

Abstract
Iris species, commonly known as rainbow flowers because of their attractive flowers, are extensively grown in landscape gardens. A few species, including Belamcanda chinensis, the synonym of I. domestica and I. tectorum, are known for their medicinal properties. However, research on the genomes and evolutionary relationships of Iris species is scarce. In the current study, the complete chloroplast (CP) genomes of I. tectorum, I. dichotoma, I. japonica, and I. domestica were sequenced and compared for their identification and relationship. The CP genomes of the four Iris species were circular quadripartite with similar lengths, GC contents, and codon usages. A total of 113 specific genes were annotated, including the ycf1 pseudogene in all species and rps19 in I. japonica alone. All the species had mononucleotide (A/T) simple sequence repeats (SSRs) and long forward and palindromic repeats in their genomes. A comparison of the CP genomes based on mVISTA and nucleotide diversity (Pi) identified three highly variable regions (ndhF-rpl32, rps15-ycf1, and rpl16). Phylogenetic analysis based on the complete CP genomes concluded that I. tectorum is a sister of I. japonica, and the subgenus Pardanthopsis with several I. domestica clustered into one branch is a sister of I. dichotoma. These findings confirm the feasibility of superbarcodes (complete CP genomes) for Iris species authentication and could serve as a resource for further research on Iris phylogeny.
1. Introduction
Iris (L.) is a genus of flowering plants, including 300 species of the Iridaceae family classified into six subgenera (subg.) [1, 2]. These species, commonly called rainbow flowers, are found in the northern hemisphere's temperate regions and are widely used in landscape gardens because of their beautiful and colorful flowers [3]. Most Iris species can adapt to dry environments, such as deserts, semideserts, or rocky habitats, and a few live in mesic and wetland areas [4]. Iris species are also used as medicinal plants. Several pharmacological studies have shown that the rhizome extracts of Iris species have anticancer, anti-inflammatory, and α-glucosidase inhibitory effects and can reduce human infarct volume [5–7]. Few species are used to treat throat-swelling diseases [8]. The dried rhizomes of I. tectorum and I. domestica, referred to as “Chuan She Gan” and “She Gan,” respectively, are used in traditional Chinese medicine, but “She Gan” is often adulterated with the dried rhizomes of I. dichotoma and I. japonica. Therefore, identifying these four species is needed for clinical safety.
Iris species are characterized by fan-shaped leaves, three colorful outer perianth segments, three inner perianth segments, three petaloid stigmas with a bifid crest, and underground tuberous organs [9]. However, these species have similar leaf shapes, flower shapes, and rhizome morphological characteristics. Therefore, identification based on morphological features alone is complicated, especially during the nonflowering period. The development of I. domestica and I. dichotoma hybrids has also made species identification challenging owing to the similarities between the hybrids and female parents [10]. Molecular phylogeny combined with palynology suggested that I. tectorum is far away from I. japonica [11], which is inconsistent with classical taxonomy that shows the two species with a close relationship. I. tectorum is a species of section (sect.) Lophiris of subg. Limniris sect. Lophiris contains 13 species distributed in Eastern Asia; Dykes included this rank in sect. Evansia [12], but this rank was later amended by Lawrence to subsection Evansia [13], by Rodionenko to subg. Crossiris [14], and finally by Mathew to sect. Lophiris of subg. Limniris [2]. Molecular phylogeny placed I. domestica in subg. Pardanthopsis [15–17] with high support rates. Goldblatt and Mabberley confirmed that Belamcanda chinensis is a synonym of Iris domestica based on molecular, karyotype, and type specimen analyses [18]. Furthermore, karyotype analysis of Iris species revealed that their chromosomal genetics are abundant because of their complex origin [10, 19–23]. A few taxa of Iris species were identified using DNA barcodes [24–27]. Wilson [28–30] made considerable progress on molecular identification and phylogeny in Iris species. However, taxonomy of the Iris species still remains complicated [10, 11, 31, 32].
Angiosperms have a circular tetramerous chloroplast (CP) genome, consisting of a pair of inverted repeats (IRs), a small single copy (SSC) region, and a large single copy (LSC) region [33, 34]. The CP genomes serve as promising tools in identifying species and analyzing phylogeny owing to their small and simple structure, conserved sequences, and moderate nucleotide substitution rate [35–37]. Few researchers analyzed the molecular phylogenies of Iris based on CP or nuclear DNA fragments; however, studies based on complete CP genomes are limited. Approximately 20 complete CP genomes about Iris species were documented in NCBI. However, the data need to be enriched to provide detailed information on the phylogeny [26, 38–45].
The current study sequenced the complete CP genomes of I. tectorum, I. dichotoma, I. japonica, and I. domestica. The study's major objectives were to (1) characterize the complete CP genome structure and functional genes, (2) analyze the codon usage, (3) identify the SSRs and long repeats, and (4) compare the whole CP genomes of Iris species to screen highly variable regions. The genomes were further used to uncover the phylogeny relationship among Iris species. The findings will lay a foundation for classifying the species and elucidating the phylogeny in Iridaceae.
2. Materials and Methods
2.1. Sample Collection
Leaves (fresh) from I. tectorum, I. dichotoma, and I. domestica were collected from the Institute of Medicinal Plant Development (IMPLAD), Beijing (40°2′5″N, 116°16′14″E), and those of I. japonica were from the Chengdu University of Traditional Chinese Medicine, Chengdu (30°24′36″N, 103°28′48″E). The leaves were stored in a −80°C freezer, and Professor Yulin Lin identified the species. Voucher specimens were deposited in the herbarium of IMPLAD, the Chinese Academy of Medical Sciences, and the Peking Union Medical College.
2.2. DNA Extraction and Sequencing
Total DNA was extracted from the leaf samples by using the DNeasy Plant Mini Kit (Qiagen Co., Hilden, Germany). DNA quality was detected by agarose gel (1%) electrophoresis. The libraries (insert size average, 350 bp) were generated from total DNA and sequenced on an Illumina NovaSeq 6000 system.
2.3. CP Genome Assembly and Annotation
Filtered reads (low quality) from raw data were generated by Fastp version 0.23.2 [46], and clean data were assembled to generate the CP genome in GetOrganelle version 1.7.5.1 [47]. The genes were annotated using GeSeq version 2.03 [48], followed by manual correction. The genome circular map was drawn by OrganellarGenomeDRAW version 1.3.1 [49]. The whole CP genome sequences of I. japonica (OK448493), I. tectorum (MW201731), I. dichotoma (OK448492), and I. domestica (B. chinensis; OK448491) were submitted to NCBI.
2.4. Genome Structure and Codon Usage Analyses
Furthermore, MEGA X [50] was used to examine the GC content of the genome. CodonW version 1.4.2 was used to calculate the codon usage using the relative synonymous codon usage (RSCU) value as follows: there is no preference in codon usage (RSCU = 1), the codon usage frequency is less than expected (RSCU > 1), and the codon usage frequency is more than expected (RSCU < 1) [51, 52].
2.5. SSR and Long Repeat Sequence Analyses
The SSRs were examined by using the Microsatellite Identification tool version 2.1 [53, 54], with the parameters mentioned by Cui et al. [55]. In addition, the forward (F), palindromic (P), reverse (R), and complement (C) types of long repeat sequences with different sizes in the CP genomes were searched by using REPuter version 3.0 [56] with 30 bp as the minimum repeat size and 3 as the hamming distance.
2.6. Comparative Genome Analysis
The CP genomes from I. tectorum, I. dichotoma, I. japonica, and I. domestica were aligned using the mVISTA program [57]. The sequences of the shared genes in the four Iris species and the complete CP genomes were further aligned using MAFFT version 7 [58]. Nucleotide diversity (Pi) was calculated using DnaSP version 6 [59] to identify the divergence hotspot regions among the four species.
2.7. Phylogenetic Analysis
Twenty-two CP genomes of Iris species were downloaded from NCBI to conduct a phylogenetic tree abided by the maximum likelihood (ML) method in IQ-TREE version 2 with 1000 bootstrap replicates. Sisyrinchium angustifolium (NC_056184) was used as the outgroup (Table S5). The optimum model of nucleotide substitution, TVM+F+R3, determined by ModelFinder [60] in IQ-TREE [61] was used for the ML analysis.
3. Results and Discussion
3.1. CP Genomes of Four Iris Species
Generally, sequences are chosen for molecular taxonomy, and fast (slow) molecular changes correspond to recent (old) evolution time [62]. The structure and components of the genome contribute to the nucleotide substitution rate [63, 64]. The whole CP genome is appropriate to relate species identification and relationship because of its moderate molecular changes [65]. The current study sequenced and analyzed the CP genomes of the four Iris species for their authentication and relationship. Illumina NovaSeq 6000 system sequencing generated 8.5, 5.3, 8.4, and 8.9 Gb of raw data for I. tectorum, I. japonica, I. dichotoma, and I. domestica, respectively. The overall lengths of the complete CP genomes were 152,443–153,736 bp as shown in Table 1. The genomes exhibited a quadripartite structure, including an SSC region (18,150–18,562 bp), an LSC region (82,833–83,237 bp), and a pair of IRs (50,716–52,428 bp; Table 1, Figure 1, and Figures S1–S3). The CP genomes of I. tectorum, I. japonica, I. dichotoma, and I. domestica had GC contents of 37.89%, 37.85%, 37.87%, and 37.85%, respectively (Table 1, Table S1) and were distributed unevenly across the four parts. The GC content illustrated in dark gray in Figure 1 was the highest in the IR region (42.97%–43.05%). This finding is probably due to the rRNA genes (rrn4.5, rrn5, rrn16, and rrn23) with less duplicated AT nucleotides [66, 67]. The LSC (35.97%–36.16%) and SSC (31.40%–31.49%) regions followed IR in terms of GC content; therefore, IR is highly conserved. Moreover, the protein-coding regions (CDS) had lengths of 78,507–79,059 bp and GC contents of 38.02%–38.15% (Table 1). The AT content at the third codon position (69.36%–69.73%) was higher than that at the second (61.75%–61.81%) and first positions (54.42%–54.48%, Table 1). These characteristics of CP genomes are different from those of nuclear and mitochondrial genomes. Moreover, these CP genome characteristics are consistent with earlier reports on I. tectorum [42], I. dichotoma [26], and I. domestica [26, 45]. Thus, the sequencing conducted in the current study has enriched the CP genome data of Iris species and could serve as an essential source for species identification and phylogeny.
Table 1.
Length and composition of the CP genomes of I. tectorum, I. japonica, I. dichotoma, and I. domestica.
| Types/species | I. tectorum | I. japonica | I. dichotoma | I. domestica |
|---|---|---|---|---|
| Accession number | MW201731 | OK448493 | OK448492 | OK448491 |
| Total length (bp) | 153,253 | 152,443 | 153,658 | 153,736 |
| SSC (bp) | 18,562 | 18,490 | 18,150 | 18,168 |
| LSC (bp) | 82,833 | 83,237 | 83,116 | 83,140 |
| IRs (bp) | 51,858 | 50,716 | 52,392 | 52,428 |
| CDS (bp) | 78,957 | 78,507 | 79,050 | 79,059 |
| Total GC (%) | 37.89 | 37.85 | 37.87 | 37.85 |
| GC of SSC (%) | 31.42 | 31.40 | 31.49 | 31.46 |
| GC of LSC (%) | 36.16 | 36.13 | 36.00 | 35.97 |
| GC of IRa (%) | 42.97 | 43.03 | 43.04 | 43.05 |
| GC of IRb (%) | 42.97 | 43.03 | 43.04 | 43.05 |
| GC of CDS (%) | 38.15 | 38.08 | 38.03 | 38.02 |
| AT at the 1st position (%) | 54.42 | 54.48 | 54.43 | 54.44 |
| AT at the 2nd position (%) | 61.77 | 61.81 | 61.75 | 61.77 |
| AT at the 3rd position (%) | 69.36 | 69.46 | 69.73 | 69.72 |
Figure 1.
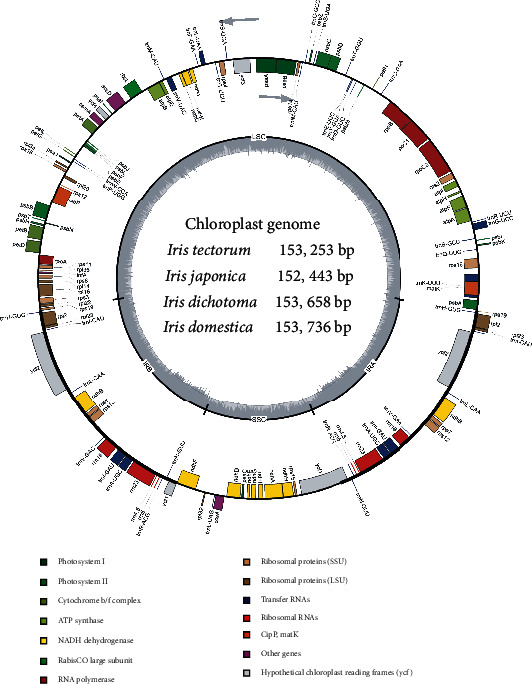
Chloroplast genome map of Iris tectorum. Arrows represent the transcription direction of genes. The dark (GC) and light (AT) gray areas are nucleotide contents.
A total of 113 specific genes were annotated in each CP genome, including 79 CDS genes, 30 tRNA genes, and 4 rRNA genes (Table 2). The pseudogene ycf1 was found in all these species, whereas the pseudogene rps19 was found only in I. japonica. In these species, 19 genes (18 in I. japonica), including 7 (6 in I. japonica) CDS genes, 8 tRNA genes, and 4 rRNA genes, were repeated twice in IRs. Moreover, 15 genes, including 9 CDS and 6 tRNA genes, contained 1 intron, whereas 3 genes contained 2 introns (Table 2). The CDS lengths of I. tectorum, I. japonica, I. dichotoma, and I. domestica were 78,957, 78,507, 79,050, and 79,059 bp, respectively, and accounted for 51.52%, 51.50%, 51.45%, and 51.43% of the genome, respectively. In I. tectorum, the rRNAs were 9,050 bp long (5.91%), and the tRNAs were 2,878 bp long (1.88%). The lengths and proportions of rRNAs and tRNAs in I. japonica, I. dichotoma, and I. domestica are shown in Table S2. In addition, the noncoding regions, including introns, intergenic spacers (IGSs), and pseudogenes, constituted 40.69%, 40.67%, 40.79%, and 40.81% of the CP genomes of I. tectorum, I. japonica, I. dichotoma, and I. domestica, respectively (Tables 1 and 2 and Table S2). These observations revealed the similarities in genomic features among these four species, indicating a close relationship.
Table 2.
Genes in the CP genomes of I. tectorum, I. japonica, I. dichotoma, and I. domestica.
| Functional group | Genes | Number of genes |
|---|---|---|
| Photosystem I | psaA, psaB, psaC, psaI, psaJ | 5 |
| Photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ | 15 |
| Cytochrome b/f complex | petA, petB∗, petD∗, petG, petL, petN | 6 |
| ATP synthase | atpA, atpB, atpE, atpF∗, atpH, atpI | 6 |
| NADH dehydrogenase | ndhA ∗, ndhB∗ (×2), ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK | 12 |
| RubisCO large subunit | rbcL | 1 |
| RNA polymerase | rpoA, rpoB, rpoC1∗, rpoC2 | 4 |
| Ribosomal proteins (SSU) | rps2, rps3, rps4, rps7 (×2), rps8, rps11, rps12∗∗ (×2), rps14, rps15, rps16∗, rps18, rps19Ψ (×2) | 15 |
| Ribosomal proteins (LSU) | rpl2 ∗ (×2), rpl14, rpl16∗, rpl20, rpl22, rpl23 (×2), rpl32, rpl33, rpl36 | 11 |
| Other genes | accD, clpP∗∗, matK, ccsA, cemA, infA | 6 |
| Proteins of unknown function | ycf1Ψ, ycf2 (×2), ycf3∗∗, ycf4 | 6 |
| Transfer RNAs | 38 tRNAs (8 in the IRs (×2), 6 contain one intron) | 38 |
| Ribosomal RNAs | rrn4.5 (×2), rrn5 (×2), rrn16 (×2), rrn23 (×2) | 8 |
×2 indicates two gene copies. ∗ and ∗∗ indicate genes that contain 1 and 2 introns, respectively. Ψ indicates a pseudogene.
3.2. Codon Usage
The CP genomes from I. tectorum, I. japonica, I. dichotoma, and I. domestica comprised 26,319, 26,169, 26,350, and 26,353 amino acid codons, respectively. The analysis of 64 codons encoding 20 amino acids (Figure 2 and Table S3) revealed that six codon types encoded leucine (Leu), serine (Ser), and arginine (Arg); these amino acids had maximum codons. However, one codon type encoded methionine (Met) and tryptophan (Try), and these amino acids had the least number of codons. Leucine was the most frequently coded amino acid (I. tectorum, 2696, 10.24%; I. japonica, 2661, 10.17%; I. dichotoma, 2692, 10.22%; and I. domestica, 2692, 10.22%), whereas cysteine (Cys) was the least coded (I. tectorum, 305, 1.16%; I. japonica, 303, 1.16%; I. dichotoma, 304, 1.15%; and I. domestica, 305, 1.16%).
Figure 2.
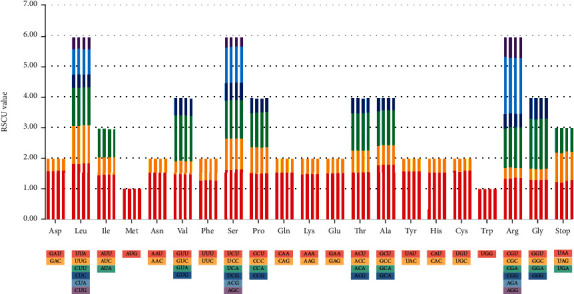
Codon usage of 20 amino acids and stop codons of the CDS in the CP genomes of Iris species. The four histograms from left to right in each amino acid represent I. tectorum, I. japonica, I. dichotoma, and I. domestica.
Furthermore, the RSCU value was measured to determine nonuniform synonymous codon usage [51]. Most codons demonstrated preferences except for AUG (Met) and UGG (Try), which had RSCU values of 1. RSCU analysis revealed the presence of A or U at the third position of the preferred synonymous codons in the four Iris species. Other than the UGA stop codon, the CUA of leucine, and the AUA of isoleucine (Ile), the codons with A or U at the third position had RSCU values greater than 1, indicating the preferential usage of A or U. The RSCU values of the UUA of leucine were 1.84, 1.83, 1.86, and 1.86, in the CP genomes of I. tectorum, I. japonica, I. dichotoma, and I. domestica, respectively. Similarly, the RSCU values of the AGA of arginine (Arg) were 1.88, 1.83, 1.83, and 1.83, and those of the GCU of alanine (Ala) were 1.79, 1.81, 1.81, and 1.81 in I. tectorum, I. japonica, I. dichotoma, and I. domestica, respectively (Table S3). Thus, the preferential codon usage patterns were similar among these four species, which was probably due to the codon usage bias toward A/T. These similarities in codon choice also reveal the related relationship in the four species. The observed codon pattern is consistent with the CP genomes of Amomum [68], Panax [69], Dipterygium and Cleome [70], and various other species [71–73].
3.3. SSR and Long Repeat Sequences
CP SSRs have been used as molecular markers in species authentication, population genetics, and phylogeny analysis owing to their high substitution rates [74–76]. A total of 59, 42, 58, and 56 SSRs were detected in the CP genomes of I. tectorum, I. japonica, I. dichotoma, and I. domestica, respectively (Table 3 and Table S4), including 38, 22, 35, and 33 mononucleotide SSRs; 11, 10, 13, and 12 dinucleotide SSRs; 4, 4, 3, and 3 trinucleotide SSRs; 3, 4, 4, and 4 tetranucleotide SSRs; 3, 1, 2, and 3 pentanucleotide SSRs; and 0, 1, 1, and 1 hexanucleotide SSRs, respectively (Table S4 and Figure 3). The mononucleotide repeats of I. tectorum and I. japonica had no C/G type. All four species had one AACTT/AAGTT pentanucleotide repeat. Additionally, an AAAAT/ATTTT pentanucleotide repeat was present in I. tectorum and I. domestica, whereas none was seen in I. japonica and I. dichotoma. Moreover, I. tectorum, I. dichotoma, and I. domestica had one specific pentanucleotide (AAAAC/GTTTT, ACTAT/AGTAT, and AATAT/ATATT, respectively). The hexanucleotide repeat (AACAAG/CTTGTT) was found in all species except I. tectorum (Table 3). The analysis uncovered that A/T mononucleotide repeats were mostly SSRs and account for 100.0% in I. tectorum and I. japonica, 97.1% in I. dichotoma, and 97.0% in I. domestica. Moreover, A or T base was the most frequent in the SSRs, which is similar to the base preference observed in the CP genomes of Symplocos [77], Achnatherum [78], and other species [79, 80]. These previous studies were all researched between close taxa. Therefore, the SSRs identified in this study might address the relationship among closely related Iris species.
Table 3.
SSRs in the CP genomes of four Iris species.
| SSR types | Repeat units | Number | Proportion (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| ① | ② | ③ | ④ | ① | ② | ③ | ④ | ||
| Mono | A/T | 38 | 22 | 34 | 32 | 100.0 | 100.0 | 97.1 | 97.0 |
| C/G | — | — | 1 | 1 | — | — | 2.9 | 3.0 | |
| Di | AT/AT | 9 | 8 | 11 | 10 | 81.8 | 80.0 | 84.6 | 83.3 |
| AG/CT | 2 | 2 | 2 | 2 | 18.2 | 20.0 | 15.4 | 16.7 | |
| Tri | AAG/CTT | 2 | 2 | 2 | 2 | 50.0 | 50.0 | 66.7 | 66.7 |
| AAT/ATT | 2 | 2 | 1 | 1 | 50.0 | 50.0 | 33.3 | 33.3 | |
| Tetra | AAAT/ATTT | 2 | 3 | 3 | 3 | 66.7 | 75.0 | 75.0 | 75.0 |
| AATG/ATTC | 1 | 1 | 1 | 1 | 33.3 | 25.0 | 25.0 | 25.0 | |
| Penta | AACTT/AAGTT | 1 | 1 | 1 | 1 | 33.3 | 100.0 | 50.0 | 33.3 |
| AAAAT/ATTTT | 1 | — | — | 1 | 33.3 | — | — | 33.3 | |
| AAAAC/GTTTT | 1 | — | — | — | 33.3 | — | — | — | |
| AATAT/ATATT | — | — | — | 1 | — | — | — | 33.3 | |
| ACTAT/AGTAT | — | — | 1 | — | — | — | 50.0 | — | |
| Hexa | AACAAG/CTTGTT | — | 1 | 1 | 1 | — | 100.0 | 100.0 | 100.0 |
①: I. tectorum; ②: I. japonica; ③: I. dichotoma; ④ I. domestica; —: the absence of a particular type.
Figure 3.
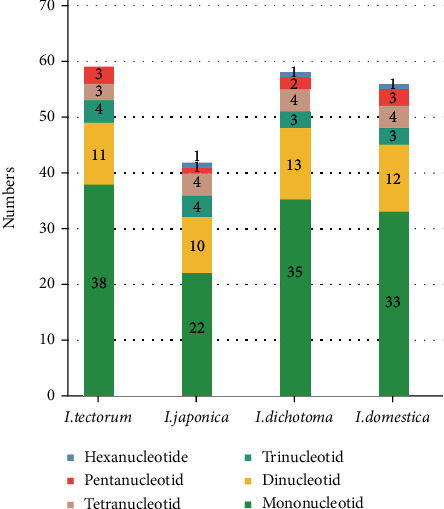
Distribution of the six types of SSRs in the CP genomes of four Iris species.
Long repeat sequences (F, P, R, and C types) are ≥30 bp long sequences and are generally located in the IGS and intron; these repeat sequences are responsible for CP genome rearrangement and genetic diversity in populations and used as sources to uncover phylogeny relationships [81, 82]. The current study analyzed the number of long repeats within Iris species (Figure 4). A total of 38, 34, 43, and 67 long repeats were identified in I. tectorum, I. japonica, I. dichotoma, and I. domestica, respectively. Most of the long repeats were F and P types, accounting for 97.37% in I. tectorum, 100.00% in I. japonica, 88.37% in I. dichotoma, and 77.61% in I. domestica. The 30–39 bp long F and P types were the majority in the Iris species: >50% for I. tectorum, I. japonica, and I. domestica and 44% for I. dichotoma. Moreover, the repeats with ≥70 bp were all F and P types. None of the species had a C repeat, and I. japonica had no R repeat. In addition, I. tectorum, I. dichotoma, and I. domestica had 1, 5, and 15 R types, respectively. The distribution of repeats in the Iris species was similar to that of Camellia [83], Saraca [84], and various other species [85–87]. These repeats, one of the CP genome's various origins, are used in elucidating the phylogeny relationships of Iris species.
Figure 4.
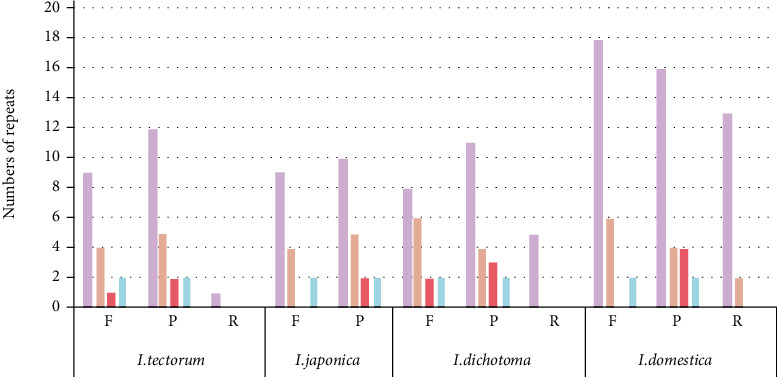
Number of long repeat sequences in the CP genomes of four Iris species. F, P, R, and C indicate the forward, palindromic, reverse, and complement repeats. Histograms in different colors represent repeats with different lengths.
3.4. Inverted Repeat Expansion and Contraction
The comparison of boundaries in the CP genomes from I. tectorum, I. japonica, I. dichotoma, and I. domestica revealed highly conserved LSC/IR/SSC conjunctional regions in the four species; however, variations were detected in the rps19, ndhF, and ycf1 genes (Figure 5). The rps19 gene was located 45, 34, and 45 bp away from the LSC/IRb boundary in I. tectorum, I. dichotoma, and I. domestica, respectively. In I. japonica, the rps19 gene extended into the IRb region (72 bp), creating the rps19 pseudogene in the IRa region. The ndhF gene crossed the SSC/IRb boundary in all species. Moreover, the ycf1 gene was located in the SSC/IRa boundary, resulting in a pseudogene 895 bp long in I. tectorum, 892 bp in I. japonica, and 893 bp in I. domestica and I. dichotoma in the IRb region. These observations suggest that the incomplete duplications at the boundaries probably knocked down the coding potential of the rps19 gene in I. japonica and the ycf1 gene in all four Iris species; these expansions in IR boundaries are consistent with those in Passiflora [88], Lagerstroemia [89], and various other species [90, 91]. Divergence variations due to IR expansion among interspecies will help distinguish closely related Iris species.
Figure 5.
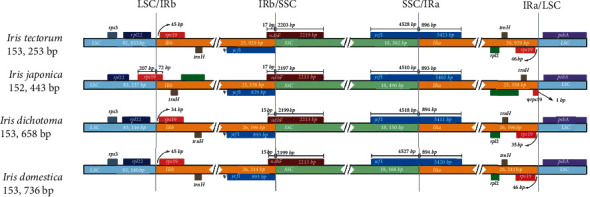
Junctions of the LSC, IR, and SSC regions in the CP genomes of the four Iris species. Numbers above genes indicate the distance between the gene ends and border sites. These features are not to scale. Ψ indicates a pseudogene.
3.5. Identification of Highly Variable Regions
The complete CP genomes of the four Iris species were compared by using the mVISTA [57] program with those available sequences of I. tectorum (MT103435), I. dichotoma (NC_056172), I. domestica (MW039136), I. domestica (NC_050833), and I. domestica (MK593156) downloaded from GenBank. The annotated genome sequence of I. tectorum (MW201731) was used as the reference (Figure 6). I. domestica had the biggest genome (153,736 bp), and I. japonica had the smallest genome (152,443 bp). The reference I. tectorum genome (153,253 bp) was the third in size. The coding regions had less divergence than the noncoding sequence regions owing to the variable regions [92–94]. The IR regions were more conserved, whereas the LSC and SSC regions were more divergent.
Figure 6.
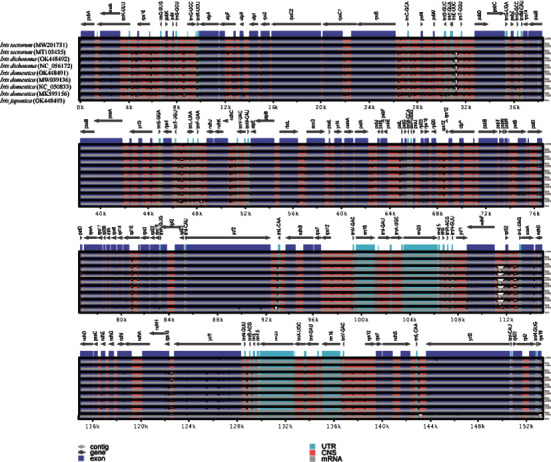
Alignment of the complete CP genomes of nine Iris species with the reference I. tectorum (MW201731) using the mVISTA program. White plots show various regions among species. The genomic regions are color coded. The vertical scale represents the percent identity ranging from 50% to 100%.
Furthermore, the average Pi values [95, 96] were calculated separately for the shared genes and IGS to compare the DNA polymorphisms and identify the highly variable regions (Figure 7). The average Pi value of the gene regions was 0.00733 (Figure 7(a)), and that of the IGSs was 0.01629 (Figure 7(b)). LSC and SSC were higher than the IR regions in Pi values, similar to other plants, such as Handroanthus [97], Speirantha [98], and Combretaceae [99]. Consistent with earlier reports on other species, 13 mutational hotspots and highly divergent loci were examined in the SSC and LSC regions (Pi > 0.03 for IGS and Pi > 0.015 for gene regions), which is helpful for species authentication. The most remarkable divergent loci were trnG-UCC-trnR-UCU (Pi = 0.10078) and rpl16 (Pi = 0.0178) in the IGS and gene regions, respectively. Finally, the combination of the mVISTA plots (divergent regions indicated in white) and the Pi values screened two IGSs, ndhF-rpl32 (Figure 7(b), 11) and rps15-ycf1 (Figure 7(b), 13), and the rpl16 gene (Figure 7(a), 4). These regions with large white plots and high Pi values will serve as potential DNA barcodes for Iris species authentication.
Figure 7.
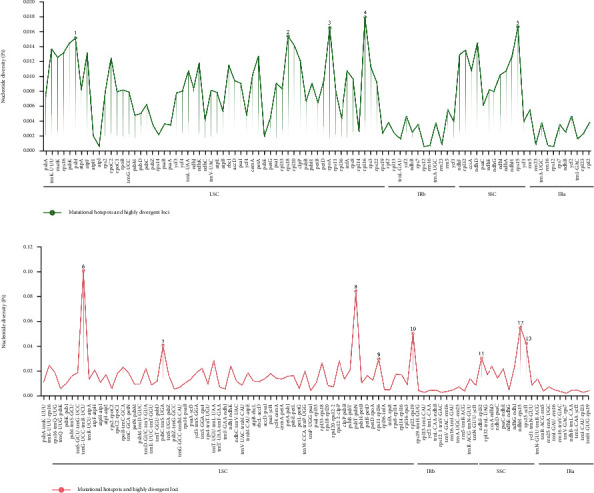
Nucleotide diversity of the gene (a) and intergenic spacer regions (b) on the whole CP genomes of four Iris species (Pi > 0, length > 100 bp).
3.6. Phylogenetic Analysis
CP genomes have been used to determine evolutionary relationships [100–104]. In the present study, a ML tree was constructed using 27 whole CP genome sequences to determine the evolutionary relationships of I. tectorum, I. japonica, I. dichotoma, and I. domestica with S. angustifolium as the outgroup (Figure 8). The phylogenetic analysis revealed the relationships between I. tectorum and I. japonica and between I. domestica and I. dichotoma. Subg. Limniris was divided into two clades: I (sect. Limniris) and IV (sect. Lophiris). Here, sect. Limniris showed a sister relationship with three clades, comprising subg. Pardanthopsis (clade II), subg. Iris (clade III), and sect. Lophiris (clade IV), including I. tectorum and I. japonica. These three monophyletic clades (clades I, II, and IV) were highly supported (bootstrap 100%). Moreover, subg. Pardanthopsis was a sister to subg. Iris, including I. gatesii of sect. Oncocyclus (bootstrap value of 100%); I. domestica and I. dichotoma in clade II were closely related sister species. Additionally, I. domestica (OK448491, B. chinensis) was clustered with the other three I. domestica sequences. This finding was consistent with the findings of Goldblatt and Mabberley [18], Mavrodiev et al. [105], and Wilson [28] who indicated that B. chinensis is a synonym of I. domestica. In addition, two I. dichotoma sequences (previous and present) were clustered into a branch, similar to the two sequences of I. tectorum. These results mutually corroborated the accuracy of the sequences. Notably, the four species were separated into distinct groups. Thus, for the first time, the present study deduced the relationship among the four Iris species based on complete CP genomes following the ML method. These results are consistent with the molecular phylogeny by Wilson [28], Guo and Wilson [11], Kang et al. [26], and Xiao et al. [106] based on different plastid fragments. Thus, the phylogenetic analysis uncovers that the CP genomes could be used to verify the subdivisions of Iris species, especially at the subgenus and section ranks.
Figure 8.
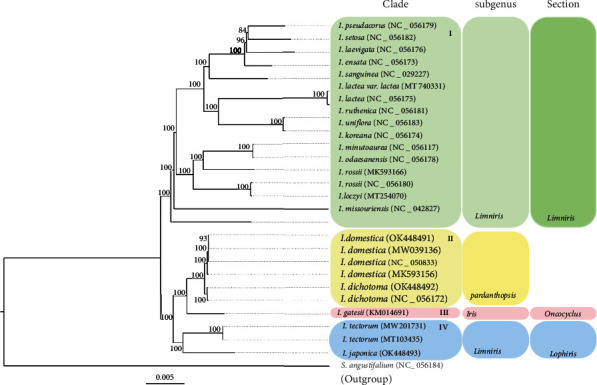
ML tree constructed based on the complete CP genomes of 26 Iris species and S. angustifolium (outgroup). Bootstrap support value is shown at each node.
The ML tree based on common protein-coding sequences (Figure S4) was similar to that based on the complete CP genomes (Figure 8), except for two branches, i.e., branch of I. pseudacorus, I. setosa, I. laevigata, and I. ensata species and branch of I. domestica and I. dichotoma species. In detain, I. ensata, in both trees, was the most primitive taxon among four species, but the I. pseudacorus, I. setosa, and I. laevigata demonstrated different relationships in these two trees. Meanwhile, I. domestica could be distinguished from I. dichotoma in the tree based on the complete chloroplast genomes, but the tree based on common protein-coding sequences could not differentiate I. domestica from I. dichotoma. The complete chloroplast genome has been commonly used as superbarcoding for species identification in researches, such as Dipterygium and Cleome [70] and Zantedeschia [91]. In the present study, the result of species authentication based on complete CP genomes among four medicinal Iris species also proved the efficacy of superbarcoding. The usage of complete CP genomes was more efficient than the usage of common protein-coding sequences for Iris species identification, probably derived from more variant regions contained in intergenic regions of the complete chloroplast genome [98, 104].
4. Conclusions
The present research sequenced and analyzed the complete CP genomes of four Iris species, namely, I. tectorum, I. dichotoma, I. japonica, and I. domestica. CP genome sizes, GC contents, codon usages, SSRs, and long repeats were examined, and the genome conservation and differences among the four Iris species were compared. Furthermore, comparing these species' genomes with other Iridaceae species revealed a few variable regions; however, the use of these markers in DNA barcoding needs to be tested. The study also generated an ML phylogenetic tree that depicted the evolutionary relationship of Iris species and confirmed that B. chinensis is a synonym of I. domestica; however, the whole CP genomes of the 13 taxa of sect. Lophiris need to be included in one robust phylogenetic analysis. The study's findings confirm that CP genomes are a worthy genetic resource for identifying Iridaceae species and analyzing their phylogeny.
Acknowledgments
This work was supported by the National Science & Technology Fundamental Resources Investigation Program of China (grant number 2018FY100700).
Abbreviations
- CP:
Chloroplast
- CDS:
Protein-coding genes
- SSR:
Simple sequence repeat
- Pi:
Nucleotide diversity
- subg:
Subgenera
- sect:
Section
- SSC:
Small single copy
- LSC:
Large single copy
- IR:
Inverted repeat
- NCBI:
National Center for Biotechnology Information
- RSCU:
Relative synonymous codon usage
- ML:
Maximum likelihood
- IGS:
Intergenic spacers.
Contributor Information
Yu-lin Lin, Email: linyulin2022@163.com.
Hui Yao, Email: scauyaoh@sina.com.
Data Availability
The data supporting the study's findings are publicly available in NCBI under the accession numbers MW201731, OK448491, OK448492, and OK448493. The associated data are available in Sequence Read Archive (SRA) under the BioSample, BioProject, and SRA numbers of Iris tectorum (SAMN17169715, PRJNA688136, and SRR13311445), Iris domestica (SAMN25087045, PRJNA798580, and SRR17692213), Iris dichotoma (SAMN25087046, PRJNA798580, and SRR17692212), and Iris japonica (SAMN25087047, PRJNA798580, and SRR17692211). The sequence data are available from https://dataview.ncbi.nlm.nih.gov/object/SRR13311445, https://dataview.ncbi.nlm.nih.gov/object/SRR17692213, https://dataview.ncbi.nlm.nih.gov/object/SRR17692212 and https://dataview.ncbi.nlm.nih.gov/object/SRR17692211. The accession numbers of others used in the present study are shown in Table S5, and these were released from NCBI.
Conflicts of Interest
The authors report no conflict of interest.
Authors' Contributions
Project conception was realized by Yu-lin Lin and Hui Yao. Experiment design and data analysis were conducted by Jing-lu Feng. Plant material collection and identification were done by Bao-li Li and Yu-lin Lin, respectively. Experiment was performed by Jing-lu Feng and Yun-jia Pan. Bioinformatic analysis was carried out by Li-wei Wu and Qing Wang. Manuscript draft was prepared by Jing-lu Feng. All authors approved the manuscript. Jing-lu Feng and Li-wei Wu contributed equally to this work and share the first authorship.
Supplementary Materials
Figure S1: CP genome map of Iris japonica. Figure S2: CP genome map of Iris dichotoma. Figure S3: CP genome map of Iris domestica. Figure S4: ML tree constructed based on common protein-coding genes of 26 Iris species and S. angustifolium (outgroup). Bootstrap support value is shown at each node. Table S1: gene content and gene order in the chloroplast genomes of four Iris species. Table S2: gene catalog of I. tectorum, I. japonica, I. dichotoma, and I. domestica. Table S3: codon usage of four Iris species. Table S4: simple sequence repeats in the complete chloroplast genomes of I. tectorum, I. japonica, I. dichotoma, and I. domestica. Table S5: GenBank accession numbers of the complete chloroplast genome sequences used in the phylogenetic analysis.
References
- 1.Linnaeus C. Species plantarum . London: Ray Society; 1957. [Google Scholar]
- 2.Mathew B. The Iris . London: Batsford; 1989. [Google Scholar]
- 3.B I S S Group. A Guide to Species Irises: Their Identification and Cultivation . Cambridge University Press; 1997. [Google Scholar]
- 4.Goldblatt P., Manning J. C. The Iris Family: Natural History & Classification . Portland: Timber Press; 2008. [Google Scholar]
- 5.Amin A., Wani S. H., Mokhdomi T. A., et al. Investigating the pharmacological potential of Iris kashmiriana in limiting growth of epithelial tumors. Pharmacognosy Journal . 2013;5(4):170–175. doi: 10.1016/j.phcgj.2013.07.003. [DOI] [Google Scholar]
- 6.Mocan A., Zengin G., Mollica A., et al. Biological effects and chemical characterization of Iris schachtii Markgr. extracts: a new source of bioactive constituents. Food and Chemical Toxicology . 2018;112:448–457. doi: 10.1016/j.fct.2017.08.004. [DOI] [PubMed] [Google Scholar]
- 7.Jalsrai A., Reinhold A., Becker A. Ethanol Iris tenuifolia extract reduces brain damage in a mouse model of cerebral ischaemia. Phytotherapy Research . 2018;32(2):333–339. doi: 10.1002/ptr.5981. [DOI] [PubMed] [Google Scholar]
- 8.Committee N. P. Pharmacopoeia of People’s Republic of China . Beijing: China Medical Science Press; 2020. [Google Scholar]
- 9.Wilson C. A., Padiernos J., Sapir Y. The royal irises (Iris subg. Iris sect. Oncocyclus): plastid and low-copy nuclear data contribute to an understanding of their phylogenetic relationships. Taxon . 2016;65(1):35–46. doi: 10.12705/651.3. [DOI] [Google Scholar]
- 10.Lian X., Luo G., Li H., Xu W., Xiao Y., Bi X. Reciprocal difference of interspecific hybridization between three different colours of Iris dichotoma and I. domestica. The Journal of Horticultural Science and Biotechnology . 2016;91(5):483–490. doi: 10.1080/14620316.2016.1173525. [DOI] [Google Scholar]
- 11.Guo J., Wilson C. A. Molecular phylogeny of crested Iris based on five plastid markers (Iridaceae) Systematic Botany . 2013;38(4):987–995. doi: 10.1600/036364413X674724. [DOI] [Google Scholar]
- 12.Dykes W. R. The Genus Iris . New York: Dover Publications; 1974. [Google Scholar]
- 13.Lawrence G. H. M. A reclassification of the genus Iris. Gentes Herbarum . 1953;8:346–371. [Google Scholar]
- 14.Rodionenko G. I. The Genus Iris L: (Questions of Morphology, Biology Evolution and Systematics) Mitchell Beazley; 1987. [Google Scholar]
- 15.Tillie N., Chase M. W., Hall T. Molecular studies in the genus Iris L.: a preliminary study. Annali di Botanica . 2000;58 doi: 10.4462/annbotrm-9068. [DOI] [Google Scholar]
- 16.Wilson C. A. Phylogeny of Iris based on chloroplast matK gene and trnK intron sequence data. Molecular Phylogenetics and Evolution . 2004;33(2):402–412. doi: 10.1016/j.ympev.2004.06.013. [DOI] [PubMed] [Google Scholar]
- 17.Wilson C. A. Patterns in evolution in characters that define Iris subgenera and sections. Aliso: A Journal of Systematic Evolutionary Botany . 2006;22(1):425–433. doi: 10.5642/aliso.20062201.34. [DOI] [Google Scholar]
- 18.Goldblatt P., Mabberley D. J. Belamcanda included in Iris, and the new combination I. domestica (Iridaceae: Irideae) Novon . 2005;15(1):128–132. [Google Scholar]
- 19.Anderson E. The species problem in Iris. Annals of the Missouri Botanical Garden . 1936;23(3):457–509. doi: 10.2307/2394164. [DOI] [Google Scholar]
- 20.Park Y. W., Kim D. M., Hwang Y. J., Lim K. B., Kim H. H. Karyotype analysis of three Korean native Iris species. Horticulture, Environment and Biotechnology . 2006;47(1):51–54. [Google Scholar]
- 21.Maugini E., Maleci L. B. Further investigations on the karyotype of several 40 chromosome dwarf bearded irises (I. chamaeiris Bertol. S.L.) Caryologia . 1974;27(1):117–127. doi: 10.1080/00087114.1974.10796567. [DOI] [Google Scholar]
- 22.Yu X. F., Zhang H. Q., Yuan M., Zhou Y. H. Karyotype studies on ten Iris species (Iridaceae) from Sichuan, China. Caryologia . 2009;62(3):253–260. doi: 10.1080/00087114.2004.10589690. [DOI] [Google Scholar]
- 23.Jozghasemi S., Rabiei V., Soleymani A., Khalighi A. Karyotype analysis of seven Iris species native to Iran. Caryologia . 2016;69(4):351–361. doi: 10.1080/00087114.2016.1239162. [DOI] [Google Scholar]
- 24.Castro D. O., Guacchio D. E., Iorio D. E., et al. Barcoding helps threatened species: the case of Iris marsica (Iridaceae) from the protected areas of the Abruzzo (Central Italy) Plant Biosystems . 2020;154(6):961–972. doi: 10.1080/11263504.2020.1762786. [DOI] [Google Scholar]
- 25.Lu Y., Liu S., Ju B., Wang J. Genetic analysis of the Bletillae Rhizoma and its common adulterants using internal transcribed spacer 2 molecular barcoding. European J Med Plants . 2017;20(2):1–8. doi: 10.9734/EJMP/2017/35791. [DOI] [Google Scholar]
- 26.Kang Y. J., Kim S., Lee J., Won H., Nam G. H., Kwak M. Identification of plastid genomic regions inferring species identity from de novo plastid genome assembly of 14 Korean-native Iris species (Iridaceae) PLoS One . 2020;15(10, article e0241178) doi: 10.1371/journal.pone.0241178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li M., Cao H., But P. P. H., Shaw P. C. Identification of herbal medicinal materials using DNA barcodes. Journal of Systematics and Evolution . 2011;49(3):271–283. doi: 10.1111/j.1759-6831.2011.00132.x. [DOI] [Google Scholar]
- 28.Wilson C. A. Subgeneric classification in Iris re-examined using chloroplast sequence data. Taxon . 2011;60(1):27–35. doi: 10.1002/tax.601004. [DOI] [Google Scholar]
- 29.Wilson C. A. Sectional relationships in the Eurasian bearded Iris (subgen. Iris) based on phylogenetic analyses of sequence data. Systematic Botany . 2017;42(3):392–401. doi: 10.1600/036364417X695970. [DOI] [Google Scholar]
- 30.Wilson C. A. Two new species in Iris series Chinenses (Iridaceae) from south-central China. PhytoKeys . 2020;161:41–60. doi: 10.3897/phytokeys.161.55483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu R., Gao Y., Ruan L., Fan Z., Li C. Variation of flower opening and closing times in hybrids of evening flowering species Iris dichotoma and daytime flowering species Iris domestica. Plant Breeding . 2018;137(6):920–927. doi: 10.1111/pbr.12654. [DOI] [Google Scholar]
- 32.Guo J. Comparative micromorphology and anatomy of crested sepals in Iris (Iridaceae) International Journal of Plant Sciences . 2015;176(7):627–642. doi: 10.1086/682135. [DOI] [Google Scholar]
- 33.Bautista M. A. C., Tao W., Zheng Y., Deng Y., Chen T., Miao S. Chloroplast genome organization and phylogeny of Gynochthodes cochinchinensis (DC.) Razafim. & B. Bremer (Rubiaceae) Mitochondrial DNA Part B . 2021;6(1):261–262. doi: 10.1080/23802359.2020.1862716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Khayi S., Gaboun F., Pirro S., et al. Complete chloroplast genome of Argania spinosa: structural organization and phylogenetic relationships in Sapotaceae. Plants (Basel) . 2020;9(10):p. 1354. doi: 10.3390/plants9101354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bai H. R., Oyebanji O., Zhang R., Yi T. S. Plastid phylogenomic insights into the evolution of subfamily Dialioideae (Leguminosae) Plant Divers . 2021;43(1):27–34. doi: 10.1016/j.pld.2020.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alzahrani D. A., Yaradua S. S., Albokhari E. J., Abba A. Complete chloroplast genome sequence of Barleria prionitis, comparative chloroplast genomics and phylogenetic relationships among Acanthoideae. BMC Genomics . 2020;21(1):p. 393. doi: 10.1186/s12864-020-06798-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Khan A. L., Asaf S., Lubna A. A.-R., Al-Harrasi A. Decoding first complete chloroplast genome of toothbrush tree (Salvadora persica L.): insight into genome evolution, sequence divergence and phylogenetic relationship within Brassicales. BMC Genomics . 2021;22(1):p. 312. doi: 10.1186/s12864-021-07626-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wilson C. A. The complete plastid genome sequence of Iris gatesii (section Oncocyclus), a bearded species from southeastern Turkey. Aliso: A Journal of Systematic Evolutionary Botany . 2014;32(1):47–54. doi: 10.5642/aliso.20143201.03. [DOI] [Google Scholar]
- 39.Lee H. J., Nam G. H., Kim K., Lim C. E., Yeo J. H., Kim S. The complete chloroplast genome sequences of Iris sanguinea donn ex Hornem. Mitochondrial DNA A DNA Mapp Seq Anal . 2017;28(1):15–16. doi: 10.3109/19401736.2015.1106521. [DOI] [PubMed] [Google Scholar]
- 40.Joyce E. M., Crayn D. M., Lam V. K. Y., Gerelle W. K., Graham S. W., Nauheimer L. Evolution of Geosiris (Iridaceae): historical biogeography and plastid-genome evolution in a genus of non-photosynthetic tropical rainforest herbs disjunct across the Indian Ocean. Australian Systematic Botany . 2018;31(6):504–522. doi: 10.1071/SB18028. [DOI] [Google Scholar]
- 41.Choi T. Y., Oh S. H., Jang C. G., Kim H. W., Kim A., Lee S. R. The complete chloroplast genome sequences of the Iris loczyi kanitz (Iridaceae) Mitochondrial DNA B Resour . 2020;5(3):2876–2877. doi: 10.1080/23802359.2020.1790312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu Z., Yu X., Cui P., Tian X. The complete chloroplast genome of Iris tectorum (Iridaceae) Mitochondrial DNA Part B . 2020;5(2):1561–1562. doi: 10.1080/23802359.2020.1742599. [DOI] [Google Scholar]
- 43.Cai X., Zhang B., Wang S., Cheng Y., Wang H. Characterization and phylogenetic analysis of the chloroplast genome of Iris lactea var. chinensis. Mitochondrial DNA B Resour . 2021;6(4):1490–1491. doi: 10.1080/23802359.2020.1847611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Feng J., Pan Y., Lin Y., Yao H. The complete chloroplast genome sequence of Iris tectorum. Mitochondrial DNA B Resour . 2021;6(12):3331–3332. doi: 10.1080/23802359.2021.1895002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li C., Hu S., Ding Y., Bi G., Su C., Xia Z. The complete chloroplast genome of Chinese medicinal herb Belamcanda chinensis (L.) Redouté (Iridaceae) Mitochondrial DNA B Resour . 2021;6(2):331–332. doi: 10.1080/23802359.2020.1866455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen S., Zhou Y., Chen Y., Gu J. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics . 2018;34(17):i884–i890. doi: 10.1093/bioinformatics/bty560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jin J. J., Yu W. B., Yang J. B., et al. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biology . 2020;21(1):p. 241. doi: 10.1186/s13059-020-02154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tillich M., Lehwark P., Pellizzer T., et al. GeSeq - versatile and accurate annotation of organelle genomes. Nucleic Acids Research . 2017;45(1):W6–W11. doi: 10.1093/nar/gkx391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Greiner S., Lehwark P., Bock R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Research . 2019;47(1):W59–W64. doi: 10.1093/nar/gkz238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kumar S., Stecher G., Li M., Knyaz C., Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Molecular Biology and Evolution . 2018;35(6):1547–1549. doi: 10.1093/molbev/msy096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sharp P. M., Li W. H. The codon adaptation index--a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Research . 1987;15(3):1281–1295. doi: 10.1093/nar/15.3.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Butt A. M., Nasrullah I., Qamar R., Tong Y. Evolution of codon usage in Zika virus genomes is host and vector specific. Emerging Microbes & Infections . 2016;5(10, article e107) doi: 10.1038/emi.2016.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Beier S., Thiel T., Münch T., Scholz U., Mascher M. MISA-web: a web server for microsatellite prediction. Bioinformatics . 2017;33(16):2583–2585. doi: 10.1093/bioinformatics/btx198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thiel T., Michalek W., Varshney R. K., Graner A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.) Theoretical and Applied Genetics . 2003;106(3):411–422. doi: 10.1007/s00122-002-1031-0. [DOI] [PubMed] [Google Scholar]
- 55.Cui Y., Nie L., Sun W., et al. Comparative and phylogenetic analyses of ginger (Zingiber officinale) in the family Zingiberaceae based on the complete chloroplast genome. Plants (Basel) . 2019;8(8):p. 283. doi: 10.3390/plants8080283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kurtz S., Choudhuri J. V., Ohlebusch E., Schleiermacher C., Stoye J., Giegerich R. REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Research . 2001;29(22):4633–4642. doi: 10.1093/nar/29.22.4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Frazer K. A., Pachter L., Poliakov A., Rubin E. M., Dubchak I. VISTA: computational tools for comparative genomics. Nucleic Acids Research . 2004;32:W273–W279. doi: 10.1093/nar/gkh458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Katoh K., Rozewicki J., Yamada K. D. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Briefings in Bioinformatics . 2019;20(4):1160–1166. doi: 10.1093/bib/bbx108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rozas J., Ferrer-Mata A., Sánchez-DelBarrio J. C., et al. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Molecular Biology and Evolution . 2017;34(12):3299–3302. doi: 10.1093/molbev/msx248. [DOI] [PubMed] [Google Scholar]
- 60.Kalyaanamoorthy S., Minh B. Q., Wong T. K. F., von Haeseler A., Jermiin L. S. ModelFinder: fast model selection for accurate phylogenetic estimates. Nature Methods . 2017;14(6):587–589. doi: 10.1038/nmeth.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Minh B. Q., Schmidt H. A., Chernomor O., et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Molecular Biology and Evolution . 2020;37(5):1530–1534. doi: 10.1093/molbev/msaa015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Besse P. Molecular Plant Taxonomy . Springer; 2021. [DOI] [Google Scholar]
- 63.Gaut B. S., Muse S. V., Clegg M. T. Relative rates of nucleotide substitution in the chloroplast genome. Molecular Phylogenetics and Evolution . 1993;2(2):89–96. doi: 10.1006/mpev.1993.1009. [DOI] [PubMed] [Google Scholar]
- 64.Golenberg E. M., Clegg M. T., Durbin M. L., Doebley J., Ma D. P. Evolution of a noncoding region of the chloroplast genome. Molecular Phylogenetics and Evolution . 1993;2(1):52–64. doi: 10.1006/mpev.1993.1006. [DOI] [PubMed] [Google Scholar]
- 65.Tan W., Gao H., Zhang H., et al. The complete chloroplast genome of Chinese medicine (Psoralea corylifolia): molecular structures, barcoding and phylogenetic analysis. Plant Gene . 2020;21, article 100216 doi: 10.1016/j.plgene.2019.100216. [DOI] [Google Scholar]
- 66.Wang T., Kuang R. P., Wang X. H., et al. Complete chloroplast genome sequence of Fortunella venosa (Champ. ex Benth.) C.C.Huang (Rutaceae): comparative analysis, phylogenetic relationships, and robust support for its status as an independent species. Forests . 2021;12(8):p. 996. doi: 10.3390/f12080996. [DOI] [Google Scholar]
- 67.Wu L., Nie L., Wang Q., et al. Comparative and phylogenetic analyses of the chloroplast genomes of species of Paeoniaceae. Scientific Reports . 2021;11(1):p. 14643. doi: 10.1038/s41598-021-94137-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang L., Feng C., Cai M. M. C., Chen J. H., Ding P. Complete chloroplast genome sequence of Amomum villosum and comparative analysis with other Zingiberaceae plants. Chinese Herbal Medicines . 2020;12(4):375–383. doi: 10.1016/j.chmed.2020.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kim K. J., Lee H. L. Complete chloroplast genome sequences from Korean ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Research . 2004;11(4):247–261. doi: 10.1093/dnares/11.4.247. [DOI] [PubMed] [Google Scholar]
- 70.Alzahrani D., Albokhari E., Yaradua S., Abba A. Complete chloroplast genome sequences of Dipterygium glaucum and Cleome chrysantha and other Cleomaceae species, comparative analysis and phylogenetic relationships. Saudi Journal of Biological Sciences . 2021;28(4):2476–2490. doi: 10.1016/j.sjbs.2021.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Alzahrani D. A., Albokhari E. J., Yaradua S. S., Abba A. Comparative analysis of chloroplast genomes of four medicinal Capparaceae species: genome structures, phylogenetic relationships and adaptive evolution. Plants (Basel) . 2021;10(6):p. 1229. doi: 10.3390/plants10061229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wanga V. O., Dong X., Oulo M. A., et al. Complete chloroplast genomes of Acanthochlamys bracteata (China) and Xerophyta (Africa) (Velloziaceae): comparative genomics and phylogenomic placement. Frontiers in Plant Science . 2021;12, article 691833 doi: 10.3389/fpls.2021.691833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nguyen H. Q., Nguyen T. N. L., Doan T. N., et al. Complete chloroplast genome of novel Adrinandra megaphylla Hu species: molecular structure, comparative and phylogenetic analysis. Scientific Reports . 2021;11(1):p. 11731. doi: 10.1038/s41598-021-91071-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tiwari K. K., Thakkar N. J., Dharajiya D. T., et al. Genome-wide microsatellites in amaranth: development, characterization, and cross-species transferability. Biotech . 2021;11(9):p. 395. doi: 10.1007/s13205-021-02930-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liu L., Low S. L., Sakaguchi S., et al. Development of nuclear and chloroplast polymorphic microsatellites for Crossostephium chinense (Asteraceae) Molecular Biology Reports . 2021;48(9):6259–6267. doi: 10.1007/s11033-021-06590-9. [DOI] [PubMed] [Google Scholar]
- 76.Leontaritou P., Lamari F. N., Papasotiropoulos V., Iatrou G. Exploration of genetic, morphological and essential oil variation reveals tools for the authentication and breeding of Salvia pomifera subsp. calycina (Sm.) Hayek. Phytochemistry . 2021;191, article 112900 doi: 10.1016/j.phytochem.2021.112900. [DOI] [PubMed] [Google Scholar]
- 77.Kim S. C., Lee J. W., Choi B. K. Seven complete chloroplast genomes from Symplocos: genome organization and comparative analysis. Forests . 2021;12(5):p. 608. doi: 10.3390/f12050608. [DOI] [Google Scholar]
- 78.Wei X., Li X., Chen T., et al. Complete chloroplast genomes of Achnatherum inebrians and comparative analyses with related species from Poaceae. FEBS Open Bio . 2021;11(6):1704–1718. doi: 10.1002/2211-5463.13170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang W., Wang H., Dong J., Zhang T., Xiao H. Comparative chloroplast genomes and phylogenetic analysis of Aquilegia. Applications in Plant Sciences . 2021;9(3, article e11412) doi: 10.1002/aps3.11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Qian S., Zhang Y., Lee S. Y. Comparative analysis of complete chloroplast genome sequences in Edgeworthia (Thymelaeaceae) and new insights into phylogenetic relationships. Frontiers in Genetics . 2021;12, article 643552 doi: 10.3389/fgene.2021.643552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li D. M., Zhu G. F., Xu Y. C., Ye Y. J., Liu J. M. Complete chloroplast genomes of three medicinal Alpinia species: genome organization, comparative analyses and phylogenetic relationships in family Zingiberaceae. Plants (Basel) . 2020;9(2):p. 286. doi: 10.3390/plants9020286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hu G., Wang Y., Wang Y., Zheng S., Dong W., Dong N. New insight into the phylogeny and taxonomy of cultivated and related species of Crataegus in China, based on complete chloroplast genome sequencing. Horticulturae . 2021;7(9):p. 301. doi: 10.3390/horticulturae7090301. [DOI] [Google Scholar]
- 83.Li L., Hu Y., He M., et al. Comparative chloroplast genomes: insights into the evolution of the chloroplast genome of Camellia sinensis and the phylogeny of Camellia. BMC Genomics . 2021;22(1):p. 138. doi: 10.1186/s12864-021-07427-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ali M. A., Pan T. K., Gurung A. B., et al. Plastome of Saraca asoca (Detarioideae, Fabaceae): annotation, comparison among subfamily and molecular typing. Saudi Journal of Biological Sciences . 2021;28(2):1487–1493. doi: 10.1016/j.sjbs.2020.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dong F., Lin Z., Lin J., Ming R., Zhang W. Chloroplast genome of rambutan and comparative analyses in Sapindaceae. Plants (Basel) . 2021;10(2):p. 283. doi: 10.3390/plants10020283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Guo S., Guo L., Zhao W., et al. Complete chloroplast genome sequence and phylogenetic analysis of Paeonia ostii. Molecules . 2018;23(2):p. 246. doi: 10.3390/molecules23020246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Huo Y., Gao L., Liu B., et al. Complete chloroplast genome sequences of four Allium species: comparative and phylogenetic analyses. Scientific Reports . 2019;9(1):p. 12250. doi: 10.1038/s41598-019-48708-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rabah S. O., Shrestha B., Hajrah N. H., et al. Passiflora plastome sequencing reveals widespread genomic rearrangements. Journal of Systematics and Evolution . 2019;57(1):1–14. doi: 10.1111/jse.12425. [DOI] [Google Scholar]
- 89.Zheng G., Wei L., Ma L., Wu Z., Gu C., Chen K. Comparative analyses of chloroplast genomes from 13 Lagerstroemia (Lythraceae) species: identification of highly divergent regions and inference of phylogenetic relationships. Plant Molecular Biology . 2020;102(6):659–676. doi: 10.1007/s11103-020-00972-6. [DOI] [PubMed] [Google Scholar]
- 90.Darshetkar A. M., Maurya S., Lee C., et al. Plastome analysis unveils inverted repeat (IR) expansion and positive selection in sea lavenders (Limonium, Plumbaginaceae, Limonioideae, Limonieae) PhytoKeys . 2021;175:89–107. doi: 10.3897/phytokeys.175.61054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.He S., Yang Y., Li Z., Wang X., Guo Y., Wu H. Comparative analysis of four Zantedeschia chloroplast genomes: expansion and contraction of the IR region, phylogenetic analyses and SSR genetic diversity assessment. PeerJ . 2020;8, article e9132 doi: 10.7717/peerj.9132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang L., Liang J., Sa W., Wang L. Sequencing and comparative analysis of the chloroplast genome of Ribes odoratum provide insights for marker development and phylogenetics in Ribes. Physiology and Molecular Biology of Plants . 2021;27(1):81–92. doi: 10.1007/s12298-021-00932-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Luo C., Li Y., Budhathoki R., et al. Complete chloroplast genomes of Impatiens cyanantha and Impatiens monticola: insights into genome structures, mutational hotspots, comparative and phylogenetic analysis with its congeneric species. PLoS One . 2021;16(4, article e0248182) doi: 10.1371/journal.pone.0248182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gu L., Su T., An M. T., Hu G. X. The complete chloroplast genome of the vulnerable Oreocharis esquirolii (Gesneriaceae): structural features, comparative and phylogenetic analysis. Plants (Basel) . 2020;9(12):p. 1692. doi: 10.3390/plants9121692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Loeuille B., Thode V., Siniscalchi C., Andrade S., Rossi M., Pirani J. R. Extremely low nucleotide diversity among thirty-six new chloroplast genome sequences from Aldama (Heliantheae, Asteraceae) and comparative chloroplast genomics analyses with closely related genera. PeerJ . 2021;9, article e10886 doi: 10.7717/peerj.10886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Choi S. J., Kim Y., Choi C. Chloroplast genome-based hypervariable markers for rapid authentication of six Korean Pyropia species. Diversity . 2019;11(12):p. 220. doi: 10.3390/d11120220. [DOI] [Google Scholar]
- 97.Sobreiro M. B., Vieira L. D., Nunes R., et al. Chloroplast genome assembly of Handroanthus impetiginosus: comparative analysis and molecular evolution in Bignoniaceae. Planta . 2020;252(5):p. 91. doi: 10.1007/s00425-020-03498-9. [DOI] [PubMed] [Google Scholar]
- 98.Raman G., Park S. The complete chloroplast genome sequence of the Speirantha gardenii: comparative and adaptive evolutionary analysis. Agronomy . 2020;10(9):p. 1405. doi: 10.3390/agronomy10091405. [DOI] [Google Scholar]
- 99.Zhang Y., Li H. L., Zhong J. D., Wang Y., Yuan C. C. Chloroplast genome sequences and comparative analyses of Combretaceae mangroves with related species. BioMed Research International . 2020;2020 doi: 10.1155/2020/5867673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cui Y., Chen X., Nie L., et al. Comparison and phylogenetic analysis of chloroplast genomes of three medicinal and edible Amomum species. International Journal of Molecular Sciences . 2019;20(16):p. 4040. doi: 10.3390/ijms20164040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chen X., Cui Y., Nie L., et al. Identification and phylogenetic analysis of the complete chloroplast genomes of three ephedra herbs containing ephedrine. BioMed Research International . 2019;2019 doi: 10.1155/2019/5921725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Schafran P. W., Zimmer E. A., Taylor W. C., Musselman L. J. A whole chloroplast genome phylogeny of diploid species of Isoëtes (Isoëtaceae, Lycopodiophyta) in the southeastern United States. Castanea . 2018;83(2):224–235. doi: 10.2179/17-132. [DOI] [Google Scholar]
- 103.Alanazi K. M., Ali M. A., Kim S. Y., et al. The cp genome characterization of Adenium obesum: gene content, repeat organization and phylogeny. Saudi Journal of Biological Sciences . 2021;28(7):3768–3775. doi: 10.1016/j.sjbs.2021.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhang Y., Wang Z., Guo Y., Chen S., Xu X., Wang R. Complete chloroplast genomes of Leptodermis scabrida complex: comparative genomic analyses and phylogenetic relationships. Gene . 2021;791, article 145715 doi: 10.1016/j.gene.2021.145715. [DOI] [PubMed] [Google Scholar]
- 105.Mavrodiev E. V., Martínez-Azorín M., Dranishnikov P., Crespo M. B. At least 23 genera instead of one: the case of Iris L. s.l. (Iridaceae) PLoS One . 2014;9(8, article e106459) doi: 10.1371/journal.pone.0106459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Xiao Y. E., Yu F. Y., Zhou X. F. A new natural hybrid of Iris (Iridaceae) from Chongqing, China. PhytoKeys . 2021;174:1–12. doi: 10.3897/phytokeys.174.62306. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: CP genome map of Iris japonica. Figure S2: CP genome map of Iris dichotoma. Figure S3: CP genome map of Iris domestica. Figure S4: ML tree constructed based on common protein-coding genes of 26 Iris species and S. angustifolium (outgroup). Bootstrap support value is shown at each node. Table S1: gene content and gene order in the chloroplast genomes of four Iris species. Table S2: gene catalog of I. tectorum, I. japonica, I. dichotoma, and I. domestica. Table S3: codon usage of four Iris species. Table S4: simple sequence repeats in the complete chloroplast genomes of I. tectorum, I. japonica, I. dichotoma, and I. domestica. Table S5: GenBank accession numbers of the complete chloroplast genome sequences used in the phylogenetic analysis.
Data Availability Statement
The data supporting the study's findings are publicly available in NCBI under the accession numbers MW201731, OK448491, OK448492, and OK448493. The associated data are available in Sequence Read Archive (SRA) under the BioSample, BioProject, and SRA numbers of Iris tectorum (SAMN17169715, PRJNA688136, and SRR13311445), Iris domestica (SAMN25087045, PRJNA798580, and SRR17692213), Iris dichotoma (SAMN25087046, PRJNA798580, and SRR17692212), and Iris japonica (SAMN25087047, PRJNA798580, and SRR17692211). The sequence data are available from https://dataview.ncbi.nlm.nih.gov/object/SRR13311445, https://dataview.ncbi.nlm.nih.gov/object/SRR17692213, https://dataview.ncbi.nlm.nih.gov/object/SRR17692212 and https://dataview.ncbi.nlm.nih.gov/object/SRR17692211. The accession numbers of others used in the present study are shown in Table S5, and these were released from NCBI.