

Abstract
SARS‐CoV‐2 has led to a worldwide pandemic, catastrophically impacting public health and the global economy. Herein, a new class of lipid‐modified polymer poly (β‐amino esters) (L‐PBAEs) is developed via enzyme‐catalyzed esterification and further formulation of the L‐PBAEs with poly(d,l‐lactide‐coglycolide)‐b‐poly(ethylene glycol) (PLGA‐PEG) leads to self‐assembly into a “particle‐in‐particle” (PNP) nanostructure for gene delivery. Out of 24 PNP candidates, the top‐performing PNP/C12‐PBAE nanoparticles efficiently deliver both DNA and mRNA in vitro and in vivo, presenting enhanced transfection efficacy, sustained gene release behavior, and excellent stability for at least 12 months of storage at −20 °C after lyophilization without loss of transfection efficacy. Encapsulated with spike encoded plasmid DNA and mRNA, the lipid‐modified polymeric PNP COVID‐19 vaccines successfully elicit spike‐specific antibodies and Th1‐biased T cell immune responses in immunized mice even after 12 months of lyophilized storage at −20 °C. This newly developed lipid‐polymer hybrid PNP nanoparticle system demonstrates a new strategy for both plasmid DNA and mRNA delivery with the capability of long‐term lyophilized storage.
Keywords: COVID‐19 vaccines, nanoparticles, gene delivery, mRNA therapeutics
A “particle‐in‐particle” (PNP) nanostructure gene delivery platform is developed. The top‐performing PNP nanoparticle‐based delivery vector shows enhanced DNA and mRNA transfection efficacy in vitro and in vivo, sustained gene release behavior, and capability of long‐term lyophilization for storage. Furthermore, the lyophilized PNP COVID‐19 vaccines elicit strong immune responses in a BALB/c mouse model even after 12 months of storage.

1. Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2), otherwise known as coronavirus disease‐2019 (COVID‐19), is the catalyst of a catastrophic global pandemic which have infected over 500 million people, resulting in over 6.1 million deaths worldwide to date (according to the World Health Organization as of April 2022). Through global public and private sector collaboration, the next‐generation nucleic acid‐based vaccines have been developed and deployed in record time including two mRNA‐based lipid nanoparticle (LNP) vaccines: the Pfizer‐BioNTech (BNT162b2),[ 1 , 2 ] and Moderna (mRNA‐1273),[ 3 , 4 ] COVID‐19 vaccines. These next‐generation LNP‐mRNA vaccines show great potential for preventing COVID‐19 and elicit a high level of neutralizing antibody titers and appear to be safe with up to 95% effectiveness in preventing COVID‐19 disease.[ 2 , 3 ] However, the current LNP‐mRNA COVID‐19 vaccines from Pfizer‐BioNTech and Moderna are limited by short shell life and a lack of stability which requires them to be kept between −60 °C and −80 °C, and −15 °C and −25 °C,[ 5 ] respectively with ultracold‐chain storage and transportation causing an exponential increase in the cost of transportation and distribution.[ 6 ] Lyophilization has been used to extend the shelf life of a broad array of therapeutics including small molecule drugs, vaccines, and protein‐based medicines, usually with the help of cytoprotectants and mitigates the hydrolysis‐based degradation of vaccines. The removal of the aqueous solution gives the material the ability to be stored as a dry powder for a long‐term basis.[ 7 , 8 ] Therefore, lyophilization may potentially solve the issue of vaccine storage and transportation since the current COVID‐19 vaccines are all stored in the aqueous phase, which increases the chance of degradation of both mRNA and LNPs leading to instability of vaccines.[ 5 , 6 ] Previous studies have demonstrated that polymeric nanoparticle (NP)‐based gene delivery systems such as poly (β‐amino ester) (PBAE) exhibit enhanced gene transfection efficacy, outstanding therapeutic outcomes,[ 9 , 10 , 11 , 12 ] excellent nanoparticle stability, and capability of long‐term storage for 2 years at −20 °C after lyophilization.[ 10 ] Owing to these advantageous characteristics of polymeric NP gene delivery platforms, PBAE NP‐based DNA/mRNA COVID‐19 vaccines capable of lyophilization are poised to solve current stability and storage issues with LNP‐mRNA vaccines and need to be studied to this effect to fill the vacancies in polymeric NP‐based nucleic acid COVID‐19 vaccines in clinical trials.
Our previous report demonstrated that the unique “nanoparticle depot” structure improved gene delivery, exemplified in a PLGA‐PEG hybrid poly (β‐amino ester) system which could efficiently deliver plasmid GFP (pGFP) with sustained gene release behavior over 8 days, lowered cellular toxicity and enhanced transfection efficiency.[ 13 ] However, this “nanoparticle depot” system was not compatible with mRNA delivery due to low transfection efficacy compared to plasmid DNA.[ 13 , 14 ] Therefore, we sought to chemically modify the cationic PBAE‐447 to create a universal and excellent gene delivery vector for both DNA and mRNA with enhanced NP stability for long‐term storage. It has been reported that incorporating hydrophobic alkyl side chains could enhance transfection potency, decrease aggregation of PBAE and improve the hydrophobicity of PBAE to enhance nanoparticle stability in physiological conditions.[ 15 ] Moreover, well‐known cationic lipids such as C12‐200,[ 16 ] G0‐C14,[ 17 , 18 ] DLin‐MC3‐DMA,[ 19 , 20 ] and other lipid derivatives[ 21 , 22 ] show outstanding siRNA and mRNA delivery efficacy benefiting from saturated or unsaturated hydrocarbon lipid chains which facilitate the fusion of nanoparticles into the negatively charged cell membrane and lead to cellular uptake via endocytosis to improve the transfection efficacy.[ 19 , 23 ] Therefore, we hypothesized that the introduction of a lipid moiety to PBAE would improve the transfection performance for both DNA and mRNA, enhance and maintain the NP stability after long‐term storage. We rationally designed a new nanoparticle formulation via: 1) chemically grafting saturated or unsaturated lipid moiety on PBAE to improve DNA/mRNA transfection efficacy and NP stability;[ 15 ] 2) incorporating a PLGA‐PEG polymeric vector to achieve sustained gene payload release,[ 13 , 24 ] and to enhance the colloidal stability in presence of serum and protect the inside nanocomplexes;[ 25 ] 3) lyophilizing the NP to further enhance its long‐term stability at low temperatures, avoiding hydrolysis and degradation.[ 5 , 8 ]
In this study, we introduced chemical modifications to the PBAE‐447 polymer and synthesized a new class of lipid‐modified PBAEs (L‐PBAEs) using different commercially available lipid acids via Candida antarctica Lipase B (CALB) enzyme‐assisted esterification. The resulting L‐PBAEs presented much more enhanced hydrophobicity and stability in aqueous phase than the PBAE‐447 and were further formulated with the PLGA‐PEG polymeric vector to form a unique “particle‐in‐particle” (PNP) nanostructure and generate a library of PNP/L‐PBAE NPs for gene delivery. Significantly, all the PNP/L‐PBAE NPs show drastically enhanced plasmid DNA and mRNA transfection efficacy in vitro and in vivo over the unmodified PNP/PBAE formulations. The top‐performing PNP/C12‐PBAE formulation was screened out from a PNP/L‐PBAE library and exhibited a much more explicit PNP nanostructure than the original “nanoparticle depot” formulation. Moreover, the PNP/C12‐PBAE NP can be lyophilized for long‐term storage and the transfection efficacy, as well as particle size and zeta potential, remained comparable to fresh PNPs even after 12 months at −20 °C. Consequently, PNP/C12‐PBAE NPs were further encapsulated with SARS‐CoV‐2 spike protein encoded plasmid DNA (DNA‐PNP) and mRNA (mRNA‐PNP) to develop lipid‐polymer hybrid nanoparticle‐based COVID‐19 vaccines. The immunogenicity in BALB/c mice was evaluated after 12‐month storage of lyophilized PNPs at −20°C, and the results showed that the PNP vaccines induced robust SARS‐CoV‐2 spike protein‐specific antibodies in mice sera and specific Th1‐biased T cell immune responses. Our studies demonstrated that the lipid‐modified PNP can be explored as a lipid‐polymer hybrid nanoparticle‐based gene delivery platform capable of lyophilization for storage at −20 °C for at least 12 months.
2. Results
2.1. Design, Synthesis, and Characterization of PNP/L‐PBAE NPs
We have designed a library of lipid‐modified PBAEs (termed L‐PBAEs) based on the rational hypothesis that the grafting of lipid chains to the PBAE backbone would: 1) facilitate the fusion of the generated nanoparticles with the cell membrane and increase cellular uptake via endocytosis,[ 19 , 23 , 26 ] 2) increase the nanoparticle‐mediated gene delivery efficacy and transfection potency, and 3) improve the hydrophobicity of PBAE polymer and the PNPs’ consequent integrity and stability in physiological conditions.[ 15 ] The L‐PBAEs were synthesized via Candida antarctica Lipase B (CALB) enzyme‐assisted esterification[ 27 ] between the hydroxyl groups of PBAE‐447[ 10 , 11 , 12 ] and the carboxylic acid from different commercially‐available saturated and unsaturated lipid acids (Figure 1A). Varying the 8 different lipid acids (Figure 1B) and 3 different molecular ratios of lipid: PBAE (1:10, 1:5, and 1:2) in the PBAE backbone, we synthesized and characterized a library of 24 different cationic L‐PBAE polymers. The new L‐PBAE polymers were synthesized successfully under CALB enzyme‐assisted esterification and characterized by gel permeation chromatography (GPC), Fourier‐transform infrared spectroscopy (FTIR), and nuclear magnetic resonance (NMR). GPC results were used to monitor the changing of molecular weight (MW) after the lipid conjugation on PBAE. For example, the GPC results for the molar ratio of C12‐lipid: PBAE varying from 1:10, 1:5 to 1:2 showed that the MW of C12‐PBAE increased from 4173 initially (without C12‐lipid modification) to 5435, 6241, and 8653, respectively (Figure S1, Supporting Information). Based on the increased MW of PBAE, we calculated that there would be around six C12‐lipids, twelve C12‐lipids, and twenty C12‐lipids grafted on the PBAE backbone via the hydroxyl groups under the above conditions. In general, the increase in molecular weight of the hydrophilic PBAE after lipid tail grafting would lead to a decrease in solubility as proven by the precipitation in aqueous solution (Figure 1I). Additionally, the conjugation of lipids on the PBAE backbone generated the relative characteristic peaks shown in FTIR and NMR spectroscopy, respectively (Figures S2 and S3, Supporting Information). Then, we prepared a library of gene‐loaded PNP/L‐PBAE NPs, where the PLGA‐PEG and L‐PBAEs formed to be PNPs through self‐assembly and the gene payloads were encapsulated inside L‐PBAEs via electrostatic interactions to form L‐PBAE/gene nanocomplexes which were embedded within a PLGA‐PEG polymeric vector (Figure 1C).[ 13 ] The resulting PNP/L‐PBAE (1:10) NPs presented with an average hydrodynamic diameter ≈160 nm with a narrow size distribution of ≈0.15 PDI (Figure S4, Supporting Information). It is important to note that the increased hydrophobicity of the L‐PBAE/gene nanocomplexes can significantly increase the encapsulating efficiency and PNP stability due to the hydrophobic‐hydrophobic interaction between the grafted lipid tails and hydrophobic moiety of the amphiphilic PLGA‐PEG copolymer. As the C12‐lipid: PBAE ratios increased from 1:10 to 1:5 to 1:2, the PNP/L‐PBAEs (1:5) and PNP/L‐PBAEs (1:2) increased in size from 180 to 210 nm (Figure 1H), respectively most likely due to more L‐PBAE nanocomplexes being embedded throughout the PLGA‐PEG vector. Additionally, all the PNP/L‐PBAE NPs showed an increased positive charge of around +26 mV compared to the PNP/PBAE control (+17 mV). The TEM images showed that the NPs have a uniform compact spherical shape with a diameter ≈160 nm which is in accordance with the DLS result. Importantly, we observed a much clearer example of “Particle‐in‐Particle” nanostructures within these PNP/L‐PBAE NPs, owing to the newly developed L‐PBAEs. Several L‐PBAE/gene nanocomplexes are clearly observed to be embedded throughout the PLGA‐PEG vectors from the TEM images (Figure 1D,E) and we did not observe this same phenomenon in PLGA‐PEG NPs without L‐PBAEs incorporated (Figure 1F). To further confirm, we characterized the size and morphology of L‐PBAEs/gene nanocomplexes, which present with a diameter of 20–50 nm using TEM (Figure 1G), which is consistent with the observed nanostructures in the combined PNP/L‐PBAE NPs.
Figure 1.
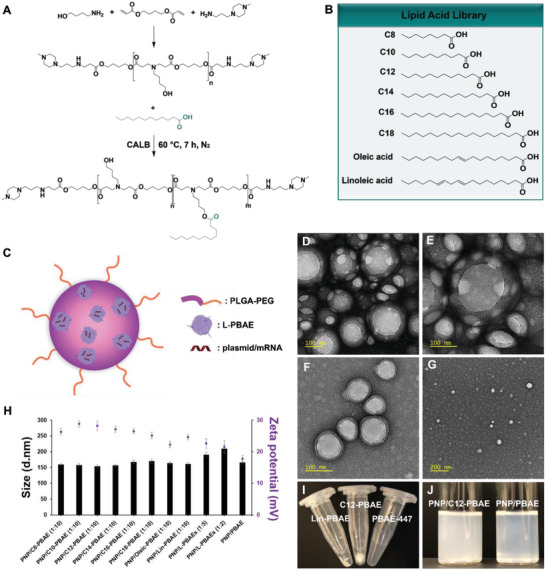
Synthesis and characterization of PNP/L‐PBAE NPs. A) The chemical synthesis route of L‐PBAEs under CALB enzyme‐assisted esterification. B) The library of lipid acids for synthesis of L‐PBAEs. C) A general schematic of PNP/L‐PBAE NPs consisting of three components: i) L‐PBAE/gene nanocomplexes embedded throughout PLGA‐PEG NPs, ii) The PLGA layer which provides sustained release and protection of embedded L‐PBAE/gene nanocomplexes, iii) An outer PEG surface. D) Representative TEM image of PNP/L‐PBAE NPs and E) a magnified section of the representative nanoparticle image. F) Representative TEM image of the PLGA‐PEG NPs without L‐PBAEs incorporated and G) Representative TEM image of L‐PBAE/gene nanocomplexes. H) The size and zeta potential of PNP/L‐PBAE NPs determined by dynamic light scattering. I) Lipid‐modified PBAEs show obvious hydrophobicity compared to the unmodified PBAE‐447 in aqueous phase. J) An image of PNP/C12‐PBAE NP solution and PNP/PBAE NP solution. The data were represented as means ± SD (standard deviation).
2.2. High‐throughput Screening to Select the Top Performing PNP/L‐PBAE NPs
An ideal nanoparticle‐mediated gene delivery vector should be biodegradable and biocompatible, with limited cytotoxicity and high transfection efficacy. We first prepared a series of NPs with different lipids and lipid moiety ratios, then evaluated the cytotoxicity of the library of PNP/L‐PBAE NP candidates. All of the 1:10 (molar ratios of lipid acid to PBAE) groups of the PNP/L‐PBAEs showed dose‐dependent cytotoxicity as tested in Hek 293 cells. At 0.2–0.3 mg mL−1 nanoparticle concentrations, the PNP/L‐PBAE NPs exhibited at least 90% cell viability as demonstrated by CCK‐8 kit, and the cell viabilities decreased to under 75% with increasing NP concentrations (Figure 2A). Next, we evaluated the 1:5 and 1:2 (molar ratios of lipid acid to PBAE) groups of PNP/L‐PBAEs. The data showed that the cell viability decreased with increasing lipid moiety ratios, due the increasing charge density.[ 28 ] Additionally, all the PNP/L‐PBAE NPs presented lower cell viability than the unmodified PNP/PBAE NPs control group due to the increasing positive charge of the new lipid‐modified PNP/L‐PBAE NPs, which is more cytotoxic to the cells. Therefore, we selected the PNP/L‐PBAE NPs with the 1:10 lipid ratio for the following experiments due to their favorable cell viability. To screen out the optimized transfection efficacy of the PNP/L‐PBAE NPs, we encapsulated firefly luciferase encoded plasmid (pLuc) and mRNA (mLuc) into PNP/L‐PBAE NPs and evaluated the plasmid and mRNA delivery efficacy of the NPs by measuring luciferase expression using the commercial Bright‐Glo Luciferase Assay System (Promega). For the pLuc transfection, the PNP/C12‐PBAE was shown to be the best transfection reagent, as it presented much higher luciferase expression than the other formulations. The luciferase expression of PNP/L‐PBAE NPs reached its peak on the third day, post‐transfection (Figure 2B) due to the sustained release behavior of the PNP system. Remarkably, all the PNP/L‐PBAE NPs groups exhibited a higher transfection efficacy than the unmodified PNP/PBAE control group. Next, we tested the transfection efficacy of mLuc‐loaded PNP/L‐PBAE NPs. The PNP/C12‐PBAE was also the leading formulation and presented a much higher transfection efficacy than the unmodified PNP/PBAE control group (Figure 2C). To summarize, high‐throughput screening demonstrated that out of all the PNP/L‐PBAE NPs, the PNP/C12‐PBAE NPs with a 1:10 ratio of lipid to PBAE was the top‐performing candidate and was selected as the optimized nanoparticle formulation for further in vitro and in vivo studies.
Figure 2.
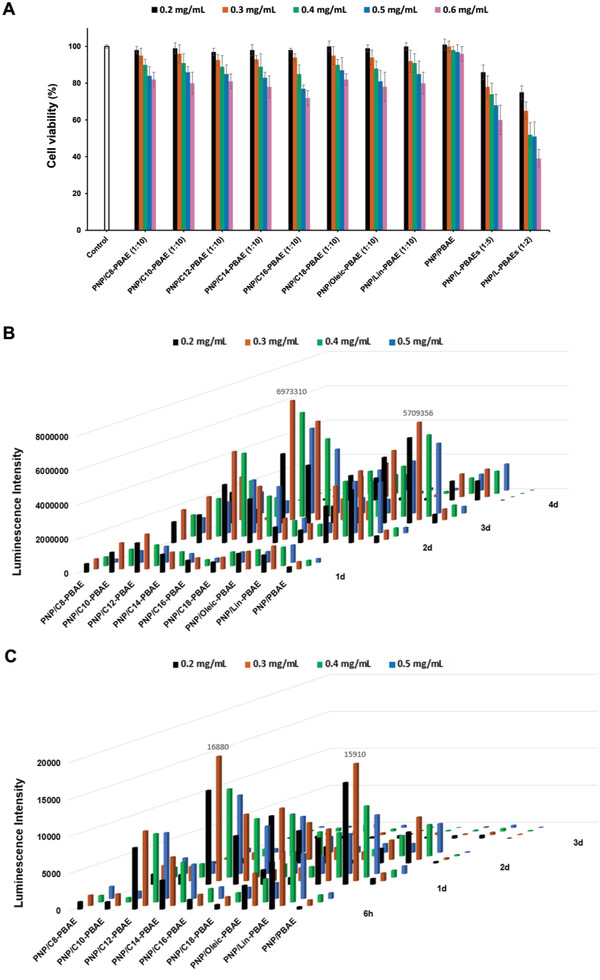
Screening for the optimal PNP/L‐PBAE NPs. A) Cell viability of each PNP/L‐PBAE NP formulation was measured by CCK‐8 kit. B) Transfection efficacy of luciferase plasmid‐loaded PNP/L‐PBAE NPs at different dosages and on different days determined by luminescence intensity. C) Transfection efficacy of luciferase mRNA‐loaded PNP/L‐PBAE NPs with different dosages and on different days determined by luminescence intensity. All experiments were repeated in triplicate to generate data using the mean values.
2.3. Comprehensive Study of the Top Performing PNP/C12‐PBAE NPs
We then performed a comprehensive evaluation of the top‐performing PNP/C12‐PBAE NPs in terms of transfection efficacy in vitro and in vivo. First, we evaluated the PNP/C12‐PBAE NPs’ transfection efficacy using green fluorescent protein encoded plasmid (pGFP) in the Hek 293 cell line. The GFP‐expressing cells were visualized by a fluorescence microscope and analyzed by fluorescence‐activated cell sorting (FACS) (Figure 3A). According to the FACS results, the population of GFP‐positive cells reached ≈95% at 96 h post‐transfection. Comparatively, the PNP/PBAE and ALC‐0315 LNPs (Pfizer/BioNTech COVID‐19 vaccine formulation) showed the percentage of GFP‐positive cells was 50% and 36%, respectively (Figure 3B). The mean fluorescence intensity (MFI) results showed that the PNP/C12‐PBAE NPs outperformed other formulations, increasing for up to 4 days (Figure 3C). Second, we tested the mCherry encoded mRNA (mCherry) transfection using PNP/C12‐PBAE NPs. The FACS results showed that the PNP/C12‐PBAE formulation has a much better mRNA transfection efficacy than the unmodified PNP/PBAE and transfected a comparable population of cells to the ALC‐0315 LNPs at ≈68% as well as similar MFI (Figure 3D,E,F). Next, we evaluated the plasmid DNA and mRNA release behaviors of PNP/C12‐PBAE NPs in vitro via PicoGreen dsDNA assay and RiboGreen RNA assay, respectively. One of the main advantages of the PLGA NP delivery vector is the sustained release of encapsulated payload, which is usually governed by diffusion and degradation processes.[ 17 , 24 , 29 , 30 ] The cumulative release profiles of both plasmid and mRNA using PNP/C12‐PBAE NPs indeed showed sustained release behavior compared and reached a peak of up to 96% gene released over 8 days after a quick release within the first 24 h (Figure 3G,H). These results clearly indicate that the PNP/C12‐PBAE NP displays a prolonged gene release behavior for both plasmid DNA and mRNA compared with C12‐PBAE formulations which have no PLGA‐PEG polymeric vector. The agarose gel electrophoresis results also demonstrated that the PNP/C12‐PBAE NPs could bind both plasmid DNA and mRNA efficiently at NP concentrations as low as 0.1 mg mL−1 (Figure 3I).
Figure 3.
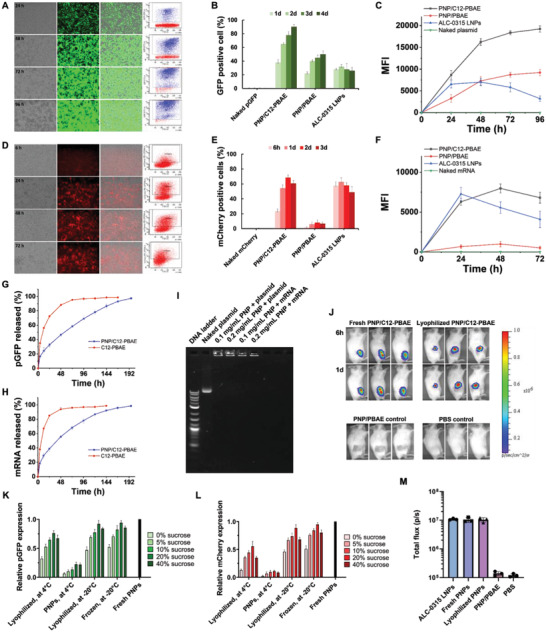
Comprehensive evaluation of PNP/C12‐PBAE NPs in vitro and in vivo. A) Fluorescence imaging and FACS results of Hek 293 cells transfected by GFP plasmid (pGFP) loaded PNP/C12‐PBAE NPs at different time points. B) A summary of GFP positive cell population post‐transfection and C) Mean fluorescence intensity (MFI) of GFP positive cells according to FACS results. D) Fluorescence imaging and FACS results of Hek 293 cells transfected by mCherry mRNA (mCherry) loaded PNP/C12‐PBAE NPs at different time points. E) A summary of mCherry positive cell population post‐transfection and F) MFI of mCherry positive cells according to FACS results. G) The cumulative release profile of pGFP and H) mRNA from PNP/C12‐PBAE NPs compared with C12‐PBAE NPs. I) Agarose gel electrophoresis binding analysis of plasmid and mRNA‐loaded PNP NPs with different nanoparticle volume and constant gene loading. J) In vivo bioluminescence images of BALB/c mice after injection of luciferase mRNA (mLuc) loaded PNPs (n = 3) at specific time points with comparison to control groups. K) Evaluation after long‐term storage of PNP/C12‐PBAE NPs using pGFP and L) mCherry as reporter genes, summarized by FACS results and normalized to fresh PNP gene expression. M) The luciferase expression between the PNP formulations and ALC‐0315 LNPs in a BALB/c mouse model was recorded after 1 day post‐injection. All experiments were repeated in triplicate and the data were represented as means ± SD.
We next investigated long‐term storage conditions and capabilities, and selected sucrose as the cryoprotectant for PNP NP formulation to stabilize the nanoparticle's structure according to previous reports.[ 7 , 10 , 11 , 31 ] The PNP/C12‐PBAE NPs were formulated with pGFP or mCherry and included different concentrations of sucrose solutions with a final concentration ranging from 0 to 40 wt.%. Then the PNP NP formulations were either lyophilized as a powder or frozen following storage at either 4 °C or −20 °C for 12 months, where the fresh PNP/C12‐PBAE was used as a positive control. The PNP pGFP transfection results after 12 months of storage at −20 °C showed that the PNPs stored under either lyophilized or frozen conditions with 20 wt.% sucrose has comparable transfection efficacy with fresh NPs. Moreover, PNP pGFP transfection results at 4 °C showed that the lyophilized PNPs have much better transfection efficacy over the PNPs stored in aqueous solution since the PNPs were easily degraded in the aqueous condition over long‐term storage (Figure 3K). Similarly, the mCherry formulated PNPs showed that the most favorable storage condition was lyophilization with 20 wt.% sucrose at −20 °C which showed efficacy comparable to fresh PNPs (Figure 3L). The lyophilized PNP/C12‐PBAE NPs remained stable in size and distribution after recovery in aqueous solution and only a minor change in particle size from 155 to 176 nm, with PDI of 0.15 to 0.21, respectively, and a minor change of zeta potential from +26.6 to +23.8 mV compared with the fresh NP formulations (Figure S5, Supporting Information). Consequently, the most favorable long‐term storage condition for PNPs was determined to be the lyophilized formulation at −20 °C for 12 months with no loss of transfection efficacy (Figure S5, Supporting Information) and therefore this storage condition was applied for the following in vivo studies.
Then, we explored the PNP transfection efficacy in vivo by injecting luciferase mRNA (mLuc) loaded PNP/C12‐PBAE NPs into BALB/c mice. The PNPs were administered by either intramuscular or subcutaneous injection to mimic typical clinical vaccination approaches.[ 32 ] Extraordinarily, both the fresh and 12‐month lyophilized PNP/C12‐PBAE successfully delivered mRNA in vivo and induced luciferase expression in mice (Figure 3J), whereas the PNP/PBAE control group and PBS group did not generate luciferase expression in mice. Consistent with our rational design and in vitro data, the lipid‐modified PNP/C12‐PBAE NPs enhanced the mRNA transfection efficacy in vivo as well. Additionally, we performed a comparison study between the PNP formulations and ALC‐0315 LNPs (which are used in Pfizer/BioNTech COVID‐19 vaccines) in terms of the luciferase expression in vivo. The results showed that both fresh PNPs and 12‐month lyophilized stored PNPs have a comparable luciferase expression intensity with ALC‐0315 LNPs in a BALB/c mouse model at 1d post‐injection (Figure 3M). Furthermore, the histopathology and immunofluorescence assays of the proinflammatory biomarker IL‐6 in muscle tissues surrounding the PNP injection site were evaluated after 5 days post‐injection and the results showed no observable PNP nanoparticle‐mediated inflammation when compared with the control PBS injected group (Figures S6 and S7, Supporting Information).[ 29 , 33 ]
2.4. Design and Characterization of DNA‐PNP and mRNA‐PNP COVID‐19 Vaccines In Vitro
After a solid and comprehensive study of PNP/C12‐PBAE NPs, we then moved toward developing nucleic acid COVID‐19 vaccines using the PNP platform. The full‐length SARS‐CoV‐2 spike (S) glycoprotein is comprised of 1273 amino acids (Figure S9, Supporting Information) including S1 and S2 subunits with an RRAR furin‐like cleavage motif in the S1/S2 cleavage site,[ 34 ] and is the major component of the coronavirus envelope. The S protein mediates virus entry into host cells by binding to the angiotensin‐converting enzyme 2 (ACE2) receptor through the receptor‐binding domain (RBD) located in the S1 subunit, which subsequently causes fusion between the viral envelope and the host cell membrane through the S2 subunit.[ 35 ] The full‐length S protein induces potent neutralizing antibodies and T cell‐mediated host immune responses; therefore, the S protein has been widely selected as a promising antigen for COVID‐19 vaccine development.[ 36 ]
To develop polymeric PNP vaccines, we formulated PNP/C12‐PBAE NPs with plasmid DNA and mRNA encoding the wild‐type full‐length S protein (Wuhan‐Hu‐1 strain, NCBI reference sequence: NC_045512.2). These were termed “DNA‐PNP” and “mRNA‐PNP” vaccines, respectively, and were stored following lyophilization at −20 °C for 12 months with 20 wt.% sucrose before use in vaccination experiments. The S protein expression was evaluated in vitro by Western blot (WB) and immunofluorescence assay (IFA). The WB analysis of lysates from DNA‐PNP or mRNA‐PNP transfected Hek 293 cells revealed clear bands at ≈180 kDa, showing the expression of full‐length spike protein (Figure 4A). In immunofluorescence studies, the spike protein expression was detected by FITC‐label secondary antibodies from both DNA‐PNP and mRNA‐PNP transfected Hek 293 cells and the fluorescent images were taken under brightfield, DAPI, FITC, and channels merged after 3 days post‐transfection of DNA‐PNP and 2 days post‐transfection of mRNA‐PNP, respectively (Figure 4B). These results demonstrated that the lyophilization stored mRNA‐PNP and DNA‐PNP vaccines can generate strong spike protein expression in vitro.
Figure 4.
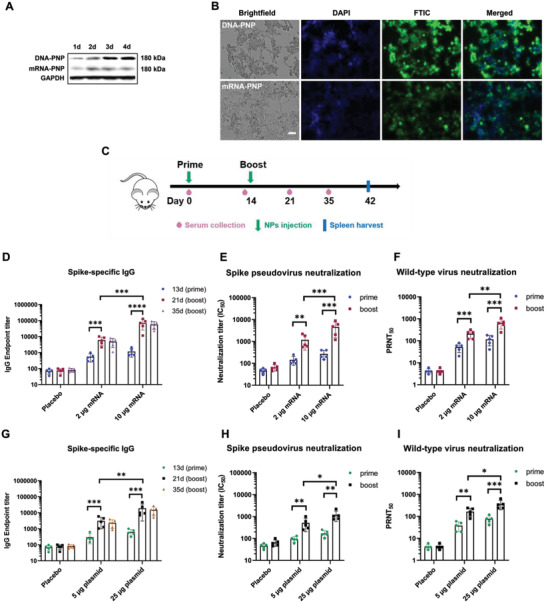
In vitro characterization of PNP vaccines and humoral immune response in vaccinated mice. A) In vitro Western blot of S protein expression after transfection with DNA‐PNP and mRNA‐PNP vaccines on Hek 293 cells. The cell lysates were probed with polyclonal anti‐SARS coronavirus 2 spike glycoprotein and anti‐GAPDH as loading control. B) In vitro immunofluorescent staining of Hek 293 cells transfected with DNA‐PNP (top row) and mRNA‐PNP (bottom row). Expression of S protein was measured with polyclonal anti‐SARS coronavirus 2 spike glycoprotein and FITC‐labeled secondary antibody. Cell nuclei were counterstained with DAPI and the fluorescent images were taken under brightfield, DAPI, FITC, and merged channels (scale bar, 50 µm). C) Schematic of mouse immunization and sample collection schedule. The prime and boost vaccine injections are separately administered at day 0 and day 14, the mice sera are collected at day 0, day 13, day 21, and day 35. The vaccinated mice are sacrificed at day 42 for spleen harvest. D) Spike‐specific IgG endpoint titers of mRNA‐PNP vaccines determined by ELISA (n = 5). (E) SARS‐CoV‐2 spike pseudovirus neutralizing antibody IC50 titers of mRNA‐PNP vaccines (n = 5). F) The plaque reduction neutralization test (PRNT) against wild‐type Wuhan SARS‐CoV‐2 strain virus was performed using mRNA‐PNP vaccinated mouse sera (n = 5). G) Spike‐specific IgG endpoint titers of DNA‐PNP vaccines determined by ELISA (n = 5). H) SARS‐CoV‐2 spike pseudovirus neutralizing antibody IC50 titers of DNA‐PNP vaccines (n = 5). I) The plaque reduction neutralization test (PRNT) against wild‐type Wuhan SARS‐CoV‐2 strain virus was performed using DNA‐PNP vaccinated mouse sera (n = 5). All data were represented as means ± SD. Significance was calculated using a one‐way ANOVA with multiple comparisons tests (ns, not significant; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
2.5. Immunogenicity of PNP Vaccines in BALB/c mice
To evaluate the immunogenicity of PNP vaccines, groups (n = 5) of female BALB/c mice were intramuscularly immunized with a 2 or 10 µg (mRNA) dose of mRNA‐PNP and 5 or 25 µg (DNA) dose of DNA‐PNP vaccines where the unloaded PNP was used as placebo. The mice were vaccinated with PNPs in a prime/boost manner spaced 14 days apart. Serum was collected at day 0, day 13, day 21, and day 35 post‐immunization, and the mice were euthanized at day 42 for spleen harvest (Figure 4C). The single prime injection of mRNA‐PNP and DNA‐PNP vaccines on day 1 induced SARS‐CoV‐2 spike‐specific IgG antibodies after 13 days post‐first injection as determined by enzyme‐linked immunosorbent assay (ELISA). The IgG geometric mean titers (GMTs) of the mRNA‐PNP vaccine are in the range of 545 to 1174 and GMTs of the DNA‐PNP vaccine were around 310 to 662, respectively. Additionally, the GMTs of spike pseudovirus neutralizing antibody 50% inhibitory concentration (IC50) is in the range of 144 to 286 and 92 to 178 for mRNA‐PNP and DNA‐PNP vaccines, respectively. On day 14, each group of mice was immunized with the second nanoparticle booster dose and the sera were collected at day 21 and day 35 after the initial vaccination. Notably, the SARS‐CoV‐2 spike‐specific IgG antibodies were significantly elevated and presented a dose‐dependent response for both mRNA‐PNP and DNA‐PNP vaccines after the booster injection whereas the empty PNP control group showed no antibodies detected from sera. 21 days after the priming dose (7 days post booster dose), the GMTs of spike‐specific IgG had increased to 5548 (2 µg) and 71178 (10 µg) for the mRNA‐PNP vaccine, and the DNA‐PNP vaccine GMTs increased to 2730 (5 µg) and 18900 (25 µg), respectively. 35 days after the priming dose (21 days post booster dose), the spike‐specific IgG GMTs slightly decreased compared with day 21, with values of 4868 (2 µg) to 57048 (10 µg) of mRNA‐PNP and 2375 (5 µg) to 14970 (25 µg) for DNA‐PNP vaccines, respectively (Figure 4D,G). Meanwhile, the GMTs of spike pseudovirus 50% neutralizing antibody IC50 after the booster injection also exhibited a dose‐dependent increase (7 days after booster dose) and approached 1080 (2 µg) to 4570 (10 µg) of mRNA‐PNP and 460 (5 µg) to 1180 (25 µg) of DNA‐PNP vaccines, respectively (Figure 4E,H). Additionally, the wild‐type Wuhan SARS‐CoV‐2 strain neutralizating antibodies were evaluated by a 50% plaque reduction neutralization test (PRNT50), and the results of mRNA‐PNP and DNA‐PNP vaccinated mouse sera were shown in Figure 4F,I, respectively. The spike‐specific IgG GMTs were strongly correlated with spike pseudovirus 50% neutralizing antibody IC50 GMTs, and mRNA‐PNP vaccines could elicit stronger spike‐specific antibody responses than that of DNA‐PNP vaccines in vivo, even though the DNA‐PNP vaccines had much stronger S protein expression in vitro. These results demonstrated that immunization with 12‐month lyophilization stored mRNA‐PNP and DNA‐PNP vaccines in BALB/c mice elicited anti‐spike antibodies as well as SARS‐CoV‐2 spike neutralizing antibodies after the priming dose and the antibody titers significantly increased after a booster vaccination in a dose‐dependent manner demonstrated by the significant increase of GMTs compared to the placebo group.
2.6. PNP Vaccines Induce T‐Cell Responses in Immunized Mice
We further evaluated whether mRNA‐PNP or DNA‐PNP vaccines could induce spike‐specific T cell immune responses after prime and booster doses in mice. The antigen‐specific T cell responses were measured with FACS through both intracellular cytokine staining (ICS) and IFN‐γ enzyme‐linked immunosorbent spot (ELISpot) with splenocytes isolated from mRNA‐PNP, DNA‐PNP, and placebo vaccinated mice on day 42. The T cells from splenocytes are ex vivo stimulated with peptide pools covering the immunodominant sequence domains of the SARS‐CoV‐2 spike glycoprotein. Flow cytometric analysis showed that the population of CD4+ T cells and CD8+ T cells expressing interferon‐gamma (IFN‐γ), tumor necrosis factor‐alpha (TNF‐α), and interleukin‐2 (IL‐2) in splenocytes from mRNA‐PNP (Figure 5A,B) and DNA‐PNP (Figure 5C,D) immunized groups were significantly higher than the placebo groups (Flow cytometry gating strategy was shown in Figure S8, Supporting Information). We also found that the frequency of IFN‐γ+, TNF‐α+, and IL‐2+ cytokine‐secreting CD4+ and CD8+ T cells in mRNA‐PNP vaccinated mice was higher than in DNA‐PNP vaccinated groups. Additionally, there was no significant difference in type 2 cytokine IL‐4 secretion between mRNA‐PNP, DNA‐PNP immunized groups, and placebo‐vaccinated mice. Furthermore, the ELISpot assay showed that secretion of IFN‐γ in splenocytes from both mRNA‐PNP and DNA‐PNP immunized mice were significantly higher than in the placebo vaccination group (Figure 5E,F) and the IFN‐γ spots were measured to be 734 (2 µg) to 1098 (10 µg) per 106 splenocytes for mRNA‐PNP vaccines and 544 (5 µg) to 722 (25 µg) for DNA‐PNP vaccines, respectively.
Figure 5.
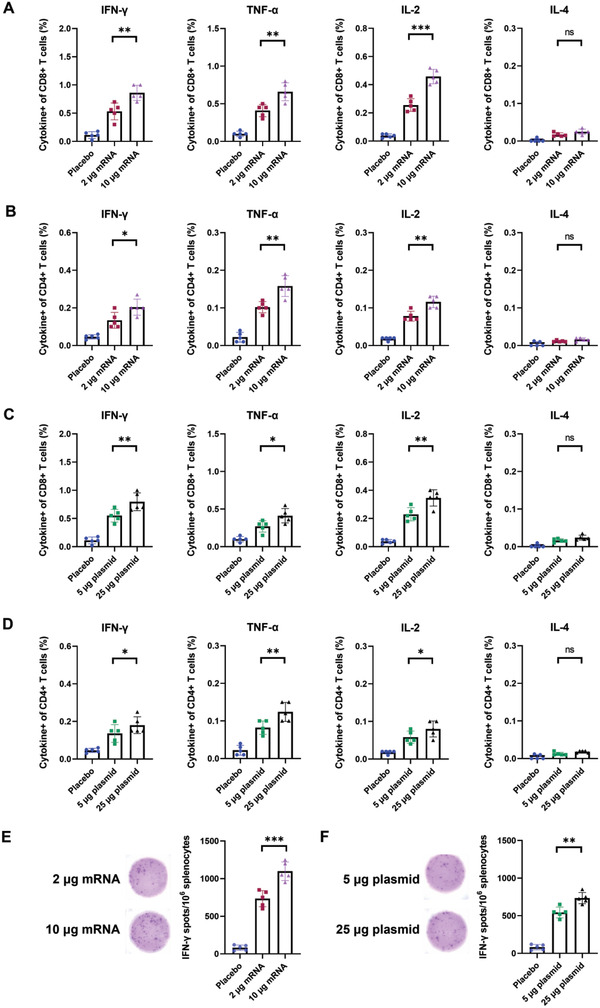
Cytokine analysis of SARS‐CoV‐2 spike‐specific T cell immune response in PNP vaccinated mice. A) The percentage of CD8+ and B) CD4+ T cells expressing IFN‐γ, TNF‐α, IL‐2, and IL‐4 from splenocytes of mRNA‐PNP vaccinated mice were determined by flow cytometry after ex vivo spike peptide pool stimulation. C) The percentage of CD8+ and D) CD4+ T cells producing cytokines from splenocytes of DNA‐PNP vaccinated mice. E) ELISpot assay for IFN‐γ in splenocytes from the mRNA‐PNP group and F) the DNA‐PNP group after ex vivo stimulation and representative images of plate wells. Data were shown as mean ± SD and n = 5 per group. Significance was calculated using unpaired t‐test (ns, not significant, *p < 0.05, **p < 0.01, ***p < 0.001).
3. Discussion
In this PNP system, L‐PBAEs were used as the biodegradable cationic polymer to condense negatively charged gene payloads, forming L‐PBAE/gene nanocomplexes. These nanocomplexes are embedded throughout the larger PLGA‐PEG vector,[ 13 ] providing protection and enhancing the colloidal stability in presence of serum as well as providing sustained release behavior. Grafting a lipid moiety onto PBAE is a rational design approach: first, incorporating hydrophobic lipid chains and improving its hydrophobicity to enhance the nanoparticle integrity and stability in physiological conditions, avoiding degradation caused by hydrolysis in aqueous solution. Second, the lipid moiety facilitates the fusion of PNPs into the phospholipid cell membrane and leads to cellular uptake via endocytosis to enhance the transfection efficacy.[ 19 , 23 , 26 ] This is exemplified in that all the PNP/L‐PBAE NPs showed an obvious increase of positive charge, associated slight increase in cytotoxicity, and increased transfection efficacy in serum circumstance over the unmodified PNP/PBAE control group. Out of the 24 PNP/L‐PBAE candidates, PNP/C12‐PBAE was screened as the top‐performing universal transfection formulation for both DNA and mRNA via high‐throughput luciferase assay and exhibited a much more explicit “Particle‐in‐Particle” nanostructure than the original PNP/PBAE formulation owing to the self‐assembly between the lipid‐modified C12‐PBAE and the PLGA‐PEG vector as shown under TEM. We demonstrated that the PNP/C12‐PBAE NP could effectively transfect fluorescence or luminescence encoded plasmid or mRNA and exhibited very limited cytotoxicity. Unlike the ALC‐0315 LNPs which were exclusively developed for mRNA delivery,[ 22 , 37 ] the PNP/C12‐PBAE NPs deliver both DNA and mRNA efficiently in vitro and in vivo.
Previous studies indicate that in an aqueous solution, mRNA is susceptible to hydrolysis causing instability and degradation. The existing research on the long‐term stability of naked mRNA suggests that mRNA needs to be frozen or dried in order to stay stable for longer periods of time,[ 5 , 6 ] Additionally, even though grafting a lipid moiety on PBAE results in more hydrophobicity and excellent stability, the L‐PBAEs are still a class of biodegradable cationic polymer which would be degraded in aqueous solution, decreasing the transfection efficacy by hydrolysis during long‐term storage. Therefore, lyophilization would be a logical improvement for the long‐term stability of the PNP delivery system and improves the feasibility of handling conditions without ultracold chain storage. During the lyophilization process, the nanoparticle structures are exposed to stress, therefore cryoprotectants such as sucrose need to be supplied in the formulation to stabilize the colloidal nanoparticles’ structure. We demonstrated that the lyophilized PNPs with 20 wt.% sucrose can be stored for at least 12 months at −20°C without loss of transfection efficacy compared to fresh PNP formulations in vitro and in vivo. Remarkably, the particle size and zeta potentials remain stable after lyophilized storage. Sucrose was previously reported to be the most effective cryoprotectant due to its very high viscosity, low molecular mobility after drying, and ability to form an amorphous, glassy matrix,[ 38 ] high glass transition temperature (T g) to maintain the temperature during lyophilization below Tg.[ 39 ] Moreover, sucrose molecules act as stabilizers to preserve the spacing between the NPs and reduce the van der Waals interactions in the dry state to stabilize the NP structure.[ 8 , 38 ]
After a solid and comprehensive evaluation of PNP/C12‐PBAE NPs with reporter gene in vitro and in vivo, we further formulated the PNP/C12‐PBAE NPs with SARS‐CoV‐2 spike encoded plasmid DNA and mRNA to develop lipid‐modified polymeric nanoparticle‐based DNA (DNA‐PNP) and mRNA (mRNA‐PNP) COVID‐19 vaccines. Based on the excellent characteristics and unique structure of the PNP systems, the DNA‐PNP and mRNA‐PNP vaccines were lyophilized and stored at ‐20°C for 12 months before mouse vaccination studies. To this end, we evaluated the spike protein expression in vitro and immunogenicity in a BALB/c mouse model. Immunization with mRNA‐PNP and DNA‐PNP vaccines in BALB/c mice elicited anti‐spike antibodies as well as SARS‐CoV‐2 spike neutralizing antibodies after the priming dose and the antibody titers significantly increased after a booster vaccination in a dose‐dependent manner demonstrated by the significant increase of GMTs compared to the placebo group. Furthermore, the intracellular cytokine staining study detected the secretion of IFN‐γ, TNF‐α, and IL‐2, but not IL‐4, demonstrating that our PNP system is a universal polymeric nanoparticle‐based DNA/mRNA vaccine platform that successfully induces a Th1‐biased SARS‐CoV‐2 spike‐specific immune response in vaccinated mice. Moreover, we found that the mRNA‐PNP vaccine induces both higher antibody titers and IFN‐γ secretion over DNA‐PNP vaccines in the immunized mice even with high DNA dosage. This may be due to the mRNA only needing to be in the cytoplasm to be translated into protein, compared to plasmid DNA which needs to be translocated to the nucleus for function.[ 40 ]
Different from mRNA‐LNP vaccines which have a shorter shelf life and reduced stability requiring ultracold‐chain storage (up to −70°C),[ 5 ] leading to extensive logistical challenges for distribution and administration, the lipid‐modified polymeric PNP gene delivery platform can be lyophilized and stored at −20°C for at least 12 months without loss of transfection efficacy for both DNA and mRNA. According to a previous study, PBAE was reported to maintain effective transfection after over 2 years at −20°C after lyophilization,[ 10 ] and we are led to believe from our data that the lipid‐modified PNP vaccines may have the potential to be stored for an even longer time while maintaining the transfection efficacy. Additionally, we did not perform an immunogenicity comparison study with commercial LNP vaccines, since the LNP systems are a different class of materials compared to our polymeric nanoparticle‐based PNP delivery system and the commercial vaccines utilize optimized mRNA sequences with modifications. This work is mainly focused on developing a universal lipid‐polymer hybrid gene delivery system for both plasmid and mRNA, which are capable of long‐term lyophilization storage. Therefore, for comparison studies, we mainly focused on delivering luciferase encoded mRNA and characterizing the luciferase expression in vivo to compare with the Pfizer/BioNTech ALC‐0315 LNP formulations.
In this manuscript, we introduced a series of chemically modified PBAEs amenable for conjugating lipid chains and used the lipid‐modified PBAEs to formulate a “Particle‐in‐Particle” PNP system to improve the transfection efficacy. We selected the unmodified PNP/PBAE as the control to perform the comparison studies on since the PNP/L‐PBAEs are derived from PNP/PBAE. Additionally, well‐known polymeric transfection agents such as polyethyleneimine (PEI) and polyamidoamine (PAMAM) dendrimers have been widely investigated for gene delivery and gene therapeutics.[ 41 ] It would be very interesting in future research to investigate if the transfection efficacy and physicochemical properties of other cationic polymers (PEI, PAMAM, etc.) could be improved after grafting the lipid chains on their polymeric or dendrimeric backbones to yield a stronger gene delivery system.
4. Conclusion
In summary, we report a new class of lipid‐modified PBAEs (L‐PBAEs) through CALB enzymatic‐assisted esterification and further formulated with PLGA‐PEG polymeric vectors to create enhanced PNP/L‐PBAE NPs for gene delivery. The rational design of PNP/L‐PBAE NPs presents a unique “Particle‐in‐Particle” nanostructure leading to sustained gene release behavior, enhanced transfection efficacy, and stability. The top‐performing candidate, PNP/C12‐PBAE NPs, efficiently delivered both DNA and mRNA in vitro and in vivo with the capability of storage after lyophilization for 12 months at −20 °C without loss of transfection efficacy and the particle size and zeta potentials remain stable after lyophilized storage. Encapsulating either SARS‐CoV‐2 spike protein‐encoding DNA or mRNA, the lyophilized PNP COVID‐19 vaccines could still induce robust spike‐specific antibodies and T cell immune responses in immunized mice after storage for 12 months at −20 °C. The universal characteristic of the PNP delivery platform lends itself for adaptation to limitless uses in infectious diseases as well as gene therapy applications. Our strategy may provide important steps toward a solution to meet the urgent demand for a lyophilizable gene delivery platform to fight against the COVID‐19 pandemic and other seasonal infectious diseases.
5. Experimental Section
Materials
Poly (DL‐lactide‐co‐glycolide) (50:50) with terminal carboxylate groups (PLGA, inherent viscosity: 0.55–0.75 dL g−1 in HFIP) was purchased from Lactel Absorbable Polymers (Birmingham, AL, USA). Amine PEG carboxyl, HCL salt (NH2‐PEG‐COOH, MW 3500) was obtained from Jenkem Technology (Beijing, China). 4‐Amino‐1‐butanol and 1‐(3‐Aminopropyl)‐4‐methylpiperazine were obtained from Alfa Aesar (Ward Hill, MA, USA). 1,4‐Butanediol diacrylate, Octanoic acid (C8), Decanoic acid (C10), Dodecanoic acid (C12), Myristic acid (C14), Palmitic acid (C16), Stearic acid (C18), Oleic acid, Cell Counting Kit‐8 (CCK‐8), Sodium acetate buffer (NaOAc, pH = 5), Cholesterol, Pur‐A‐Lyzer Midi Dialysis Kits (MWCO 3.5 kDa) and Candida Antarctica Lipase B (CALB) were obtained from Sigma‐Aldrich (St. Louis, MO, USA). Linoleic acid was purchased from Acros Organics (Fair Lawn, NJ, USA). ALC‐0315 was purchased from BroadPharm. 1,2‐distearoyl‐sn‐glycero‐3‐phosphocholine (DSPC) and 1,2‐dimyristoyl‐rac‐glycero‐3‐methoxypolyethylene glycol‐2000 (DMG‐PEG) were purchased from Avanti Polar Lipids, Inc. (AL, USA). Dulbecco's Modified Eagle Medium (DMEM, with 4.5 g L−1 D‐Glucose, L‐Glutamine, and 110 mg L−1 Sodium Pyruvate), Opti‐MEM reduced Serum Medium, Heat‐Inactivated Fetal Bovine Serum, Penicillin‐Streptomycin and 0.25% Trypsin‐EDTA (1X) were purchased from Gibco (Paisley, UK). Firefly luciferase encoded mRNA (mLuc) and mCherry encoded mRNA (mCherry) were purchased from TriLink Biotechnologies, Inc. (San Diego, CA). Bright‐Glo luciferase assay substrate was from Promega (Madison, WI). All lab supplies and analytical grade reagents were from VWR (Radnor, PA, USA) and Sigma‐Aldrich (St. Louis, MO, USA).
Synthesis of PLGA‐b‐PEG and poly (β‐amino ester) (PBAE‐447)
Copolymer PLGA‐b‐PEG was synthesized by the conjugation of COOH‐PEG‐NH2 to PLGA‐COOH as previously reported.[ 13 , 42 ] In brief, PLGA‐COOH (500 mg) was dissolved for 1 h in 3 mL DCM with 1‐(3‐Dimethylaminopropyl)‐3‐ethylcarbodiimide hydrochloride (EDC·HCL, 23 mg, 0.12 mmol) to activate the carboxylic acid of PLGA. Excess N‐hydroxysuccinimide (NHS, 13.5 mg, 0.11 mmol) was added to such solution to obtain PLGA‐NHS. PLGA‐NHS was precipitated with 20 mL of an ice‐cold mixture of ethyl ether and methanol (1: 1, vol: vol) and repeatedly washed using the same mixture two times to remove residual EDC and NHS. After drying under vacuum, PLGA‐NHS (100 mg, 0.0059 mmol) was dissolved in 3 mL chloroform followed by the addition of NH2‐PEG‐COOH (25 mg, 0.0071 mmol) and N,N‐diisopropylethylamine (DIEA, 2.8 mg, 0.022 mmol). The co‐polymer was precipitated with an ice‐cold mixture of ethyl ether and methanol (1: 1, vol: vol) after overnight reaction and washed with the same solvent two times to remove unreacted PEG. The resulting PLGA‐PEG block co‐polymer was dried under a vacuum and used for NP preparation without further treatment.
The unmodified cationic polymer poly (β‐amino ester) (PBAE‐447) was synthesized via a two‐step reaction with slight modifications as previously described.[ 11 , 13 ] Briefly, 1,4‐Butanediol diacrylate (4 g, 18.2 mmol) was polymerized with 4‐Amino‐1‐butanol (1.5 g, 16.5 mmol) for 24 h at 90 °C without any solvent with stirring. For the second step of the synthesis, the diacrylate‐terminated backbone reactants were dissolved in 15 mL anhydrous tetrahydrofuran (THF) and combined with 1‐(3‐Aminopropyl)‐4‐ methylpiperazine (1.57 g, 9.8 mmol) overnight at room temperature with stirring. After synthesis, the polymer PBAE‐447 THF was divided into four 15 mL tubes and then purified via precipitation in diethyl ether following centrifugation at 4000 rpm for 5 min to remove excess monomers in each tube. The supernatant was decanted to collect PBAE‐447 and repeated two times for washing with excess diethyl ether. Then the PBAE‐447 in each tube was used for modification with different lipid acids after drying under vacuum for 48 h.
Synthesis of Lipid‐Modified Poly (β‐amino esters) (L‐PBAEs)
The lipid‐modified poly (β‐amino esters) (L‐PBAEs) were synthesized through a one‐step esterification reaction between hydroxyl groups from PBAE‐447 and carboxylic acids from lipid acids library using CALB‐immobilized on acrylic resin as a catalyst.[ 27 ] Briefly, the PBAE‐447 (4.2 mmol) in each tube was dissolved in 8 mL THF and transferred into a 100 mL two‐neck round bottom flask followed by the addition of 0.42 mmol (1:10), 0.84 mmol (1:5) and 2.1 mmol (1:2) of lipid acids dissolved in 2 mL THF and 0.5 g CALB as esterification catalyst. The mixture was stirred at 55–60 °C under nitrogen protection for 7 h. After 7 h reaction, the mixture was centrifuged at 4000 rpm for 5 min and the collected supernatant was centrifuged again to make sure the CALB was removed. The clear L‐PBAEs THF supernatant was precipitated in cold diethyl ether following centrifugation at 4000 rpm for 5 min under cold temperature conditions. Then the supernatant was decanted and L‐PBAEs were washed with cold diethyl ether. The L‐PBAEs such as C8‐PBAE, C10‐PBAE, C12‐PBAE, C14‐PBAE, C16‐PBAE, C18‐PBAE, Oleic‐PBAE, and Lin‐PBAE were used directly to prepare PNP/L‐PBAE NPs after drying under vacuum for 48 h.
The molecular weight and polydispersities (PDI) of L‐PBAEs were determined by Gel Permeation Chromatography System (GPC, Malvern) with number‐averaged (Mn) and weight‐averaged (Mw) molecular weights. 1H nuclear magnetic resonance (NMR) spectra were recorded on a Bruker 500 MHz NMR spectrometer. FTIR spectra were collected using a Nicolet IS‐10 (Thermo Fisher Scientific).
Preparation of PNP/L‐PBAE Nanoparticle Formulations
The PNP/L‐PBAE NPs were prepared through self‐assembly of polymeric PLGA‐PEG and cationic L‐PBAEs hybrid systems using a double‐emulsion solvent evaporation method with slight modifications to a previously described method.[ 13 ] Briefly, 5–10 mg copolymer PLGA‐PEG and 10 mg L‐PBAEs were co‐dissolved in 1 mL methylene chloride (DCM). UltraPure Distilled Water (DNase and RNase free, Invitrogen) was added drop‐wisely into 1 mL of PLGA‐PEG and L‐PBAEs solution and emulsified by probe sonification (Qsonica Sonicatiors, Newtown, CT, USA) to form the first emulsion. Next, the emulsified mixture was added into 3 mL of an aqueous solution containing 1.67 wt.% PVA, followed by probe sonification to form the double emulsion. The final emulsion solution was added drop‐wisely into 7 mL of DI water and stirred for 2.5 h at 800 rpm to allow the DCM solvent to evaporate and the particles to harden. The resulting NPs were then concentrated to a final volume of 0.5 or 1 mL (for animal experiments and cell experiments, respectively) using a 50 mL Amicon Ultra Centrifugal Filter (MWCO 100 kDa, Millipore) for 50 min at 1600 rpm (515 g) in a centrifuge (Eppendorf, 5810 R). The concentrated gene‐unloaded PNP/L‐PBAE NPs could either be mixed with the desired amount of plasmid DNA or mRNA via pipette with 25–30 min incubation at RT to be fresh formulations or stored at −20 °C. For long‐term storage studies, the PNPs were supplied with different sucrose solutions to be either frozen or lyophilized and stored at 4 °C or −20 °C for 12 months.[ 7 , 31 ]
Nanoparticle Characterization and Morphology Analysis
The NP size and zeta potential were measured using a Zeta Sizer dynamic light‐scattering detector (15‐mW laser, incident beam of 676 nm; Malvern, UK) at 25 °C and at a scattering angle of 90°. The intensity‐weighted mean value was recorded as the average of three measurements.
The morphology of gene‐loaded PNP/C12‐PBAE NPs, PLGA‐PEG NPs, and C12‐PBAE NPs were observed under transmission electron microscope (JEM‐F200 TEM, USA). Samples were prepared by placing 5 µL of concentrated NPs on TEM grids and removed by filter paper after 1 min. The sample was stained with 5 µL of 2% uranyl acetate solution for 30 s, then the uranyl acetate was removed by filter paper. The staining procedures were repeated another 2 times in the same manner. After staining, the samples were placed in the fume hood for 10 min drying and then directly observed using TEM.
Gel Electrophoresis
The binding efficiency of PNP/C12‐PBAE NPs with plasmid or mRNA was investigated by agarose gel electrophoresis assay. Briefly, 10 uL plasmid or mRNA‐loaded PNP samples (with different concentrations) were mixed with 2 uL RNA loading dye (New England Biolab) and then loaded on a 1% agarose gel for 30 min at 110 V. The gel images were acquired using an ChemiDoc Gel Imaging System (Bio‐Rad Laboratories).
Spike Encoded mRNA Synthesis
SARS‐CoV‐2 full‐length spike encoded mRNA was purchased from System Biosciences. Briefly, wild‐type full‐length spike mRNA was synthesized by using in vitro transcription (IVT) reactions templated by PCR amplicons with non‐modified nucleosides and did not involve any modified nucleosides such as pseudouridine. To promote efficient translation and boost RNA half‐life in the cytoplasm, a 3′‐O‐Me‐m7G (5′) ppp (5′) G ARCA cap analog was incorporated into the IVT reactions (TriLink Biotechnologies). Within IVT templates, the open reading frame (ORF) of spike was flanked by a 5′ untranslated region (UTR) containing a strong Kozak translational initiation signal and an alpha‐globin 3″ UTR terminating with an oligo (dT) sequence for templated addition of a polyA tail.
Animals and Cells
All animal procedures were performed with ethical compliance and approval by the Animal Care and Use Committee at Rutgers‐New Jersey Medical School (Protocol No.202000177). Female BALB/c mice (6–8 weeks) were obtained from the Jackson Laboratory and housed in Rutgers‐New Jersey Medical School animal facility.
The Hek 293 cell line was kindly given by Dr. Lei Bu from NYU Langone Medical Center as a gift. The ACE2‐293T stable cell line was a gift by Dr. Abraham Pinter from Rutgers‐New Jersey Medical School. These cells were cultured in high glucose Dulbecco's modified Eagle's medium (DMEM; Gibco Life Technologies, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS; Sigma‐Aldrich), and 1% penicillin–streptomycin (Gibco Life Technologies, Carlsbad, CA). The splenocytes from vaccinated mice were cultured in RPMI‐1640 (Gibco Life Technologies) supplemented with 10% FBS and 1% penicillin–streptomycin. All cells were cultured at 37 °C in a humidified incubator with 5% CO2.
In vitro Transfection and High‐throughput Screening Out
The Hek 293 cells were seeded into round‐bottom, black, and clear 96‐well plates (Corning) at 2 × 104 cells per well in 100 µL complete culture medium and incubated at 37 °C overnight prior to transfection. For PNP/L‐PBAE NPs cell transfection, 0.1 µg luciferase encoded mRNA (mLuc) or plasmid (pLuc) was diluted in 5 µL NaOAc buffer (pH 5, 25 × 10−3 m) (or Opti‐MEM serum reduced medium) and mixed with 1.5 µL, 2 µL, 2.5 µL or 3 µL concentrated NPs (1 mL final volume) and the NPs concentration varies from 0.2 to 0.5 mg mL−1 in each well, respectively. Incubate 25 min for gene encapsulation and add the gene‐loaded NPs directly into a 96‐well plate. After 4 h culture, the transfection medium was replaced with 100 µL complete culture medium (optional), and the cells were incubated sequentially for 6, 24, 48, 72, and 96 h post‐transfection. The luciferase transfection efficiency and the cell viability were simultaneously measured using Bright Glo Luciferase Assay System (Promega) and CCK‐8 kit according to manufacturer's instructions. The luminescence and absorbance were quantified using the Tecan Infinite M200 Pro plate reader (Tecan).
Evaluation of In Vitro Transfection by Fluorescence Microscopy and Flow Cytometry
The transfection efficacy of PNP/L‐PBAE NPs was evaluated in the Hek 293 cell line using GFP encoded plasmid (pGFP) and mCherry encoded mRNA as reporter genes. 1 × 105 Hek 293 cells were seeded in 0.5 mL complete culture medium in each well of a 24‐well plate and incubated at 37 °C overnight. For PNP/L‐PBAE NPs preparation, 1 µg pGFP or mCherry was diluted in 20 µL NaOAc buffer (or Opti‐MEM medium), then mixed with 10 µL concentrated NPs for 25 min and added into 24‐well plate. ALC‐0315 LNPs were prepared as ALC‐0315/DSPC/cholesterol/DMG‐PEG = 46/10/42/2, ALC‐0315/mRNA = 10/1 (wt./wt.) with 1 µg pGFP or mCherry in 25 × 10−3 m NaOAc buffer (pH = 5).[ 5 ]
After 4 h incubation, the transfection medium was replaced with 0.5 mL complete culture medium (optional), and the cells were observed by All‐in‐One Fluorescence Microscope (BZ‐X710, Keyence, Japan) at 6, 24, 48, 72, and 96 h with brightfield, fluorescent and merged pictures using 10× PanFluor lens (Nikon, Japan). After observation and taking images, the cells were trypsinized by 0.25% Trypsin–EDTA, followed by the addition of 0.5 mL PBS and centrifuged to decant supernatant to get cell pellets. The cell pellets were resuspended in 0.5 mL PBS for flow cytometric analysis using a BD LSR II flow cytometer (BD Biosciences, San Jose, CA) and the data were analyzed using FACSDiva software (BD Biosciences, San Jose, CA). Data were acquired using a 488 nm laser with a 530/30 BP filter for the detection of GFP positive cells under a voltage of 250 V and a 561 nm laser with a 610/20 BP filter for the detection of mCherry positive cells under a voltage of 400 V. 10 000 events were collected for each measurement.
In vitro Gene Release
The in vitro plasmid or mRNA release from PNP/C12‐PBAE NPs was measured over 8 days using separate samples for each time point according to the following procedures.[ 30 ] Briefly, the concentrated PNP/C12‐PBAE NPs were diluted by a factor of 10 using 1X PBS buffer. 200 µL of NPs solution was loaded in 1.5 mL Eppendorf tubes and then shaken horizontally at 37 °C and 300 rpm (Eppendorf Thermomixer R). At predetermined time intervals, the NPs were centrifuged at 10,000 g for 5 min (Eppendorf centrifuge 5418) and the supernatants were collected for analysis. The amount of plasmid (pGFP) and mRNA (mCherry) released from PNP/C12‐PBAE NPs was evaluated by Quant‐iT PicoGreen dsDNA Assay Kit and Quant‐iT RiboGreen RNA Assay Kit according to the manufacturer's protocol, respectively. Background readings were corrected using the centrifugation supernatants from the control group which was from PNP/C12‐PBAEs NPs without gene loaded.
Western Blot
The Western blot was prepared following a previous protocol. Briefly, the Hek 293 cells after transfection by spike plasmid or spike mRNA loaded PNP/C12‐PBAE were harvested using cold cell lysis buffer (Boster Bio Tech, CA, USA) at preset time points and stored at −20 °C until use. After measuring protein concentration of each sample by Bio‐Rad protein assay, the protein samples were mixed with loading dye following heated at 95 °C for 5 min, and loaded onto a 5% stacking/12% resolving SDS‐polyacrylamide gel (Bio‐Rad). The proteins were then electrophoretically transferred onto a polyvinylidene difluoride membrane (Bio‐Rad) in a 4 °C cool room for 4 h and the membranes were blocked overnight at 4 °C using 3% BSA PBS solution. Next day, the membranes were incubated for 2 h with the following primary antibodies: rabbit anti‐spike (1:1000; NR‐52947, BEI Resources), rabbit anti‐GAPDH (1:2000; Boster Bio‐Tech, CA, USA). The membranes were submerged in Tris‐buffered saline Tween 20 (TBST), washed 3 times, and incubated for 1 h with HRP conjugated anti‐rabbit (IgG) secondary antibody (1:5000; Amersham, Cytiva) at room temperature following 3 times washed by TBST. The proteins were visualized by Western Lightning Plus‐ECL, Enhanced Chemiluminescence Substrate (PerkinElmer). Exposure was done using ChemiDoc Imaging System with Image Lab software (Bio‐Rad).
Immunofluorescence Assay
Immunofluorescence assay was performed for staining of spike protein expressed on Hek 293 cells. Briefly, 2 × 105 Hek 293 cells were seeded on cover glass, placed in a 12‐well plate overnight. The next day, the cells were transfected by spike plasmid or spike mRNA‐loaded PNP/C12‐PBAE NPs following culture for 3 and 2 days, respectively. The cells were fixed with cold 4% Paraformaldehyde (PFA) for 10 min and cold methanol for 10 min, then washed using PBS. Before staining, the cells seeded on cover glass were blocked with 3% BSA PBS solution for 30 min at RT. Cells were stained with rabbit anti‐spike antibody (1:500; NR‐52947, BEI Resources) for 2 h at RT. The cover glasses were washed 3 times with PBS and then stained with Alexa Fluor 488 conjugated goat anti‐rabbit IgG (1:1000; Abcam) for 1 h at RT. The cover glasses were washed again and the nucleus was stained with one drop of DAPI (Invitrogen). After sealing the slides, images were taken by All‐in‐One Fluorescence Microscope (BZ‐X710, Keyence) with brightfield, fluorescent (FITC, DAPI), and merged pictures using 20× PanFluor lens (Nikon).
In Vivo Nanoparticle Administration and Bioluminescence
Female, 6–8 weeks old BALB/c mice from Jackson Laboratory (n = 3 per group) were used for the vivo bioluminescence imaging study. 10 µg (≈0.5 mg/kg) luciferase mRNA (mLuc) was diluted in 50 µL NaOAc buffer and then formulated with 50 µL of the 0.5 mL concentrated fresh PNP/C12‐PBAE NPs for 25–30 min at RT as the fresh formulations (n = 3). The 12 months stored lyophilized PNPs included 20 wt.% sucrose at −20 °C was prepared in the same way and resuspended before injection (n = 3). The unmodified PNP/PBAE encapsulated with 10 µg mLuc (n = 3) and 50 µL PBS (n = 3) were injected into mice via intramuscular route as control groups. ALC‐0315 LNPs were prepared as ALC‐0315/DSPC/cholesterol/DMG‐PEG = 46/10/42/2, ALC‐0315/mRNA = 10/1 (wt./wt.) with 10 µg mLuc in 25 × 10−3 m, pH 5 NaOAc buffer (n = 3).[ 5 ] At indicated time points, animals were injected intraperitoneally with 150 µL D‐Luciferin potassium salt (30 mg mL−1, PerkinElmer). After reaction for 15–20 minutes, luminescence signals were collected by IVIS spectrum instrument (IVIS‐200, Xenogen) with an exposure time of 30 s.
Histology and Tissue Immunofluorescence Assay
The mice were sacrificed and muscle tissue around the PNP injection site was collected and submerged in 4% paraformaldehyde (PFA) overnight for fixation and following 30% sucrose solution for dehydration. Then the tissues were collected in a cryomold, embedded with Tissue‐Tek O.C.T. compound (VWR), frozen at −80 °C, sectioned (15 µm thick) using a Leica CM3050 S cryostat. For histology study, the sections were stained with hematoxylin and eosin (H&E) (Sigma‐Aldrich) for histological examination according to the manufacturer's instructions. For immunofluorescence assay, the sections were first blocked for 1 h at 25 °C in 1xPBS containing 5% goat serum and 3% BSA. Then, the sections were permeabilized by 1xPBS containing 1% TritonX‐100 for 30 min at RT. The sections were then incubated with rabbit anti‐IL‐6 (1:1000, ab290735, Abcam, Cambridge, MA) overnight at 4 °C. After washing three times, the sections were incubated with goat anti‐rabbit IgG (Alexa Fluor 488) (1:1000) for 2 h at RT. After washing three times, the tissues were mounted using ProLong Glass Antifade Mountant with NucBlue Stain on the glass slide with a coverslip. The images were taken by All‐in‐One Fluorescence Microscope (BZ‐X710, Keyence).
Mouse Vaccination Design
Female, 6–8 weeks old BALB/c mice (n = 5 biologically independent animals per group) from Jackson Laboratory were used for immunogenicity studies. Different doses of lyophilized DNA‐PNP and mRNA‐PNP (with 20 wt.% sucrose) were stored for 12 months at −20°C in the same preparation described above and resuspended in 100 µL NaOAc buffer before injection. Mice were inoculated intramuscularly in the left tibialis anterior for both prime and boost injections at day 0 and day 14, respectively. For control groups, mice received 50 µL empty NPs.[ 43 ] The blood samples were collected on day 0, day 13, day 21, and day 35 via the orbital sinus under isoflurane anesthesia, and mice were sacrificed on day 42 to collect spleens for cellular immune response analysis. Blood samples were allowed to clot at room temperature and centrifuged at 2500 g for 10 min at 4 °C, then the serum samples were heat‐inactivated at 56 °C for 30 min and stored at −20 °C until use.
ELISA
SARS‐CoV‐2 spike‐specific IgG titers were determined by ELISA. Briefly, 96‐well microtiter ELISA plates (MaxiSorp, Thermo Fisher Scientific) were coated with 50 µL per well of a 2 µg ml−1 SAR‐CoV‐2 spike S1+S2 protein (40589‐V08B1, Sino Biological) suspended in 1x PBS overnight at 4 °C. Next day, the spike protein‐coated ELISA plates were washed three times with 200 µL wash buffer (ELISA Buffer Kit, Thermo Fisher Scientific) following 200 µL assay buffer as blocking solution for 1 h at room temperature. 100 µL of serial dilutions of mice heat‐inactivated sera samples in assay buffer were added to the plates and incubated at RT for 2 h. After three washes, the plates were added with 50 µL of HRP‐conjugated goat anti‐mouse IgG (1:3000, Novus Biologicals) and incubated at RT for 1 h with three washes. The binding antibody was detected by 50 µL TMB substrate solution for 15 min following 50 µL Stop Solution. The absorbance at 450 nm was measured using Tecan Infinite M200 Pro plate reader (Tecan). The endpoint titers were determined using a 4‐parameter logistic curve fit in GraphPad Prism and defined as the highest reciprocal serum dilution that yielded an absorbance≥2.1 fold over negative control.[ 44 ]
SARS‐CoV‐2 Pseudovirus Neutralization Assay
HIV‐based luciferase expressing lentivirus pseudotyped with SARS‐CoV‐2 full‐length spike protein (Creative Biogene Inc) was used for the pseudovirus neutralization assay. Serial dilutions of heat‐inactivated sera were incubated with SARS‐CoV‐2 pseudovirus for 60 min at 37 °C before addition to ACE2‐293T cells previously seeded in the 96‐well microplate. Cells were lysed using a Bright‐Glo Luciferase Assay System kit (Promega) according to the manufacturer's instructions 48 h post‐infection and the luciferase activity in relative light units (RLU) was measured by Tecan plate reader. Neutralization half‐maximal inhibitory concentration (IC50) titers were calculated as the serum dilution at which RLU was reduced by 50% compared with the RLU in pseudovirus‐only control wells after subtraction of background RLU in cell control wells.
Plaque Reduction Neutralization Tests (PRNT) Assay
The Vero cells were seeded in 24‐well plates with around 90% confluent after overnight incubation. The serial diluted serum samples were mixed with an equal volume of wild‐type Wuhan SARS‐CoV‐2 strain virus to obtain the mixtures containing ≈200 PFU mL−1 of viruses and incubated for 1 h at 37 °C. The mixed samples were added to the 24‐well plates of Vero cell monolayers. The plates were then incubated at 37 °C for 1 h with intermittent rocking every 20 min. Then, the mixtures were removed and cells were overlaid with 1% agarose in DMEM containing 2% FBS. After further incubation at 37 °C for 2 days, the cells were fixed with 4% PFA and stained with 0.2% crystal violet. Plaque numbers were recorded after rinsing the plates with DI water. The 50% neutralization titer of plaque reduction neutralization tests (PRNT50) was calculated and defined as the reciprocal serum dilution at the neutralization curve crossed the 50% threshold.[ 45 ]
ELISpot
Spleens from vaccinated and placebo mice were collected individually, homogenized into single cell suspensions by 70 µm cell strainer following ACK lysis buffer (Quality Biological) to eliminate red blood cells, and then cultured in RPMI1640 media supplemented with 10% FBS and 1% penicillin/streptomycin (R10). SAR‐CoV‐2 specific cellular immune responses in mice were evaluated by Mouse IFN‐γ ELISpot kit (Mabtech, Sweden) according to the manufacturer's instructions. Briefly, the well plate was washed 3 times using PBS and blocked by R10 medium for 2 h. 2 × 105 splenocytes from vaccinated mice were seeded into each well and stimulated by 2 µg mL−1 of the peptide pool consisting of 15‐mer sequences with 11 amino acids overlap, covering the spike glycoprotein (Miltenyi Biotec) in a 37 °C cell incubator for 24 h. The well plate was washed 5 times by PBS and incubated with biotin‐conjugated detection antibody for 2 h at RT, washed, incubated with streptavidin‐ALP for 1 h at RT, washed, and developed with BCIP/NBT‐plus substrate for 20 min. The plate was then washed extensively in DI water to stop color development following drying in the dark at RT, and the spots were imaged and analyzed by CTL ImmunoSpot Analyzer. Concanavalin A (ConA, 2 µg mL−1) was used as positive control and a complete medium only was used as the negative control. The results were expressed as the number of IFN‐γ spots per 1 × 106 splenocytes.
Intracellular Cytokine Staining
Cytokine‐producing T cells were evaluated by intracellular cytokine stating. Briefly, 1 × 106 of splenocytes were stimulated with or without 2 µg mL−1 of the peptide pool covering the spike glycoprotein (Miltenyi Biotec) for 6 h at 37 °C in the presence of BD GolgiPlug to inhibit the intracellular protein transport process during the last 4 h. Cells were washed and stained for viability by LIVE/DEAD Fixable Near‐IR Dead Cell Stain Kit (1:1000, Invitrogen) following a mixture of antibodies against surface markers: PerCP‐Cy5.5 anti‐CD3 (clone 17A2, 1:100, BD Biosciences), Alexa Fluor 700 anti‐CD4 (clone RM4‐5, 1:100, BD Biosciences) and FITC anti‐CD8a (Clone 53–6.7, 1:100, BD Biosciences). After washes, the cells were fixed and permeabilized by BD Cytofix/Cytoperm (BD Biosciences) according to the manufacturer's instructions. The cells were washed with Perm/Wash buffer (BD Biosciences) and stained with PE anti‐IFN‐γ (clone XMG1.2, 1:100, BD Biosciences), PE‐Cy7 anti‐TNF (clone MP6‐XT22, 1:100, BD Biosciences), BV421 anti‐IL‐2 (clone JES6‐5H4, 1:100, BD Biosciences) and APC anti‐IL‐4 (clone 11B11, 1:100, BD Biosciences). The cells were washed with Perm/Wash buffer and resuspended in PBS for flow cytometric analysis using a BD LSR II Flow Cytometer (BD Biosciences) and the data were acquired by FACS Diva software with at least 150 000 events collected (BD Biosciences, San Jose, CA).
Statistical Analysis
The mean value was plotted in all graphs and the error bars represent the standard deviation. All statistical analyses and significance were performed by GraphPad Prism 8 software (La Jolla, CA) or Microsoft Excel. A one‐way ANOVA with multiple comparisons was used for over two groups and an unpaired t‐test was used to compare between two groups. Statistical significance among different groups and the data were considered statistically significant if p < 0.05 (ns, not significant; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). The experiments were not randomized, and the investigators were not blinded to allocation during experiments and outcome assessment.
Conflict of Interest
The authors declare no conflict of interest.
Author Contributions
Z.L., X.Z., and X.X. conceived the project and designed the experiments. Z.L. designed, synthesized, and characterized the materials and nanoparticles. Z.L. performed most experiments and prepared the figures. Z.L., W.H., X.B., D.K.J., F.L., R.K., A.K., S.S., H.Z., X.Z., and X.X. analyzed and discussed the results. Z.L. and X.X. wrote the manuscript. Z.L., W.H., X.Z., and X.X. reviewed and edited the manuscript. Z.L. X.Z. and X.X. were primarily responsible for the data analysis and interpretation. Z.L. and X.Z. contributed equally to this work. All authors have given approval to the final version of the manuscript.
Supporting information
Supporting Information
Acknowledgements
This work was supported by American Heart Association grant #19AIREA34380849 (X.X.). X.X. also acknowledges support from the National Science Foundation (2001606) and the Gustavus and Louise Pfeiffer Research Foundation Award. X.‐Q.Z. acknowledges support from the Interdisciplinary Program of Shanghai Jiao Tong University [project number ZH2018ZDA36 (19X190020006)], Shanghai Jiao Tong University Scientific and Technological Innovation Funds (2019TPA10), and the Foundation of National Facility for Translational Medicine (Shanghai) (TMSK‐2020‐008).
Li Z., Zhang X.‐Q., Ho W., Bai X., Jaijyan D. K., Li F., Kumar R., Kolloli A., Subbian S., Zhu H., Xu X., Lipid‐Polymer Hybrid “Particle‐in‐Particle” Nanostructure Gene Delivery Platform Explored for Lyophilizable DNA and mRNA COVID‐19 Vaccines. Adv. Funct. Mater. 2022, 32, 2204462. 10.1002/adfm.202204462
Contributor Information
Xue‐Qing Zhang, Email: xueqingzhang@sjtu.edu.cn.
Xiaoyang Xu, Email: xiaoyang@njit.edu.
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.
References
- 1. Sahin U., Muik A., Derhovanessian E., Vogler I., Kranz L. M., Vormehr M., Baum A., Pascal K., Quandt J., Maurus D., Nature 2020, 586, 594. [DOI] [PubMed] [Google Scholar]
- 2. Polack F. P., Thomas S. J., Kitchin N., Absalon J., Gurtman A., Lockhart S., Perez J. L., Marc G. P., Moreira E. D., Zerbini C., N. Engl. J. Med. 2020, 383, 2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Baden L. R., El Sahly H. M., Essink B., Kotloff K., Frey S., Novak R., Diemert D., Spector S. A., Rouphael N., Creech C. B., N. Engl. J. Med. 2021, 384, 403.33378609 [Google Scholar]
- 4. Widge A. T., Rouphael N. G., Jackson L. A., Anderson E. J., Roberts P. C., Makhene M., Chappell J. D., Denison M. R., Stevens L. J., Pruijssers A. J., N. Engl. J. Med. 2021, 384, 80.33270381 [Google Scholar]
- 5. Schoenmaker L., Witzigmann D., Kulkarni J. A., Verbeke R., Kersten G., Jiskoot W., Crommelin D. J., Int. J. Pharm. 2021, 601, 120586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Uddin M. N., Roni M. A., Vaccines 2021, 9, 1033.34579270 [Google Scholar]
- 7. Ball R. L., Bajaj P., Whitehead K. A., Int. J. Nanomed. 2017, 12, 305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen C., Han D., Cai C., Tang X., J. Controlled Release 2010, 142, 299. [DOI] [PubMed] [Google Scholar]
- 9. Pack D. W., Hoffman A. S., Pun S., Stayton P. S., Nat. Rev. Drug Discovery 2005, 4, 581. [DOI] [PubMed] [Google Scholar]
- 10. Guerrero‐Cázares H., Tzeng S. Y., Young N. P., Abutaleb A. O., Quiñones‐Hinojosa A., Green J. J., ACS Nano 2014, 8, 5141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mangraviti A., Tzeng S. Y., Kozielski K. L., Wang Y., Jin Y., Gullotti D., Pedone M., Buaron N., Liu A., Wilson D. R., ACS Nano 2015, 9, 1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tzeng S. Y., Guerrero‐Cázares H., Martinez E. E., Sunshine J. C., Quiñones‐Hinojosa A., Green J. J., Biomaterials 2011, 32, 5402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li Z., Ho W., Bai X., Li F., Chen Y.‐j., Zhang X.‐Q., Xu X., J. Controlled Release 2020, 322, 622. [DOI] [PubMed] [Google Scholar]
- 14. Karlsson J., Rhodes K. R., Green J. J., Tzeng S. Y., Expert Opinion on Drug Delivery 2020, 17, 1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Eltoukhy A. A., Chen D., Alabi C. A., Langer R., Anderson D. G., Adv. Mater. 2013, 25, 1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Love K. T., Mahon K. P., Levins C. G., Whitehead K. A., Querbes W., Dorkin J. R., Qin J., Cantley W., Qin L. L., Racie T., Proc. Natl. Acad. Sci. USA 2010, 107, 1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xu X., Xie K., Zhang X.‐Q., Pridgen E. M., Park G. Y., Cui D. S., Shi J., Wu J., Kantoff P. W., Lippard S. J., Proc. Natl. Acad. Sci. USA 2013, 110, 18638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tao W., Yurdagul A., Kong N., Li W., Wang X., Doran A. C., Feng C., Wang J., Islam M. A., Farokhzad O. C., Sci. Transl. Med. 2020, 12, 1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Semple S. C., Akinc A., Chen J., Sandhu A. P., Mui B. L., Cho C. K., Sah D. W., Stebbing D., Crosley E. J., Yaworski E., Nat. Biotechnol. 2010, 28, 172. [DOI] [PubMed] [Google Scholar]
- 20. Sinegra A. J., Evangelopoulos M., Park J., Huang Z., Mirkin C. A., Nano Lett. 2021, 21, 6584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu S., Cheng Q., Wei T., Yu X., Johnson L. T., Farbiak L., Siegwart D. J., Nat. Mater. 2021, 20, 701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hou X., Zaks T., Langer R., Dong Y., Nat. Rev. Mater. 2021, 6, 1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hafez I., Maurer N., Cullis P., Gene Ther. 2001, 8, 1188. [DOI] [PubMed] [Google Scholar]
- 24. Chen Q., Gao M., Li Z., Xiao Y., Bai X., Boakye‐Yiadom K. O., Xu X., Zhang X.‐Q., J. Controlled Release 2020, 323, 179. [DOI] [PubMed] [Google Scholar]
- 25. Kaczmarek J. C., Kauffman K. J., Fenton O. S., Sadtler K., Patel A. K., Heartlein M. W., DeRosa F., Anderson D. G., Nano Lett. 2018, 18, 6449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stamatatos L., Leventis R., Zuckermann M. J., Silvius J. R., Biochemistry 1988, 27, 3917. [DOI] [PubMed] [Google Scholar]
- 27. Tsou Y. H., Zhang X. Q., Bai X., Zhu H., Li Z., Liu Y., Shi J., Xu X., Adv. Funct. Mater. 2018, 28, 1802607. [Google Scholar]
- 28. Fröhlich E., Int. J. Nanomed. 2012, 7, 5577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tian X., Zhu H., Du S., Zhang X.‐Q., Lin F., Ji F., Tsou Y.‐H., Li Z., Feng Y., Ticehurst K., The Journal of Pain 2021, 22, 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Perez C., Sanchez A., Putnam D., Ting D., Langer R., Alonso M., J. Controlled Release 2001, 75, 211. [DOI] [PubMed] [Google Scholar]
- 31. Zhao P., Hou X., Yan J., Du S., Xue Y., Li W., Xiang G., Dong Y., Bioactive materials 2020, 5, 358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Miao L., Li L., Huang Y., Delcassian D., Chahal J., Han J., Shi Y., Sadtler K., Gao W., Lin J., Nat. Biotechnol. 2019, 37, 1174. [DOI] [PubMed] [Google Scholar]
- 33. Wang B., Tang M., Yuan Z., Li Z., Hu B., Bai X., Chu J., Xu X., Zhang X.‐Q., Bioactive materials 2022, 16, 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Coutard B., Valle C., de Lamballerie X., Canard B., Seidah N., Decroly E., Antiviral Res. 2020, 176, 104742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tian J.‐H., Patel N., Haupt R., Zhou H., Weston S., Hammond H., Lague J., Portnoff A. D., Norton J., Guebre‐Xabier M., Nat. Commun. 2021, 12, 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Polack F. P., Thomas S. J., Kitchin N., Absalon J., Gurtman A., Lockhart S., Perez J. L., Pérez Marc G., Moreira E. D., Zerbini C., N. Engl. J. Med. 2020, 383, 2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.a) Jayaraman M., Ansell S. M., Mui B. L., Tam Y. K., Chen J., Du X., Butler D., Eltepu L., Matsuda S., Narayanannair J. K., Angew. Chem. 2012, 124, 8657; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang Y., Sun C., Wang C., Jankovic K. E., Dong Y., Chem. Rev. 2021, 121, 12181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Stark B., Pabst G., Prassl R., Eur. J. Pharm. Sci. 2010, 41, 546. [DOI] [PubMed] [Google Scholar]
- 39. del Pozo‐Rodríguez A., Solinís M., Gascón A., Pedraz J., Eur. J. Pharm. Biopharm. 2009, 71, 181. [DOI] [PubMed] [Google Scholar]
- 40. Liu M. A., Vaccines 2019, 7, 37. [Google Scholar]
- 41.a) Zhang X.‐Q., Intra J., Salem A. K., J. Microencapsulation 2008, 25, 1; [DOI] [PubMed] [Google Scholar]; b) Zhang X.‐Q., Intra J., Salem A. K., Bioconjugate Chem. 2007, 18, 2068. [DOI] [PubMed] [Google Scholar]
- 42. Cheng J., Teply B. A., Sherifi I., Sung J., Luther G., Gu F. X., Levy‐Nissenbaum E., Radovic‐Moreno A. F., Langer R., Farokhzad O. C., Biomaterials 2007, 28, 869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Corbett K. S., Edwards D. K., Leist S. R., Abiona O. M., Boyoglu‐Barnum S., Gillespie R. A., Himansu S., Schäfer A., Ziwawo C. T., DiPiazza A. T., Nature 2020, 586, 567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wu S., Zhong G., Zhang J., Shuai L., Zhang Z., Wen Z., Wang B., Zhao Z., Song X., Chen Y., Nat. Commun. 2020, 11, 4081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang N.‐N., Li X.‐F., Deng Y.‐Q., Zhao H., Huang Y.‐J., Yang G., Huang W.‐J., Gao P., Zhou C., Zhang R.‐R., Cell 2020, 182, 1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.