visser K, van der Heijde DM.. Risk and management of liver toxicity during methotrexate treatment in rheumatoid and psoriatic arthritis: a systematic review of the literature. Clin Exp Rheumatol 2009;27:1017–25. [PubMed] [Google Scholar]
- 22. Mease P, Hall S, FitzGerald O. et al. Tofacitinib or adalimumab versus placebo for psoriatic arthritis. N Engl J Med 2017;377:1537–50. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual and trial-level data (analysis datasets), as well as other information (e.g. protocols and clinical study reports), provided the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. These clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research, and will be provided following review and approval of a research proposal and statistical analysis plan and execution of a Data Sharing Agreement.
Data requests can be submitted at any time and the data will be accessible for 12 months, with possible extensions considered. For more information on the process or to submit a request, visit https://www.abbvie.com/our-science/clinical-trials/clinical-trials-data-and-information-sharing/data-and-information-sharing-with-qualified-researchers.html.