

Abstract

Chemisorption on organometallic-based adsorbents is crucial for the controlled separation and long-term storage of gaseous molecules. The formation of covalent bonds between the metal centers in the adsorbents and the targeted gases affects the desorption efficiency, especially when the oxidation state of the metal is low. Herein, we report a pressure-responsive nickel(0)-based system that is able to reversibly chemisorb carbon monoxide (CO) at room temperature. The use of N-heterocyclic carbene ligands with hemi-labile N-phosphine oxide substituents facilitates both the adsorption and desorption of CO on nickel(0) via ligand substitution. Ionic liquids were used as the reaction medium to enhance the desorption rate and establish a reusable system. These results showcase a way for the sustainable chemisorption of CO using a zero-valent transition-metal complex.
Introduction
Carbon monoxide (CO) is an essential feedstock that is widely used in the synthesis of commodity chemicals such as alcohols, carboxylic acids, and polycarbonates.1 CO has also been used for metal-refining processes, e.g., the Mond process, which consists of the carbonylation of crude Ni(0) at around 50 °C and thermolysis of gaseous Ni(CO)4 at 180–280 °C.2,3 Huge amounts of high-purity CO are thus produced during the removal of contaminants such as H2, N2, CO2, and CH4 from crude materials obtained from the gasification processes of hydrocarbon resources1 and the steel production industry.4 In these cases, cryogenic distillation technology is typically applied for CO purification, although technologies based on adsorption, absorption, and membranes have also been explored intensively.4,5 In terms of the purity of the produced CO, repeatable adsorption/desorption sequences based on the coordination/dissociation of CO on metal ions such as Fe(II),6−8 Co(II),8,9 Ni(II),10 Cu(I),11 Cu(II),12 and Ir(III)13 incorporated in solid-state adsorbents have shown exceptional results (Figure 1A). For example, Kirchner et al. reported the use of the crystalline solid of a Fe(II)-carbonyl complex that bears a PNP pincer-type ligand for the reversible chemisorption of CO.6 In this reaction, the adsorption proceeded smoothly under ambient conditions, while the efficient desorption required heating (100 °C) under reduced pressure. The crystalline coordination polymers known as metal–organic frameworks (MOFs) have also been used for the purification of CO.5,14 Matsuda et al. demonstrated the separation of CO from a gaseous mixture including N2, which is the most competitive gas for CO in physisorption-based separation processes due to its similar molecular size, using a Cu(II)-based nanoporous crystalline material; this separation was enabled by the coordination of CO to Cu(II) ions under cryogenic conditions.12 In this report, desorption was carried out by raising the temperature to 27 °C in a closed system that had undergone a single degassing cycle. Similarly, Long et al. proposed the application of Fe(II)-, Co(II)-, and Ni(II)-based MOFs for the purification of CO based on their high susceptibility to adsorb CO.10,15 Various metal-containing sorbents dispersed in activated carbons, zeolite, and silica have also been proven to be potential materials for CO purification;5,16−19 however, these are usually associated with higher costs and lower metal density compared to molecular-based systems and MOFs. Given the significant impact of purification processes on capital and operating costs in industry (accounting for 40–70%) and on global energy consumption (accounting for 10–15%),20 the establishment of a novel strategy for less-energy-consuming and sustainable chemisorption systems is desirable.
Figure 1.
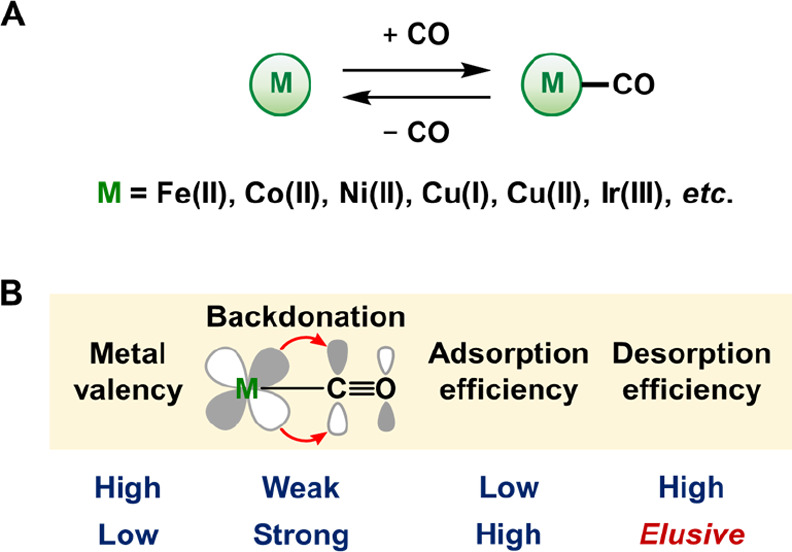
Reversible chemisorption of CO using organometallic-based adsorbents. (A) Simplified scheme of the adsorption/desorption of CO on transition metals (M). (B) General comparison of the strength of M-to-CO backdonation and the efficiency of adsorption/desorption for high- and low-valent metals.
To design such systems for CO purification, the affinity between the metal and CO is a central point of consideration (Figure 1B). In principle, more rapid and selective adsorption of CO can be achieved by the introduction of low- (or zero-) valent, d-electron-rich metals as such metals can form thermodynamically favorable metal–CO interactions through their stronger metal-to-CO π-backdonation compared to higher-valent metals.21,22 However, the strong interaction between low-valent metals and CO greatly affects the desorption efficiency. In fact, the reversible chemisorption of CO with zero-valent transition metals has been achieved under the extreme conditions used in the Mond process (vide supra). Thus, hitherto reported adsorption technologies have predominantly relied on the chemisorption of CO by higher-valent metals to minimize the influence of metal-to-CO π-backdonation. Temperature-swing operations, i.e., the use of a higher operation temperature during CO desorption than during CO adsorption, are frequently applied, and desorption is often carried out under reduced pressure (pressure-swing operation).6,8,9,12,13,15 Against this background, we envisioned the development of a process for the reversible chemisorption of CO using a zero-valent transition metal that could potentially exhibit strong metal-to-CO π-backdonation to showcase a novel strategy for the controlled separation and long-term storage of CO.
Herein, we present a method for the reversible pressure-swing chemisorption of CO on a Ni(0) complex at room temperature (rt, indicating a temperature of around 22–27 °C in this work) via ligand substitution (Figure 2B). This mechanism for CO adsorption/desorption is based on using a multifunctional carbene ligand with a hemi-labile coordination site, whereas previously reported systems rely on the simple coordination/dissociation of CO on coordinatively unsaturated metal centers (Figure 2A).6,9,10,13 Furthermore, we demonstrate that the use of an ionic liquid (IL) as the reaction medium, i.e., as a dispersant and/or solvent for the adsorbents enhances the desorption effectively, which stands in sharp contrast to the typical use of ILs for the absorption of CO.23,24
Figure 2.

Comparison of the design strategies between (A) previous systems using high-valent metals and (B) the Ni(0)-based system in this work.
Results and Discussion
For the design of the Ni(0)-based system, the choice of ancillary ligand is critical. This ligand should be equipped with a hemi-labile coordination moiety that can compete with the coordination of CO to the Ni(0) centers even in the solid (crystalline) state. We thus focused on the use of N-phosphine-oxide-substituted imidazolylidenes (PoxIms; 1a and 1b) and the corresponding imidazolinylidene (SPoxIm; 1c) as the N-phosphinoyl group can serve as a hemi-labile ligand to coordinate Ni(0) in addition to the diaminocarbene moiety (Figure 3A).25,26 To date, (S)PoxIms have demonstrated various coordination modes toward metals, including coordination by only the carbene atom (κ-C),27,28 by only the N-phosphinoyl oxygen atom (κ-O),29,30 and by both the carbene and oxygen atoms (κ-C,O);31,32 however, dynamic coordination exchange between the κ-C and κ-C,O modes remains unknown.
Figure 3.

Selective preparation of Ni(κ-C,O-1)(CO)2 (2) and Ni(κ-C-1)(CO)3 (3). (A) Synthesis of 2 and 3. Isolated yields are shown. aNMR yield confirmed in situ. (B) Molecular structures of 2a, 2c, 3a, and 3c with thermal ellipsoids at 30% probability; H atoms (except those bounded to C4 and C5 atoms) are omitted for clarity. Selected bond lengths/interatomic distances (Å) and angles (°) for 2a: Ni–C1 1.937 (2), Ni–C3 1.742 (3), Ni–O1 2.269 (2), C1-N2-P-O1 5.8 (2); 3a: Ni–C1 1.986 (3), Ni–C2 1.794(4), Ni–C3 1.789(5), Ni···O1 3.148(2), C1-N2-P-O1 1.3(3); 2c: Ni–C1 1.929(2), Ni–C3 1.757(2), Ni–O1 2.227(1), C1-N2-P-O1 8.5(1); 3c: Ni–C1 1.977(2), Ni–C2 1.807(3), Ni–C3 1.792(3), Ni···O1 3.099(2), C1-N2-P-O1 6.5(2).
Initially, we explored a method that can selectively afford Ni(κ-C,O-1)(CO)2 (2) or Ni(κ-C-1)(CO)3 (3) (Figure 3A). Treatment of a THF solution of PoxIm 1a, which bears an N-2,6-diisopropylphenyl (Dipp) group, and Ni(cod)2 with 2.2 equiv of ex situ-generated CO at rt resulted in the formation of 2a, which was isolated in 85% yield. The selective preparation of 3a was achieved by using an excess of CO (ca. 8.0 equiv) at rt in toluene, and 3a was isolated in 92% yield. The yield of 3a slightly decreased to 70% when the reaction was carried out in THF. Similarly, di-/tri-carbonyl complexes 2b/3b, which bear PoxIm 1b with an N-mesityl group, and dicarbonyl complex 2c, which bears SPoxIm 1c with a Dipp group, were prepared in excellent yields, whereas Ni(κ-C-1c)(CO)3 (3c) was isolated in 85% after recrystallization due to its rapid conversion to 2c during the removal of volatile species in vacuo. These compounds were unambiguously characterized using multinuclear NMR and IR spectroscopy as well as single-crystal X-ray diffraction (SC-XRD) analysis. For example, in the 31P NMR spectra, the resonance of the N-phosphinoyl moiety in 2a is observed at δp 66.9, which represents a significant downfield shift compared to that of 3a (δp 58.4). The A1-symmetrical carbonyl stretching frequencies of 3a/3c (2048–2049 cm–1; in CH2Cl2) are nearly identical but slightly lower than the values of Ni(κ-C-1d/1e)(CO)3 (3d/3e, 2052 cm–1),33 where 1d is 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene and 1e is 1,3-bis(2,6-diisopropylphenyl)imidazolidin-2-ylidene, indicating a negligible difference in the electron density on their Ni(0) centers.34
The molecular structures of 2a, 2c, 3a, and 3c obtained from SC-XRD analysis are shown in Figure 3B. In these cases, the C1 and O1 atoms adopt a syn-orientation with respect to the N–P bonds (C1-N2-P-O1 torsion angle: 5.8(2)° in 2a; 8.5(1)° in 2c; 1.3(3)° in 3a; 6.5(2)° in 3c), indicating that the complexation proceeded via the rotation of the N-phosphinoyl group in free 1, wherein the C1 and O1 atoms adopt an anti-orientation (C-N-P-O: 175.9(1)° in 1a; 179.7(2)° in 1c).25 The interatomic distances between Ni and O1 suggest the absence of a bonding interaction between these atoms in 3a (3.148(2) Å) and 3c (3.099(2) Å), while the formation of Ni–O1 bonds is clearly confirmed in 2a (2.269(2) Å) and 2c (2.227(1) Å). Stronger interactions are expected between the Ni and C1 atoms in 2a (1.937(2) Å) and 2c (1.929(2) Å) compared to those in 3a (1.986(3) Å) and 3c (1.977(2) Å), which can be rationalized in terms of the decreased number of π-acidic CO ligands. Ni K-edge X-ray absorption spectroscopy confirmed that the electronic states and local structures around the Ni centers are almost identical for 3a and 3c (Figures S52–S54). In their entirety, these results demonstrate the first example of the selective formation of Ni(0) complexes that bear two or three carbonyl ligands in the presence of a single N-heterocyclic carbene (NHC) ligand.
During the preparation of the aforementioned complexes, we noticed the partial formation of 2c when a solution of 3c was concentrated in vacuo. In fact, stirring the crystalline powder of 3c at rt for 10 h in vacuo (0.3 mmHg) resulted in the formation of 2c in 50% yield (Figure 4A). Prolonging the reaction time resulted in a slight improvement in the efficiency of CO desorption from 3c (20 h, 59%). Nevertheless, further desorption was not expected, as the solids adhered to the inner surface of the reaction vessel, limiting the surface area of 3c exposed to the reduced pressure even under stirring conditions (Figure S22). To promote the desorption, we explored the use of a dispersant. Dispersing 3c into tetradecane (C14H30) in the reaction flask (V = 50 mL) resulted in a significant improvement of the desorption, and 2c was obtained in >99% yield after 2 h at rt with concomitant loss of the crystallinity (Figure 4A and Figure S24); however, ca. 2 wt% of C14H30 was removed under the applied reaction conditions. Desorption also proceeded quantitatively within 30 min when 3c was fully dissolved in 1,3-dimethoxybenzene (DMB), albeit that the partial removal of DMB (ca. 2 wt%) was again inevitable (Figure S26).
Figure 4.

Reversible chemisorption of CO on Ni(0) complexes. (A) Effect of C14H30 as a dispersant and DMB as a solvent. (B) Effect of the structure and amount of the IL. General procedure for the CO desorption: 3c (3.0 × 10–2 mmol) was dispersed in the IL in the reaction vial under reduced pressure (0.3 mmHg) followed by the addition of THF-d8 for NMR analysis to estimate the yield of 2c. (C) Effect of the ligand. For the reaction from 3f, a mixture of 3f/2f (86/14) was employed. (D) Adsorption of CO by 2. General procedure: a mixture of 2 (3.0 × 10–2 mmol) and IL-2 (350 mg) was stirred in the reaction vial (V = 2.0 mL) at rt in the presence of a CO source followed by the addition of THF-d8 for NMR analysis to estimate the yield of 3. (E) Reaction between 2g/2h and CO. (F) Reaction between 1i/1j and Ni(cod)2 in the presence of CO (2.2 equiv for 1i; >17 equiv for 1j).
To achieve a fully reusable and reversible chemisorption system, the concomitant removal of reaction media should be avoided. We thus turned our attention to the use of ionic liquids (ILs), which exhibit negligible vapor pressure. ILs including Cu(I) ions have been explored as potential CO absorbents;24 however, these have not yet been used as the dispersant/solvent in the desorption process, probably because ILs can occupy the pores of nanoporous materials. The dispersion of the crystalline powder of 3c in imidazolium-based IL-1 with the anion OSO2CF3– (OTf–) (350 mg) under reduced pressure resulted in obvious improvement of the desorption to afford 2c in 90% yield (run 1, Figure 4B). It should be noted that the yield of 2c was calculated via NMR analysis after the addition of THF-d8 to the resulting mixture. We experimentally confirmed that the addition of THF-d8 causes negligible changes to 2c over a short period at rt; however, after 24 h, 2c partially decomposed to give 1c·HOTf (36%) via deprotonation of the proton at the C2 position in IL-1 (Figure S34). In contrast, 3c did not show any decomposition under identical conditions.
Subsequently, we explored the optimization of the desorption conditions. The use of C2-methylated IL-2 with the anion NTf2– resulted in the formation of 2c in 98% yield by preventing the aforementioned decomposition (run 2). IL-3 with the anion PF6– also exhibited good compatibility with the applied conditions, although a slight decrease in the desorption efficiency was observed (90%; run 3). In contrast, a black precipitate was immediately generated after mixing 3c and IL-4 with the anion CH3SO4–, and 2c was not formed (run 4). Thus, IL-2 was used in the following experiments. It should be noted that up to 2.1 × 10–2 M 3c can be dissolved in IL-2 at 25 °C, which corresponds to a 20% loading of 3c, while 2c shows a higher solubility (6.7 × 10–2 M at 25 °C) (Figure S32). Thus, a significant amount of solid 3c remained during the initial stage of the CO desorption, while little solid was observed after the reaction had completed, as most of the formed 2c was dissolved in IL-2 (vide infra).
The loading amount of IL-2 was optimized by comparing the average yields of 2c obtained in five independent experiments under each loading condition (runs 5–8). When 350 mg of IL-2 was used, an average yield of 65(3)% was confirmed (run 5), while using 100 mg of IL-2 (run 6) furnished a yield of 54(3)%; however, this difference was not confirmed to be statistically significant. Nevertheless, the desorption efficiency significantly decreased when 500 mg (39(3)%; run 7) or 1000 mg (45(3)%; run 8) of IL-2 was employed, even though more 3c could be dissolved in IL-2 under these conditions. Based on the aforementioned results, the CO desorption should occur predominantly on the dispersed solids of 3c, and the amount of IL should influence the efficiency of its dispersion. The amount of IL should be optimized based on the reaction apparatus; accordingly, we employed 350 mg of IL-2 in the reaction vial (V = 2.0 mL) in subsequent experiments.
Under the optimal desorption conditions using IL-2 as the reaction medium, 3c was converted into 2c in 97% yield after 4 h at rt under reduced pressure (Figure 4C). Interestingly, only 9% desorption of CO from 3a proceeded under identical conditions,35 even though the geometric and electronic features of 3a and 3c are almost identical (vide supra). This significant difference in the desorption rate could be interpreted in terms of the structural flexibility of the ancillary ligands. Complex 3b was also subjected to identical desorption conditions, but the resulting yield of 2b was only 19%. No reaction occurred for 3d or 3e. To evaluate the role of the N-phosphinoyl oxygen atom in 3c, we synthesized Ni(κ-C-1f)(CO)3 (3f), which underwent desorption of CO to generate Ni(κ-C,P-1f)(CO)2 (2f) in 39% yield, where 1f is a N-phosphanyl-substituted imidazolidin-2-ylidene. These results demonstrate that the hemi-labile behavior of the N-phosphinoyl moiety (3c vs 3e and 3f) and the structural flexibility derived from the ethylene moiety in the imidazolidin-2-ylidene ring (3c vs 3a) are both essential to achieve the efficient desorption of CO from the Ni(0) center under the applied conditions.
Then, we explored the adsorption of CO by 2 in the presence of IL-2 at rt; this adsorption should predominantly occur in the solvated state, given the sufficient solubility of complexes 2 in IL-2 (Figure 4D). Stirring 2a/2c and 350 mg of IL-2 under a CO/N2 (1 atm each) atmosphere afforded 3a/3c in 98% yields via the selective adsorption of CO with concomitant precipitation of fine crystals of 3 (3c is shown as an example in Figure 6). The reversible coordination of the N-phosphinoyl oxygen atom was again confirmed to be effective as treatment of 2f with CO/N2 resulted in the formation of 3f in 47% after 30 min. In addition, CO was directly stored in 3c from gaseous mixtures of CO/CH4/N2 (1 atm each) and CO/H2/N2 (1 atm each) in excellent yields through adsorption by 2c. Thus, the present system could also be used for the purification of CH4 and H2 through the removal of the accompanying CO. Ni(0) dicarbonyl complexes 2g and 2h, which bear a bidentate carbene (1g) or phosphine (1h) ligand, respectively, did not react with excess CO even after being dissolved in THF-d8 (Figure 4E). We also explored the preparation of Ni(0) dicarbonyl complexes that bear the bidentate ligands, 1i and 1j, with a phosphine oxide group. As a result, Ni(κ-C-1i)2(CO)2 (4i) and Ni(κ-P-1j)2(CO)2 (4j) were obtained even in the presence of excess CO (Figure 4F, Figure S11, and Scheme S7). The results of these experiments show that our strategy based on the use of (S)PoxIm ligands and IL-based media is effective for the precise separation of CO from gaseous mixtures including N2, H2, and CH4.
Figure 6.

Photographs and micrographs of reaction samples during the first cycle of CO desorption from 3c to give 2c and subsequent CO adsorption to again furnish 3c. The scale-bar shown in red is equivalent to 3.0 mm.
We then investigated the reusability of the present chemisorption system and found a significant acceleration of the rate of CO desorption between the first and second cycles (Figure 5). In fact, 2c was afforded in 87–88% yield within 2 h from 3c prepared under the optimized conditions via either the adsorption of CO on 2c or sequential CO desorption–adsorption reactions from crystalline 3c, whereas 2c was obtained in 65(3)% yield from crystalline 3c (run 5, Figure 4B). This result can be explained by the increase in the total surface area of 3c exposed to the reduced pressure as the crystals of 3c that were re-precipitated after CO adsorption were significantly smaller than those used in the first desorption process (Figure 6). Furthermore, CO was effectively desorbed from 3c even after five desorption–adsorption cycles. It should be noteworthy that 3c can be used for the purpose of CO-storage as solid-state 3c and the mixture of 3c/IL-2 were stable for at least 7 days at rt and −30 °C, respectively (Figure S36). In the aforementioned five cycles, the mixture of 3c/IL-2 was stored at −30 °C for 14–16 h after each desorption/adsorption cycle was completed. These results shed light on the key features of the Ni(0)-based reversible chemisorption of CO, i.e., this system can be reused without the removal/addition of the IL, and crystallization is not essential for the preparation of the adsorbents.
Figure 5.

Repeated use of the present CO chemisorption system. Five cycles of repeated CO adsorption/desorption with IL-2 (350 mg).
To clarify the reason for the obvious difference in the CO desorption rates of 3a and 3c, density functional theory (DFT) calculations were carried out at the ωB97X-D/Def2-TZVPD//M06-L/Def2-SVPD (for Ni and O) and Def2-SVP for others//gas phase level of theory. First, we identified two plausible pathways that connect 3c and 2c; in the first, the C2≡O2 moiety at the distal position with respect to the N-phosphinoyl oxygen atom dissociates from 3c, while in the second, the C3≡O3 moiety at the proximal position dissociates (for atomic labels, see Figure 7B. For the details of these two pathways, see Figure S40). A significant difference in the activation energy barriers (ΔG‡) of these pathways was observed (+13.3 kcal mol–1 for the former; +17.0 kcal mol–1 for the latter), indicating that the dissociation of CO from 3c should proceed via cleavage of the C2–Ni bond (Figure 7A). In the optimized structure of TS1c shown in Figure 7B, the interatomic distance between Ni and O1 is shortened to 2.53 Å from the 3.14 Å found in the optimized structure of 3c, while the distance between Ni and C2 is elongated to 2.73 Å from 1.82 Å in 3c. Although these results are based on the structures optimized in the gas phase, the transformation of 3c to 2c should proceed via ligand substitution even under the applied experimental conditions. This ligand substitution results in the formation of the intermediate [2c···CO] (ΔG° = +12.9 kcal mol–1 with respect to 3c). The 2c moiety in [2c···CO] exhibits a geometry that is nearly identical to that of the optimized 2c, e.g., the Ni–O1 lengths are 2.31 Å in [2c···CO] and 2.30 Å in 2c.
Figure 7.

Theoretical studies. (A) Plausible reaction pathways for the interconversion between 3 and 2, calculated at the ωB97X-D/Def2-TZVPD//M06-L/Def2-SVPD (for O and Ni) and Def2-SVP (for all other atoms) level of theory (gas-phase, 298.15 K, 1 atm). Relative Gibbs energies (kcal mol–1) are given with respect to 3 (+0.0 kcal mol–1). %Vbur values calculated using the program SambVca (r = 3.5 Å; d = 2.0 Å; bondi radii scaled by 1.17; H atoms are omitted) based on the structural parameters optimized by DFT calculations: 3c, 40.5; TS1c, 43.6; [2c···CO], 43.8; 2c, 45.2; 3a, 40.2; TS1a, 43.5; [2a···CO], 44.0; 2a, 42.7; 3f, 39.1; TS1f, 43.6; [2f···CO], 46.6; 2f, 46.9. aΔ%Vbur is the difference between the maximum and minimum %Vbur values obtained for 3, TS1, and [2···CO]. (B) Optimized gas-phase structures of 3c, TS1c, and [2c···CO]. Selected bond lengths (Å) are shown. (C) Comparison of the geometric deviations generated during the transformations from 3c to TS1c (left) and from 3a to TS1a (right). The structures for each 3/TS1 pair are overlaid with respect to their N1-Ni-N2 planes. The deviation distances (Å) for the Ni, O1, and P atoms are also shown.
Next, the activation energy barriers for the dissociation of CO from the Ni(0) centers of 3a, 3c, and 3f were compared (Figure 7A). The values of ΔG‡ (with respect to that of 3) increase in the order TS1c (+13.3 kcal mol–1) < TS1f (+14.2 kcal mol–1) < TS1a (+14.9 kcal mol–1). This trend is consistent with the experimental results that show that the efficiency of CO desorption increases in the order 3a (9%) < 3f (39%) < 3c (97%) under the applied experimental conditions (Figure 4C). The presence of the N-phosphinoyl oxygen atom in 3c minimizes the change in the spatial environment around the Ni(0) center during the CO substitution process, which was evaluated using the percent buried volume (%Vbur) calculated based on the geometrical parameters obtained from DFT calculations.36,37 The change in %Vbur (Δ%Vbur)26 was found to be 3.3 when the %Vbur values of the 1c moieties in 3c, TS1c, and [2c···CO] were compared; this value is obviously smaller than the Δ%Vbur of 7.5 calculated for the transformation of 3f into [2f···CO] via TS1f, thus rationalizing the faster interconversion in the former case compared to the latter.
A comparison of the coordinates of the Ni, C5, C6, P, and O1 atoms between 3 and TS1 reveals that larger deviations are generated in these atoms during the transformation from 3c to TS1c than during that from 3a to TS1a, highlighting the enhanced flexibility of the skeleton of 1c (Figure 7C). The C2, Ni, and O1 atoms in 3c can thus smoothly adopt a suitable orientation for the ligand substitution by reducing the Ni···O1 distance by 0.61 Å to reach TS1c. In contrast, in the case of the formation of TS1a from 3a, the Ni···O1 distance must be shortened by 0.69 Å under more structurally restricted conditions, resulting in a larger ΔG‡ to reach TS1a.
The reported monodentate NHCs yielded either nickel dicarbonyl (e.g., 2k and 2l) or tricarbonyl (e.g., 3d and 3e and 3m–p) complexes, depending on their steric demand when a single molecule of NHC was treated with a Ni(0) species (Figure 8).33,38−40 Interestingly, %Vbur values of around 39.5–40.0 seem to represent a plausible boundary that determines whether di- or tri-carbonyl complexes are generated as isolable species. In this context, (S)PoxIms 1a–c and N-phosphanyl-substituted 1f demonstrate unprecedented reactivity to afford both di- and tri-carbonyl complexes and realize their interconversion beyond the possible boundary of %Vbur by effectively scaling the spatial volume around the Ni center.
Figure 8.

Plausible %Vbur boundary for the formation of Ni(0) di- or tri-carbonyl complexes that bear NHCs. The %Vbur values were calculated based on the geometric parameters obtained from the SC-XRD analysis, reported in this work (2a–c, 2f, 3a–c, and 3f) and the previous works (2k, 2l, 3d, 3e, and 3m–p).33,38−40
The presented preliminary results serve as a proof-of-concept for a reusable and reversible chemisorption system for CO based on the use of zero-valent transition-metal complexes at room temperature driven only by pressure-swing manipulation. We believe that the strategy shown in this work, i.e., (i) the construction of a ligand system that functions even in the solid state for the reversible CO substitution using flexible multifunctional ligands and (ii) the use of an ionic liquid as the reaction medium, will pave the way for the design of an unprecedented molecular-based chemisorption system that can effectively purify (or remove) CO in a low-energy-consuming and sustainable manner.
Acknowledgments
This project was supported by Grants-in-Aid for Scientific Research (C) (JSPS KAKENHI Grant 21K05070 for Y.H.) and for Young Scientists (JSPS KAKENHI Grant 20K15279 for Y.U.), the Environment Research and Technology Development Fund (JPMEERF20211R01) of the Environmental Restoration and Conservation Agency of Japan (Y.H.), a Mitsubishi Gas Chemical Award in Synthetic Organic Chemistry, Japan (Y.U.), and the RIKEN-Osaka University Science and Technology Hub Collaborative Research Program from RIKEN and Osaka University (Y.U.). A part of this work was supported by JST SPRING, Grant Number JPMJSP2138 (Y.Y.). Ni K-edge XAS measurements were performed at the BL14B2 beamline of SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (proposal nos. 2021A1630 and 2021B1717). Part of the computation was performed using Research Center for Computational Science, Okazaki, Japan (Project: 21-IMS-C105).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.2c02870.
Full details pertaining to the experimental methods, identification of the compounds, and DFT calculations (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Häring H.-W. Ed., Industrial Gases Processing (Wiley–VCH: Weinheim, 2008). [Google Scholar]
- Mond L.; Langer C.; Quincke F. L. Action of Carbon Monoxide on Nickel. J. Chem. Soc., Trans. 1890, 57, 749–753. 10.1039/CT8905700749. [DOI] [Google Scholar]
- Morrison A.; Leitch J. J.; Szymanski G.; Moula G.; Barlow B.; Burgess I. J.; Shobeir B.; Huang H.; Lipkowski J. Electrochim. Acta 2018, 260, 684–694. 10.1016/j.electacta.2017.12.016. [DOI] [Google Scholar]
- Ramírez-Santos Á. A.; Castel C.; Favre E. A review of gas separation technologies within emission reduction programs in the iron and steel sector: Current application and development perspectives. Sep. Purif. Technol. 2018, 194, 425–442. 10.1016/j.seppur.2017.11.063. [DOI] [Google Scholar]
- Evans A.; Luebke R.; Petit C. The use of metal–organic frameworks for CO purification. J. Mater. Chem. A 2018, 6, 10570–10594. 10.1039/C8TA02059K. [DOI] [Google Scholar]
- Benito-Garagorri D.; Puchberger M.; Mereiter K.; Kirchner K. Stereospecific and Reversible CO Binding at Iron Pincer Complexes. Angew. Chem., Int. Ed. 2008, 47, 9142–9145. 10.1002/anie.200803665. [DOI] [PubMed] [Google Scholar]
- Benito-Garagorri D.; Lagoja I.; Veiros L. F.; Kirchner K. A. Reactivity of coordinatively unsaturated iron complexes towards carbon monoxide: to bind or not to bind?. Dalton Trans. 2011, 40, 4778–4792. 10.1039/c0dt01636e. [DOI] [PubMed] [Google Scholar]
- Tate C. W.; de Mello A.; Gee A. D.; Kealey S.; Vilar R.; White A. J. P.; Long N. J. Hemilabile and reversible carbon monoxide binding properties of iron(II), cobalt(II) and nickel(II) complexes containing a new tridentate P–S–N ligand. Dalton Trans. 2012, 41, 83–89. 10.1039/C1DT11248A. [DOI] [PubMed] [Google Scholar]
- Gallagher A. T.; Malliakas C. D.; Harris T. D. CO Binding at a Four-Coordinate Cobaltous Porphyrin Site in a Metal–Organic Framework: Structural, EPR, and Gas Adsorption Analysis. Inorg. Chem. 2017, 56, 4654–4661. 10.1021/acs.inorgchem.7b00292. [DOI] [PubMed] [Google Scholar]
- Bloch E. D.; Hudson M. R.; Mason J. A.; Chavan S.; Crocellà V.; Howe J. D.; Lee K.; Dzubak A. L.; Queen W. L.; Zadrozny J. M.; Geier S. J.; Lin L.-C.; Gagliardi L.; Smit B.; Neaton J. B.; Bordiga S.; Brown C. M.; Long J. R. Reversible CO Binding Enables Tunable CO/H2 and CO/N2 Separations in Metal–Organic Frameworks with Exposed Divalent Metal Cations. J. Am. Chem. Soc. 2014, 136, 10752–10761. 10.1021/ja505318p. [DOI] [PubMed] [Google Scholar]
- Tate C. W.; Gee A. D.; Vilar R.; White A. J. P.; Long N. J. Reversible carbon monoxide binding at copper(I) P–S–X (X = N, O) coordination polymers. J. Organomet. Chem. 2012, 715, 39–42. 10.1016/j.jorganchem.2012.05.032. [DOI] [Google Scholar]
- Sato H.; Kosaka W.; Matsuda R.; Hori A.; Hijikata Y.; Belosludov R. V.; Sakaki S.; Takata M.; Kitagawa S. Self-Accelerating CO Sorption in a Soft Nanoporous Crystal. Science 2014, 343, 167–170. 10.1126/science.1246423. [DOI] [PubMed] [Google Scholar]
- Sun L.-Y.; An Y.-Y.; Ma L.-L.; Han Y.-F. Single-Crystalline Organoiridium Complex for Gas-Triggered Chromogenic Switches and Its Applications on CO Detection and Reversible Scavenging. Chin. J. Chem. 2019, 37, 763–768. 10.1002/cjoc.201900151. [DOI] [Google Scholar]
- Islamoglu T.; Chen Z.; Wasson M. C.; Buru C. T.; Kirlikovali K. O.; Afrin U.; Mian M. R.; Farha O. K. Metal–Organic Frameworks against Toxic Chemicals. Chem. Rev. 2020, 120, 8130–8160. 10.1021/acs.chemrev.9b00828. [DOI] [PubMed] [Google Scholar]
- Reed D. A.; Xiao D. J.; Gonzalez M. I.; Darago L. E.; Herm Z. R.; Grandjean F.; Long J. R. Reversible CO Scavenging via Adsorbate-Dependent Spin State Transitions in an Iron(II)–Triazolate Metal–Organic Framework. J. Am. Chem. Soc. 2016, 138, 5594–5602. 10.1021/jacs.6b00248. [DOI] [PubMed] [Google Scholar]
- Hirai H.; Wada K.; Komiyama M. Active Carbon-Supported Copper(I) Chloride as Solid Adsorbent for Carbon Monoxide. Bull. Chem. Soc. Jpn. 1986, 59, 2217–2223. 10.1246/bcsj.59.2217. [DOI] [Google Scholar]
- Moragues M. E.; Esteban J.; Ros-Lis J. V.; Martínez-Mánez R.; Marcos M. D.; Martínez M.; Soto J.; Sancenón F. Sensitive and Selective Chromogenic Sensing of Carbon Monoxide via Reversible Axial CO Coordination in Binuclear Rhodium Complexes. J. Am. Chem. Soc. 2011, 133, 15762–15772. 10.1021/ja206251r. [DOI] [PubMed] [Google Scholar]
- Balkus K. J. Jr.; Kortz A.; Drago R. S. Carbon Monoxide Binding by Copper(I) Complexes Supported on Polystyrene. Inorg. Chem. 1988, 27, 2955–2958. 10.1021/ic00290a013. [DOI] [Google Scholar]
- Dulebohn J. I.; Haefner S. C.; Berglund K. A.; Dunbar K. R. Reversible Carbon Monoxide Addition to Sol-Gel Derived Composite Films Containing a Cationic Rhodium(I) Complex: Toward the Development of a New Class of Molecule-Based CO Sensors. Chem. Mater. 1992, 4, 506–508. 10.1021/cm00021a006. [DOI] [Google Scholar]
- Materials for Separation Technologies: Energy and Emission Reduction Opportunities (Oak Ridge National Laboratory, U. S. Department of Energy, 2005).
- Sunderlin L. S.; Wang D.; Squires R. R. Metal Carbonyl Bond Strengths in Fe(CO)n– and Ni(CO)n–. J. Am. Chem. Soc. 1992, 114, 2788–2796. 10.1021/ja00034a004. [DOI] [Google Scholar]
- Zhou M.; Andrews L.; Bauschlicher C. W. Jr. Spectroscopic and Theoretical Investigations of Vibrational Frequencies in Binary Unsaturated Transition-Metal Carbonyl Cations, Neutrals, and Anions. Chem. Rev. 2001, 101, 1931–1962. 10.1021/cr990102b. [DOI] [PubMed] [Google Scholar]
- Raeissi S.; Florusse L. J.; Peters C. J. Purification of Flue Gas by Ionic Liquids: Carbon Monoxide Capture in [bmim][Tf2N]. AIChE J. 2013, 59, 3886–3891. 10.1002/aic.14125. [DOI] [Google Scholar]
- Tu Z.-H.; Zhang Y.-Y.; Wu Y.-T.; Hu X.-B. Self-enhancement of CO reversible absorption accompanied by phase transition in protic chlorocuprate ionic liquids for effective CO separation from N2. Chem. Commun. 2019, 55, 3390–3393. 10.1039/C9CC00089E. [DOI] [PubMed] [Google Scholar]
- Hoshimoto Y.; Kinoshita T.; Ohashi M.; Ogoshi S. A Strategy to Control the Reactivation of Frustrated Lewis Pairs from Shelf-Stable Carbene Borane Complexes. Angew. Chem., Int. Ed. 2015, 54, 11666–11671. 10.1002/anie.201505974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshimoto Y.; Ogoshi S. Development of Metal Complexes Equipped with Structurally Flexible Carbenes. Bull. Chem. Soc. Jpn. 2021, 94, 327–338. 10.1246/bcsj.20200293. [DOI] [Google Scholar]
- Branzi L.; Baron M.; Armelao L.; Rancan M.; Sgarbossa P.; Graiff C.; Pöthig A.; Biffis A. Coordination chemistry of gold with N-phosphine oxide-substituted imidazolylidenes (POxIms). New J. Chem. 2019, 43, 17275–17283. 10.1039/C9NJ04911H. [DOI] [Google Scholar]
- Asada T.; Hoshimoto Y.; Ogoshi S. Rotation-Triggered Transmetalation on a Heterobimetallic Cu/Al N-Phosphine-Oxide-Substituted Imidazolylidene Complex. J. Am. Chem. Soc. 2020, 142, 9772–9784. 10.1021/jacs.0c03252. [DOI] [PubMed] [Google Scholar]
- Kinoshita T.; Sakuraba M.; Hoshimoto Y.; Ogoshi S. Complexation between MOTf (M = Li and Na) and N-Phosphine Oxide-substituted Imidazolylidenes via Coordination of the N-Phosphoryl Groups. Chem. Lett. 2019, 48, 230–233. 10.1246/cl.180930. [DOI] [Google Scholar]
- Asada T.; Hoshimoto Y.; Kawakita T.; Kinoshita T.; Ogoshi S. Axial Chirality around N–P Bonds Induced by Complexation between E(C6F5)3 (E = B, Al) and an N-Phosphine Oxide-Substituted Imidazolinylidene: A Key Intermediate in the Catalytic Phosphinoylation of CO2. J. Org. Chem. 2020, 85, 14333–14341. 10.1021/acs.joc.9b03210. [DOI] [PubMed] [Google Scholar]
- Tao W.; Akita S.; Nakano R.; Ito S.; Hoshimoto Y.; Ogoshi S.; Nozaki K. Copolymerisation of ethylene with polar monomers by using palladium catalysts bearing an N-heterocyclic carbene–phosphine oxide bidentate ligand. Chem. Commun. 2017, 53, 2630–2633. 10.1039/C7CC00002B. [DOI] [PubMed] [Google Scholar]
- Branzi L.; Franco D.; Baron M.; Armelao L.; Rancan M.; Sgarbossa P.; Biffis A. Palladium(II) Complexes with N-Phosphine Oxide-Substituted Imidazolylidenes (PoxIms): Coordination Chemistry and Catalysis. Organometallics 2019, 38, 2298–2306. 10.1021/acs.organomet.9b00185. [DOI] [Google Scholar]
- Dorta R.; Stevens E. D.; Scott N. M.; Costabile C.; Cavallo L.; Hoff C. D.; Nolan S. P. Steric and Electronic Properties of N-Heterocyclic Carbenes (NHC): A Detailed Study on Their Interaction with Ni(CO)4. J. Am. Chem. Soc. 2005, 127, 2485–2495. 10.1021/ja0438821. [DOI] [PubMed] [Google Scholar]
- We evaluated the σ-donor/π-acceptor ability of (S)PoxIms using several spectroscopic techniques (for details, see the Supporting Information). The results imply that (i) the σ-donor/π-acceptor ability of SPoxIm is higher than that of PoxIm (e.g., 1c vs 1a) and (ii) (S)PoxIms exhibit higher π-accepting properties and slightly lower σ-donating properties compared to commonly used N-heterocyclic carbenes such as 1d and 1e.
- When the desorption of CO was carried out from the fully dissolved states of 3a and 3f using a far larger amount of IL-2 (>4 g), 2a and 2f were obtained in 20 and 47% yield, respectively. In the case of full-dispersion experiments using C14H30 (200 mg), 2a and 2f were obtained in 10 and 73% yield, respectively. These results support the conclusion that the CO desorption from 3a and 3f predominantly proceeds in their solid states under the applied conditions.
- Gómez-Suárez A.; Nelson D. J.; Nolan S. P. Quantifying and understanding the steric properties of N-heterocyclic carbenes. Chem. Commun. 2017, 53, 2650–2660. 10.1039/C7CC00255F. [DOI] [PubMed] [Google Scholar]
- Falivene L.; Cao Z.; Petta A.; Serra L.; Poater A.; Oliva R.; Scarano V.; Cavallo L. Towards the online computer-aided design of catalytic pockets. Nat. Chem. 2019, 11, 872–879. 10.1038/s41557-019-0319-5. [DOI] [PubMed] [Google Scholar]
- Dorta R.; Stevens E. D.; Hoff C. D.; Nolan S. P. Stable, Three-Coordinate Ni(CO)2(NHC) (NHC = N-Heterocyclic Carbene) Complexes Enabling the Determination of Ni–NHC Bond Energies. J. Am. Chem. Soc. 2003, 125, 10490–10491. 10.1021/ja0362151. [DOI] [PubMed] [Google Scholar]
- Collado A.; Balogh J.; Meiries S.; Slawin A. M. Z.; Falivene L.; Cavallo L.; Nolan S. P. Steric and Electronic Parameters of a Bulky yet Flexible N-Heterocyclic Carbene: 1,3-Bis(2,6-bis(1-ethylpropyl)phenyl)imidazol-2-ylidene (IPent). Organometallics 2013, 32, 3249–3252. 10.1021/om400168b. [DOI] [Google Scholar]
- Balogh J.; Slawin A. M. Z.; Nolan S. P. Bulky N-Heterocyclic Carbene IPr* in Selected Organo- and Transition Metal-Mediated Catalytic Applications. Organometallics 2012, 31, 3259–3263. 10.1021/om300104j. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.