

Abstract
Several traditional observational studies suggested an association between COVID‐19 and leukocyte telomere length (LTL), a biomarker for biological age. However, whether there was a causal association between them remained unclear. We aimed to investigate whether genetically predicted COVID‐19 is related to the risk of LTL, and vice versa. We performed bidirectional Mendelian randomization (MR) study using summary statistics from the genome‐wide association studies of critically ill COVID‐19 (n = 1 388 342) and LTL (n = 472 174) of European ancestry. The random‐effects inverse‐variance weighted estimation method was applied as the primary method with several other estimators as complementary methods. Using six single‐nucleotide polymorphisms (SNPs) of genome‐wide significance as instrumental variables for critically ill COVID‐19, we did not find a significant association of COVID‐19 on LTL (β = 0.0075, 95% confidence interval [CI]: −0.018 to 0.021, p = 0.733). Likewise, using 97 SNPs of genome‐wide significance as instrumental variables for LTL, we did not find a significant association of LTL on COVID‐19 (odds ratio = 1.00, 95% CI: 0.79–1.28, p = 0.973). Comparable results were obtained using MR‐Egger regression, weighted median, and weighted mode approaches. We did not find evidence to support a causal association between COVID‐19 and LTL in either direction.
Keywords: bidirectional two‐sample Mendelian randomization, COVID‐19, telomere length.
1. INTRODUCTION
The current COVID‐19 pandemic, because of severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) infection, has led to enormous health and economic consequences worldwide. The majority of individuals who died from COVID‐19 are the aged, those with cardiovascular disease (CVD), diabetes, chronic obstructive pulmonary disease (COPD), and cancer alike. 1 , 2 , 3 In contrast, young adults, infants, and children typically had milder clinical symptoms. 4 , 5 , 6 Respiratory failure and multiorgan dysfunction resulting from impaired immune response and uncontrolled inflammatory process are the leading causes of death in COVID‐19 patients. 7 , 8 , 9 The severe infection, occasionally along with a “cytokine‐storm” symptom in which the level of oxidative stress increase significantly, 10 , 11 may alter host cell physiology and cellular functions. 12 , 13 , 14 At the population level, SARS‐CoV‐2 and COVID‐19 may lead to aging‐related diseases or accelerate the aging process.
As specialized structures at the ends of chromosomes, telomeres maintain genome stability. 15 As cells divide and DNA replicates, telomeres become progressively shorter because of the “end replication problem.” 16 , 17 Telomeres have been proposed as one of the biomarkers of aging for a long. 18 In addition to aging, environmental factors, such as exposure to pollutants, smoking, and oxidative stress, may shorten telomeres, and lead to an increased risk of aging‐related diseases. 19 , 20 , 21 , 22 Several studies reported that COVID‐19 was associated with shorter leukocyte telomere length (LTL). Mongelli et al., 23 Sanchez‐Vazquez et al., 24 Benetos et al., 25 Dos Santos et al., 26 and Froidure et al. 27 conducted case‐control studies and discovered that severe post‐COVID‐19 survivors had shorter telomeres compared with milder post‐COVID‐19 survivors or COVID‐19‐free people, which suggested significant correlations of LTL with COVID‐19 severity. However, the observed association between COVID‐19 and LTL in observational studies might be biased because of confounding factors. 28 , 29 Thus, it is not clear whether COVID‐19 infection is the cause or consequence of LTL shortening.
Mendelian randomization (MR) is an instrumental variable method that uses the single‐nucleotide polymorphism (SNP) as an instrument variable (IV) to infer the causal relationship between two traits and has the advantage of minimizing bias due to confounding factors and reverse causality. 30 , 31 , 32 , 33 , 34 In this study, we employed a bidirectional two‐sample MR design to examine the potential bidirectional relationship between COVID‐19 and LTL.
2. MATERIALS AND METHODS
2.1. Study design description
Figure 1 shows a brief description of this bidirectional MR design between COVID‐19 and LTL. We performed a total of two MR analyses using summary statistics from a genome‐wide association study (GWAS) to investigate the bidirectional association between critically ill COVID‐19 and LTL. The forward MR analyses considered critically ill COVID‐19 as the exposure and LTL as the outcome, while the reverse MR analyses LTL as the exposure and critically ill COVID‐19 as the outcome. The core MR assumptions are shown in Figure 1. This study is based on publicly available summary statistics, therefore no ethical approval is required.
Figure 1.
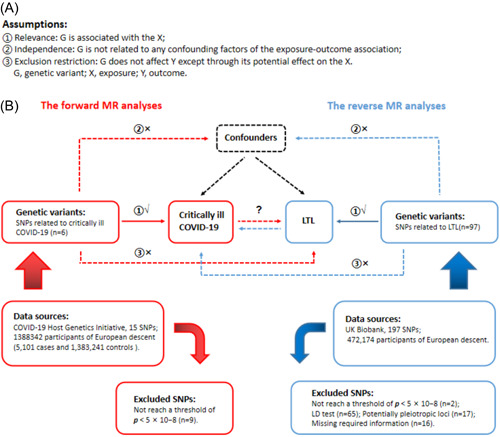
Description of the study design in this bidirectional MR study. (A) MR analyses depend on three core assumptions. (B) Sketch of the study design. The red represented the forward MR analyses, with critically ill COVID‐19 as exposure and LTL as the outcome. The blue represented the reverse MR analyses, with LTL as exposure and critically ill COVID‐19 as the outcome. LD, linkage disequilibrium; LTL, leukocyte telomere length; MR, Mendelian randomization; SNPs, single‐nucleotide polymorphisms.
2.2. Selection of instrumental variables for MR analyses
Appropriate instrumental variables for MR analyses were selected from two different GWAS summary results. First, SNPs were selected at a threshold of genome‐wide significance (p < 5 × 10−8). Second, appropriate SNPs were kept based on linkage disequilibrium as measured by r 2 > 0.01 in the European 1000 Genome reference panel. Then, those associating with the outcome with p < 5 × 10−8 were excluded. When harmonizing exposure and outcome data, palindromic SNPs with intermediate allele frequencies were removed. We estimated the F‐statistics to evaluate the instrument strength. F < 10 indicates weak instrument strength. 35 , 36
2.3. Data sources and instrumental variables selection for critically ill COVID‐19
We used a centralized meta‐analysis data comprised of 15 European cohorts for critically ill COVID‐19, 37 which was obtained from the COVID‐19 Host Genetics Initiative (https://www.covid19hg.org/results/r5/), consisting of 5101 cases and 1 383 241 controls (Supporting Information: Table 1). The critically ill COVID‐19 cases included patients who were hospitalized because of symptoms associated with laboratory‐confirmed SARS‐CoV‐2 infection and required respiratory support or whose primary cause of death was COVID‐19. 37 The control groups were selected as genetically ancestry‐matched samples without known SARS‐CoV‐2 infection if that information was available. 37 Fifteen SNPs associated with critically ill COVID‐19 were identified in this GWAS and were selected as instrumental variables. Among the 15 SNPs, 9 SNPs with p > 5 × 10−8 were excluded. Thus, we finally included 6 variants as genetic instruments in the MR analyses (Figure 1, Supporting Information: Table 2).
2.4. Data sources and instrumental variables selection for LTL
Data sources for LTL were taken from the UK Biobank (https://figshare.com/s/caa99dc0f76d62990195), including 472 174 UKB participants 38 (Supporting Information: Table 1). For reverse MR analyses, we selected the appropriate instrumental variables among 197 independent loci associated with LTL. Two SNPs with p > 5 × 10−8 were excluded, 65 SNPs were removed by using PLINK clumping with r 2 > 0.01, and 17 SNPs were removed because of its potentially pleiotropic loci, and 16 SNPs were excluded as the required information for MR analyses was missing. Finally, 97 instrumental variables were selected for MR analyses (Figure 1, Supporting Information: Table 3).
2.5. Statistical analysis
The random‐effects inverse‐variance weighted (IVW) method was used as the main statistical method to estimate the potential bidirectional causal associations between critically ill COVID‐19 and LTL. The IVW method is based on a hypothesis that all core assumptions of MR are valid. However, IVs influence the outcome through other pathways indicating that there exist potential horizontal pleiotropic effects, and the causal estimate from IVW could be biased. Therefore, we performed the sensitivity analyses by using the MR‐Egger 39 and weighted median 40 methods, from which we can estimate the causal relationship accurately even though the existence of invalid SNPs.
As MR depends on three core IV assumptions for the main analyses (Figure 1), we reported the methods applied to assess the assumptions or justify their validity. For the relevance assumption, R 2 was calculated to represent the proportion of variance in the exposure variable explained by the genetic variants. We estimated the F‐statistics to evaluate the instrument strength of the association between IVs and risk of exposure of interest. F < 10 indicates weak instrument strength. 35 , 36 For the exclusion restriction assumption, the MR‐Egger regression intercept, and its 95% confidence intervals (CIs) were used to investigate the degree of bias in casual estimates due to directional pleiotropy. 39 , 41 Besides this, the horizontal pleiotropy was assessed by employing the Mendelian randomization pleiotropy residual sum and outlier (MR‐PRESSO) global test and the outlying SNPs were excluded through the MR‐PRESSO outlier test. 42 As well, after removing outlying IVs, we also investigated whether there was a statistically significant difference compared with the former one. We also tested heterogeneity for IVW and MR‐Egger methods via Cochran's Q statistics and funnel plots. 43 Finally, several sensitivity analyses, such as leave‐one‐out analysis and single SNP analysis, were applied to identify whether a single SNP influenced the main causal relationship. 41 Power calculation for this MR study was obtained via an online web tool (https://sb452.shinyapps.io/power/).
For binary outcome, an odds ratio (OR) and 95% CI were applied to estimate the degree of a causal relationship. The causal estimate for both the binary and continuous outcomes, p‐value, β, and its standard error were also presented. All p‐values are two‐tailed. All analyses were performed by applying the TwoSampleMR and MendelianRandomization packages in R (version 4.2.0, www.r-project.org/).
3. RESULTS
3.1. The casual effect of critically ill COVID‐19 on LTL
As shown in Table 1 and the scatter plot (Figure 2A), the IVW result suggested that genetically predicted critically ill COVID‐19 was not significantly associated with LTL (β and 95% CI: 0.0075, −0.018 to 0.021; p = 0.733). No significant association was identified in other models. Although MR‐Egger regression analyses showed no incidence of potential directional pleiotropy, the MR PRESSO result indicated notable horizontal pleiotropy across SNPs in the causal estimates (Table 2, global test p = 0.004). However, even after removing instrumental variables which had horizontal pleiotropy, the p‐value was still not significant (Table 2, corrected p = 0.174). In addition, by combining Cochran's Q p‐value in IVW and MR‐Egger methods (Table 2, all p‐value of Cochran's Q < 0.001) with the funnel plot (Figure 2D), it was suggested that the observed association was along with obvious heterogeneity. However, even using the weighted median method, no significant association was observed (Table 1, beta and 95% CI: −0.0095, −0.018 to 0.009; p = 0.490).
Table 1.
Telomere length and its association with critically ill COVID‐19 in the MR analyses
| Exposure | Outcome | No. of SNPs | Methods | OR (95% CI) | β (SE) | p |
|---|---|---|---|---|---|---|
| The forward MR analyses | ||||||
| Critically ill COVID‐19 | LTL | 6 | IVW | 0.0075 (0.0220) | 0.734 | |
| MR‐Egger | −0.0006 (0.0571) | 0.992 | ||||
| Weighted median | −0.0095 (0.0141) | 0.497 | ||||
| Weighted mode | −0.0199 (0.0142) | 0.220 | ||||
| The reverse MR analyses | ||||||
| LTL | Critically ill COVID‐19 | 97 | IVW | 1.00 (0.79–1.28) | 0.004 (0.125) | 0.973 |
| MR‐Egger | 1.19 (0.78–1.81) | 0.174 (0.213) | 0.415 | |||
| Weighted median | 1.28 (0.86–1.90) | 0.249 (0.202) | 0.217 | |||
| Weighted mode | 1.29 (0.87–1.94) | 0.258 (0.205) | 0.211 | |||
Abbreviations: CI, confidence interval; IVW, inverse variance weighted; LTL, leukocyte telomere length; MR, Mendelian randomization; OR, odds ratio; SNPs, single‐nucleotide polymorphisms.
Figure 2.
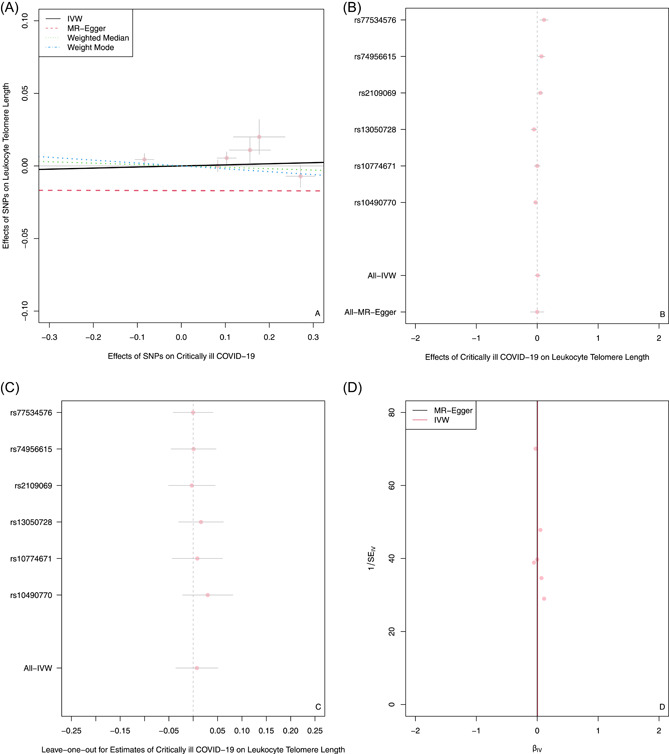
The forward MR analyses: Casual effect of critically ill COVID‐19 on LTL. (A) Scatter plot of the association between critically ill COVID‐19 and LTL. The four methods applied in the current manuscript were all depicted. Lines in black, red, green, and blue represent IVW, MR‐Egger, weighted median, and weight mode methods. (B) A forest plot was used to show the MR estimate and 95% CI values (gray line segment) for each SNP, which also shows the IVW and MR‐Egger results at the bottom. (C) Leave‐one‐out analyses to evaluate whether any single instrumental variable was driving the causal effect. (D) A funnel plot was applied to detect whether the observed association was along with obvious heterogeneity. CI, confidence interval; IVW, inverse variance weighted; LTL, leukocyte telomere length; MR, Mendelian randomization; SNPs, single‐nucleotide polymorphisms.
Table 2.
Pleiotropy and heterogeneity analyses
| Exposure | Outcome | No. of SNPs | MR‐Egger regression | MR PRESSO | Heterogeneity analyses | ||||
|---|---|---|---|---|---|---|---|---|---|
| Intercept | p_intercept | Global test p | Correct p * | Method | Q | Q_pval | |||
| Critically ill COVID‐19 | LTL | 6 | 0.001 | 0.885 | 0.004 | 0.174 | IVW | 29.853 | <0.001 |
| MR‐Egger | 29.677 | <0.001 | |||||||
| The reverse MR analyses | |||||||||
| LTL | Critically ill COVID‐19 | 97 | −0.006 | 0.352 | 0.485 | NA | IVW | 90.434 | 0.497 |
| MR‐Egger | 89.465 | 0.496 | |||||||
Abbreviations: IVW, inverse variance weighted; LTL, leukocyte telomere length; MR PRESSO, Mendelian randomization pleiotropy residual sum and outlier; SNPs, single‐nucleotide polymorphisms.
If MR PRESSO global test detects the horizontal pleiotropy and there is a significant difference before and after removing the outlier, correct p is calculated by removing instruments variants that have horizontal pleiotropy.
To increase statistical power, we additionally performed a secondary analysis by selecting SNPs that were of a significance level of p < 5 × 10−7 (Supporting Information: Table 4). The results showed that COVID‐19 was not significantly associated with LTL (Table 3).
Table 3.
Genetically predicted telomere length and its association with critically ill COVID‐19 using 15 instrumental variables
| Exposure | Outcome | No. of SNPs | Methods | β (SE) | p |
|---|---|---|---|---|---|
| Critically ill COVID‐19 | LTL | 15 | IVW | 0.0010 (0.0050) | 0.841 |
| MR‐Egger | −0.0035 (0.0140) | 0.806 | |||
| Weighted median | −0.0093 (0.0047) | 0.045 | |||
| Weighted mode | −0.0106 (0.0060) | 0.102 |
Abbreviations: IVW, inverse variance weighted; LTL, leukocyte telomere length; MR, Mendelian randomization; SNPs, single‐nucleotide polymorphisms.
3.2. The causal effect of LTL on critically ill COVID‐19
As shown in Table 1, the scatter plot (Figure 3A) and forest plots (Figure 3B), the IVW result showed LTL was not causally related to critically ill COVID‐19 (OR and 95% CI: 1.00, 0.79–1.28; p = 0.973). The other three results were in line with the IVW results. Both MR‐Egger regression analyses (Table 2, intercept = −0.006, p = 0.352) and the MR PRESSO result (Table 2, global test p = 0.485) showed no incidence of potential pleiotropy. The Cochran's Q test in the IVW method (Table 2, Q = 90.434, p = 0.497) and MR‐Egger method (Table 2, Q= 89.465, p = 0.496) suggested that there was an absence of heterogeneity in the IVs. Furthermore, even if removing any single SNP, the observed association didn't significantly change in leave‐one‐out analyses (Figure 3C), suggesting the stability of the results.
Figure 3.
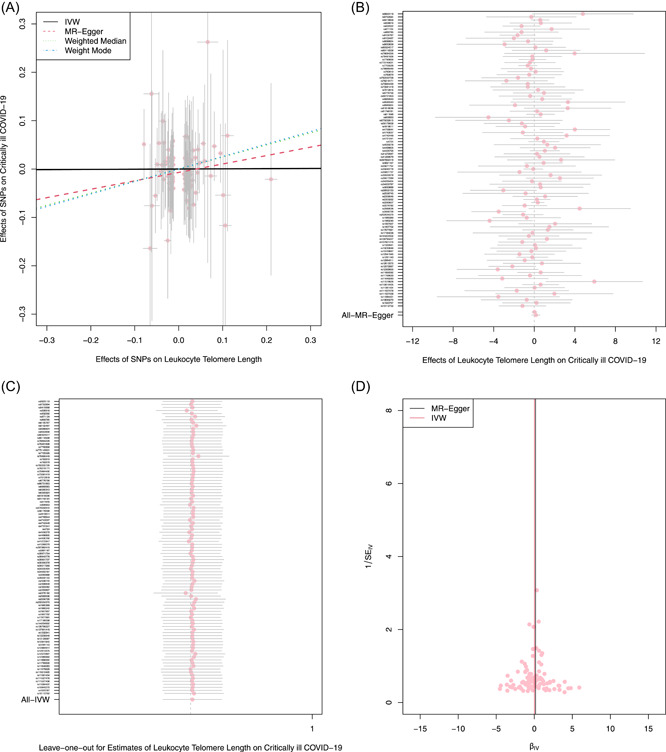
The reverse MR analyses: Casual effect of LTL on critically ill COVID‐19. (A) Scatter plot of the association between critically ill COVID‐19 and LTL. The four methods applied in the current manuscript were all depicted. Lines in black, red, green, and blue represent IVW, MR‐Egger, weighted median, and weight mode methods. (B) A forest plot was used to show the MR estimate and 95% CI values (gray line segment) for each SNP, also show the IVW and MR‐Egger MR results at the bottom. (C) Leave‐one‐out analyses to evaluate whether any single instrumental variable was driving the causal effect. (D) A funnel plot was applied to detect whether the observed association was along with obvious heterogeneity. CI, confidence interval; IVW, inverse variance weighted; LTL, leukocyte telomere length; MR, Mendelian randomization; SNPs, single‐nucleotide polymorphisms.
4. DISCUSSION
In this bidirectional two‐sample MR study, we did not find an association between critically ill COVID‐19 and LTL from MR analyses.
As mentioned previously, several previous observational studies have suggested that shorter TL was likely to be independently associated with critically ill COVID‐19, which might be biased by potential confounding factors and reverse causality, for example, they all had measured TL after SRAS‐CoV‐2 infection. Thus, we are unable to interpret whether shorter TL aggravated virus infection or telomere shortening (TS) was due to a “cytokine‐storm” symptom in response to virus infection. However, in our study, neither the forward MR analyses nor the reverse direction suggested a significant association between critically ill COVID‐19 and TL. In addition, our results of the reverse MR analyses were in line with a one‐sample MR study which used a set of 130 genome‐wide significant (p < 5 × 10−8) genetic variants measured several years before SARS‐CoV‐2 infection to reduce bias. 44
We have not obtained significant results; however, several hypotheses have been put forward. According to an “Amplifier/Rheostat Hypothesis,” shorter TL along with genomic instability as well as a decrease in cell proliferative capacity, might cause cell senescence, the decline in B and T lymphopoiesis, negative consequences on the immune competence and induce cytokine storm, driving to more severe SARS‐CoV‐2 infection as well as promoting telomeres shorten even more. 13 , 45 , 46 We can't dismiss these biological mechanisms. Of note, TL is influenced by genetic and environmental factors. 13 , 47 The majority of individuals who died from COVID‐19 were adults with age‐related dysfunction and chronic diseases, such as CVD, diabetes, COPD, and so on. 1 , 2 , 3 Those diseases are all risk factors for TS, which may play an important role in the biological mechanisms. For example, several studies demonstrated that smokers have shorter telomeres than nonsmokers. 48 , 49 Therefore, the results we obtained in this study drive us to make a hypothesis that shorter TL observed in severe post‐COVID‐19 survivors attributed to the interaction between SRAS‐CoV‐2 infection and initial physical condition. There is a complicated association between critically ill COVID‐19 and TL, instead of pure causal relevance.
Compared with traditional observational studies, the greatest advantage in this study is that the causal estimate obtained by MR avoids reverse causality and confounding bias. As well, applying comprehensive GWASs data for MR analyses can improve the accuracy of the estimated effect. However, several limitations can't be avoided in our study. First, based on three core assumptions in the MR study, instrument strength of the association between IVs and risk of exposure of interest is expected to be high, while relatively low in our study. Secondly, through Cochran's Q value in the forward MR analyses, obvious heterogeneity can't be eliminated. Although the results were robust in all methods, a small number of genetic instruments, a small sample size, or potential sample overlap between exposure and outcome might lead to bias. In addition, the results can't be generalized to other ethnicities and races as the population we used was Europeans.
In conclusion, our bidirectional MR study indicated neither the forward nor the reverse direction showed a causal association between critically ill COVID‐19 and LTL. Facing the fact that SARS‐CoV‐2 infection has caused high mortality in the elderly, further investigations are encouraged to explore the relevance between critically ill COVID‐19 and TL.
AUTHOR CONTRIBUTIONS
Writing the first draft of the manuscript, statistical analysis, data curation, writing review, and editing: Danqi Huang. Investigation, statistical analysis, and editing: Siqi Lin and Junting He. Critically reviewing, editing, and partial funding: Qi Wang. Conceptualization, designed the study, supervised, editing, and reviewed, revised the manuscript, and funding: Yiqiang Zhan. All authors approved the final version.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ETHICS STATEMENT
The present Mendelian randomization analysis was based on summary data from previous studies that had gained written informed consent and ethics approval. No ethical permit is required for the secondary analysis of summary data.
Supporting information
Supplementary information.
ACKNOWLEDGMENTS
This study was supported by the Innovation and Entrepreneurship Training Program for College Students of Sun Yat‐Sen University (No. 202211634); the Start‐up Grant of Sun Yat‐Sen University; the Fundamental Research Funds for the Central Universities, Sun Yat‐sen University (No. 22qntd4306); and the National Natural Science Foundation of China (No. 72061137006).
Huang D, Lin S, He J, Wang Q, Zhan Y. Association between COVID‐19 and telomere length: a bidirectional Mendelian randomization study. J Med Virol. 2022;94:5345‐5353. 10.1002/jmv.28008
Contributor Information
Qi Wang, Email: wangqi_tj@hust.edu.cn.
Yiqiang Zhan, Email: zhanyq8@mail.sysu.edu.cn.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available in COVID‐19 Host Genetics Initiative. These data were derived from the following resources available in the public domain: Telomere GWAS.
REFERENCES
- 1. Wu Z, McGoogan JM. Characteristics of and important lessons from the coronavirus disease 2019 (COVID‐19) outbreak in China: summary of a report of 72 314 cases from the Chinese center for disease control and prevention. JAMA. 2020;13:1239‐1242. 10.1001/jama.2020.2648 [DOI] [PubMed] [Google Scholar]
- 2. World Health Organization . Report of the WHO‐China joint mission on coronavirus disease 2019 (COVID‐19) [Pdf]; 2020.
- 3. Grasselli G, Zangrillo A, Zanella A, et al. Baseline characteristics and outcomes of 1591 patients infected with SARS‐CoV‐2 admitted to ICUs of the Lombardy Region, Italy. JAMA. 2020;323(16):1574‐1581. 10.1001/jama.2020.5394. Erratum in: JAMA. 2021;325(20):2120. [DOI] [PMC free article] [PubMed]
- 4. Dong Y, Mo X, Hu Y, et al. Epidemiological characteristics of 2143 pediatric patients with 2019 coronavirus disease in China. Pediatrics. 2020;145(6):e20200702.32179660 [Google Scholar]
- 5. CDC COVID‐19 Response Team . Coronavirus disease 2019 in children – United States, February 12–April 2, 2020. MMWR Morb Mortal Wkly Rep. 2020;69(14):422‐426. 10.15585/mmwr.mm6914e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lu Q, Shi Y. Coronavirus disease (COVID‐19) and neonate: what neonatologist need to know. J Med Virol. 2020;92(6):564‐567. 10.1002/jmv.25740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hasan SS, Capstick T, Ahmed R, et al. Mortality in COVID‐19 patients with acute respiratory distress syndrome and corticosteroids use: a systematic review and meta‐analysis. Expert Rev Respir Med. 2020;14(11):1149‐1163. 10.1080/17476348.2020.1804365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sheervalilou R, Shirvaliloo M, Dadashzadeh N, et al. COVID‐19 under spotlight: a close look at the origin, transmission, diagnosis, and treatment of the 2019‐nCoV disease. J Cell Physiol. 2020;235(12):8873‐8924. 10.1002/jcp.29735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Diao B, Wang C, Tan Y, et al. Reduction and functional exhaustion of T cells in patients with coronavirus disease 2019 (COVID‐19). Front Immunol. 2020;11:827. 10.3389/fimmu.2020.00827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Coperchini F, Chiovato L, Croce L, Magri F, Rotondi M. The cytokine storm in COVID‐19: an overview of the involvement of the chemokine/chemokine‐receptor system. Cytokine Growth Factor Rev. 2020;53:25‐32. 10.1016/j.cytogfr.2020.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Storci G, Bonifazi F, Garagnani P, Olivieri F, Bonafè M. The role of extracellular DNA in COVID‐19: clues from inflamm‐aging. Ageing Res Rev. 2021;66:101234. 10.1016/j.arr.2020.101234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. V'kovski Kratzel P, Steiner A, Stalder S, Thiel H, Coronavirus V. Coronavirus biology and replication: implications for SARS‐CoV‐2. Nat Rev Microbiol. 2021;19(3):155‐170. 10.1038/s41579-020-00468-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tay MZ, Poh CM, Rénia L, MacAry PA, Ng LFP. The trinity of COVID‐19: immunity, inflammation and intervention. Nat Rev Immunol. 2020;20(6):363‐374. 10.1038/s41577-020-0311-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Garcia G, Jr. , Sharma A, Ramaiah A, et al. Antiviral drug screen identifies DNA‐damage response inhibitor as potent blocker of SARS‐CoV‐2 replication. Cell Rep. 2021;35(1):108940. 10.1016/j.celrep.2021.108940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Blackburn EH. Structure and function of telomeres. Nature. 1991;350(6319):569‐573. 10.1038/350569a0 [DOI] [PubMed] [Google Scholar]
- 16. Olovnikov AM. A theory of marginotomy. The incomplete copying of template margin in enzymic synthesis of polynucleotides and biological significance of the phenomenon. J Theor Biol. 1973;41(1):181‐190. 10.1016/0022-5193(73)90198-7 [DOI] [PubMed] [Google Scholar]
- 17. Watson JD. Origin of concatemeric T7 DNA. Nat New Biol. 1972;239(94):197‐201. 10.1038/newbio239197a0 [DOI] [PubMed] [Google Scholar]
- 18. López‐Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;53(6):1194‐1217. 10.1016/j.cell.2013.05.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tichy ED, Ma N, Sidibe D, et al. Activation in muscle stem cells induces proliferation‐independent telomere shortening. Cell Rep. 2021;35(6):109098. 10.1016/j.celrep.2021.109098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ibironke O, Carranza C, Sarkar S, et al. Urban air pollution particulates suppress human T‐Cell responses to mycobacterium tuberculosis. Int J Environ Res Public Health. 2019;16(21):4112. 10.3390/ijerph16214112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brouilette S, Singh RK, Thompson JR, Goodall AH, Samani NJ. White cell telomere length and risk of premature myocardial infarction. Arterioscler Thromb Vasc Biol. 2003;23(5):842‐846. 10.1161/01.ATV.0000067426.96344.32 [DOI] [PubMed] [Google Scholar]
- 22. Cheng F, Carroll L, Joglekar MV, et al. Metabolic disease, and telomere length. Lancet Diabetes Endocrinol. 2021;9(2):117‐126. 10.1016/S2213-8587(20)30365-X [DOI] [PubMed] [Google Scholar]
- 23. Mongelli A, Barbi V, Gottardi Zamperla M, et al. Evidence for biological age acceleration and telomere shortening in COVID‐19 survivors. Int J Mol Sci. 2021;22(11):6151. 10.3390/ijms22116151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sanchez‐Vazquez R, Guío‐Carrión A, Zapatero‐Gaviria A, Martínez P, Blasco MA. Shorter telomere lengths in patients with severe COVID‐19 disease. Aging. 2021;13(1):1‐15. 10.18632/aging.202463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Benetos A, Lai TP, Toupance S, et al. The nexus between telomere length and lymphocyte count in seniors hospitalized with COVID‐19. J Gerontol A Biol Sci Med Sci. 2021;6(8):e97‐e101. 10.1093/gerona/glab026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dos Santos GA, Pimenta R, Viana NI, et al. Shorter leukocyte telomere length is associated with severity of COVID‐19 infection. Biochem Biophys Rep. 2021;27:101056. 10.1016/j.bbrep.2021.101056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Froidure A, Mahieu M, Hoton D, et al. Short telomeres increase the risk of severe COVID‐19. Aging. 2020;12(20):19911‐19922. 10.18632/aging.104097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Victor J, Deutsch J, Whitaker A, et al. SARS‐CoV‐2 triggers DNA damage response in Vero E6 cells. Biochem Biophys Res Commun. 2021;579:141‐145. 10.1016/j.bbrc.2021.09.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kirkland JL, Tchkonia T. Cellular senescence: a translational perspective. EBioMedicine. 2017;21:21‐28. 10.1016/j.ebiom.2017.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Emdin CA, Khera AV, Kathiresan S. Mendelian randomization. JAMA. 2017;318(19):1925‐1926. 10.1001/jama.2017.17219 [DOI] [PubMed] [Google Scholar]
- 31. Lawlor DA, Harbord RM, Sterne JA, Timpson N, Smith GD. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat Med. 2008;27(8):1133‐1163. 10.1002/sim.3034 [DOI] [PubMed] [Google Scholar]
- 32. Davies NM, Holmes MV, Davey Smith G. Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ. 2018;362:k601. 10.1136/bmj.k601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet. 2014;23(R1):R89‐R98. 10.1093/hmg/ddu328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ziegler A, Mwambi H, König IR. Mendelian randomization versus path models: making causal inferences in genetic epidemiology. Hum Hered. 2015;79(3‐4):194‐204. 10.1159/000381338 [DOI] [PubMed] [Google Scholar]
- 35. Burgess S, Thompson SG. Bias in causal estimates from Mendelian randomization studies with weak instruments. Stat Med. 2011;30(11):1312‐1323. 10.1002/sim.4197 [DOI] [PubMed] [Google Scholar]
- 36. Burgess S, Thompson SG, CRP CHD Genetics Collaboration . Avoiding bias from weak instruments in Mendelian randomization studies. Int J Epidemiol. 2011;40(3):755‐764. 10.1093/ije/dyr036 [DOI] [PubMed] [Google Scholar]
- 37. COVID‐19 Host Genetics Initiative . Mapping the human genetic architecture of COVID‐19. Nature. 2021;600(7889):472‐477. 10.1038/s41586-021-03767-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Codd V, Wang Q, Allara E, et al. Polygenic basis and biomedical consequences of telomere length variation. Nat Genet. 2021;53(10):1425‐1433. 10.1038/s41588-021-00944-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44(2):512‐525. 10.1093/ije/dyv080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bowden J, Smith GD, Haycock PC, Burgess S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. 2016;40(4):304‐314. 10.1002/gepi.21965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Burgess S, Thompson SG. Interpreting findings from Mendelian randomization using the MR‐Egger method. Eur J Epidemiol. 2017;32(5):377‐389. 10.1007/s10654-017-0255-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Verbanck M, Chen CY, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet. 2018;50(5):693‐698. 10.1038/s41588-018-0099-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bowden J, Spiller W, Del Greco MF, et al. Improving the visualization, interpretation and analysis of two‐sample summary data Mendelian randomization via the radial plot and radial regression. Int J Epidemiol. 2018;47(4):1264‐1278. 10.1093/ije/dyy101. Erratum in: Int J Epidemiol. 2018;47(6):2100. [DOI] [PMC free article] [PubMed]
- 44. Wang Q, Codd V, Raisi‐Estabragh Z, et al. Shorter leukocyte telomere length is associated with adverse COVID‐19 outcomes: a cohort study in UK Biobank. EBioMedicine. 2021;70:103485. 10.1016/j.ebiom.2021.103485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wissler Gerdes EO, Vanichkachorn G, Verdoorn BP, et al. Role of senescence in the chronic health consequences of COVID‐19. Transl Res. 2022;241:96‐108. 10.1016/j.trsl.2021.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mahmoodpoor A, Sanaie S, Roudbari F, Sabzevari T, Sohrabifar N, Kazeminasab S. Understanding the role of telomere attrition and epigenetic signatures in COVID‐19 severity. Gene. 2022;811:146069. 10.1016/j.gene.2021.146069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Broer L, Codd V, Nyholt DR, et al. Meta‐analysis of telomere length in 19,713 subjects reveals high heritability, stronger maternal inheritance and a paternal age effect. Eur J Hum Genet. 2013;21(10):1163‐1168. 10.1038/ejhg.2012.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Astuti Y, Wardhana A, Watkins J, Wulaningsih W. PILAR research network. cigarette smoking and telomere length: a systematic review of 84 studies and meta‐analysis. Environ Res. 2017;158:480‐489. 10.1016/j.envres.2017.06.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Maremanda KP, Sundar IK, Li D, Rahman I. Age‐dependent assessment of genes involved in cellular senescence, telomere, and mitochondrial pathways in human lung tissue of smokers, COPD, and IPF: associations with SARS‐CoV‐2 COVID‐19 ACE2‐TMPRSS2‐Furin‐DPP4 axis. Front Pharmacol. 2020;11:584637. 10.3389/fphar.2020.584637 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information.
Data Availability Statement
The data that support the findings of this study are available in COVID‐19 Host Genetics Initiative. These data were derived from the following resources available in the public domain: Telomere GWAS.