

Abstract

Homologation of carbon monoxide is central to the heterogeneous Fischer–Tropsch process for the production of hydrocarbon fuels. C–C bond formation has been modeled by homogeneous systems, with [CnOn]2– fragments (n = 2–6) formed by two-electron reduction being commonly encountered. Here, we show that four- or six-electron reduction of CO can be accomplished by the use of anionic aluminum(I) (“aluminyl”) compounds to give both topologically linear and branched C4/C6 chains. We show that the mechanism for homologation relies on the highly electron-rich nature of the aluminyl reagent and on an unusual mode of interaction of the CO molecule, which behaves primarily as a Z-type ligand in initial adduct formation. The formation of [C6O6]4– from [C4O4]4– shows for the first time a solution-phase CO homologation process that brings about chain branching via complete C–O bond cleavage, while a comparison of the linear [C4O4]4– system with the [C4O4]6– congener formed under more reducing conditions models the net conversion of C–O bonds to C–C bonds in the presence of additional reductants.
Introduction
The assembly of complex molecules from carbon monoxide via C–C bond formation represents a fundamental chemical challenge that has key relevance to the production of hydrocarbon fuels via the heterogeneous Fischer–Tropsch process.1 Although employed industrially since 1925, the mechanism for the conversion of mixtures of CO and H2 to short-to-medium-chain alkane products through the formation of C–C bonds remains the subject of significant debate.2 C=C bond formation to yield alkenes represents a competing homologation process, and the product mixture typically also includes oxygenated species along with methane—an undesirable reduction product produced without the accompanying C–C bond formation. The idealized ratio of H2/CO (ca. 2:1; eq 1, typically with 10 < n < 20) reflects the importance of the reductant (H2) in generating the desired alkane products. The much lower proportion of dihydrogen in the coal-derived synthesis gas (“Syn Gas”) feedstock is typically rectified through application of the water gas shift reaction.3
| 1 |
The amenability of homogeneous systems to small-molecule characterization techniques has driven the investigation of organometallic compounds capable of modeling the homologation of CO via C–C bond formation.4 Prominent examples include d-block metal compounds (mirroring the use of heterogeneous transition-metal catalysts such as iron and cobalt in the Fischer–Tropsch process),5−17 with examples derived from low-valent s-, p-,18−33 and f-block compounds34−50 having also been reported (e.g., I–VII, Figure 1). Prevalent among the homologation products formed via reduction processes are cyclic systems of the type [CnOn]2– (n = 3, 4, and 6) and the related ethynediolate system [C2O2]2–,6−8,11,18,20−23,29,40,41,44−48 formed by a formal two-electron reduction process, although a number of systems featuring longer linear chains of carbon atoms have also been reported (Figure 1).14,17,22,25,31,33,34,36,37,42,49 Within this sphere, a small number of studies have emerged, which demonstrate the potential for control of homologation processes at a constant oxidation level: the selective formation of [CnOn]2– (n = 2, 3, or 4) by the cooperative action of U(III) or Mg(I) centers has been shown to be influenced by the steric bulk of ancillary ligands,21,48 and the stepwise growth of C3 chains by the addition of CO has been demonstrated at d-block metal systems derived from metal carbonyl precursors.17 Very recently, the formation of C4 or C5 chains using aluminum(I) systems has been demonstrated, with the product formed appearing to reflect the nuclearity of the metal reagent.33
Figure 1.

Selected examples of CO homologation relevant to the current study.
We have been developing the chemistry of electron-rich aluminum (“aluminyl”) compounds—stabilized by NON supporting ligands (4,5-bis(2,6-diisopropylanilido)-2,7-di-tert-butyl-9,9-dimethylxanthene; Figure 2).51,52 Such systems have been shown to be capable of the cleavage of C–H,51 C–C,53 and C–O bonds in arene substrates54 and to allow access to reactive terminal imide species which can cleave the CO bond in carbon monoxide.55 Systems such as K2[(NON)Al]2 ([1-K]2) have also shown strongly reducing capabilities toward oxygen-containing substrates,56 and here, we report on the reactivity of aluminyl compounds toward CO. We show that the homologation to give the known [C4O4]4– fragment occurs with both mono- and dinuclear systems, a finding rationalized computationally through a rate-determining C=C bond-forming dimerization process. The initial interaction of the CO molecule at aluminum is unusual, in that it behaves primarily as a Z-type ligand, reflecting the highly electron-rich nature of the aluminyl reagent. Subsequent formation of [C6O6]4– from [C4O4]4– shows for the first time a solution-phase CO homologation process that brings about chain branching via complete C–O bond cleavage. In addition, comparison of the linear [C4O4]4– system with the (unprecedented) [C4O4]6– congener formed under more reducing conditions models the conversion of C–O bonds to C–C bonds in the presence of additional reductants.
Figure 2.

Aluminyl compounds central to the current study: dimeric potassium aluminyl compound K2[(NON)Al]2, [1-K]2, and monomeric lithium analogue (Et2O)2LiAl(NON), [1-Li(OEt2)2].
Results and Discussion
Synthesis of [C4O4]4– and [C6O6]4– via CO Homologation
The reaction of potassium aluminyl compound [1-K]251 with CO (ca. 1.5 atm) in benzene over 16 h at ca. 35 °C (followed by recrystallization from toluene) leads to the isolation of the orange crystalline product [K(C7H8)]2[{(NON)Al}2(C4O4)], [2-K(C7H8)]2, which has been characterized by standard spectroscopic and micro-analytical methods (Scheme 1 and Figure 3). [2-K(C7H8)]2 can be shown by X-ray crystallography to feature a centrosymmetric structure in which two [(NON)Al] units are bridged by a [C4O4] fragment and two K+ counterions (which each interact with the π system of a Dipp group of one NON ligand and an additional toluene molecule; Figure 3).
Scheme 1. Homologation of CO by Aluminyl Compounds Yielding Products Containing [C4O4]4– or [C6O6]4– Fragments.

Four-electron reduction of CO by [1-K]2 or 2 equiv of [1-Li(OEt2)2] to give [2-K(C7H8)]2/[2-Li(OEt2)]2, featuring the topologically linear [C4O4]4– fragment; onward reaction of [2-K(C7H8)]2 with CO to yield the [C6O6]4– system, 3.
Figure 3.
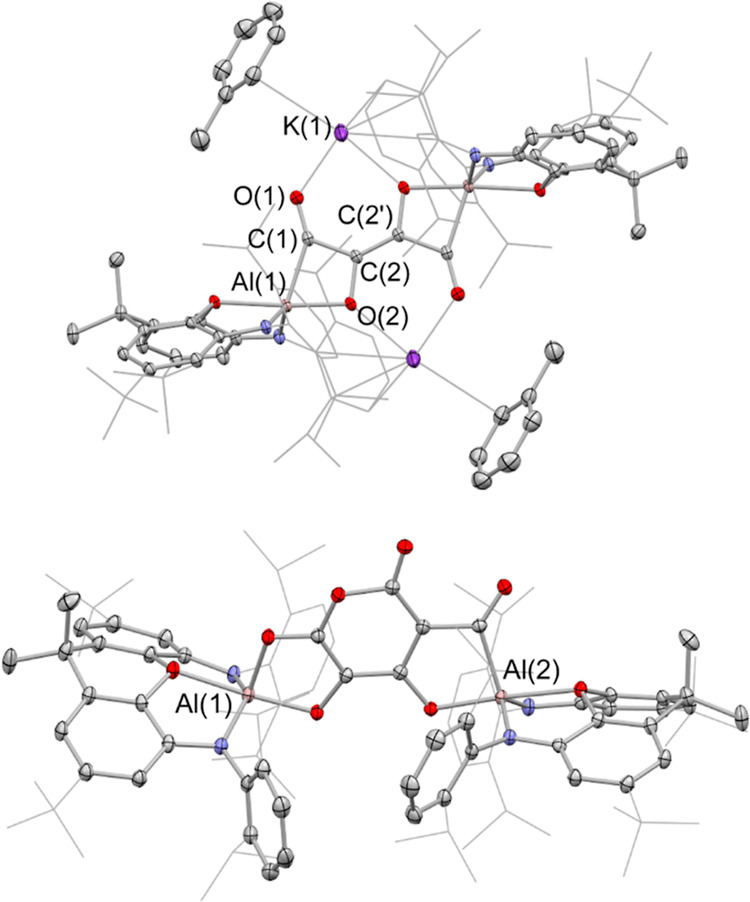
Molecular structures of (upper) [K(C7H8)]2[{(NON) Al}2(C4O4)], [2-K(C7H8)]2, and (lower) the anionic component of the asymmetric unit of K[K(THF)2][{(NON)Al}2(C6O6)], 3, in the solid state as determined by X-ray crystallography. Hydrogen atoms omitted and selected groups represented in the wireframe format for clarity; thermal ellipsoids drawn at the 50% probability level. Key metrical parameters are listed in Table 1.
Overall charge balance implies that the bridging unit is [C4O4]4–, a finding consistent with the presence in the (dimeric) aluminyl starting material of two Al(I) reductants, that is, with four-electron reduction of four molecules of CO. To a first approximation, C–C distances within the [C4O4]4– unit reflect the presence of two C–C single bonds (1.507(1) Å) and a central C=C double bond (1.373(1) Å), while the C–O distances (1.383(1)/1.233(1) Å) are consistent with C–O single bonds linked to the central carbon atoms [i.e., C(2)] and C=O double bonds involving the Al-bound carbons C(1).
Four-carbon homologation has some literature precedent. In terms of a metal-bound ligand system, the C/O skeleton bears some resemblance connectivity-wise to the enedione diolate [R2C4O4]2– system reported by Marks and co-workers, derived from the insertion of two molecules of CO into a thorium(IV) alkyl bond (IV)34,36 and to the mixed d-block/Al systems reported by Crimmin et al. (VI).17 Structurally, however, it is most closely related to Stephan’s B/N-functionalized system Cy2N{C4O4(BCy2)2}NCy2 derived from the trapping of CO by a combination of LiNCy2 and ClBCy2, and which also features a C(=O)–C(−O)=C(−O)–C(=O) skeleton (V).31 In addition, a similar C4 system was reported very recently by Coles and co-workers (VII).33
The isolated yield of [2-K(C7H8)]2 obtained from [1-K]2 and CO is low (ca. 10%), and in situ 1H NMR monitoring is consistent with the formation of other (NON)Al-containing species under these reaction conditions. Hypothesizing that this is due to the formation of other homologation products of the type [CnOn]4–, we sought (i) to probe the reactivity of isolated samples of [2-K(C7H8)]2 with CO under more forcing conditions, to see if homologation could be driven toward longer C–C-bonded chains and (ii) to investigate if greater control of the delivery of the carbon monoxide (and with it the product distribution) could be achieved by the use of alternative CO-containing reagents. Targeting the second of these objectives, we employed Fe(CO)5 as a labile (and readily soluble) source of the CO molecule in reactions with [1-K]2. Reaction with 4 equiv of Fe(CO)5 in benzene-d6 in the presence of the crown ether 12-crown-4 (to aid crystallization) leads to the formation of [2-K(12-c-4)]2, the solid-state structure of which is very closely related to that of [2-K(C7H8)]2 (see the Supporting Information). While the potential involvement of the iron center in Fe(CO)5 in this chemistry cannot be ruled out, it is noteworthy that conversion to the same [C4O4]4– fragment via this approach is markedly higher (20–30%) than that obtained using gaseous CO.
The possibility for assembling [CnOn]4– chains with n > 4 was probed via the reaction of [2-K(C7H8)]2 with CO at higher temperatures/pressures. Use of a 2 atm pressure in THF-d8 at ca. 65 °C over 4 d leads to quantitative conversion by 1H NMR (through at least three intermediate species) to a single product featuring two inequivalent NON ligands, which can be shown by X-ray crystallography to be K[K(THF)2] [{(NON)Al}2(C6O6)], (3; Figure 3). 3 features a [C6O6]4– fragment formed by the assimilation of two further equivalents of CO, which presents distinct (O,O and C,O) coordination modes at the two aluminum centers. The geometry of the [C6O6]4– group is unique, featuring a six-membered pyran core, with the mono-branched nature of the six-carbon chain implying that C–C bond formation is also accompanied by complete cleavage of one of the C–O bonds. While the homologation of CO by highly reducing molecular species to give linear or cyclic products has a literature precedent, to our knowledge, the formation of branched carbon chains in this fashion, with the accompanying cleavage of the exceptionally strong C–O bond, is unprecedented. Examples of branched-chain carbon skeletons have been reported previously—but only via subsequent derivatization reactions of linear CO-derived chains with electrophiles such as CO2 or MeI.50
Mechanistic Studies: CO Coordination and C–C/C=C Bond Formation
While the two-electron reduction of CO by a number of main group compounds to give systems of the type [CnOn]2– has been reported,18,20−23,29 four-electron processes yielding the topologically linear [C4O4]4– fragment have a more limited literature precedent.31,33 We were therefore keen to probe the mechanism of this transformation through both experimental and quantum chemical methods. As a key mechanistic principle, we first set out to determine whether the dimeric nature of the aluminyl reagent, [1-K]2, is critical in driving the formation of the (four-electron reduction) product, [C4O4]4–.
[1-K]2 has been shown by DOSY NMR measurements to retain a dinuclear structure in solution in arene solvents. With this in mind, we targeted the reactivity of CO with related aluminyl compounds known to possess a mononuclear structure. The reaction of the (monomeric) lithium aluminyl complex (Et2O)2LiAl(NON) ([1-Li(OEt2)2])57 with CO was therefore probed under similar conditions. Intriguingly, this chemistry leads to the formation of a very similar dinuclear product, [2-Li(OEt2)]2, featuring an analogous [C4O4]4– fragment, and two encapsulated Li+ counterions each ligated by two of the oxygen atoms of the [C4O4]4– unit and an additional molecule of diethyl ether. The structural metrics relating to the Al2(C4O4) unit (Table 1 and the Supporting Information) are very similar to those measured for [2-K(C7H8)]2, being consistent with a centrosymmetric four-carbon chain incorporating two C–C and one C=C bonds. Moreover, the formation of [2-Li(OEt2)]2 from [1-Li(OEt2)2] in this manner (in greater conversion to [2-K(C7H8)]2, 40 vs 10%) suggests that the state of aggregation of the aluminyl reagent is not critical in determining the stoichiometry of this carbon-containing product. This observation is consistent with the mechanism proposed by Marks and co-workers for the formation of [Cp*2Th{OC(CH2tBu)C(O)}]2, in which the central C=C bond is postulated as being constructed in the final mechanistic step through the dimerization of two mono-metallic carbene units (themselves accessed by bending of a coordinated ketene ligand).36 Coles has also proposed that the dimerization of a dioxocarbene moiety is the final step in the formation of an ethenetraolate ligand, [C2O4]4–.58
Table 1. Key Metrical Parameters for Aluminum Complexes Containing [C4O4]n− Ions (n = 2, 4, and 6).
| [C4O4]4– |
[C4O4]6– | [C4O4]2– | |||
|---|---|---|---|---|---|
| d/Å | [2-K(C7H8)]2 | [2-K(THF)]2 | [2-Li(OEt2)]2 | [4-K2(BEt3)]2 | 5 |
| C–C | 1.507(1) | 1.525(4) | 1.482(5) | 1.481(3) | 1.438(2) |
| 1.373(1) | 1.489(3) | 1.357(4) | 1.377(4) | 1.461(2) | |
| 1.376(3) | |||||
| C–O | 1.233(1) | 1.213(4) | 1.246(5) | 1.382(3) | 1.257(2) |
| 1.383(1) | 1.247(3) | 1.391(4) | 1.440(3) | 1.265(2) | |
| 1.373(3) | |||||
| 1.381(3) | |||||
| Al–C | 2.038(1) | 2.053(2) | 2.063(4) | 2.008(2) | n/a |
| 2.041(3) | |||||
| Al–O | 1.871(1) | 1.872(2) | 1.877(2) | 1.874(2) | 1.933(1) |
| 1.860(2) | 1.994(1) | ||||
In the case of the (NON)Al-derived systems presented here, this hypothesis is consistent with the results of mechanistic calculations carried out using density functional theory—for a model system in which the backbone tBu and Dipp iPr groups are replaced by Me and the cation is omitted for computational efficiency (Figure 4). The rate-determining step for the lowest energy pathway is found to involve dimerization of two carbene fragments, with these each being derived from the coupling of two molecules of CO. Kinetically, the transition state energy for the C=C forming step is located ca. 34 kcal mol–1 above the starting materials (and ca. 37 kcal mol–1 above the preceding singlet carbene intermediate). This barrier height reflects, at least in part, the sterically encumbered (and anionic) nature of the two NON-supported fragments being brought together and presumably underpins the relatively low yields associated with systems such as [2-K(C7H8)]2 (and the possibility for forming longer C-based chains via carbene/CO coupling).33
Figure 4.

Proposed mechanism for CO homologation. The lowest energy pathway (blue) involves iso-carbonyl binding of one molecule of CO (I2); the alternative (higher barrier) pathway shown in red involves binding of the second CO through carbon (I2′) and then isomerization to I2. (Calculations carried out at the B3LYP/def2-TZVP//B3LYP/def2-SVP level with solvation modeled with CPCM (benzene) and tBu and iPr groups replaced by Me for computational efficiency.)
In terms of the initial interaction of the CO molecule(s) with the (model) [(NON′)Al]− unit, we find that the end-on approach of CO perpendicular to the AlN2 plane in a manner analogous to that determined for CO-adducts of isoelectronic silylene systems, X2Si·CO,59 does not correspond to a local minimum on the potential energy surface. Rather, reflecting the nucleophilic nature of the aluminyl reagent, the approach of the carbon monoxide unit gives rise to a bent Al–C–O unit, and the predominant orbital interaction involves electron donation from Al to the π* orbital of CO. For the model C-bound bis (carbonyl) adduct [(NON′)Al(CO)2]−, the HOMO (Figure 5) defines a three-center interaction involving the aluminum-centered lone pair and the π* orbitals of the two carbonyl ligands. Moreover, extended transition state with natural orbitals for chemical valence (ETS-NOCV) analysis determines that this three-center “back-bonding” interaction accounts for the majority (261 kcal mol–1) of the orbital interaction energy (ΔEorb).60 By contrast, the complementary three-center interaction involving donation from the CO lone pairs to the Al-centered pπ-orbital, accounts for only 25 kcal mol–1. By means of comparison, a related ETS-NOCV analysis on the mono-carbonyl adduct of a cationic borole (in which the CO ligand behaves primarily as a σ-donor) yields 26 kcal mol–1 (back-bonding to CO) and 106 kcal mol–1 (sigma donation from CO).61 As such, the primary role of the CO donors in [(NON′)Al(CO)2]− is strongly suggested to be as Z-type ligands.
Figure 5.
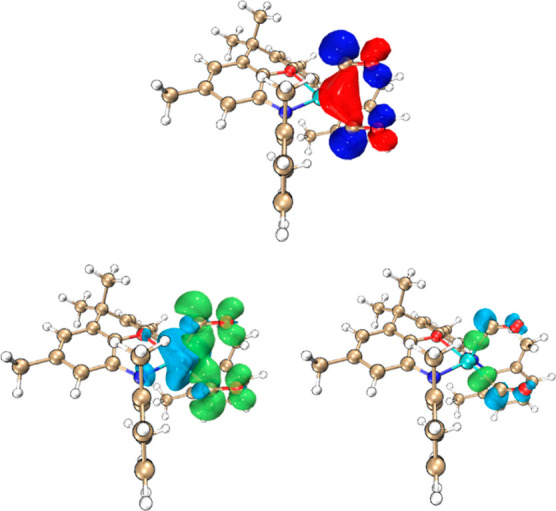
(Upper) HOMO of the model C-bound bis(carbonyl) adduct [(NON′)Al(CO)2]− (shading denotes phasing of the wavefunction); (lower) predominant orbital interactions determined by ETS-NOCV methods for the binding of the two carbonyl ligands (shading denotes deformation densities: blue/green—depletion/enhancement of electron density).
While the bis-C-ligated adduct [(NON′)Al(CO)2]− (I2′) represents the lowest energy form of the dicarbonyl species, onward transformation into carbene intermediate I3 necessarily involves rotation of one of the CO ligands to generate the isomeric species [(NON′)Al(CO)(OC)]− (I2) featuring one isocarbonyl donor.62 The barrier to this isomerization within the coordination sphere of aluminum (ca. 40.2 kcal mol–1) is found to be significantly higher than that for simple dissociation/re-association. Moreover, while I2 is ca. 17.5 kcal mol–1 higher in energy than I2′, it is capable of undergoing a very facile C–C coupling to generate carbene intermediate I3, which is lower in energy than either of the carbonyl adducts.
Interestingly, although C=C bond formation represents the key thermodynamic driving force for the overall homologation reaction (with the final dimerization process being energetically downhill by ca. 70 kcal mol–1), experimental evidence for the lability of the central C=C bond in [C4O4]4– systems can be obtained from the reactions of isolated samples of [2-K(C7H8)]2 with alkali metal hydroborate reagents. In the case of the reaction with Li[sBu3BH] in benzene/THF, simple cation metathesis is observed, generating [Li(THF)]2 [{(NON)Al}2(C4O4)], [2-Li(THF)]2, the structure of which closely resembles both [2-K(C7H8)]2 and [2-Li(OEt2)]2 (see the Supporting Information). However, thermolysis with K[Et3BH] at ca. 65–70 °C in the same solvent system leads to 70% conversion over a period of 14 d to a new species, the apparently analogous potassium salt [2-K(THF)]2. The structure of this species, however, can be shown by X-ray crystallography to feature the alternative syn disposition of substituents about the central C=C double bond (Scheme 2 and Figure 6).33 The C=C/C–C and C=O/C–O bond lengths within the carbon chain are not significantly different from the anti-form, but the [2-K(THF)]2 units are linked in the solid state via bridging K+ cations into tetra-aluminum “dimer of dimers” (see the Supporting Information). The lower solubility of this aggregate presumably drives the anti-to-syn isomerization process.
Scheme 2. Isomerization of [C4O4]4–: anti to syn Isomerization of the [C4O4]4– Fragment in the Presence of K[Et3BH] in Benzene/THF at ca. 60 °C.

Figure 6.

Molecular structure of [K(THF)]2[{(NON)Al}2(C4O4)], [2-K(THF)]2, in the solid state as determined by X-ray crystallography, showing dimeric unit analogous to [2-K(C7H8)]2. Further aggregation into “dimer of dimers” shown in Figure S16 (Supporting Information). Hydrogen atoms omitted and selected groups represented in a wireframe format for clarity; thermal ellipsoids drawn at the 50% probability level. Key metrical parameters are listed in Table 1.
Kinetically, both heat and the presence of additional K+ cations can be shown to be necessary for the transformation to occur. C=O/C=C conjugation within the [C4O4]4– unit (manifested through resonance structures featuring a central C(2)–C(2′) single bond) presumably lowers the barrier to rotation about the central C=C double bond and allows mechanistically for the anti-to-syn isomerization process.
[C4O4]6– and [C4O4]2– Systems: Synthesis and Structural Comparisons
While isolated samples of [2-K(C7H8)]2 are resistant to further reduction by the hydride component of Li[sBu3BH] or K[Et3BH], the addition of K[Et3BH] to a mixture of [1-K]2/CO in benzene/THF in situ allows a more reduced product to be obtained. In this case, the reaction leads to evolution of dihydrogen (as judged by 1H NMR spectroscopy) and the formation of the further reduced species K4[{(NON)Al}2(C4O4)(BEt3)2] ([4-K2(BEt3)]2), containing an unprecedented formally hexa-anionic [C4O4]6– chain (Scheme 3 and Figure 7). Mechanistically, the fact that isolated samples of [2-K(C7H8)]2 are not reduced (either chemically or electrochemically) suggests that the formation of [4-K2(BEt3)]2 involves a reduction event which occurs prior to the dimerization of the carbene intermediate I3.
Scheme 3. Synthesis of a Dialuminum System Containing the [C4O4]6– Fragment: Synthesis of K4[{(NON)Al}2(C4O4) (BEt3)2], [4-K2(BEt3)]2, via the Reaction of [1-K]2 with CO in the Presence of the Hydride Source K[Et3BH].

Figure 7.

Molecular structure of [4-K2(BEt3)]2 in the solid state as determined by X-ray crystallography. Hydrogen atoms omitted and selected groups represented in a wireframe format for clarity; thermal ellipsoids drawn at the 50% probability level. Key metrical parameters are listed in Table 1.
[4-K2(BEt3)]2 is extremely reactive, particularly in solution, and has limited solubility in compatible solvents; as such, it can be characterized by 1H NMR spectroscopy and X-ray crystallography only. The centro-symmetric structure revealed crystallographically reveals four K+ cations encapsulated by a combination of O-atom and aryl π system ligation. Two molecules of triethylborane are also incorporated into the molecular framework, bound to the terminal O-atoms of the [C4O4]6– chain. The central C–C distance is consistent with a single bond (1.481(3) Å), and the two terminal CC linkages feature bond lengths (1.377(4) Å) reflective of a C=C bond within an enolate functionality. The C–O distances[(1.382(3) and 1.440(3) Å] are consistent with single bonds. As such, the structural metrics for [4-K2(BEt3)]2 are consistent with a system based around a butadiene-tetraolate skeleton, C(−O)=C(−O)–C(−O)=C(−O).
To allow for a complete structural comparison of the series of systems of the type [C4O4]n− (n = 2, 4, 6) within the same aluminum coordination sphere, we also targeted the corresponding [C4O4]2– (squarate) system. Such systems have been synthesized in the past by homologation of CO using metal centers (or combinations of metal centers) capable of effecting a two-electron reduction.20,44 In our hands, however, dimeric Al(II) systems such as (NON)Al–Al(NON)51 do not react with CO under either thermal or photolytic conditions, presumably reflecting the stronger nature of the metal–metal bond compared, for example, to dimeric Mg(I) reductants.20−22 For structural comparison, we therefore examined alternative synthetic approaches involving the reactions of pre-formed squarate salts M2[C4O4] with the readily available Al(III) iodide precursor (NON)AlI. In the event, (NON)Al(C4O4)Al(NON) (5) is most easily accessed by combining the disilver salt of squaric acid, Ag2[C4O4], with (NON)AlI in Et2O. 5 has been characterized by standard spectroscopic and analytical methods and by X-ray crystallography (Figure 8). Its molecular structure features a planar carbocyclic [C4O4]2– ligand bridging between the two aluminum centers. The carbon-containing fragment closely resembles those reported previously,20,44 featuring C–C and C–O distances of 1.438(2)/1.461(2) and 1.257(2)/1.265(2) Å, respectively, consistent with the presence of a delocalized π system involving all four CO units, albeit with the fourfold symmetry of the free squarate ion being disrupted by chelation to the aluminum centers. The two different Al–O distances [1.933(1) and 1.994(1) Å] reflect a geometry at aluminum, which is between square pyramidal and trigonal bipyramidal (τ = 0.55), with the longer bond length being associated with the axial O-donor (assuming the TBP limit).
Figure 8.

Molecular structure of the dialuminum-supported [C4O4]2– compound {(NON)Al}2(C4O4), 5, in the solid state as determined by X-ray crystallography. Hydrogen atoms omitted and selected groups represented in a wireframe format for clarity; thermal ellipsoids drawn at the 50% probability level. Key metrical parameters are listed in Table 1.
The availability of structural data for 5, [2-K(C7H8)]2, and [4-K2(BEt3)]2 provides a unique opportunity to probe the effects on the carbon/oxygen aggregate of stepwise (formal) reduction of the carbocyclic fragment (Figure 9)—notwithstanding the fact that (experimentally) both chemical oxidation and electrochemical oxidation or reduction of [2-K(C7H8)]2 are not facile. The two π-electron squarate dianions have been described as possessing a “moderate” aromaticity,63 a factor which underpins the prevalence of this planar carbocycle in four-carbon systems derived from two-electron reduction processes of CO.20,44 In the case of 5, DFT calculations imply that an alternative (linear) isomer featuring a chain of four −C(=O)– units (akin to a doubly oxidized form of [2-K(C7H8)]2) is not an energetic minimum, rearranging to give a system featuring a bridging ethynediolate fragment ([OCCO]2–) and two aluminum-bound CO ligands (which even then lies ca. 10.8 kcal mol–1 above the squarate form; see the Supporting Information). The topologically linear butenedione diolate structure associated with the more heavily reduced [C4O4]4– tetra-anion avoids the formation of a Hückel 4π anti-aromatic carbocycle derived from the addition of an additional pair of electrons to the LUMO of the squarate system. An alternative carbocyclic isomer featuring a more pronounced rectangular “squarate” core (akin to the Jahn–Teller distorted structure of the related 4π electron system cyclobutadiene)64 is found to lie significantly higher in energy (by >48 kcal mol–1).
Figure 9.

Comparison of the structures of the [C4O4]n− fragments within 5, [2-K(C7H8)]2, and [4-K2(BEt3)]2: (upper) important resonance structures of [C4O4]2–, [C4O4]4–, and [C4O4]6– fragments; (lower) Key frontier orbitals: HOMO-1 and LUMO of [2-K(C7H8)]2 and HOMO of [4-K2(BEt3)]2.
The HOMO-1 of [2-K(C7H8)]2 [Figure 9 (lower)] corresponds to a π-bonding orbital across the central CC linkage of the [C4O4]4– ligand. The formal addition of a further pair of electrons would then generate the [C4O4]6– hexa-anion, which adopts a butadienetetraolate structure via reduction of the enedione component to the corresponding bis(enolate). The structural changes associated with the CC and CO linkages on transitioning from [C4O4]4– to [C4O4]6– are consistent with the form of the LUMO of [2-K(C7H8)]2 (and the HOMO of [4-K2(BEt3)]2), which features out-of-phase π interactions in the terminal CO and central CC units (Figure 9).
The overall transition from [C4O4]2– to [C4O4]4– to [C4O4]6– is characterized by net bond cleavage (C–C/C–O bonds: 11 to 10 to 9), consistent (at a simplistic level) with successive filling of anti-bonding molecular orbitals. A comparison between the closely related [C4O4]4– and [C4O4]6– fragments in [2-K(C7H8)]2 and [4-K2(BEt3)]2, respectively, reflects the net conversion of C–O bonds to C–C bonds in the presence of an additional reductant. The roles of the [Et3BH]− reagent in bringing about this transformation (acting as a source of both electrons and of the BEt3 fragment bound at C–O–) suggest that it acts as a surrogate for H2 in the Fischer–Tropsch process.2
Conclusions
We have shown here that four- or six-electron reduction of CO can be accomplished by the use of anionic aluminum(I) (“aluminyl”) compounds of the type Mn[(NON)Al]n to give topologically linear or branched C4/C6 chains depending on the reaction conditions. The mechanism for homologation to the [C4O4]4– system proceeds via the rate-limiting formation of the central C=C double bond from two carbene fragments and rationalizes the synthesis of this system from both monomeric (M = Li) and dimeric (M = K) precursors. Initial adduct formation with CO relies on the highly electron-rich nature of the aluminyl reagent, which drives an unusual (primarily Z-type) mode of interaction of the CO molecule with the metal center. The onward formation of [C6O6]4– from [C4O4]4– demonstrates for the first time a homogeneous process which brings about chain branching via complete C–O bond cleavage. A comparison of the linear [C4O4]4– system with the [C4O4]6– congener formed under more reducing conditions reflects the net conversion of C–O bonds to C–C bonds in the presence of an additional reductant.
Experimental Section
Complete details of the synthetic methods and characterizing data, crystallographic information, and details of quantum chemical studies are provided in the Supporting Information. Starting materials NONAlI,51a K2[NONAl]2,51a [Li(OEt2)2] [NONAl],57 and Ag2(squarate)65 were prepared according to literature methods. All other reagents were used as received.
[K(C7H8)]2[{(NON)Al}2(C4O4)], [2-K(C7H8)]2
To a 25 mL reaction bomb were added [1-K]2 (250 mg, 0.34 mmol) and benzene (7.5 mL). The resulting mixture was degassed twice using the freeze–pump–thaw method and sealed under vacuum. The bomb was then warmed to 307 K in an oil bath, and after a few minutes, the headspace was charged with CO (1.5 atm, ca. 1.2 mmol). After sealing the vessel, the mixture was vigorously shaken for 30 s and subsequently allowed to stand undisturbed at 307 K for 16 h. Recrystallization from toluene led to the formation of small orange crystals of [2-K(C7H8)]2·4(C7H8); isolated yield of single crystals: 37 mg, 11%. Anal. Calcd for C98H124Al2K2N4O6 + 1.5 toluene: C, 75.57; H, 7.95; N, 3.25. Found: C, 75.52; H, 8.04; N, 3.22. 1H NMR (400 MHz, THF-d8): δ 7.07–6.95 (m, 6H, Dipp-Ar-CH), 6.42 (d, J = 1.9 Hz, 2H, XA-CH1), 5.62 (d, J = 1.9 Hz, 2H, XA-CH3), 3.30–3.14 (m, 4H, CHMe2), 1.73 (s, 3H, C(CH3)2), 1.56 (s, 3H, C(CH3)2), 1.11 (d, J = 6.8 Hz, 6H, CH(CH3)2), 1.04 (s, 18H, C(CH3)3), 0.86 (d, J = 6.8 Hz, 6H, CH(CH3)2), 0.68 (d, J = 6.8 Hz, 6H, CH(CH3)2), 0.43 (d, J = 6.8 Hz, 6H, CH(CH3)2). 13C NMR (126 MHz, THF-d8): δ 148.2 (Dipp-o-C), 147.8 (CtBu), 147.6 (Dipp-o-C), 146.8 (XA-CN), 144.8 (Dipp-i-C), 142.5 (XA-CO), 133.8 (CCMe2), 131.3 (C(=O)CO), 129.2 (C6H6), 125.4 (Dipp-Ar-CH), 125.2 (Dipp-Ar-CH), 123.6 (Dipp-m-CH), 111.0 (XA-C3H), 105.3 (XA-C1H), 37.9 (CCMe2), 35.5 (CMe3), 32.6 (C(CH3)2), 32.2 (C(CH3)3), 29.1 (CH(CH3)2), 28.5 (CH(CH3)2), 26.3 (CH(CH3)2), 25.5* (CH(CH3)2), 24.7 (CH(CH3)2), 24.5 (CH(CH3)2), 23.8 (C(CH3)2). *Overlapped with solvent signal—located in HSQC and HMBC. Due to quadrupolar broadening, the resonance associated with the Al–C(=O) group was not observed in the range −30 to 300 ppm. The corresponding signal is measured for the related derivative [2-K(12-crown-4)]2 at 296.2 ppm.
[K(12-c-4)]2[{(NON)Al}2(C4O4)], [2-K(12-c-4)]2
(Method 1): To a suspension of [2-K(C7H8)]2 (15 mg, 0.009 mmol) in benzene-d6 (0.5 ml) was added a solution of 12-crown-4 in toluene (0.18 mL, 0.1 M, 0.018 mmol, 2.05 equiv), and the reaction mixture was heated at 358 K for 2 h. Volatiles were removed under vacuum, and the product was obtained as an orange powder. Yield 13 mg, 80% (quantitative by NMR). (Method 2): To a solution of [1-K]2 (0.120 g, 0.163 mmol) in toluene (5 mL) was added a solution of 12-crown-4 in toluene (3.2 mL of a 0.1 M solution, 0.320 mmol). The resulting red solution was stirred at room temperature for 5 min before addition of Fe(CO)5 (22 μL, 0.161 mmol). The resulting dark red solution was stirred overnight. Removal of volatiles yields a yellow oil, which can be dissolved in minimal benzene and stored at room temperature overnight to yield single crystals of [2-K(12-c-4)]2·4(C6H6) suitable for X-ray crystallography. Isolated yield of single crystals ca. 5 mg, 20% (30% by NMR). 1H NMR (600 MHz, THF-d8): δ 7.01 (t, J = 7.0 Hz, 2H, Dipp-p-CH), 6.96 (d, J = 7.0 Hz, 4H, Dipp-m-CH), 6.40 (d, J = 2.0 Hz, 2H, XA-CH1), 5.59 (d, J = 1.9 Hz, 2H, XA-CH3), 3.60 (s, 16H, 12-crown-4), 3.42–3.34 (m, 2H, CHMe2), 3.14 (sept., J = 6.9 Hz, 2H, CHMe2), 1.73 (s, 3H, C(CH3)2), 1.58 (s, 3H, C(CH3)2), 1.24 (d, J = 6.7 Hz, 6H, CH(CH3)2), 1.03 (s, 18H, C(CH3)3), 0.79 (d, J = 6.8 Hz, 6H, CH(CH3)2), 0.77 (d, J = 6.8 Hz, 6H, CH(CH3)2), 0.33 (d, J = 6.8 Hz, 6H, CH(CH3)2). 13C NMR (151 MHz, THF-d8): δ 296.2 (AlCO), 147.7 (Dipp-o-C), 147.5 (Dipp-o-C), 147.4 (CtBu), 146.9 (XA-CN), 145.2 (Dipp-i-C), 142.7 (XA-CO), 134.1 (CCMe2), 130.8 (C(=O)CO), 125.9 (Dipp-m-CH), 125.1 (Dipp-p-CH), 123.1 (Dipp-m-CH), 111.4 (XA-C3H), 105.3 (XA-C1H), 70.5 (OCH2), 37.8 (CMe2), 35.4 (C(CH3)3), 32.2 (C(CH3)3), 32.0 (C(CH3)2), 29.4 (CHMe2), 28.7 (CHMe2), 26.5 (CH(CH3)2), 25.0 (CH(CH3)2), 24.6 (CH(CH3)2), 24.3 (CH(CH3)2), 23.6 (C(CH3)2).
[Li(OEt2)][{(NON)Al}2(C4O4)], [2-Li(OEt2)]2 and [Li(THF)]2[{(NON)Al}2(C4O4)], [2-Li(THF)]2
Ether Adduct
(Method 1) A solution of [1-Li] (15 mg, 0.017 mmol) in C6D6 (0.5 mL) was degassed twice using the freeze–pump–thaw method. The headspace was then charged with CO (2 bar) and briefly shaken. 1H NMR reveals the formation of a mixture of products, including [2-Li(OEt2)]2 in ca. 40% yield. Upon concentration (to ca. 1/3 volume) and standing for 2 days, X-ray quality crystals of [2-Li(OEt2)]2 formed.
(Method 2) [2-K(C7H8)]2 (30 mg, 0.018 mmol) and LiI (5 mg, 2.1 equiv) were dissolved in Et2O (15 mL) and briefly heated to 318 K in a sealed ampoule, leading to a color change from orange to yellow. The mixture was allowed to cool to room temperature, and the resulting cloudy mixture was filtered onto benzene (8 mL). After mixing, the solution was concentrated until it turned turbid (ca. 5 mL). At this point, a sample (0.2 mL) was taken and diluted with C6D6 (0.3 mL), and a 1H NMR spectrum was measured. The resulting spectrum shows the presence of a single species, which has the same spectroscopic signals as those obtained using method 1 in a J. Young’s NMR tube. 1H NMR (400 MHz, C6D6): δ 6.67 (d, J = 2.0 Hz, 2H, XA-CH1), 6.00 (d, J = 2.0 Hz, 2H, XA-CH3), 3.72 (sept, J = 6.9 Hz, 2H), 1.70 (s, 3H, C(CH3)2), 1.61 (s, 3H, C(CH3)2), 1.52 (d, J = 6.9 Hz, 6H, CH(CH3)2), 1.19 (s, 18H, C(CH3)3), 0.36 (d, J = 6.9 Hz, 6H, CH(CH3)2). Samples of [2-Li(OEt2)]2 lose Et2O under continuous vacuum. Dissolution in THF-d8, however, yields spectroscopic signals indistinguishable from samples of [2-Li(THF)]2 prepared as set out below.
THF Adduct
A solution containing [2-K(C7H8)]2 (ca. 20 mg, 0.012 mmol) was prepared as described above, and Li[HBsBu3] (1 mL of a 1 M solution in THF) was added. The mixture was heated at 307 K for 16 h, affording small yellow crystals. Analytically pure samples can be obtained by recrystallization from minimal benzene or THF and washing with THF. Isolated yield of single crystals: 7 mg, 30% over last step. Anal. Calcd for C106H140Al2Li2N4O8 + 1.75 THF: C, 75.73; H, 8.66; N, 3.13. Found: C, 75.13; H, 8.66; N, 3.21. 1H NMR (400 MHz, C6D6): δ 7.25 (m, 6H, Dipp-Ar-CH), 6.70 (d, J = 2.0 Hz, 2H, XA-CH1), 6.05 (d, J = 2.0 Hz, 2H, XA-CH3), 3.77 (sept, J = 6.9 Hz, 2H, CHMe2), 3.70 (s, m, 4H, THF), 3.41 (sept, J = 6.9 Hz, 2H, CHMe2), 1.69 (s, 3H, C(CH3)2), 1.60 (s, 3H, C(CH3)2), 1.58 (d, J = 6.9 Hz, 6H, CH(CH3)2), 1.32 (m, 4H, THF), 1.21 (s, 18H, C(CH3)3), 1.15 (overlapping d, J = 6.9 Hz, 12H, CH(CH3)2), 0.36 (d, J = 6.9 Hz, 6H, CH(CH3)2). 13C NMR (151 MHz, C6D6): δ 147.8 (CtBu), 147.2 (Dipp-o-C), 146.3 (Dipp-o-C), 145.7 (XA-C4), 143.6 (Dipp-i-C), 141.4 (XA-CO), 133.1 (CCMe2), 125.8 (Dipp-p-C and Dipp-m-C), 123.0 (Dipp-m-C), 111.3 (XA-C3H), 106.4 (XA-C1H), 68.6 (THF-CO), 37.0 (CMe2), 35.0 (CMe3), 32.1 (C(CH3)2), 31.9 (C(CH3)3), 29.5 (CHMe2), 28.2 (CHMe2), 26.3 (CH(CH3)2), 25.4 (THF), 24.1 (CH(CH3)2), 23.9 (CH(CH3)2), 23.2 (C(CH3)2), 23.2 (CH(CH3)2). Due to quadrupolar broadening (and overlap with the solvent), the resonances associated with the [C4O4]4– fragment were not observed in the range −30 to 300 ppm. The corresponding signals are measured for the related derivative [2-K(12-crown-4)]2 at 130.8 and 296.2 ppm. 7Li NMR (156 MHz, C6D6) δ 0.61.
K[K(THF)2][{(NON)Al}2(C6O6)], 3
[2-K(C7H8)]2 (25 mg, 0.015 mmol) was suspended in THF-d8 (0.5 mL) in a J. Young’s NMR tube and degassed twice using the freeze–pump–thaw method. The headspace was then charged with CO (2 atm) and heated at 339 K for 4 d. During this time, [2-K(C7H8)]2 dissolved, and a color change from yellow to red to orange was observed. 1H NMR monitoring shows quantitative conversion to a single product. After concentrating the solution to a quarter of its original, small pale orange plates formed at the surface of the solution. Anal. Calcd for C108H140Al2K2N4O10 + 2 THF: C, 72.16; H, 8.14; N, 2.90. Found: C, 72.13; H, 8.06; N, 2.55. 1H NMR (600 MHz, THF-d8): δ 7.43 (t, J = 7.6 Hz, 2H, Dipp-p-CH), 7.32 (t, J = 7.6 Hz, 2H, Dipp-p-CH), 7.25 (dd, J = 7.8, 1.6 Hz, 2H, Dipp-m-CH), 7.16 (dd, J = 7.6, 1.6 Hz, 2H, Dipp-m-CH), 7.09 (dd, J = 7.8, 1.6 Hz, 2H, Dipp-m-CH), 7.00 (dd, J = 7.7, 1.6 Hz, 2H, Dipp-m-CH), 6.49 (d, J = 1.9 Hz, 2H, XA-C1H), 6.42 (d, J = 1.9 Hz, 2H, XA-C1H), 5.36 (d, J = 1.9 Hz, 2H, XA-C3H), 5.34 (d, J = 1.9 Hz, 2H, XA-C3H), 3.46–3.36 (overlapping sept., 4H, CHMe2), 3.32 (sept., J = 7.2 Hz, 2H, CHMe2), 3.16 (sept., J = 6.9 Hz, 2H, CHMe2), 1.75 (s, 3H, C(CH3)2), 1.73 (s, 3H, C(CH3)2), 1.57 (s, 3H, C(CH3)2), 1.54 (s, 3H, C(CH3)2), 1.20 (d, J = 7.0 Hz, 6H, CH(CH3)2), 1.03 (s, 18H, C(CH3)3), 1.00 (s, 18H, C(CH3)3), 0.95 (d, J = 6.9 Hz, 6H, CH(CH3)2), 0.87 (d, J = 6.7 Hz, 6H, CH(CH3)2), 0.83 (d, J = 6.8 Hz, 6H, CH(CH3)2), 0.80 (d, J = 6.7 Hz, 6H, CH(CH3)2), 0.73 (d, J = 6.7 Hz, 6H, CH(CH3)2), 0.68 (d, J = 6.8 Hz, 6H, CH(CH3)2), 0.51 (d, J = 6.8 Hz, 6H, CH(CH3)2). 13C NMR (151 MHz, THF-d8): δ 258.6 (C6O6), 173.5 (C6O6), 169.0 (C6O6), 156.2 (C6O6), 149.4 (Dipp-o-C), 149.1 (Dipp-o-C), 148.0 (Dipp-o-C), 147.6 (CtBu), 147.5 (Dipp-o-C), 147.3 (CtBu), 146.1 (XA-C4), 145.8 (Dipp-i-C), 145.7 (Dipp-i-C), 145.6 (XA-C4), 142.7 (XA-CO), 141.5 (XA-CO), 133.7 (CCMe2), 133.3 (CCMe2), 129.2 (free C6H6), 127.4 (Dipp-p-CH), 127.3 (Dipp-p-CH), 127.2 (Dipp-m-CH), 126.2 (Dipp-m-CH), 124.4 (Dipp-m-CH), 123.8 (Dipp-m-CH), 118.2 (C6O6), 111.3 (XA-C3H), 110.5 (C6O6), 106.5 (XA-C1H), 105.9 (XA-C1H), 37.9 (CMe2), 37.8 (CMe2), 35.5 (CMe3), 35.4 (CMe3), 32.2 (C(CH3)3), 32.1 (C(CH3)3), 32.1 (C(CH3)2), 32.0 (C(CH3)2), 29.2 (CHMe2), 28.8 (CHMe2), 28.1 (CHMe2), 28.0 (CHMe2), 26.2 (CH(CH3)2), 26.1 (CH(CH3)2), 26.0 (CH(CH3)2), 25.3 (CH(CH3)2), 25.2 (CH(CH3)2), 24.9 (CH(CH3)2), 22.9 (C(CH3)2), 22.9 (C(CH3)2).
K4[{(NON)Al}2(C4O4) (BEt3)2], [4-K2(BEt3)]2
To a stirred solution containing [1-K]2 (220 mg, 0.30 mmol), benzene (6.6 mL), and CO (ca. 1.5 bar) in a 25 mL reaction bomb was added K[HBEt3] (1.5 mL of a 1 M solution in THF, 1.5 mmol). After 24 h, the solution was concentrated to a fifth of its original volume under reduced pressure. Prolonged standing led to the formation of colorless extremely sensitive crystals of [4-K2(BEt3)]2. Yield 12 mg, 4%. 1H NMR (400 MHz, THF-d8): δ 7.27 (br s, 6H, Dipp-Ar-CH), 6.36 (br s, 2H, XA-CH1), 5.28 (s, 2H, XA-CH3), 3.79 (br s, 2H, CHMe2), 3.47 (sept, J = 6.6 Hz, 2H, CHMe2), 1.68 (s, 6H, C(CH3)2), 1.28 (br s, 6H, CH(CH3)2), 1.00 (s, 18H, C(CH3)3), 0.80 (d, J = 6.6 Hz, 13H), 0.57 (t, J = 7.7 Hz, 9H, BCH2CH3), 0.06 (q, J = 7.7 Hz, 6H, BCH2CH3). Due to the low solubility and stability of this compound in solution, no satisfactory 11B and 13C spectra could be obtained.
[(NON)Al]2(C4O4), 5
(NON)AlI (500 mg, 0.60 mmol, 2.0 equiv) and Ag2(C4O4) (110 mg, 0.33 mmol) were suspended in Et2O (10 mL), and the reaction mixture was refluxed at 308 K for 48 h in the dark. Volatiles were removed in vacuo, and the residue was extracted into toluene (15 mL), filtered, and layered with hexane (15 mL). Upon standing for several days, crystals of 5 suitable for single-crystal X-ray diffraction were obtained. Yield 180 mg, 39%. Anal. Calcd for C98H124Al2N4O6 + 0.5 hexane: C, 78.21; H, 8.51; N, 3.61. Found 78.08; H, 8.76; N, 3.34. 1H NMR (400 MHz, C6D6): δ 7.20 (m, 2H, Dipp-p-CH), 7.14 (m, 4H, Dipp-p-CH), 6.72 (d, J = 1.9 Hz, 2H, XA-CH1), 5.98 (d, J = 1.9 Hz, 2H, XA-CH3), 3.54 (sept, J = 6.8 Hz, 4H, CHMe2), 1.58 (s, 6H, C(CH3)2), 1.15 (s, 18H, C(CH3)3), 1.06 (d, J = 6.8 Hz, 12H, CH(CH3)2), 0.91–0.83 (br s, 12H, CH(CH3)2). 13C NMR (126 MHz, C6D6): δ 194.0 (C4O4), 148.9 (CtBu), 147.0 (Dipp-o-C), 144.4 (XA-CN), 141.0, 140.8 (Dipp-i-C, XA-CO), 133.3 (CCMe2), 127.0 (Dipp-p-C), 125.1 (Dipp-m-C), 112.1 (XA-C3H), 108.2 (XA-C1H), 37.2 (CMe2), 35.1(CMe3), 31.7 (C(CH3)3), 28.4 (CHMe2), 26.4 (br, C(CH3)2), 25.4 (CH(CH3)2), 24.7(CH(CH3)2).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.2c05228.
Complete details of synthetic procedures and characterizing data; representative spectra; details of quantum chemical calculations; and xyz coordinates for optimized structures (PDF)
Author Present Address
† Research School of Chemistry, Australian National University, Acton, ACT, 2061, Australia
Author Present Address
‡ Matthew Roy, Catalysis Research Center, Technical University of Munich, Ernst-Otto-Fischer Straße 1, 85748, Garching bei München, Germany.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was supported by the Leverhulme Trust (RP-2018-246, studentship to AH; post-doctoral fellowship to JH), NSERC (post-doctoral fellowship to MDDR), and the EPSRC Centre for Doctoral Training in Inorganic Chemistry for Future Manufacturing (OxICFM, EP/S023828/1, studentships to LPG and AC).
The authors declare no competing financial interest.
Supplementary Material
References
- Schulz H. Short history and present trends of Fischer–Tropsch synthesis. Appl. Catal., A 1999, 186, 3–12. 10.1016/S0926-860X(99)00160-X. [DOI] [Google Scholar]
- van Santen R. A.; Ciobîcă I. M.; van Steen E.; Ghouri M. M.. Mechanistic Issues in Fischer–Tropsch Catalysis, 1st ed.; Elsevier Inc.: Amsterdam, 2011; Vol. 54, pp 127–187. [Google Scholar]
- Kaneko T.; Derbyshire F.; Makino E.; Gray D.; Tamura M.; Li K.. Coal Liquefaction. Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH: Weinheim, 2012. [Google Scholar]
- For a recent review, seeKong R. Y.; Crimmin M. R. Cooperative strategies for CO homologation. Dalton Trans. 2020, 49, 16587–16597. 10.1039/D0DT01564D. [DOI] [PubMed] [Google Scholar]
- Manriquez J. M.; McAlister D. R.; Sanner R. D.; Bercaw J. E. Reduction of carbon monoxide promoted by alkyl and hydride derivatives of permethylzirconocene. J. Am. Chem. Soc. 1978, 100, 2716–2724. 10.1021/ja00477a025. [DOI] [Google Scholar]
- Bianconi P. A.; Williams I. D.; Engeler M. P.; Lippard S. J. Reductive coupling of two carbon monoxide ligands to form a coordinated alkyne. J. Am. Chem. Soc. 1986, 108, 311–313. 10.1021/ja00262a030. [DOI] [Google Scholar]
- Bianconi P. A.; Vrtis R. N.; Rao C. P.; Williams I. D.; Engeler M. P.; Lippard S. J. Reductive coupling of carbon monoxide ligands to form coordinated bis(trimethylsiloxy)ethyne in seven-coordinate niobium(I) and tantalum(I) [M(CO)2(dmpe)2Cl] complexes. Organometallics 1987, 6, 1968–1977. 10.1021/om00152a023. [DOI] [Google Scholar]
- Coffin V. L.; Brennen W.; Wayland B. B. Thermodynamic studies of competitive adduct formation: single- and double-insertion reactions of carbon monoxide with rhodium octaethylporphyrin dimer. J. Am. Chem. Soc. 1988, 110, 6063–6069. 10.1021/ja00226a022. [DOI] [PubMed] [Google Scholar]
- Vrtis R. N.; Rao C. P.; Bott S. G.; Lippard S. J. Synthesis and stabilization of tantalum-coordinated dihydroxyacetylene from two reductively coupled carbon monoxide ligands. J. Am. Chem. Soc. 1988, 110, 7564–7566. 10.1021/ja00230a062. [DOI] [Google Scholar]
- Wayland B. B.; Sherry A. E.; Coffin V. L. Selective reductive coupling of carbon monoxide. J. Chem. Soc., Chem. Commun. 1989, 662–663. 10.1039/C39890000662. [DOI] [Google Scholar]
- Protasiewicz J. D.; Lippard S. J. Vanadium-promoted reductive coupling of carbon monoxide and facile hydrogenation to form cis-disiloxyethylenes. J. Am. Chem. Soc. 1991, 113, 6564–6570. 10.1021/ja00017a030. [DOI] [Google Scholar]
- Cummins C. C.; Van Duyne G. D.; Schaller C. P.; Wolczanski P. T. Carbonylation of zirconium complex [tert-Bu3SiNH]3ZrH and x-ray structure study of [tert-Bu3SiNH]3ZrCH3. Organometallics 1991, 10, 164–170. 10.1021/om00047a044. [DOI] [Google Scholar]
- Miller A. J. M.; Labinger J. A.; Bercaw J. E. Reductive coupling of carbon monoxide in a rhenium carbonyl complex with pendant Lewis acids. J. Am. Chem. Soc. 2008, 130, 11874–11875. 10.1021/ja805108z. [DOI] [PubMed] [Google Scholar]
- Watanabe T.; Ishida Y.; Matsuo T.; Kawaguchi H. Reductive coupling of six carbon monoxides by a ditantalum hydride complex. J. Am. Chem. Soc. 2009, 131, 3474–3475. 10.1021/ja9007276. [DOI] [PubMed] [Google Scholar]
- Buss J. A.; Agapie T. Four-electron deoxygenative reductive coupling of carbon monoxide at a single metal site. Nature 2016, 529, 72–75. 10.1038/nature16154. [DOI] [PubMed] [Google Scholar]
- Sharpe H. R.; Geer A. M.; Taylor L. J.; Gridley B. M.; Blundell T. J.; Blake A. J.; Davies E. S.; Lewis W.; McMaster J.; Robinson D.; Kays D. L. Selective reduction and homologation of carbon monoxide by organometallic iron complexes. Nat. Commun. 2018, 9, 3757. 10.1038/s41467-018-06242-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kong R. Y.; Crimmin M. R. Carbon Chain Growth by Sequential Reactions of CO and CO2 with [W(CO)6] and an aluminum(I) Reductant. J. Am. Chem. Soc. 2018, 140, 13614–13617. 10.1021/jacs.8b09761. [DOI] [PubMed] [Google Scholar]; See also; b Kong R. Y.; Batuecas M.; Crimmin M. R. Reactions of aluminium(I) with transition metal carbonyls: scope, mechanism and selectivity of CO homologation. Chem. Sci. 2021, 12, 14845–14854. 10.1039/D1SC04940B. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Batuecas M.; Kong R. Y.; White A. J. P.; Crimmin M. R. Functionalization and Hydrogenation of Carbon Chains Derived from CO**. Angew. Chem., Int. Ed. 2022, 61, e202202241 10.1002/anie.202202241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalrempuia R.; Kefalidis C. E.; Bonyhady S. J.; Schwarze B.; Maron L.; Stasch A.; Jones C. Activation of CO by hydrogenated magnesium(I) dimers: sterically controlled formation of ethenediolate and cyclopropanetriolate complexes. J. Am. Chem. Soc. 2015, 137, 8944–8947. 10.1021/jacs.5b06439. [DOI] [PubMed] [Google Scholar]
- Anker M. D.; Hill M. S.; Lowe J. P.; Mahon M. F. Alkaline-earth-promoted CO homologation and reductive catalysis. Angew. Chem., Int. Ed. 2015, 54, 10009–10011. 10.1002/anie.201505851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuvaraj K.; Douair I.; Paparo A.; Maron L.; Jones C. Reductive trimerization of CO to the deltate dianion using activated magnesium(I) compounds. J. Am. Chem. Soc. 2019, 141, 8764–8768. 10.1021/jacs.9b04085. [DOI] [PubMed] [Google Scholar]
- Yuvaraj K.; Douair I.; Jones D. D. L.; Maron L.; Jones C. Sterically controlled reductive oligomerisations of CO by activated magnesium(I) compounds: deltate vs. ethenediolate formation. Chem. Sci. 2020, 11, 3516–3522. 10.1039/D0SC00836B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paparo A.; Yuvaraj K.; Matthews A. J. R.; Douair I.; Maron L.; Jones C. Reductive hexamerization of CO involving cooperativity between magnesium(I) reductants and [Mo(CO)6]: synthesis of well-defined magnesium benzenehexolate complexes. Angew. Chem., Int. Ed. 2021, 60, 630–634. 10.1002/anie.202009523. [DOI] [PubMed] [Google Scholar]
- Liu H.-Y.; Schwamm R. J.; Neale S. E.; Hill M. S.; McMullin C. L.; Mahon M. F. Reductive dimerization of CO by a Na/Mg(I) Diamide. J. Am. Chem. Soc. 2021, 143, 17851–17856. 10.1021/jacs.1c09467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Zhu Z.; Peng Y.; Lei H.; Fettinger J. C.; Power P. P. Room-temperature reaction of carbon monoxide with a stable diarylgermylene. J. Am. Chem. Soc. 2009, 131, 6912–6913. 10.1021/ja9017286. [DOI] [PubMed] [Google Scholar]
- Braunschweig H.; Dellermann T.; Dewhurst R. D.; Ewing W. C.; Hammond K.; Jimenez-Halla J. O. C.; Kramer T.; Krummenacher I.; Mies J.; Phukan A. K.; Vargas A. Metal-free binding and coupling of carbon monoxide at a boron–boron triple bond. Nat. Commun. 2013, 5, 1025–1028. 10.1038/nchem.1778. [DOI] [PubMed] [Google Scholar]
- Majumdar M.; Omlor I.; Yildiz C. B.; Azizoglu A.; Huch V.; Scheschkewitz D. Reductive cleavage of carbon monoxide by a disilenide. Angew. Chem., Int. Ed. 2015, 54, 8746–8750. 10.1002/anie.201503455. [DOI] [PubMed] [Google Scholar]
- Anker M. D.; Kefalidis C. E.; Yang Y.; Fang J.; Hill M. S.; Mahon M. F.; Maron L. Alkaline earth-centered CO homologation, reduction, and amine carbonylation. J. Am. Chem. Soc. 2017, 139, 10036–10054. 10.1021/jacs.7b04926. [DOI] [PubMed] [Google Scholar]
- Shi X.; Hou C.; Zhou C.; Song Y.; Cheng J. A molecular barium hydrido complex stabilized by a super-bulky hydrotris(pyrazolyl)borate ligand. Angew. Chem., Int. Ed. 2017, 56, 16650–16653. 10.1002/anie.201709344. [DOI] [PubMed] [Google Scholar]
- Protchenko A. V.; Vasko P.; Do D. C. H.; Hicks J.; Fuentes M. Á.; Jones C.; Aldridge S. Reduction of carbon oxides by an acyclic silylene: reductive coupling of CO. Angew. Chem., Int. Ed. 2019, 58, 1808–1812. 10.1002/anie.201812675. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Kostenko A.; Hadlington T. J.; Luecke M.-P.; Yao S.; Driess M. Silicon-mediated selective homo- and heterocoupling of carbon monoxide. J. Am. Chem. Soc. 2019, 141, 626–634. 10.1021/jacs.8b11899. [DOI] [PubMed] [Google Scholar]
- Xu M.; Qu Z.-W.; Grimme S.; Stephan D. W. Lithium Dicyclohexylamide in Transition-Metal-Free Fischer-Tropsch Chemistry. J. Am. Chem. Soc. 2021, 143, 634–638. 10.1021/jacs.0c11482. [DOI] [PubMed] [Google Scholar]
- Yuvaraj K.; Jones C. Reductive coupling of CO with magnesium anthracene complexes: formation of magnesium enediolates. Chem. Commun. 2021, 57, 9224–9227. 10.1039/D1CC03890G. [DOI] [PubMed] [Google Scholar]
- Evans M. J.; Gardiner M. G.; Anker M. D.; Coles M. P. Extending chain growth beyond C1 → C4 in CO homologation: aluminyl promoted formation of the [C5O5]5– ligand. Chem. Commun. 2022, 58, 5833. 10.1039/D2CC01554D. [DOI] [PubMed] [Google Scholar]
- Fagan P. J.; Manriquez J. M.; Marks T. J.; Day V. W.; Vollmer S. H.; Day C. S. Carbon monoxide activation by f-element organometallics. An unusually distorted, carbene-like dihaptoacyl and CO tetramerization. J. Am. Chem. Soc. 1980, 102, 5393–5396. 10.1021/ja00536a048. [DOI] [Google Scholar]
- Fagan P. J.; Moloy K. G.; Marks T. J. Carbon monoxide activation by organoactinides. Migratory carbon monoxide insertion into metal-hydrogen bonds to produce polynuclear formyls. J. Am. Chem. Soc. 1981, 103, 6959–6962. 10.1021/ja00413a032. [DOI] [Google Scholar]
- Evans W. J.; Wayda A. L.; Hunter W. E.; Atwood J. L. Organolanthanoid activation of carbon monoxide: single and multiple insertion of CO into t-butyl lanthanoid bonds; X-ray crystallographic identification of a new bonding mode for a bridging enedione diolate ligand formed by formal coupling of four CO molecules. J. Chem. Soc., Chem. Commun. 1981, 706–708. 10.1039/C39810000706. [DOI] [Google Scholar]
- Evans W. J.; Grate J. W.; Hughes L. A.; Zhang H.; Atwood J. L. Reductive homologation of CO to a ketenecarboxylate by a low-valent organolanthanide complex—synthesis and X-ray crystal-structure of [(C5Me5)4Sm2(O2CCCO)(THF)]2. J. Am. Chem. Soc. 1985, 107, 3728–3730. 10.1021/ja00298a060. [DOI] [Google Scholar]
- Radu N. S.; Engeler M. P.; Gerlach C. P.; Tilley T. D.; Rheingold A. L. Isolation of the first d0 metalloxy ketene complexes via “double insertion” of carbon monoxide into thorium-silicon bonds. J. Am. Chem. Soc. 1995, 117, 3621–3622. 10.1021/ja00117a035. [DOI] [Google Scholar]
- Ferrence G. M.; McDonald R.; Takats J. Stabilization of a discrete lanthanide(II) hydrido complex by a bulky hydridotris(pyrazolyl)borate ligand. Angew. Chem., Int. Ed. 1999, 38, 2233–2237. . [DOI] [PubMed] [Google Scholar]
- Summerscales O. T.; Cloke F. G. N.; Hitchcock P. B.; Green J. C.; Hazari N. Reductive cyclotrimerization of carbon monoxide to the deltate dianion by an organometallic uranium complex. Science 2006, 311, 829–831. 10.1126/science.1121784. [DOI] [PubMed] [Google Scholar]
- Summerscales O. T.; Cloke F. G. N.; Hitchcock P. B.; Green J. C.; Hazari N. Reductive cyclotetramerization of CO to squarate by a U(III) complex: the X-ray crystal structure of [(U(η-C8H6{(SiPr3)-Pr-i-1,4}2)(η-C5Me4H)]2(μ–η2:η2-C4O4). J. Am. Chem. Soc. 2006, 128, 9602–9603. 10.1021/ja063222r. [DOI] [PubMed] [Google Scholar]
- Evans W. J.; Lee D. S.; Ziller J. W.; Kaltsoyannis N. Trivalent [(C5Me5)2(THF)Ln]2(μ–η2:η2-N2) complexes as reducing agents including the reductive homologation of CO to a ketene carboxylate, (μ–η4-O2C–C=C=O)2–. J. Am. Chem. Soc. 2006, 128, 14176–14184. 10.1021/ja0640851. [DOI] [PubMed] [Google Scholar]
- Werkema E. L.; Maron L.; Eisenstein O.; Andersen R. A. Reactions of Monomeric [1,2,4-(Me3C)3C5H2]2CeH and CO with or without H2: An Experimental and Computational Study. J. Am. Chem. Soc. 2007, 129, 2529–2541. 10.1021/ja066482h. [DOI] [PubMed] [Google Scholar]
- Frey A. S.; Cloke F. G. N.; Hitchcock P. B.; Day I. J.; Green J. C.; Aitken G. Mechanistic studies on the reductive cyclooligomerisation of CO by U(III) mixed sandwich complexes; the molecular structure of [U(η-C8H6{SiiPr3-1,4}2)(η-Cp*)]2(μ-η1:η1-C2O2). J. Am. Chem. Soc. 2008, 130, 13816–13817. 10.1021/ja8059792. [DOI] [PubMed] [Google Scholar]
- Arnold P. L.; Turner Z. R.; Bellabarba R. M.; Tooze R. P. Carbon monoxide coupling and functionalisation at a simple uranium coordination complex. Chem. Sci. 2011, 2, 77–79. 10.1039/C0SC00452A. [DOI] [Google Scholar]
- Mansell S. M.; Kaltsoyannis N.; Arnold P. L. Small molecule activation by uranium tris(aryloxides): experimental and computational studies of binding of N2, coupling of CO, and deoxygenation insertion of CO2 under ambient conditions. J. Am. Chem. Soc. 2011, 133, 9036–9051. 10.1021/ja2019492. [DOI] [PubMed] [Google Scholar]
- Gardner B. M.; Stewart J. C.; Davis A. L.; McMaster J.; Lewis W.; Blake A. J.; Liddle S. T. Homologation and functionalization of carbon monoxide by a recyclable uranium complex. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 9265–9270. 10.1073/pnas.1203417109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsoureas N.; Summerscales O. T.; Cloke F. G. N.; Roe S. M. Steric effects in the reductive coupling of CO by mixed-sandwich uranium(III) complexes. Organometallics 2013, 32, 1353. 10.1021/om301045k. [DOI] [Google Scholar]
- Wang B.; Luo G.; Nishiura M.; Luo Y.; Hou Z. Cooperative trimerization of carbon monoxide by lithium and samarium boryls. J. Am. Chem. Soc. 2017, 139, 16967–16973. 10.1021/jacs.7b10108. [DOI] [PubMed] [Google Scholar]
- Simler T.; McCabe K. N.; Maron L.; Nocton G.. CO reductive oligomerization by a divalent thulium complex and CO2-induced functionalization. 2021, ChemRxiv (accessed June 22, 2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hicks J.; Vasko P.; Goicoechea J. M.; Aldridge S. Synthesis, structural and reaction chemistry of a nucleophilic aluminyl anion. Nature 2018, 557, 92–95. 10.1038/s41586-018-0037-y. [DOI] [PubMed] [Google Scholar]; b Hicks J.; Vasko P.; Goicoechea J. M.; Aldridge S. The aluminyl anion: a new generation of aluminium nucleophile. Angew. Chem., Int. Ed. 2021, 60, 1702–1713. 10.1002/anie.202007530. [DOI] [PubMed] [Google Scholar]
- a Schwamm R. J.; Anker M. D.; Lein M.; Coles M. P. Reduction vs. addition: the reaction of an aluminyl anion with 1,3,5,7-cyclooctatetraene. Angew. Chem., Int. Ed. 2019, 58, 1489–1493. 10.1002/anie.201811675. [DOI] [PubMed] [Google Scholar]; b Kurumada S.; Takamori S.; Yamashita M. An alkyl-substituted aluminium anion with strong basicity and nucleophilicity. Nat. Chem. 2020, 12, 36–39. 10.1038/s41557-019-0365-z. [DOI] [PubMed] [Google Scholar]; c Schwamm R. J.; Coles M. P.; Hill M. S.; Mahon M. F.; McMullin C. L.; Rajabi N. A.; Wilson A. S. S. A stable calcium alumanyl. Angew. Chem., Int. Ed. 2020, 59, 3928–3932. 10.1002/anie.201914986. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Koshino K.; Kinjo R. Construction of σ-Aromatic AlB2 Ring via Borane Coupling with a Dicoordinate Cyclic (Alkyl)(Amino)Aluminyl Anion. J. Am. Chem. Soc. 2020, 142, 9057–9062. 10.1021/jacs.0c03179. [DOI] [PubMed] [Google Scholar]; e Grams S.; Eyselein J.; Langer J.; Färber C.; Harder S. Boosting low-valent aluminum(I) reactivity with a potassium reagent. Angew. Chem., Int. Ed. 2020, 59, 15982–15986. 10.1002/anie.202006693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks J.; Vasko P.; Goicoechea J. M.; Aldridge S. Reversible, room-temperature C-C bond activation of benzene by an isolable metal complex. J. Am. Chem. Soc. 2019, 141, 11000–11003. 10.1021/jacs.9b05925. [DOI] [PubMed] [Google Scholar]
- Hicks J.; Vasko P.; Heilmann A.; Goicoechea J. M.; Aldridge S. Arene C-H activation at aluminium(I): meta selectivity driven by the electronics of SNAr chemistry. Angew. Chem., Int. Ed. 2020, 59, 20376–20380. 10.1002/anie.202008557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilmann A.; Hicks J.; Vasko P.; Goicoechea J. M.; Aldridge S. Carbon monoxide activation by a molecular aluminium imide: C-O Bond cleavage and C-C bond formation. Angew. Chem., Int. Ed. 2020, 59, 4897–4901. 10.1002/anie.201916073. [DOI] [PubMed] [Google Scholar]
- Hicks J.; Heilmann A.; Vasko P.; Goicoechea J. M.; Aldridge S. Trapping and reactivity of a molecular aluminium oxide ion. Angew. Chem., Int. Ed. 2019, 58, 17265–17268. 10.1002/anie.201910509. [DOI] [PubMed] [Google Scholar]
- Roy M. M. D.; Hicks J.; Vasko P.; Heilmann A.; Baston A. M.; Goicoechea J. M.; Aldridge S. Probing the extremes of covalency in M–Al bonds: lithium and zinc aluminyl compounds. Angew. Chem., Int. Ed. 2021, 60, 22301–22306. 10.1002/anie.202109416. [DOI] [PubMed] [Google Scholar]
- Anker M. D.; McMullin C. L.; Rajabi N. A.; Coles M. P. Carbon–carbon bond forming reactions promoted by aluminyl and alumoxane anions: introducing the ethenetetraolate ligand. Angew. Chem., Int. Ed. 2020, 59, 12806–12810. 10.1002/anie.202005301. [DOI] [PubMed] [Google Scholar]
- a Ganesamoorthy C.; Schoening J.; Wölper C.; Song L.; Schreiner P. R.; Schulz S. A silicon-carbonyl complex stable at room temperature. Nat. Commun. 2020, 12, 608–614. 10.1038/s41557-020-0456-x. [DOI] [PubMed] [Google Scholar]; b Reiter D.; Holzner R.; Porzelt A.; Frisch P.; Inoue S. Silylated silicon-carbonyl complexes as mimics of ubiquitous transition-metal carbonyls. Nat. Commun. 2020, 12, 1131–1135. 10.1038/s41557-020-00555-4. [DOI] [PubMed] [Google Scholar]; See also; c Fujimori S.; Inoue S. Carbon monoxide in main-group chemistry. J. Am. Chem. Soc. 2022, 144, 2034–2050. 10.1021/jacs.1c13152. [DOI] [PubMed] [Google Scholar]
- Mitoraj M. P.; Michalak A.; Ziegler T.; A combined charge and energy decomposition scheme for bond analysis. J. Chem. Theory Comput. 2009, 5, 962–975. 10.1021/ct800503d. [DOI] [PubMed] [Google Scholar]; The ETS method partitions the interaction energy between two fragments according to: ΔEint = ΔEprep + ΔEorb + ΔEPauli + ΔEelstat. ΔEorb (the orbital interaction term) represents the interactions between the occupied molecular orbitals on one fragment and the unoccupied molecular orbitals of the other fragment, as well as the mixing of occupied and virtual orbitals within the same fragment
- Heitkemper T.; Sindlinger C. P. A cationic NHC-supported borole. Chem.—Eur. J. 2020, 26, 11684–11689. 10.1002/chem.202001916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a previous example of a transformation of CO proceeding through an O-bound adduct, see for exampleBerkefeld A.; Piers W. E.; Parvez M.; Castro L.; Maron L.; Eisenstein O. Carbon monoxide activation via O-Bound CO using decamethylscandocinium–hydridoborate ion pairs. J. Am. Chem. Soc. 2012, 134, 10843–10851. 10.1021/ja300591v. [DOI] [PubMed] [Google Scholar]
- Schleyer P. v. R.; Najafian K.; Kiran B.; Jiao H. Are oxocarbon dianions aromatic?. J. Org. Chem. 2000, 65, 426–431. 10.1021/jo991267n. [DOI] [PubMed] [Google Scholar]
- Kollmar H.; Staemmler V. A theoretical study of the structure of cyclobutadiene. J. Am. Chem. Soc. 1977, 99, 3583–3587. 10.1021/ja00453a009. [DOI] [Google Scholar]
- Hill-Cousins J. T.; Pop I.-A.; Pileio G.; Stevanato G.; Håkansson P.; Roy S. S.; Levitt M. H.; Brown L. J.; Brown R. C. D. Synthesis of an isotopically labeled naphthalene derivative that supports a long-lived nuclear singlet state. Org. Lett. 2015, 17, 2150–2153. 10.1021/acs.orglett.5b00744. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.