

Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) is more than merely a respiratory disease, as it also presents with various neurological symptoms. SARS‐CoV‐2 may infect the central nervous system (CNS) and thus is neurotropic. However, the pathophysiological mechanism of coronavirus disease 2019 (COVID‐19)‐associated neuropathy remains unclear. Many studies have reported that SARS‐CoV‐2 enters the CNS through the hematogenous and neuronal routes, as well as through the main host neurological immune responses and cells involved in these responses. The neurological immune responses to COVID‐19 and potential mechanisms of the extensive neuroinflammation induced by SARS‐CoV‐2 have been investigated. Although CNS infection with SARS‐CoV‐2 was shown to lead to neuronal impairment, certain aspects of this mechanism remain controversial and require further analysis. In this review, we discussed the pathway and mechanisms of SARS‐CoV‐2 invasion in the CNS, and associated clinical manifestations, such as anosmia, headache, and hyposmia. Moreover, the mechanism of neurological damage caused by SARS‐CoV‐2 may provide potential treatment methods for patients presenting with SARS‐CoV‐2‐associated neuropathy.
Keywords: central nervous system, COVID‐19, immune response, neuropathy, SARS‐CoV‐2
1. INTRODUCTION
Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) emerged in 2019 and is the causative agent of the coronavirus disease 2019 (COVID‐19) pandemic. Although this pathogen mainly causes respiratory‐related disease, it is also associated with symptoms involving the central nervous system (CNS) in some cases, including anosmia, headache, nausea, vomiting, and memory impairment. Severe manifestations include delirium and agitation, and delirium can involve seizures caused by specific underlying encephalitis. 1 , 2 Hyperinflammation was also detected in the brains of humans infected with SARS‐CoV‐2. 1 In 2020, Taquet et al. showed that 33.62% of 236,379 (79,471) patients with COVID‐19 had developed neuropathies and mental disorders within six months after initial diagnosis. 3 According to a 2021 cohort study, 82% of 3743 (3069) patients hospitalized with COVID‐19 reported neurological symptoms. 4 These large numbers of nervous system injuries suggest that SARS‐COV‐2 can invade and damage the CNS. In addition, reports of mental health disorders were increased in the general population, and neurological and psychiatric symptoms were exacerbated in patients with chronic diseases, including in those with Alzheimer's disease and Parkinson's disease. However, although both the frequency and severity of most mental disorders worsened during the COVID‐19 pandemic, these manifestations in some other neuropsychiatric diseases (such as Wilson's disease) did not. 5 This review describes the potential pathways by which SARS‐COV‐2 invades the CNS and the pathophysiology of this invasion. Previous studies proposed various infection models for SARS‐CoV‐2 such as olfactory transmucosal and blood–brain barrier (BBB) penetration. 6 However, these models have not been systematically organized and presented. Here, we summarized the different routes of SARS‐CoV‐2 neuroinvasion. Moreover, few systematic reviews have described the pathophysiology of SARS‐CoV‐2. To construct an integrated dependency chain for COVID‐19 neuropathy, we reviewed the mechanisms of viral neuroinfection, immune response of the host, and fate of infected neurons.
2. METHODS
To review the mechanisms of SARS‐CoV‐2 neuroinfection, we performed a comprehensive literature search of PubMed using the following search terms: “severe acute respiratory syndrome coronavirus 2,” “coronavirus disease 2019,” “SARS‐CoV‐2,” “COVID‐19,” “neuropathy,” and “neurological.” All studies published after 2020 were included. A total of 193 papers was identified, some of which were excluded after reading the abstract. Included were only original articles which explored the mechanisms of SARS‐CoV‐2‐associated central and peripheral nervous system impairment and the clinical presentation of nerve impair caused by COVID‐19. Excluded the review, case report, and the articles unrelated to the mechanism of nerve injury. Ultimately, 23 papers were mainly included in our review to conclude the pathway of SARS‐CoV‐2 invasion in the CNS, 26 papers were included in the review to clarify the host immune and inflammatory responses in the CNS after SARA‐CoV‐2 infection, and 10 papers were included to summarize the nerve cell impairment induced by SARA‐CoV‐2.
3. RESULTS
In this article, we have reviewed the pathway of SARS‐CoV‐2 invasion in the CNS, including peripheral nerve pathway and cross‐BBB pathway, and we also summarized the related mechanisms, clinical presentation, and treatment strategies of SARS‐CoV‐2‐associated central and peripheral nervous system impairment (Table 1). In addition, we reviewed the host immune and inflammatory responses in the CNS and nerve cell impairment post‐SARA‐CoV‐2 infection, which provide potential therapeutic strategies for future clinical management.
TABLE 1.
Pathway of SARS‐CoV‐2 Invasion in the CNS
| Routes of SARS‐CoV‐2 neuroinvasion | Object of study | Results (Ref.) | Clinical manifestations (Ref.) | Treatment strategies (Ref.) |
|---|---|---|---|---|
| The olfactory route for SARS‐CoV‐2 to invade the central nervous system | Rhesus monkey model of COVID‐19 |
|
Anosmia 17 , 21 , 26 , 27 | |
| Autopsy material from 33 individuals with COVID‐19 |
|
|||
| 7 patients with olfactory function loss and COVID‐19 infection and 4 healthy controls; Hamster | Olfactory mucosa sampling from patients showing long‐term persistence of COVID‐19‐associated anosmia revealed the presence of virus transcripts and of SARS‐CoV‐2‐infected cells, together with protracted inflammation. SARS‐CoV‐2 persistence and associated inflammation in the olfactory neuroepithelium may account for prolonged or relapsing symptoms of COVID‐19, such as loss of smell 21 | |||
| K18‐hACE2 mice | SARS‐CoV‐2 infected cells within the nasal turbinate, eye, and olfactory bulb, suggesting SARS‐CoV‐2 entry into the brain by this route after intranasal infection. This study indicated that direct infection of CNS cells together with the induced inflammatory response in the brain resulted in the severe disease observed in SARS‐CoV‐2 infected K18‐hACE2 mice 20 | |||
| Golden Syrian hamsters |
|
|||
| Cross‐BBB Pathway of SARS‐CoV‐2 Penetration into the CNS | K18‐hACE2 transgenic mice |
|
Headache; hyposmia; hypogeusia; stroke; seizures; coma; encephalitis 18 , 45 | |
| SARS‐CoV‐2‐infected patients and hamster models |
|
|||
| Pericyte‐like cells | PLC‐containing cortical organoids (PCCOs) represent a new “assembloid” model that supports astrocytic maturation as well as SARS‐CoV‐2 entry and replication in neural tissue 6 | |||
| Human pluripotent stem cell‐derived brain organoids |
|
|||
| Human brain microvascular endothelial cells (BMVEC) and normal human astrocytes (NHA) | SARS‐COV‐2 virus protein can induce endothelial inflammation and change the integrity of the BBB through the ACE2 receptor on primary human brain microvascular endothelial cells, thereby promoting the nerve invasion of SARS‐COV‐2 37 | |||
| Mouse | The spike 1 protein (S1) of SARS‐CoV‐2 may shed from the virus and cause cytotoxicity in mice 42 , 43 |
3.1. Pathway of SARS‐CoV‐2 invasion in the CNS
SARS‐CoV‐2 belongs to lineage B of the β‐coronavirus genus in the family Coronaviridae and contains positive‐sense, single‐strand RNA. 7 Products translated from the SARS‐CoV‐2 genome include nonstructural proteins (NSP), structural proteins (SP), and accessory proteins (AP). The SPs include spike (S), envelope (E), membrane (M), and nucleocapsid (N) proteins that regulate viral assembly, stability, and invasiveness. 8 The S protein consists of a signal peptide and receptor‐binding domain that enable the virus to recognize host cells. Angiotensin‐converting enzyme 2 (ACE2) and transmembrane serine protease 2 (TMPRSS2) are crucial for SARS‐CoV‐2 infection. ACE2 is an integral membrane protein comprised of 805 amino acids and has a bilobate N‐terminal peptidase domain; the tip of each lobe interacts with the receptor‐binding domain of S protein, facilitating SARS‐CoV‐2 entry into the host cell. 7 , 9 , 10 TMPRSS2 is expressed mainly on the membrane surfaces of bronchial epithelial cells and cleaves peptide bonds to promote fusion of the viruses with host cell membranes. 11 , 12 Emerging neurological symptoms in response to SARS‐CoV‐2 infection have raised questions regarding whether SARS‐CoV‐2 infects the CNS and how it reaches the brain. Similar to other coronaviruses, SARS‐CoV‐2 is neuroinvasive, neurotropic, and neurovirulent in animals and humans. 13 For example, two types of CoV have been confirmed as invasive and persistent in the CNS; in an autopsy study, human CoV RNA was detected in the CNS of 48% of infected patients. 14 In addition, supporting evidence from animal studies have shown that SARS‐CoV enters the brain during intranasal infection in mice expressing human ACE2. 12 Compared with other human CoVs, SARS‐CoV‐2 has a higher binding affinity for the ACE2 receptor through the special receptor‐binding domain of the S protein. 15 ACE2 receptors are found in nearly all human organs and the CNS. Both SARS‐CoV and SARS‐CoV‐2 use these receptors to penetrate tissues, trigger immune responses, invade the CNS, and promote neuroinfection. 16 SARS‐CoV‐2 enters the CNS mainly via the peripheral nerve and cross‐BBB pathways.
3.1.1. Peripheral nerve pathway of SARS‐CoV‐2 CNS invasion
The peripheral nerves connect central nerves via interactions between neurotransmitters and receptors at the postsynaptic membranes. Certain viruses such as CoVs reversibly invade the CNS through nerve endings via neuronal active transport. 17 , 18 The olfactory nerve is one of the most common CNS access points for CoVs 18 , 19 , 20 (Figure 1A). Neuronal and other cells combine to form an epithelial layer composed of apical sustentacular cells, Bowman's glands, microvillous and neural stem cells, and olfactory sensory neurons. 19 , 21 Both the ACE2 and TMPRSS2 receptors are expressed in olfactory sustentacular cells and provide a convenient invasion port and replication hub for the virus. Olfactory sensory neurons are bipolar cells that express mature olfactory membrane protein and immature class III β‐tubulin (TuJ1). In autopsied CoV‐infected patients, viral particles were identified in sensory neurons that were positive for olfactory membrane protein and TuJ1. Furthermore, in patients with COVID‐19, S protein immunoreactivity was detected in TuJ1‐positive and olfactory membrane protein‐positive cells, thereby indicating that the virus is present in olfactory neurons. 19 , 21 Moreover, infected TuJ1+ sensory neurons can be engulfed by ionized calcium‐binding adapter molecule 1+ macrophages, which may promote viral metastasis. In addition, clinical symptoms related to smell and taste disturbances suggest that SARS‐CoV‐2 uses the olfactory nerve as a port into the CNS. However, the details of the infection mechanism are unclear.
FIGURE 1.
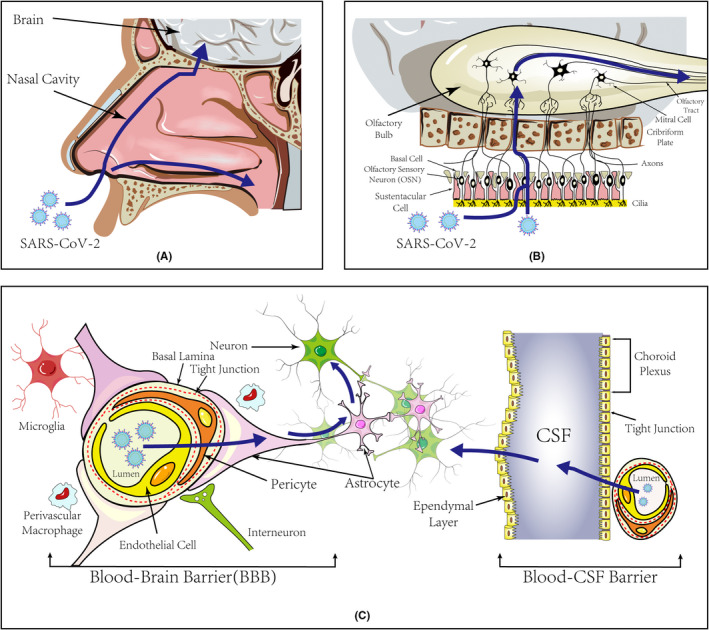
Pathways of SARS‐CoV‐2 Invasion of the CNS. (A) Two main travel pathes after SARS‐CoV‐2 entering the nasal cavity. (1) SARS‐CoV‐2 infects the nasal cavity mucosa and moves upstream to approach brain tissues. (2) The virus travels into the oropharynx with the airway, and then into the trachea. (B) Peripheral nerve pathway of SARS‐CoV‐2 CNS Invasion. Nasal mucosa is composed of apical sustentacular cells, Bowman's glands, microvillous, and olfactory sensory neurons (OSNs). The axons of OSNs ascend and traverse the cribriform plate to reach the olfactory bulb. (1) SARS‐CoV‐2 can damage the sustentacular cells, and indirectly harm olfactory sensory neurons by the ensuing inflammation. SARS‐CoV‐2 can directly infect OSNs as well. (2) With fast axonal transport and trans‐neuron strategy, the virus may move upstream to the olfactory bulb and further brain tissues. (C) Cross‐BBB Pathway of SARS‐CoV‐2 Penetration into the CNS. Given the abundant blood flow beneath the nasal mucosa and pulmonary alveolus, the virus can invade the brain by hematogenous route. Blood brain barrier (BBB) is a highly specialized structure which separates the parenchyma and the plasma. Endothelial cells with tight junctions, and astrocytes, pericytes, microglia, and other cell types collectively form the structure. SARS‐CoV‐2 can infect endothelial cells, pericytes, and astrocytes to enter the CNS. SARS‐CoV‐2 can overactivate glia cells, causing the disruption of tight junctions and an increase in BBB permeability. In addition, infection by SARS‐CoV‐2 damages the choroid plexus epithelium, causes it to leak, facilitates viral penetration into the cerebrospinal fluid (CSF), and damages the brain tissue
After penetrating the olfactory mucosa, SARS‐CoV‐2 may exploit retrograde pathways to disseminate from the olfactory nerves to different regions within the brain, causing symptoms such as headache, consciousness disorder, and seizures. A study demonstrated that human CoV OC43 uses axon transport to spread between neurons. 22 SARS‐CoV‐2 may invade and diffuse in neurons via the same propagation strategy, including endocytosis and exocytosis, to spread within the synaptic cleft and undergo fast axonal transport to move along microtubules toward the neuron cell body. 17 Recent studies have proposed certain retrograde pathways of SARS‐CoV‐2 from the cribriform plate to the brain via the olfactory bulb or vagus or trigeminal nerves. 19 , 21 , 23 After penetrating the olfactory mucosa, the virus induces apoptosis, which may facilitate penetration of other viruses into nerves and the brain. Nevertheless, definitive evidence is needed to support these hypotheses. To confirm this retrograde route, the presence of the virus in specific specimens, such as the pyriform lobe and frontal lobe, should be evaluated (Figure 1B).
Hyposmia has been reported in patients since the onset of the COVID‐19 pandemic. 1 , 24 , 25 As the epidemic spread worldwide, anosmia, a severe form of the disease, was gradually regarded as an isolated (most patient have no nasal obstruction or rhinitis) and major manifestation with a nearly 60% prevalence. 17 , 26 The disease course depends on the site of damage. Studies of animal models showed that cilia (dendrite extensions responsible for olfactory perception) in the olfactory neuroepithelium infected with SARS‐CoV‐2 either decrease significantly or temporarily retract and reduce their protein expression, resulting in a temporary loss of smell. 21 , 27 The infected area may be disorganized, and experimental results indicated that these ciliary alterations were reversible, with the olfactory mucosa becoming ciliated again to recover the perception of smell. These studies suggested the neuro‐pathology leading to hyposmia. However, histological analysis of cilia loss has not been widely performed in human samples; therefore, this pathology requires verification. In addition, a large number of dead olfactory sensory neurons was detected both in animal models and human samples. 21 Nerve cell death is typically irreparable, and thus, the noticeable cell death of olfactory sensory neurons may be the main cause of anosmia occurring through direct infection or indirect induction. Expression of cleaved‐caspase‐3 was widely detected in infected cells, suggesting that SARS‐CoV‐2 can induce cellular apoptosis, whereas positive expression was observed in non‐neuronal and even uninfected cells. 21 It has been proposed that sustentacular cells are the primary targets of the virus, whereas nerve cells can be indirectly harmed by the ensuing inflammation. 27 Thus, this dysfunction may be the synthetic effect of both direct infection and inflammation damage. A combination of the long‐term viral or particle presence plus continual inflammation may be responsible for permanent anosmia. 21
Overall, anosmia is the main clinical symptom during the peripheral nerve pathway of SARS‐CoV‐2 CNS invasion. The evidence‐based treatment for post viral olfactory dysfunction is olfactory training, involving aromatic oils and using dedicated web application to quantify the benefit of olfactory training and visual stimulation, which suggested that a significant improvement in olfaction by olfactory training and visual stimulation assisted by a dedicated web application, especially after 28 days of olfactory training. 28 In addition, numerous studies suggested that neuroinflammatory contribute to olfactory disorders after SARS‐CoV‐2 infection, therefore, targeting neuroinflammation is a potential strategy for improving olfactory dysfunction post‐COVID‐19 infection. Di Stadio et al. demonstrated that the anti‐inflammatory/neuroprotective agents, such as palmitoylethanolamide and luteolin, combined with olfactory training resulted in greater recovery of smell than olfactory training alone. 29
3.1.2. Cross‐BBB pathway of SARS‐CoV‐2 penetration into the CNS
Autopsy studies of patients with acute COVID‐19 show that macrophages, CD8+ T lymphocytes infiltrated in perivascular regions, and widespread microglial activation throughout the brain, 30 suggesting SARS‐CoV‐2 can injure the brain by some ways. Given the abundant blood flow beneath the nasal mucosa and pulmonary alveolus, the virus can invade the brain by hematogenous route. Hence, the cerebrovasculature is a potential pathway for SARS‐CoV‐2 brain invasion. To approach the brain tissue, SARS‐CoV‐2 must cross the BBB separating the parenchyma and plasma (Figure 1C). The BBB is a highly specialized structure consisting chiefly of capillary endothelial cells. Adjacent endothelial cells are closely connected by tight junctions that block harmful substances from entering the brain. Astrocytes, pericytes, microglia, and other cell types along with brain endothelial cells collectively form the neurovascular unit. The end feet of astrocytes form a substantial part of the BBB and extensively cover intracranial blood vessels. 31 CoV HEV67 uses a clathrin‐dependent endocytic/extracellular pathway for neuronal transfer. 32 Mondolfi et al. detected SARS‐CoV‐2 virus‐like particles in brain capillary endothelial cells, pericytes, and astrocytes, suggesting that SARS‐CoV‐2 infects astrocytes and enters the CNS via endocytosis. Wenzel et al. suggested that SARS‐CoV‐2 produces main protease (Mpro) that can ablate the NEMO protein, whose role is to regulate the apoptosis of cerebrovascular endothelial cells in humans. Loss of small‐diameter vessels and patchy hypoxias were also observed in the brains of mice after ablation of NEMO. 33 Wang et al. proposed that pericyte‐like cells containing cortical organoids are SARS‐CoV‐2 infection points. In humans, pericyte‐like cells trigger astrocyte maturation and the production of basement membrane components that can be transformed into a viral replication hub. SARS‐CoV‐2 may actively infect pericyte‐like cells, induce astrocyte death, and mediate an inflammatory type I interferon transcription response. 6 In addition, in infected K18‐hACE2 transgenic mice, SARS‐CoV‐2 RNA was detected in the vascular wall, perivascular space, and brain microvascular endothelial cells. Moreover, the permeability of the infected vessel was increased, and the BBB was disintegrated in infected hamsters. 34
ACE2 is relatively abundant in certain neurovascular components in brain pericytes and is expressed in mature choroid plexus cells. 35 Viral tropism of choroid plexus epithelial cells was observed for a spike pseudovirus of novel CoV and during live virus attack. 36 Thus, infection by novel CoVs damages the choroid plexus epithelium, causes it to leak, facilitates viral penetration into the cerebrospinal fluid, and damages the brain tissue. Reynolds et al. reported that primary human brain microvascular endothelial cells express ACE2 receptors, and that the expression of these receptors was increased by exposure to S protein from SARS‐CoV‐2. These data indicate that SARS‐CoV‐2 proteins can induce endothelial inflammation and alter the integrity of the BBB through the ACE2 receptor on primary human brain microvascular endothelial cells, thereby promoting nerve invasion by SARS‐CoV‐2. In addition, SARS‐CoV‐2 may cross the BBB through an S protein‐ACE2 interaction or non‐specific endocytosis, which may drive neuroinflammatory responses related to neuropathology. 37
Hyperinflammation may facilitate the movement of SARS‐CoV‐2 across the BBB. COVID‐19 can provoke severe peripheral inflammation, thereby liberating copious proinflammatory cytokines that disrupt the BBB and increase its permeability. 38 , 39 In this manner, SARS‐CoV‐2 and cytokines gain access to the brain. After SARS‐CoV‐2 enters the CNS, glial cells are activated by upregulation of proinflammatory cytokines and endothelial cell tight junctions are disintegrated, thereby disrupting the BBB and inducing neuroinflammation and neurological symptoms. 39 Studies have shown that infiltration of inflammatory cells around blood vessels in the brains of infected animals and activation of glial cells may also induce the recruitment of peripherally infected white blood cells by pro‐inflammatory cytokines around endothelial cells to spread between them, similar to a “Trojan horse”. 40 ATP may also participate in BBB disintegration. ATP activates P2X7 receptors expressed on endothelial cells and on certain glial cells that discharge numerous proinflammatory cytokines. Activated P2X7 receptors then mediate NLRP3 inflammasome activation and promote IL‐1β production, which disrupts the BBB. 41
In addition, deciduous SARS‐CoV‐2 particles also affect the CNS. The spike 1 protein (S1) of SARS‐CoV‐2 may be shed from the virus and cause cytotoxicity. Intravenously injected radioiodinated S1 easily traversed the BBB of male mice, was absorbed in the brain, and entered the parenchymal brain space. Radioiodinated S1 crosses the BBB via adsorptive transcytosis. 42 , 43
Generally, headache, hyposmia, and hypogeusia are the most common CNS symptoms of COVID‐19, and most of these symptoms are acute or mild, followed by stroke and seizures. 17 Patients with more severe disease may experience coma, encephalitis, and other conditions. As described above, some diseases, particularly those with long courses, may show persistence of the virus and inflammation. 44 Viral products can be factors or cofactors affecting irreversible cell or tissue deformation. 45 For instance, MPro from SARS‐CoV‐2 can cleave NEMO to affect the neurovascular unit, triggering disruption of the BBB and causing small‐vessel diseases. 33 Immunofluorescence staining confirmed an increase in string vessels (an MPro‐mediated vascular morphology with an empty endothelial basement) in the frontal cortex of patients with COVID‐19. 33 Cleaved NEMOs induce apoptosis and necroptosis of endotheliocytes via receptor interacting protein kinase (RIPK) signaling. 46 This pathological alteration can activate astrocytes and increase BBB permeability. 33 Subsequently, patients with COVID‐19 may develop epileptic seizures. 33 , 47 In addition, SARS‐CoV‐2 protein ORF3a can induce inflammation and cell death. 48 , 49 These factors indicate that viral persistence and the inflammatory response are inextricably linked to the occurrence and course of the disease. To better understand the virus and develop treatment strategies, studies are needed to evaluate the inflammatory process that occurs following viral infection.
3.2. Host immune and inflammatory responses in the CNS
The host immune response may function counter‐currently to viral invasion in some cases. Severe SARS‐CoV‐2 infection may cause inflammatory impairment including epithelial cell apoptosis, vascular damage, pulmonary edema, acute respiratory distress syndrome, and multi‐organ failure. 41 , 50 , 51 These symptoms indicate that SARS‐CoV‐2 can induce excessive inflammatory responses in the lungs. The virus may evoke similar responses in the CNS. Upregulation of the expression of certain soluble cerebral factors has been detected in response to SARS‐CoV‐2 infection. 19 , 21 , 23 , 41 Nevertheless, the mechanism of CNS inflammation in the CNS remains unclear. As brain‐related symptoms occur mainly in cases of severe COVID‐19 presenting with systemic inflammatory effects, SARS‐CoV‐2 pathogenicity may be related to pulmonary inflammation.
3.2.1. Innate immune response in COVID‐19
The pulmonary inflammatory response to SARS‐CoV‐2 occurs in two phases. In the first phase, the virus evades the immune system after recognition. The innate immune system detects molecules expressed on the viral surface known as pathogen‐associated molecular patterns. The innate immune system opposes viral attack by secreting interferons (IFNs) and enhances host defense by releasing chemokines that recruit white blood cells. 52 They initiate strong IFN responses such as the RIG‐I‐MAVS signaling cascade to interfere with viral replication. Cytoplasmic RIG‐I and MDA5 activate various factors and kinases and phosphorylate the “master regulators” IRF3 and IRF7, resulting in the transcription of numerous type I IFNs and interferon‐stimulated genes. 51 , 53 However, in patients and in vitro infection models, COVID‐19 was shown to be associated with low levels of IFNs and moderate interferon‐stimulated genes. 54 , 55 The second phase of the pulmonary inflammatory response to SARS‐CoV‐2 is the “cytokine storm” (CS), which leads to immunosuppression. Overreaction between the innate immune system and virus induce a CS or cytokine release syndrome. 41 , 51 , 56 The latter is characterized by upregulation of the expression of proinflammatory cytokines, chemokines, complement, and other factors. 41 , 57 Yang et al. proposed that monocyte‐derived macrophages are the major sources of proinflammatory cytokines. 58
There has been no evidence of a direct association between SARS‐CoV‐2‐induced pulmonary inflammation and cerebral inflammation. 59 Nevertheless, many cases of severe COVID‐19 present with neurological manifestations. Upregulation of the expression of proinflammatory cytokines in the peripheral blood may disrupt the BBB and increase its permeability. In this condition, the infected brain parenchyma may recruit external innate immune cells to enter the CNS and mediate the inflammatory response. External cytokines can affect the brain by crossing the BBB, enabling other substances to traverse the BBB. 60 Overall, SARS‐CoV‐2‐induced CS may indirectly affect CNS function.
3.2.2. Immune response to SARS‐CoV‐2 in the CNS
SARS‐CoV‐2 may also mediate inflammatory responses by direct neuroinvasion. We propose that P2X7 receptors and NLRP3 inflammasomes in the innate immune system and T‐cells in the acquired immune system are the main triggers of neuroinflammation in COVID‐19.
3.2.2.1. P2X7 receptors in neuroinflammation
The P2X7 receptor is generally expressed on microglia and oligodendrocytes in the CNS. 61 , 62 However, its expression on neurons and astrocytes has been controversial. 61 , 63 Neuroinflammation and glial cell dysfunction are closely related. 64 Injured brain cells release proinflammatory cytokines and ATP. Elevated extracellular ATP levels activate P2X7 receptors which, in turn, induce rapid Na+ and Ca2+ influx and K+ efflux. These cation movements cause various intracellular intermediators including phospholipase C, phospholipase A2, protein kinase C, and mitogen‐activated protein kinase (MAPK) to modulate certain cellular functions such as cell proliferation, cell death, IL secretion, and reactive oxygen species formation. 41 , 65 As described above, the P2X7 receptor can increase the permeability of the BBB, thus allowing more viruses and molecules to enter the CNS and thereby aggravating neuroinflammation.
3.2.2.2. NLRP3 inflammasomes in neuroinflammation
COVID‐19 neuroinflammation is associated with the inflammasome, 41 which is an intracellular innate immune system sensor and receptor comprised of NLRP1, NLRP3, NLRC4, and AIM2. The NLRP3 inflammasome is expressed in the neurons, microglia, astrocytes, and other cells, and plays important roles in the pathogenesis of certain CNS diseases such as Alzheimer's disease and ischemic stroke. 66 , 67
Induction of the NLRP3 inflammasome requires priming and activation signals. Resting microglia do not contain sufficient quantities of the inflammasome. Hence, they require a priming signal to upregulate the expression of its components. 68 , 69 After pathogen‐associated molecular patterns and pattern recognition receptors are combined or cytokines and their associated receptors (TNFR, IL‐1R1, and others) interact, 66 , 69 nuclear factor κB is activated to upregulate the expression of NLRP3, caspase‐1, and pro‐IL‐1β. 41 , 66 , 68 , 70
A study demonstrated that the N protein of SARS‐CoV‐2 is involved in the process of NLRP3 inflammasome induction. 71 First, the N protein of SARS‐CoV‐2 can specifically trigger the expression of NLRP3 protein. Second, the N protein participates in activation and assembly of the NLRP3 inflammasome. The N protein of SARS‐CoV‐2 then notably induces the secretion of IL‐1β and maturation of caspase‐1. Interestingly, NEK7 was not found to be required for NLRP3 inflammasome activation but its expression is induced by the N protein. In addition, several other proteins such as ORF3a and the S protein of SARS‐CoV‐2 are involved in this process. 72 , 73 However, most current research on the inflammasome has focused on pulmonary cells or blood‐derived cells, and whether these processes occur in glial cells is unknown.
Upregulated IL‐1β and IL‐18 are captured by IL‐1R on inactive microglia. This interaction then assembles NLRP3 inflammasomes that release cytokines, and SARS‐CoV‐2 may perpetually stimulate cytokine maturation and secretion. This explosive increase in specific cytokines may greatly alter brain function in a manner resembling that of a pulmonary CS. However, the downstream products of these pathways in COVID‐19 have not been evaluated, and the mechanisms require further investigation.
3.2.2.3. T‐Cells in neuroinflammation
T‐cells may also promote neuroinflammation. Schwabenland et al. observed that pronounced immune activation in the CNS along with apparent neuropathology. They detected substantial CD8+ T‐cell infiltration and an elevated number of parenchymal cells that were positive for ionized calcium‐binding adapter molecule 1 in the CNS of deceased patients with COVID‐19. 74 Twenty‐five patients who had succumbed to COVID‐19 exhibited varying degrees of neuroinflammation that manifested as extensive changes in microglia, considerable CD8 inflammatory cell infiltration, and other severe phenotypes accompanied by microglial nodule formation. Nearly 70% of patients presented with CD8+ T‐cell infiltration and ~40% of patients developed microgliosis and microglial nodules. The expression of numerous immune checkpoint molecules was detected, revealing that programmed death‐1 (PD‐1) was upregulated on SARS‐CoV‐2‐specific CD8+ T‐cells in microglial nodules. 74 These findings suggest that microglia‐T‐cell crosstalk is involved in COVID‐19 neuroinflammation. Infected microglia may activate CD8+ T cells and other immune cells to secrete profound levels of cytokines that increase axonal damage in brain neurons. Damage to neurons may be associated with tissue damage to the brain, thus presenting as various neurological diseases. This process requires immune cells to penetrate the BBB. Although the study revealed increased permeability of the BBB in patients, whether this dysfunction is related to cytotoxic CD8+ T cells or other factors remains unclear.
COVID‐19 neuroinflammation is highly complex and may be caused by simple direct infection or a combination of direct infection and indirect systemic responses. P2X7 stimulates the NLPR3 inflammasome and triggers excessive cytokine release. Patients infected with SARS‐CoV‐2 show upregulation of IL‐6, IL‐10, IL‐1β, TNF‐α, IFN‐γ, CCL2, CXCL10, CCL7, IL‐1 receptor antagonist, and IL‐2 receptor. These factors are thought to be associated with disease severity.
3.3. SARS‐COV‐2‐induced nerve cell impairment
Nerve cell death is irreparable and may result in numerous serious symptoms depending on the site of damage. As we described above, de Melo et al. reported that many neuronal and non‐neuronal cells infected by SARS‐CoV‐2 died in the olfactory mucosae of humans and hamsters. 21 These observations indicate that similar effects occur in brain tissues subjected to SARS‐CoV‐2 neuroinvasion. As this virus has high pathogenicity, it may damage brain neurons in several different ways.
3.3.1. Neuroinflammation‐associated nerve cell impairment
Several neuroinflammatory factors including secreted endogenous molecules and intracellular inflammasomes can mediate neuronal death. Distressed cells may release ATP to activate P2X7 receptors on microglial cells and astrocytes and increase Ca2+ influx and glutamic acid (Glu) release. 41 Glu triggers N‐methyl‐D‐aspartic acid receptors on nerve terminals to release increased amounts of Glu and ATP, thereby forming an auto‐regenerative loop. 41 Glu is an excitatory amino acid that acts as a neurotransmitter and neurotoxin under certain conditions. Increased Ca2+ levels in postsynaptic neurons mediate formation of the CaM (Ca2+‐calmodulin) complex and induce nitric oxide synthase. 41 Nitric oxide augmentation can injure neurons by interacting with iron–sulfur centers and blocking mitochondrial energy transfer. In response to the actions of NADH oxidase and nitric oxide synthase 2, nitric oxide interacts with reactive oxygen species and generates free radicals and nitro compounds such as peroxynitrite (ONOO−) and peroxynitrous acid (HOONO). The latter substances damage DNA, peroxidize lipids, cause other structural changes, and lead to neuronal death. 41
NLRP3 inflammasomes can induce pyroptosis, apoptosis, and necroptosis. Pyroptosis is a type of cell death associated with inflammation and plays important roles in CNS disease development and progression. It is a unique proinflammatory, caspase‐dependent cell death with distinct morphophysiological characteristics. In NLRP3 inflammasome‐mediated pyroptosis, activated caspase‐1 cleaves GSDMD and separates its gasdermin‐N domain, 75 which binds membrane lipids, inositol phosphate, and cardiolipin. 76 These compounds are translocated to the plasma membranes of infected cells where they form gasdermin pores that induce pyroptosis. Abundant endogenous substances are released by pyrolyzed cells, promote inflammation, and strengthen the host immune response. Alzheimer's disease may be associated with neuronal death regulated by NLRP1 inflammasomes and NLRP3 inflammasome‐mediated neuronal pyroptosis. 77 , 78 Hence, the pyroptosis mechanisms associated with Alzheimer's disease may also apply to neuronal damage associated with COVID‐19.
3.3.2. Cerebrovascular hypercoagulopathy‐associated nerve cell impairment
Severe COVID‐19 is characterized by frequent hemostatic abnormalities related to extensive and excessive thrombosis as well as high D‐dimer and ferritin levels within cerebral vessels; these manifestations indicate cerebrovascular hypercoagulopathy. 1 In humans, cerebral blood flow is relatively strong. When this flow is impeded or hindered, cerebral neurons are immediately damaged. Therefore, coagulation is a major factor contributing to ischemic stroke. Five individuals aged 33–49 years presented with large‐vessel stroke after SARS‐CoV‐2 infection. 79 These and other discoveries underscore the importance of studying cerebrovascular disease in COVID‐19.
Several SARS‐CoV‐2‐related factors may induce cerebrovascular hypercoagulopathy. SARS‐CoV‐2 can activate the MAPK pathway. ERK1/2/eIF4E/p38 phosphorylation was upregulated in the platelets of patients with severe COVID‐19. 80 Upregulation of downstream signaling indicates that the MAPK signaling pathway is activated in COVID‐19. 80 MAPK signaling promotes thromboxane biosynthesis which, in turn, is regulated by cytosolic phospholipase A2 activation. 81 Cytosolic phospholipases are highly upregulated in response to COVID‐19. SARS‐CoV‐2 infection upregulated MAPK signaling and increased thromboxane levels, which promoted platelet aggregation. However, how SARS‐CoV‐2 triggers the MAPK pathway requires further study. SARS‐CoV‐2 may also alter the platelet transcriptome. Manne et al. performed RNA‐sequencing to evaluate changes in platelet gene expression and functional responses in patients infected with SARS‐CoV‐2. Pathway analysis identified differentially expressed genes in all pathways related to protein ubiquitination, antigen presentation, and mitochondrial dysfunction except for ACE‐2. The latter is a receptor that binds SARS‐CoV‐2. Compared with healthy donors, patients with COVID‐19 displayed markedly elevated levels of circulating platelet neutrophils, monocytes, and T‐cell aggregates. Moreover, patients with COVID‐19 exhibited faster platelet aggregation and increased fibrinogen and collagen diffusion when compared with healthy individuals. The observed SARS‐CoV‐2‐related increase in platelet activation and aggregation may be attributed in part to MAPK pathway activation and increased thromboxane production. These results indicate that SARS‐CoV‐2 infection is associated with platelet hyperresponsiveness, possibly contributing to the COVID‐19 pathophysiology. 80
Cerebrovascular hypercoagulopathy can lead to perturbations in cerebral blood flow and brain tissue damage. Cerebral ischemia can cause cerebral infarction, neuronal cell death, and ischemic stroke. 82 The pathogenesis of cerebral ischemia is based on a series of complex cascades that occur after long‐term or permanent occlusion of at least one cerebral blood vessel. Traditional ischemic necrosis depends on type I cell death, which is caspase‐dependent apoptosis, resulting in neuronal morphological alterations and apoptotic body formation. Recent research revealed that type II (programmed) cell death is mediated by autophagy. Factors associated with cerebral neuronal autophagy in focal ischemia include upregulation of the expression of autophagy regulator Beclin‐1 and autophagy marker LC3. 83 Overall, cell death associated with cerebrovascular hypercoagulopathy is regulated by complex, systematic cascades. This discovery provides clues to cerebrovascular syndrome pathogenesis in COVID‐19. Nevertheless, it is unknown whether SARS‐CoV‐2 causes vascular disease or COVID‐19‐related cerebrovascular syndrome, which must be evaluated through vascular imaging and pathological analyses.
4. CONCLUSIONS
We summarized the association between SARS‐CoV‐2 and the neurological symptoms of COVID‐19. The olfactory mucosa is a putative port for SARS‐CoV‐2 neuroinvasion. In this pathway, specific components within the olfactory epithelium are infected, and certain retrograde routes for transsynaptic dissemination are opened. SARS‐CoV‐2 may also enter the CNS by crossing the BBB or blood‐cerebrospinal fluid barrier. Certain cytokines mediated by SARS‐CoV‐2 can disrupt BBB integrity, thereby increasing its permeability and enabling more viruses to penetrate the brain. Upon entry to the CNS, SARS‐CoV‐2 may induce a strong neuroinflammatory response in the host. Brain hyperresponsiveness may be associated with viral immunosuppression. Inflammasome activation may also play an important role in neuroinflammation observed in COVID‐19. Infection and excessive inflammation damage host tissues and induce host cell death. We review these mechanisms of SARS‐CoV‐2‐associated central and peripheral nervous system impairment, which are critical for developing diagnostic and therapeutic tools to identify and treat this growing public health problem.
Many researchers have designed treatments for COVID‐19 to reduce the events of neurological injury, such as treating inflammation with the NLRP3 inhibitor MCC950 and Food and Drug Administration‐approved oral drug. 49 Mpro inhibitors can also be used as a therapeutic option to prevent the neurological complications of COVID‐19. 33 Another potential treatment involves reducing RIPK3 or inhibiting RIPK1, and some RIPK1 inhibitors have entered clinical trials. 84
A limitation of the review is that not have a comprehensive analysis of the neuropathological studies and lack of systematic clinical diagnostic methods. Further analyses of autopsy samples are required to confirm the retrogression of SARS‐CoV‐2 from the olfactory mucosa to the CNS. Furthermore, vascular imaging and neuropathological analyses are needed to establish the correlations among virus‐induced vascular disease, vasculitis, and COVID‐19‐related cerebrovascular syndrome.
AUTHOR CONTRIBUTIONS
Yan Zhang conceived the idea for the review and wrote the first draft of the manuscript. Xue Chen edited the manuscript and provided corrections. Lin Jia edited the manuscript for the English language. Yulin Zhang acquired the funding, provided important intellectual content, and supervised the process. All authors read and approved the final manuscript.
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1111/ane.13657.
CONSENT TO PARTICIPATE AND PUBLICATION
All authors participated in this research, and read and approved the final manuscript for publication.
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (grant numbers 81571178 and 81873761); National Science and Technology Major Special Program of the 13th Five‐Year Plan of China (grant number 2018ZX10302104).
Zhang, Y. , Chen, X. , Jia, L. & Zhang, Y. (2022). Potential mechanism of SARS‐CoV‐2‐associated central and peripheral nervous system impairment. Acta Neurologica Scandinavica, 146, 225–236. 10.1111/ane.13657
Yan Zhang and Xue Chen are co‐first author.
DATA AVAILABILITY STATEMENT
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
REFERENCES
- 1. Mao L, Jin H, Wang M, et al. Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan, China. JAMA Neurol. 2020;77(6):683‐690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497‐506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Taquet M, Geddes JR, Husain M, Luciano S, Harrison PJ. 6‐month neurological and psychiatric outcomes in 236 379 survivors of COVID‐19: a retrospective cohort study using electronic health records. Lancet Psychiatry. 2021;8(5):416‐427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chou SHY, Beghi E, Helbok R, et al. Global incidence of neurological manifestations among patients hospitalized with COVID‐19‐a report for the GCS‐NeuroCOVID consortium and the ENERGY consortium. JAMA Netw Open. 2021;4(5):e2112131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lanza G, Godani M, Ferri R, Raggi A. Impact of COVID‐19 pandemic on the neuropsychiatric status of Wilson's disease. World J Gastroenterol. 2021;27(39):6733‐6736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang L, Sievert D, Clark AE, et al. A human three‐dimensional neural‐perivascular ‘assembloid’ promotes astrocytic development and enables modeling of SARS‐CoV‐2 neuropathology. Nat Med. 2021;27:1600‐1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang QQ, Xiang R, Huo S, et al. Molecular mechanism of interaction between SARS‐CoV‐2 and host cells and interventional therapy. Signal Transduct Target Ther. 2021;6(1):1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cui J, Li F, Shi ZL. Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol. 2019;17(3):181‐192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shang J, Ye G, Shi K, et al. Structural basis of receptor recognition by SARS‐CoV‐2. Nature. 2020;581(7807):221‐224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li F, Li W, Farzan M, Harrison SC. Structure of SARS coronavirus spike receptor‐binding domain complexed with receptor. Science. 2005;309(5742):1864‐1868. [DOI] [PubMed] [Google Scholar]
- 11. Stopsack KH, Mucci LA, Antonarakis ES, Nelson PS, Kantoff PW. TMPRSS2 and COVID‐19: serendipity or opportunity for intervention? Cancer Discov. 2020;10(6):779‐782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hoffmann M, Kleine‐Weber H, Schroeder S, et al. SARS‐CoV‐2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271‐280.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Song E, Zhang C, Israelow B, et al. Neuroinvasion of SARS‐CoV‐2 in human and mouse brain. J Exp Med. 2021;218(3):e20202135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Arbour N, Day R, Newcombe J, Talbot PJ. Neuroinvasion by human respiratory coronaviruses. J Virol. 2000;74(19):8913‐8921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wrapp D, Wang N, Corbett KS, et al. Cryo‐EM structure of the 2019‐nCoV spike in the prefusion conformation. Science. 2020;367(6483):1260‐1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bian J, Li Z. Angiotensin‐converting enzyme 2 (ACE2): SARS‐CoV‐2 receptor and RAS modulator. Acta Pharmaceutica Sinica B. 2021;11(1):1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zubair AS, McAlpine LS, Gardin T, Farhadian S, Kuruvilla DE, Spudich S. Neuropathogenesis and neurologic manifestations of the coronaviruses in the age of coronavirus disease 2019: a review. JAMA Neurol. 2020;77(8):1018‐1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jiao L, Yang Y, Yu W, et al. The olfactory route is a potential way for SARS‐CoV‐2 to invade the central nervous system of rhesus monkeys. Signal Transduct Target Ther. 2021;6(1):169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Meinhardt J, Radke J, Dittmayer C, et al. Olfactory transmucosal SARS‐CoV‐2 invasion as a port of central nervous system entry in individuals with COVID‐19. Nat Neurosci. 2021;24(2):168‐175. [DOI] [PubMed] [Google Scholar]
- 20. Kumari P, Rothan HA, Natekar JP, et al. Neuroinvasion and encephalitis following intranasal inoculation of SARS‐CoV‐2 in K18‐hACE2 mice. Viruses. 2021;13(1):132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. de Melo GD, Lazarini F, Levallois S, et al. COVID‐19‐related anosmia is associated with viral persistence and inflammation in human olfactory epithelium and brain infection in hamsters. Sci Transl Med. 2021;13(596):eabf8396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dube M, Le Coupanec A, Wong AH, Rini JM, Desforges M, Talbot PJ. Axonal transport enables neuron‐to‐neuron propagation of human coronavirus OC43. J Virol. 2018;92(17):e00404‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Boldrini M, Canoll PD, Klein RS. How COVID‐19 affects the brain. JAMA Psychiat. 2021;78(6):682‐683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen N, Zhou M, Dong X, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020;395(10223):507‐513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang Z, Yang B, Li Q, Wen L, Zhang R. Clinical features of 69 cases with coronavirus disease 2019 in Wuhan, China. Clin Infect Dis. 2020;71(15):769‐777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. von Bartheld CS, Hagen MM, Butowt R. Prevalence of chemosensory dysfunction in COVID‐19 patients: a systematic review and meta‐analysis reveals significant ethnic differences. ACS Chem Neurosci. 2020;11(19):2944‐2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bryche B, St Albin A, Murri S, et al. Massive transient damage of the olfactory epithelium associated with infection of sustentacular cells by SARS‐CoV‐2 in golden Syrian hamsters. Brain Behav Immun. 2020;89:579‐586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Denis F, Septans AL, Periers L, et al. Olfactory training and visual stimulation assisted by a web application for patients with persistent olfactory dysfunction after SARS‐CoV‐2 infection: observational study. J Med Internet Res. 2021;23(5):e29583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Di Stadio A, D'Ascanio L, Vaira LA, et al. Ultramicronized palmitoylethanolamide and luteolin supplement combined with olfactory training to treat post‐COVID‐19 olfactory impairment: a multi‐center double‐blinded randomized placebo‐controlled clinical trial. Curr Neuropharmacol. 2022;20. Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Matschke J, Lütgehetmann M, Hagel C, et al. Neuropathology of patients with COVID‐19 in Germany: a post‐mortem case series. Lancet Neurol. 2020;19(11):919‐929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Verkhratsky A, Nedergaard M. Physiology of astroglia. Adv Exp Med Biol. 2019;1175:45‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li Y, Bai WZ, Hirano N, et al. Neurotropic virus tracing suggests a membranous‐coating‐mediated mechanism for transsynaptic communication. J Comp Neurol. 2013;521(1):203‐212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wenzel J, Lampe J, Müller‐Fielitz H, et al. The SARS‐CoV‐2 main protease M(pro) causes microvascular brain pathology by cleaving NEMO in brain endothelial cells. Nat Neurosci. 2021;24(11):1522‐1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang L, Zhou L, Bao L, et al. SARS‐CoV‐2 crosses the blood‐brain barrier accompanied with basement membrane disruption without tight junctions alteration. Signal Transduct Target Ther. 2021;6(1):337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Brann DH, Tsukahara T, Weinreb C, et al. Non‐neuronal expression of SARS‐CoV‐2 entry genes in the olfactory system suggests mechanisms underlying COVID‐19‐associated anosmia. Sci Adv. 2020;6(31):5801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pellegrini L, Albecka A, Mallery DL, et al. SARS‐CoV‐2 infects the brain choroid plexus and disrupts the blood‐CSF barrier in human brain organoids. Cell Stem Cell. 2020;27(6):951‐961.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Reynolds J, Mahajan S. SARS‐COV2 alters blood brain barrier integrity contributing to neuro‐inflammation. J Neuroimmune Pharmacol. 2021;16(1):4‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Najjar S, Pearlman DM, Devinsky O, Najjar A, Zagzag D. Neurovascular unit dysfunction with blood‐brain barrier hyperpermeability contributes to major depressive disorder: a review of clinical and experimental evidence. J Neuroinflammation. 2013;10:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Najjar S, Najjar A, Chong DJ, et al. Central nervous system complications associated with SARS‐CoV‐2 infection: integrative concepts of pathophysiology and case reports. J Neuroinflammation. 2020;17(1):231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Erickson M, Rhea EM, Knopp RC, Banks WA. Interactions of SARS‐CoV‐2 with the blood‐brain barrier. Int J Mol Sci. 2021;22(5):2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ribeiro DE, Oliveira‐Giacomelli Á, Glaser T, et al. Hyperactivation of P2X7 receptors as a culprit of COVID‐19 neuropathology. Mol Psychiatry. 2021;26(4):1044‐1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rhea EM, Logsdon AF, Hansen KM, et al. The S1 protein of SARS‐CoV‐2 crosses the blood‐brain barrier in mice. Nat Neurosci. 2021;24(3):368‐378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Walls AC, Tortorici MA, Snijder J, et al. Tectonic conformational changes of a coronavirus spike glycoprotein promote membrane fusion. Proc Natl Acad Sci U S A. 2017;114(42):11157‐11162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Payus AO, Liew Sat Lin C, Mohd Noh M, Jeffree MS, Ali RA. SARS‐CoV‐2 infection of the nervous system: a review of the literature on neurological involvement in novel coronavirus disease‐(COVID‐19). Bosn J Basic Med Sci. 2020;20(3):283‐292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fisicaro F, di Napoli M, Liberto A, et al. Neurological sequelae in patients with COVID‐19: a histopathological perspective. Int J Environ Res Public Health. 2021;18(4):1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kondylis V, Kumari S, Vlantis K, Pasparakis M. The interplay of IKK, NF‐kappaB and RIPK1 signaling in the regulation of cell death, tissue homeostasis and inflammation. Immunol Rev. 2017;277(1):113‐127. [DOI] [PubMed] [Google Scholar]
- 47. Senatorov VV Jr, Friedman AR, Milikovsky DZ, et al. Blood‐brain barrier dysfunction in aging induces hyperactivation of TGFbeta signaling and chronic yet reversible neural dysfunction. Sci Transl Med. 2019;11(521):eaaw8283. [DOI] [PubMed] [Google Scholar]
- 48. Ren Y, Shu T, Wu D, et al. The ORF3a protein of SARS‐CoV‐2 induces apoptosis in cells. Cell Mol Immunol. 2020;17(8):881‐883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Xu H, Akinyemi IA, Chitre SA, et al. SARS‐CoV‐2 viroporin encoded by ORF3a triggers the NLRP3 inflammatory pathway. Virology. 2022;568:13‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Xu Z, Shi L, Wang Y, et al. Pathological findings of COVID‐19 associated with acute respiratory distress syndrome. Lancet Respir Med. 2020;8(4):420‐422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Schultze JL, Aschenbrenner AC. COVID‐19 and the human innate immune system. Cell. 2021;184(7):1671‐1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ribero MS, Jouvenet N, Dreux M, Nisole S. Interplay between SARS‐CoV‐2 and the type I interferon response. PLoS Pathog. 2020;16(7):e1008737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Loo YM, Gale M Jr. Immune signaling by RIG‐I‐like receptors. Immunity. 2011;34(5):680‐692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Blanco‐Melo D, Nilsson‐Payant BE, Liu WC, et al. Imbalanced host response to SARS‐CoV‐2 drives development of COVID‐19. Cell. 2020;181(5):1036‐1045 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hadjadj J, Yatim N, Barnabei L, et al. Impaired type I interferon activity and inflammatory responses in severe COVID‐19 patients. Science. 2020;369(6504):718‐724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mulchandani R, Lyngdoh T, Kakkar AK. Deciphering the COVID‐19 cytokine storm: systematic review and meta‐analysis. Eur J Clin Invest. 2021;51(1):e13429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Körtvelyessy P, Goihl A, Guttek K, Schraven B, Prüss H, Reinhold D. Serum and CSF cytokine levels mirror different neuroimmunological mechanisms in patients with LGI1 and Caspr2 encephalitis. Cytokine. 2020;135:155226. [DOI] [PubMed] [Google Scholar]
- 58. Yang D, Chu H, Hou Y, et al. Attenuated interferon and proinflammatory response in SARS‐CoV‐2‐infected human dendritic cells is associated with viral antagonism of STAT1 phosphorylation. J Infect Dis. 2020;222(5):734‐745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Karki R, Sharma BR, Tuladhar S, et al. Synergism of TNF‐alpha and IFN‐gamma triggers inflammatory cell death, tissue damage, and mortality in SARS‐CoV‐2 infection and cytokine shock syndromes. Cell. 2021;184(1):149‐168 e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Banks WA. Blood‐brain barrier transport of cytokines: a mechanism for neuropathology. Curr Pharm Des. 2005;11(8):973‐984. [DOI] [PubMed] [Google Scholar]
- 61. Kaczmarek‐Hajek K, Zhang J, Kopp R, et al. Re‐evaluation of neuronal P2X7 expression using novel mouse models and a P2X7‐specific nanobody. eLife. 2018;7:e36217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. He Y, Taylor N, Fourgeaud L, Bhattacharya A. The role of microglial P2X7: modulation of cell death and cytokine release. J Neuroinflammation. 2017;14(1):135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Anderson CM, Nedergaard M. Emerging challenges of assigning P2X7 receptor function and immunoreactivity in neurons. Trends Neurosci. 2006;29(5):257‐262. [DOI] [PubMed] [Google Scholar]
- 64. Vargas G, Medeiros Geraldo LH, Gedeão Salomão N, Viana Paes M, Regina Souza Lima F, Carvalho Alcantara Gomes F. Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) and glial cells: insights and perspectives. Brain Behav Immun Health. 2020;7:100127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kopp R, Krautloher A, Ramírez‐Fernández A, Nicke A. P2X7 interactions and signaling ‐ making head or tail of it. Front Mol Neurosci. 2019;12:183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. 2019;19(8):477‐489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Stutz A, Kolbe CC, Stahl R, et al. NLRP3 inflammasome assembly is regulated by phosphorylation of the pyrin domain. J Exp Med. 2017;214(6):1725‐1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bauernfeind FG, Horvath G, Stutz A, et al. Cutting edge: NF‐kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. 2009;183(2):787‐791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Franchi L, Eigenbrod T, Nunez G. Cutting edge: TNF‐alpha mediates sensitization to ATP and silica via the NLRP3 inflammasome in the absence of microbial stimulation. J Immunol. 2009;183(2):792‐796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Xing Y, Yao X, Li H, et al. Cutting edge: TRAF6 mediates TLR/IL‐1R signaling‐induced nontranscriptional priming of the NLRP3 inflammasome. J Immunol. 2017;199(5):1561‐1566. [DOI] [PubMed] [Google Scholar]
- 71. Pan P, Shen M, Yu Z, et al. SARS‐CoV‐2 N protein promotes NLRP3 inflammasome activation to induce hyperinflammation. Nat Commun. 2021;12(1):4664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ratajczak M, Kucia M. SARS‐CoV‐2 infection and overactivation of Nlrp3 inflammasome as a trigger of cytokine “storm” and risk factor for damage of hematopoietic stem cells. Leukemia. 2020;34(7):1726‐1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Siu KL, Yuen KS, Castano‐Rodriguez C, et al. Severe acute respiratory syndrome coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3‐dependent ubiquitination of ASC. FASEB J. 2019;33(8):8865‐8877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Schwabenland M, Salié H, Tanevski J, et al. Deep spatial profiling of human COVID‐19 brains reveals neuroinflammation with distinct microanatomical microglia‐T‐cell interactions. Immunity. 2021;54(7):1594‐1610 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Huang Y, Xu W, Zhou R. NLRP3 inflammasome activation and cell death. Cell Mol Immunol. 2021;18:2114‐2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Ding J, Wang K, Liu W, et al. Pore‐forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535(7610):111‐116. [DOI] [PubMed] [Google Scholar]
- 77. Wang S, Yuan YH, Chen NH, Wang HB. The mechanisms of NLRP3 inflammasome/pyroptosis activation and their role in Parkinson's disease. Int Immunopharmacol. 2019;67:458‐464. [DOI] [PubMed] [Google Scholar]
- 78. Tan MS, Tan L, Jiang T, et al. Amyloid‐beta induces NLRP1‐dependent neuronal pyroptosis in models of Alzheimer's disease. Cell Death Dis. 2014;5:e1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Oxley TJ, Mocco J, Majidi S, et al. Large‐vessel stroke as a presenting feature of Covid‐19 in the young. N Engl J Med. 2020;382(20):e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Manne BK, Denorme F, Middleton EA, et al. Platelet gene expression and function in patients with COVID‐19. Blood. 2020;136(11):1317‐1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Manne BK, Münzer P, Badolia R, et al. PDK1 governs thromboxane generation and thrombosis in platelets by regulating activation of Raf1 in the MAPK pathway. J Thromb Haemost. 2018;16(6):1211‐1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Shin TH, Lee DY, Basith S, et al. Metabolome changes in cerebral ischemia. Cells. 2020;9:7, 1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Rami A, Kogel D. Apoptosis meets autophagy‐like cell death in the ischemic penumbra: two sides of the same coin? Autophagy. 2008;4(4):422‐426. [DOI] [PubMed] [Google Scholar]
- 84. Martens S, Hofmans S, Declercq W, Augustyns K, Vandenabeele P. Inhibitors targeting RIPK1/RIPK3: old and new drugs. Trends Pharmacol Sci. 2020;41(3):209‐224. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.