

Abstract
The emergence of more transmissible or aggressive variants of SARS‐CoV‐2 requires the development of antiviral medication that is quickly adjustable to evolving viral escape mutations. Here we report the synthesis of chemically stabilized small interfering RNA (siRNA) against SARS‐CoV‐2. The siRNA can be further modified with receptor ligands such as peptides using CuI‐catalysed click‐chemistry. We demonstrate that optimized siRNAs can reduce viral loads and virus‐induced cytotoxicity by up to five orders of magnitude in cell lines challenged with SARS‐CoV‐2. Furthermore, we show that an ACE2‐binding peptide‐conjugated siRNA is able to reduce virus replication and virus‐induced apoptosis in 3D mucociliary lung microtissues. The adjustment of the siRNA sequence allows a rapid adaptation of their antiviral activity against different variants of concern. The ability to conjugate the siRNA via click‐chemistry to receptor ligands facilitates the construction of targeted siRNAs for a flexible antiviral defence strategy.
Keywords: Corona pandemic, SARS-CoV-2, antiviral compounds, siRNA, peptide RNA conjugates
siRNAs protect cells from SARS‐CoV‐2 and can be quickly adjusted to variants of concern. An alkyne moiety was integrated at the 3′ end of the siRNA to enable chemical modification of the siRNA via CuI‐catalysed click‐chemistry. Following this strategy, the siRNA can be conjugated to receptor ligands such as hACE2‐binding peptides, which allows target‐specific delivery.

Introduction
The severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) is the causative agent of the coronavirus disease 2019 (COVID‐19) pandemic, resulting in a high disease burden and socioeconomic distress. The current success in curbing the COVID‐19‐related death toll is based to a large extent on chemically modified messenger RNA (mRNA)‐based vaccines. [1] However, the emergence of SARS‐CoV‐2 variants of concern (VoCs) has reduced the protective efficacy of vaccination. [2] This makes the development of variant‐adjusted vaccines or of alternative anti‐viral therapies a prime global goal. One emerging concept in anti‐COVID‐19 medication involves the development of nucleic acid‐based therapeutics, which can degrade the viral genome and can be quickly adjusted to viral mutations. This fast adjustability is a major advantage of nucleic acid‐based therapies in comparison to other antiviral medications, e.g. antibodies that target the SARS‐CoV‐2 spike protein (S protein). [3] Mutations in the S protein reduce the efficacy of therapeutic, neutralizing monoclonal antibodies. [4] Small molecule‐based anti‐SARS‐CoV‐2 compounds have recently also entered the market, but their efficacy is potentially also threatened by mutations in the respective viral enzymes as well as potential side effects that impact their risk profile. [5]
Nucleic acid‐based therapeutics include small interfering RNAs (siRNAs). These are about 21 base‐pair long, non‐coding RNA duplexes that are able to induce degradation of complementary cellular RNAs. After cellular uptake, the siRNA duplex is loaded into the RNA‐induced silencing complex (RISC). Here, the duplex is processed to a single strand that binds with high specificity to complementary RNAs present in the cytosol, resulting in their cleavage. [6] siRNA therapeutics are already in clinical use, which proves the validity of the concept. [7]
SARS‐CoV‐2 is a positive‐sense, single‐stranded RNA virus, which enters the host cell via the human angiotensin‐converting enzyme 2 (hACE2) receptor. The viral RNA genome needs to be replicated for the assembly of new virus particles. In addition, the viral genomic RNA is copied into subgenomic mRNAs (sgmRNAs) for the biosynthesis of viral proteins. [8] Hence, during productive infection, the virus produces two types of single‐stranded RNAs (ssRNAs) and both the genomic RNA and the sgmRNA strands are in principle targetable by siRNA‐mediated cleavage, which creates a dual‐hit possibility. [9] Such an siRNA‐based degradation concept has the advantage that the siRNAs can be rapidly adjusted if mutations occur in the viral target RNA sequences. It also allows the simultaneous targeting of multiple viral genomic sites. Altogether, these advantages make an siRNA‐based approach a promising concept to target and control constantly evolving RNA viruses.
Results and Discussion
To investigate the feasibility of an siRNA‐based strategy against SARS‐CoV‐2, we analysed the genomic structure of the virus and designed several siRNAs (L1, R‐2 to R‐5, S‐6 to S‐9) complementary to different viral RNA target regions (Figure 1a, Figure S1a, Table S1). All of them were designed to contain two 2′‐deoxy‐thymidine (T) nucleotides at the respective 3′‐ends to increase the stability towards exonuclease‐based degradation. [10] Using this design principle, we prepared several siRNAs against the coding regions of the RNA‐dependent RNA polymerase (RdRp, Nsp12), the S protein and the 5′‐leader sequence (L) (Figure 1a). A non‐targeting (scrambled) siRNA (Ctrl.‐10) served as a negative control.
Figure 1.

Development of efficient siRNAs targeting SARS‐CoV‐2. a) Target sequences of designed siRNAs within the SARS‐CoV‐2 genome. b) Screening of antiviral efficacy of different siRNAs using a surrogate luciferase‐reporter assay. Bars show mean, error bars represent SD. c), d) Reduction of plaque forming units (PFU) per mL normalized to Ctrl.‐10‐treated Vero‐E6 cells in the context of siRNA treatment (17 nm) and SARS‐CoV‐2 infection (MOI 0.01) 72 h p.i. c) siRNA was transfected one hour before or d) one hour after virus challenge. Bars show mean, error bars represent SD. e) siRNA sequences of the most potent siRNAs, R‐2 and S‐6. f)–i) Viability of MDA‐MB‐231‐hACE2 cells depends on the SARS‐CoV‐2 variant B.1.177 (EU1) (f), B.1.1.7 (Alpha VoC) (g), B.1.351 (Beta VoC) (h), B.1.617.2 (Delta VoC) (i), virus inoculum, and specific siRNA treatment (40 nm, lipofection). Dots show the mean of two technical replicates from one representative biologically independent experiment. Details about reproducibility and statistical analysis are given in the Methods section in the Supporting Information.
The efficacy of the designed siRNAs was first assessed in a human lung adenocarcinoma cell line A549 with the help of a dual‐luciferase reporter assay (Figure S1b, Table S2). For the assay, A549 were transfected with a plasmid that encodes for Renilla luciferase and the firefly luciferase. The Renilla luciferase reporter gene was fused to small genomic target sequences of SARS‐CoV‐2. Efficient targeting of the designed siRNAs resulted in degradation of the Renilla luciferase transcript and consequently lower protein expression. As a readout, the Renilla luciferase activity was quantified relative to the firefly luciferase activity that served as an intraplasmid‐encoded reporter for normalization to balance differences in the transfection efficiency. Using this assay, we detected for the siRNAs L‐1 (target: L), R‐2 and R‐5 (target: RdRp, Nsp12) and S‐6 (target: S) high knockdown efficiencies between 80 % and 92 % relative to the non‐targeting control siRNA with a scrambled sequence (Ctrl.‐10, Figure 1b).
Following this initial pre‐screening, we measured the antiviral potency of the four most promising siRNAs in Vero‐E6 cells infected with an authentic, replication‐competent SARS‐CoV‐2 isolate of the B.3 pangolin lineage. The siRNAs were applied using lipofectamine as the transfection agent either 1 h prior to infection or 1 h post‐infection. We quantified the efficacy of the siRNAs by measuring the plaque forming units (PFU) three days post‐infection. This assay allowed us to confirm the high anti‐SARS‐CoV‐2 activity of the siRNAs R‐2 and S‐6. The siRNA R‐5, in contrast, was ineffective in this assay (Figure 1c, d), likely because the full‐length genomic SARS‐CoV‐2 RNA has a hairpin loop at the target position, [11] which probably impairs its accessibility for the siRNA‐RISC‐complex. Since only small fragments of the viral genome were fused to the Renilla luciferase gene in the dual‐luciferase reporter assay to test R‐5 (Table S2), secondary and tertiary RNA structures were different and did therefore not impair R‐5 efficacy. The siRNAs R‐2 and S‐6 reduced the viral loads up to 96 % when cells were transfected one hour before virus challenge (Figure 1c, Figure S1c) and up to 99.95 % when cells were transfected one hour after virus challenge (Figure 1d, Figure S1d). Based on these results, we selected R‐2 and S‐6 for further studies (Figure 1e).
To obtain more detailed efficacy data, we established an assay to quantify SARS‐CoV‐2‐induced cytotoxicity over a high dynamic range of virus inoculum. For these studies we used MDA‐MB‐231‐hACE2 cells, which were engineered to overexpress the hACE2 virus entry receptor. This makes them highly susceptible to SARS‐CoV‐2. [12] Titration of the SARS‐CoV‐2 inoculum on these cells resulted in a volume‐dependent increase of cell death, which was monitored by microscopy and quantified by a luminometric cell viability assay. As an antiviral positive control, the clinical RdRp inhibitor remdesivir (RDV) [13] potently blocked SARS‐CoV‐2 replication and virus‐induced cell death (Figure S2a).
We next inoculated target cells with SARS‐CoV‐2 (B.1.177 pangolin lineage, 20E.EU1) either 21 h or 4 h before, or 2 h after siRNA treatment. For siRNA delivery, we used lipofection‐mediated transfection. The depicted data show a highly effective protection of the siRNA‐treated cells from virus‐induced cytotoxicity, when R‐2 or S‐6 were applied 21 h before virus challenge (Figure S2a–c). Cells transfected with the non‐targeting scrambled siRNA Ctrl.‐10 and also untreated MDA‐MB‐231‐hACE2 cells showed, in contrast, dramatically decreased viability (Figure 1f). In experiments with Vero‐E6 cells, we observed a substantial protective effect when the siRNAs were applied either one hour prior (Figure 1c) or post‐infection (Figure 1d). This indicates cell type‐specific differences in the dynamics of siRNA delivery and antiviral potency. In the assay using MDA‐MB‐231‐hACE2, pre‐treatment with siRNAs R‐2 and S‐6 protected the cells from being killed by both VoCs Alpha (B.1.1.7, Figure 1g) and Beta (B.1.351, Figure 1h). Notably, the combination of siRNAs R‐2 and S‐6 was particularly effective (Figure 1f–h). The virus inoculum required to eliminate 50 % of the cells (cytotoxic volume (CV50)) increased relative to Ctrl.‐10, for R‐2 from 78‐ to 456‐fold, and for S‐6 to >798‐fold. With a combination of R‐2 and S‐6, we observed >80 % cell viability even at the highest virus inoculum. The protective siRNA effect with this mixture was so strong that we were unable to determine any meaningful CV50 values for the applied volumes of virus inoculum (Table S3).
We next examined the anti‐viral effect of the siRNA S‐6 using the SARS‐CoV‐2 VoC Delta (B.1.617.2, Figure 1i). Here, we detected a loss of activity of S‐6 in line with the known C to G point mutation at position 21 618, which is present directly in the binding target region of the siRNA (Figure S2d). [14] This activity loss could be partially compensated when we combined S‐6 with R‐2, but maximum efficiency could not be restored, showing that viral mutations in the siRNA‐binding region can have a dramatic effect on the antiviral activity. In order to prove that the siRNA approach allows rapid adaptation to virus mutations we adjusted the S‐6 sequence to account for the mutation in VoC Delta (S‐6δ). Indeed, when we examined the efficacy of S‐6δ, we noted that the original antiviral activity was re‐established (Figure 1i). This result demonstrates the fast adaptability of the siRNA‐based medication approach.
All siRNA‐based therapeutics that are in clinical use feature modified nucleosides with either 2′OMe or 2′F substituents (Figure 2a) next to the presence of phosphothioates. These chemical modifications establish a higher stability and resistance against endonucleases that are present in human serum. [7a] Using the design principle adapted from Givosiran, [15] we prepared a fully modified siRNA S‐6fm (Figure 2b). We performed serum stability studies over multiple time points and noted that S‐6fm showed the expected increase of stability in serum in comparison to S‐6. To our surprise, however, we observed in a subsequent siRNA efficacy study using the dual‐luciferase reporter assay a 40 % loss of activity (dual‐luciferase reporter assay, Figure 2b). Furthermore, a complete loss of activity was observed in virus challenge studies using replication‐competent SARS‐CoV‐2 (Figure S3a). Therefore, we screened various differently modified siRNAs and found that an only partially modified S‐6, with just four 2′‐OMe‐modifications (S‐6m ), featured already the required stability in serum (Figure 2c), while the antiviral activity was preserved. S‐6m had full activity in the dual‐luciferase reporter assay (Figure 2b) and brightfield microscopy of MDA‐MB‐231‐hACE2 cells, that had been pre‐treated with S‐6m and challenged with SARS‐CoV‐2, indicated a very high protective effect of S‐6m (Figure 2d).
Figure 2.
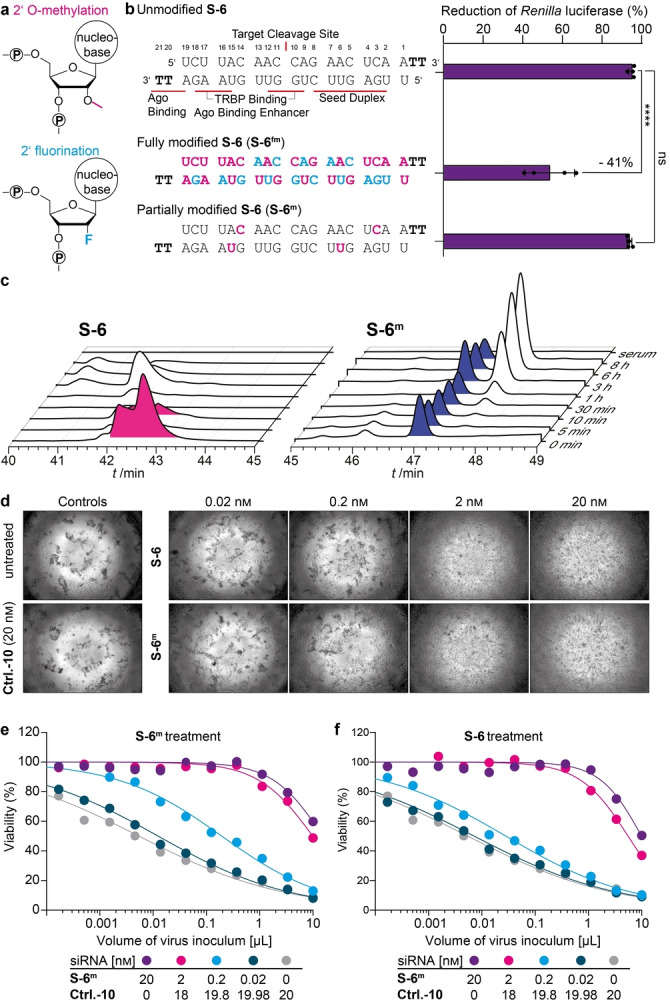
Chemical stabilization of the antiviral siRNAs. a) Methoxy‐ and fluor modifications on the 2′OH position of the ribose to improve siRNA stability. b) Chemically modified versions of S‐6 were compared to the unmodified version in the surrogate luciferase reporter assay. Ordinary one‐way ANOVA combined with Dunnett's multiple comparisons test was performed **** p adj‐value<0.0001. TRBP=TAR RNA binding protein that loads siRNA into RISC. c) Serum stability of unmodified S‐6 and S‐6m . d) Brightfield microscopy images of virus‐induced cytopathic effects on MDA‐MB‐231‐hACE2 cells and siRNA concentration‐dependent protection of cells challenged with 0.01 μL B.1.177 stock two days earlier. e), f) Viability of SARS‐CoV‐2‐infected MDA‐MB‐231‐hACE2 cells to determine the CV50 in the context of titration of virus inoculum (0.0001 to 10 μL) and of siRNA concentrations (0.02 to 20 nm, lipofection). Dots represent the mean of two technical replicates from one biologically independent experiment. In (e) S‐6m was added, whereas in (f) the unmodified S‐6 was used. For each concentration, CV50 differences between S‐6m and S‐6 were analysed using multiple t‐tests with FDR multiple comparisons control. Details about reproducibility and statistical analysis are given for all experiments in the Methods section.
Next, we performed a quantitative assessment of the activity of S‐6m . While MDA‐MB‐231‐hACE2 cells pre‐treated with Ctrl.‐10 showed the expected concentration‐dependent cytotoxicity two days after virus challenge, pre‐treatment with S‐6m (Figure 2e) prevented cell death at siRNA concentrations as low as 2 nm. Importantly, we observed a significantly higher protective effect of S‐6m compared to unmodified S‐6 (Figure 2f) at both 2 nm and 0.2 nm (Table S4). These results were confirmed in a second SARS‐CoV‐2‐susceptible cell line, A549‐hACE2 [16] (Figure S3b, c).
While RNA therapeutics and vaccines are typically delivered into cells and tissues using cationic lipid‐ or polymer‐based nanoparticles, the delivery agents often have adverse effects. In particular, delivery into epithelial cells, as required for the treatment of SARS‐CoV‐2 infection in patients, is often not possible.[ 7a , 17 ] To investigate delivery into epithelial cells, we first assessed whether our siRNAs can prevent SARS‐CoV‐2 infection and virus‐induced cell death in human 3D mucociliary lung EpiAirwayTM microtissues. These microtissues are derived from differentiated airway epithelial cells originating from the tracheobronchial tract. They feature a realistic air‐liquid interface including pseudo‐stratified structures with numerous apical cilia as well as apical mucous secretion. [18] EpiAirwayTM microtissues are hACE2‐positive. As such they are SARS‐CoV‐2‐susceptible and consequently suitable for the evaluation of antiviral approaches. [19]
We noted that all attempts to use lipid‐ or polymer‐based transfection reagents to deliver the siRNA into the microtissues were not successful. In addition, the sporadically observed effects could not be assigned to an siRNA‐specific block of infection (Figure S4a, b). In order to investigate if an siRNA delivery into these microtissues is possible without a transfection agent, we chemically modified our siRNA with receptor ligands. We prepared chimeric siRNA‐ligand conjugates that we hypothesized would facilitate cellular delivery. This approach is similar to the clinically approved N‐acetyl galactosamine‐modification of siRNAs that target liver cells. [7] For siRNA modification, we used the highly efficient and mild CuI‐catalysed click‐chemistry. [20] The initial idea was to connect the siRNA to short peptides that are known to interact with the hACE2 receptor as part of the receptor‐binding domain of the SARS‐CoV S protein [21] (Figure 3a). For the synthesis of the chimeric peptide‐siRNA, we utilized a CuI‐induced click‐ligation between an N3‐Linker‐YKYRYL (P) peptide and an siRNA, which we modified with an alkyne‐bearing dT at 3′‐end (S‐6m‐sense). [22] For synthetic simplicity, we inserted the alkyne‐bearing dT units at the 2nd position at the 3′‐ends. After the click‐reaction with the azido‐modified peptide, the peptide‐siRNA S‐6m‐P‐sense was rinsed through a size exclusion column to remove residual Cu‐salts and was then purified by HPLC. MALDI‐TOF analysis confirmed the successful synthesis of the peptide‐siRNA conjugate. The so prepared S‐6m‐P‐sense strand was finally hybridized to the corresponding S‐6m‐antisense strand to furnish the peptide‐modified siRNA chimera S‐6m‐P.
Figure 3.

RBD‐peptide siRNA conjugates suppress SARS‐CoV‐2 infection in human 3D lung microtissues. a) Depiction of the S‐6m‐P‐sense strand synthesis using click‐chemistry. b) hACE2‐peptide‐dependent delivery of S‐6m‐P‐Alexa647 compared to S‐6m‐Alexa647 (900 nm each, no transfection reagent) into human Caco‐2 cells. Lipofection of S‐6m‐Alexa647 (20 nm) served as a positive control. Scale bar: 25 μm. c), d) Ct values of SARS‐CoV‐2 RT‐qPCR in 3D lung microtissues 72 h after virus challenge (SARS‐CoV‐2 B1.177 stock) are shown. Tissues were pre‐treated with either S‐6m‐P siRNA (300 nm, no transfection reagent) or RDV (10 μm) or left untreated before virus challenge. Dots represent mean, error bars represent SEM. Two‐way ANOVA combined with Dunnett's multiple comparisons test was performed. c) RT‐qPCR on S gene of SARS‐CoV‐2. d) RT‐qPCR on viral intracellular sgmRNAs in harvested tissue, GAPDH served as a reference. e), f) SARS‐CoV‐2 RNA FISH analysis of 3D lung microtissues, either pre‐treated with S‐6m‐P (300 nm, no transfection reagent) or RDV (10 μm) or left untreated, following SARS‐CoV‐2 infection (stock B1.177). e) Representative microscopic images 72 h p.i., scale bars: 60 μm. F) Quantitative analysis of e). Nested one‐way ANOVA combined with Dunnett's multiple comparisons test was performed. g) Histopathological analysis of S‐6m‐P (300 nm, no transfection reagent), RDV‐pre‐treated (10 μm) or untreated 3D lung microtissues 72 h post SARS‐CoV‐2 infection to quantify apoptosis of epithelial cells. Bar represents mean, error bars represent SD. Ordinary one‐way ANOVA combined with Dunnett's multiple comparisons test. Representative images are displayed, scale bar is 30 μm, arrows indicate apoptotic cells. c), d), f), g) * p adj‐value<0.05, ** p adj‐value <0.01, *** p adj‐value<0.001, **** p adj‐value<0.0001. Details about reproducibility and statistical analysis are given for all experiments in the Methods section.
In a pilot experiment, we analysed peptide‐mediated uptake of the siRNA into Caco‐2 cells, which endogenously express the virus entry receptor hACE2, using Alexa647‐labelled S‐6m and S‐6m‐P. We investigated the cellular uptake of the siRNAs by confocal microscopy, which allows the recording of z‐stacks to assess whether the siRNA has been taken up by the cells or is bound to the cell surface. We indeed observed uptake of the siRNA into Caco‐2 cells and, importantly, this uptake was dependent on the conjugation to the peptide (Figure 3b). However, the microscopy analyses also revealed that the peptide‐mediated siRNA uptake into cells was in this assay considerably less efficient compared to the lipofection‐based delivery.
Next, we explored whether the peptide modified siRNA (S‐6m‐P) can prevent SARS‐CoV‐2 replication in the EpiAirwayTM microtissues. For this experiment, we pre‐treated the human 3D lung microtissues with siRNA S‐6m‐P (300 nm, 24 h pre‐treatment basal and apical, no transfection reagent). RDV was again used as an antiviral positive control (10 μm, 2 h pre‐treatment). Untreated microtissue served as an additional negative control (Figure 3c, Input). Microtissues were subsequently challenged with SARS‐CoV‐2, followed by apical washes to remove input virus. Thereafter, post‐wash samples were taken (Figure 3c, Post‐Wash) and microtissues were subsequently cultivated for three additional days. Afterwards, viral replication was quantified by RT‐qPCR, both in the apical wash of the intact microtissues (Harvest) and in lysates of dispersed tissue (Tissue). In addition, we also quantified cell‐associated levels of several SARS‐CoV‐2 transcripts. These viral transcripts (sgmRNAs, Figure 3d) are synthesized in productively infected cells to express the viral nucleocapsid (N), as well as the spike (S), membrane (M), and envelope (E) proteins. These sgmRNAs can be distinguished in the RT‐qPCR from the viral genomic RNA by the presence of the 5′‐leader sequence, which is attached to every viral transcript, but only present once at the very 5′‐end of the viral genomic RNA. We observed an siRNA‐peptide chimera‐mediated inhibitory effect on SARS‐CoV‐2, which was comparable to the small molecule inhibitor RDV. Pre‐treatment with S‐6m‐P provided a ≈25‐fold reduction of the virus load in the apical wash and ≈80‐fold reduction of cell‐associated SARS‐CoV‐2 total RNA levels (Figure 3c and Figure S4c, d). Moreover, S‐6m‐P efficiently repressed the levels of all four viral transcripts investigated in a range from 130‐ to 400‐fold reduction (Figure 3d and Figure S4e, f).
Next, we performed RNA‐fluorescence in situ‐hybridization (RNA FISH) to further quantify SARS‐CoV‐2‐positive cells in infected lung microtissues. This allowed us to confirm the protective effect of our peptide‐conjugated siRNA. In this experiment, we also observed a significant reduction of the number of infected cells after pre‐treatment with S‐6m‐P (Figure 3e, f and Figure S4g). Importantly, we detected virus‐induced apoptotic cell death in untreated SARS‐CoV‐2‐infected tissues, but not after pre‐treatment with S‐6m‐P or RDV (Figure 3g, Figure S4h). These data fully support the protective effect of S‐6m‐P.
However, we noted that despite the siRNA treatment of the lung microtissue, genetic viral material was still detectable in the supernatant by RT‐qPCR (Figure 3c). To characterize this further, we re‐investigated the virus titration experiments with the siRNA S‐6m and lipofectamine transfection. In these experiments, we noted the same discrepancy. While the cell viability data indicated a complete suppression of virus replication at 72 h post infection (Figure 4a), residual virus material was, in contrast, still detectable by RT‐qPCR (Figure 4b) with only minor Ct value differences between S‐6m ‐ and Ctrl.‐10‐pre‐treated cells. To exclude a primer‐specific artefact, we performed additional PCR quantification with RdRp, E and M gene‐targeted primers and observed that the impact of S‐6m was in fact strongest for the S gene (Figure 4c). Based on these results, we hypothesized that the application of S‐6m induced a highly specific viral RNA degradation, limited to the siRNA‐targeting region. We reasoned that this might result in secretion of defective particles that lack the S protein and are thus non‐infectious. To test this hypothesis, we performed a supernatant transfer experiment, in which the culture supernatant of siRNA‐pre‐treated SARS‐CoV‐2‐infected A549‐hACE2 cells was used to challenge MDA‐MB‐231‐hACE2 cells (Figure 4d). We compared the effects of pre‐treatment of A549‐hACE2 cells with either siRNAs R‐2, S‐6m or an equimolar combination thereof, relative to Ctrl.‐10. We found that while Ctrl.‐10‐treated A549‐hACE2 supernatants induced a titratable cytopathic effect on MDA‐MB‐231‐hACE2 cells, supernatants of R‐2‐, S‐6m ‐ and in particular R‐2+S‐6m ‐treated A549‐hACE2 cells did not show evidence for a productive SARS‐CoV‐2 infection with release of infectious virions (Figure 4e). These data suggest that the PCR‐based analysis of the antiviral effect indeed underestimates the antiviral impact of the applied siRNAs. This result is of importance for future antiviral siRNA developments.
Figure 4.
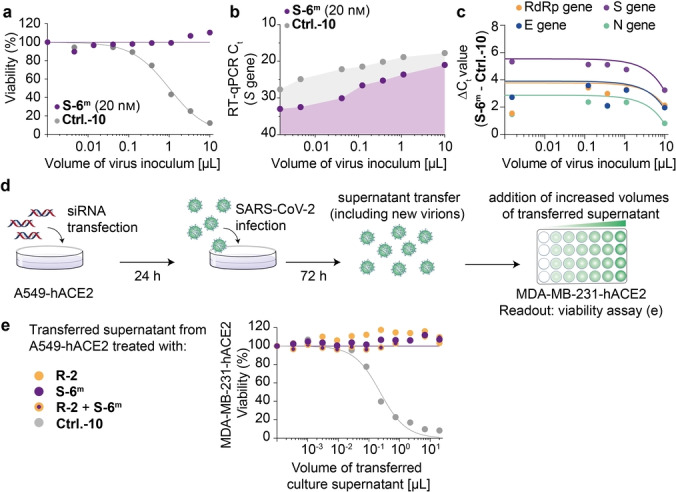
Effects of siRNA treatment on SARS‐CoV‐2 replication. a) Viability of SARS‐CoV‐2 (stock B1.177) challenged A549‐hACE2 cells 72 h post infection. Dots represent mean of two technical replicates from one representative experiment. b) SARS‐CoV‐2 RT‐qPCR to monitor SARS‐CoV‐2 RNA that was released into the supernatant of the infected A549‐hACE2 cells displayed in (a). c) Comparison of the ΔC t values including trend lines for RT‐qPCR on different SARS‐CoV‐2 coding regions. The same sample as in (b) was analysed. d) Schematic overview of transfer experiment to assess infectious virus titer in the supernatant of siRNA‐pretreated, infected A549‐hACE2 cells. An inoculum of 25 μL of B1.177 was used. e) Viability of MDA‐MB‐231‐hACE2 cells exposed to supernatants of A549‐hACE2 cells that had been pre‐treated with either R‐2 or S‐6m or a combination thereof or with Ctrl.‐10 prior to SARS‐CoV‐2 challenge.
Conclusion
In summary, the data presented here show that chemically modified and stabilized siRNAs offer a superb potential to develop antiviral therapies. A particular strength of the concept is its flexibility to rapidly adapt the siRNA sequences to newly arising mutants. Since the Omicron VoC (BA.1), similar to the Alpha and Beta VoCs, carries no mutation in the RNA regions targeted in this study, we expect that the here reported siRNA sequences will be active on such variants as well.
In contrast to small molecule‐based approaches, the siRNA‐based approach allows targeting of multiple viral sites at once, reducing the risk of escape variants. Furthermore, our study shows that fully‐modified siRNAs are not efficient in blocking virus replication. All the siRNAs that are in clinical use as of today target cellular genes that, in contrast to viruses, do not underlie a constant evolutionary pressure. Our data show that siRNAs can in principle be used to effectively suppress genetically rapidly evolving viral RNAs. Of note, our siRNA approach resulted in a stronger protective effect on virus‐induced cytotoxicity than on suppression of viral RNA species. Our data indicate that a partial 2′‐OMe‐modification is the ideal compromise for high siRNA stability and strong antiviral activity. Finally, click‐chemistry can be applied to modify the siRNA further, e.g. with receptor ligands such as peptides. This may allow the generation of potent antiviral siRNAs that can be used for cell type‐specific targeting. The future challenge is to identify high‐affinity receptor ligands that facilitate optimal siRNA internalization. Collectively, this study demonstrates that ligand‐coupled, optimized siRNA‐based therapeutics can be generated that efficiently restrict the infection by RNA viruses. For the treatment of patients with COVID‐19 inhalation of siRNA‐peptide conjugates may be a viable strategy.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
We thank Katharina Hofmann, Helga Mairhofer, Christopher Dächert, Natascha Grzimek‐Koschewa and Janett Wieseler for excellent technical support and discussion. We are grateful to Ulrike Protzer for providing access to the P3 laboratory of the Institute of Virology, TUM. We are grateful to Jochen Rech and Veit Hornung for help with qPCR analysis and laboratory access, respectively. We thank Andreas Sing for providing SARS‐CoV‐2 clinical isolates and Maximilian Münchhoff, Helmut Blum, Stefan Krebs and Alexander Graf for SARS‐CoV‐2 sequencing and analysis. We thank the Bayerische Forschungsstiftung (T.C. and O.T.K.) and Bay‐VOC (O.T.K.) for generous financial support. B.M.K was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—Project‐ID 369799452‐TRR237 and the Bundesministerium für Bildung und Forschung (BMBF), project TTU 01.806. The project has received additional funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program under grant agreement EpiR, No 741912. Further financial support was obtained from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) via SFB1309 (Project‐ID 325871075) and SPP 1784 (Project‐ID 277203618). We thank the Volkswagen Foundation for generous support through the EvoRib VW‐project. Open Access funding enabled and organized by Projekt DEAL.
F. R. Traube, M. Stern, A. J. Tölke, M. Rudelius, E. Mejías-Pérez, N. Raddaoui, B. M. Kümmerer, C. Douat, F. Streshnev, M. Albanese, P. R. Wratil, Y. V. Gärtner, M. Nainytė, G. Giorgio, S. Michalakis, S. Schneider, H. Streeck, M. Müller, O. T. Keppler, T. Carell, Angew. Chem. Int. Ed. 2022, 61, e202204556; Angew. Chem. 2022, 134, e202204556.
Contributor Information
Prof. Dr. Oliver T. Keppler, Email: Keppler@mvp.lmu.de.
Prof. Dr. Thomas Carell, Email: Thomas.carell@lmu.de.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Shrotri M., Swinnen T., Kampmann B., Parker E. P. K., Lancet Glob. Health 2021, 9, e590–e592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.X. Chen, A. S. Azman, W. Lu, R. Sun, N. Zheng, S. Ge, X. Deng, J. Yang, D. T. Leung, H. Yu, medRxiv 2021, 2021.08.26.21262699.
- 3.
- 3a. Hussain A., Hasan A., Nejadi Babadaei M. M., Bloukh S. H., Chowdhury M. E. H., Sharifi M., Haghighat S., Falahati M., Biomed. Pharmacother. 2020, 130, 110559; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b. Zost S. J., Gilchuk P., Case J. B., Binshtein E., Chen R. E., Nkolola J. P., Schäfer A., Reidy J. X., Trivette A., Nargi R. S., Sutton R. E., Suryadevara N., Martinez D. R., Williamson L. E., Chen E. C., Jones T., Day S., Myers L., Hassan A. O., Kafai N. M., Winkler E. S., Fox J. M., Shrihari S., Mueller B. K., Meiler J., Chandrashekar A., Mercado N. B., Steinhardt J. J., Ren K., Loo Y.-M., Kallewaard N. L., McCune B. T., Keeler S. P., Holtzman M. J., Barouch D. H., Gralinski L. E., Baric R. S., Thackray L. B., Diamond M. S., Carnahan R. H., Crowe J. E., Nature 2020, 584, 443–449; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3c. Taylor P. C., Adams A. C., Hufford M. M., de la Torre I., Winthrop K., Gottlieb R. L., Nat. Rev. Immunol. 2021, 21, 382–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Harvey W. T., Carabelli A. M., Jackson B., Gupta R. K., Thomson E. C., Harrison E. M., Ludden C., Reeve R., Rambaut A., Peacock S. J., Robertson D. L., Consortium C.-G. U., Nat. Rev. Microbiol. 2021, 19, 409–424; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Zhou D., Dejnirattisai W., Supasa P., Liu C., Mentzer A. J., Ginn H. M., Zhao Y., Duyvesteyn H. M. E., Tuekprakhon A., Nutalai R., Wang B., Paesen G. C., Lopez-Camacho C., Slon-Campos J., Hallis B., Coombes N., Bewley K., Charlton S., Walter T. S., Skelly D., Lumley S. F., Dold C., Levin R., Dong T., Pollard A. J., Knight J. C., Crook D., Lambe T., Clutterbuck E., Bibi S., Flaxman A., Bittaye M., Belij-Rammerstorfer S., Gilbert S., James W., Carroll M. W., Klenerman P., Barnes E., Dunachie S. J., Fry E. E., Mongkolsapaya J., Ren J., Stuart D. I., Screaton G. R., Cell 2021, 184, 2348–2361.e2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhao L., Li S., Zhong W., Front. Pharmacol. 2022, 13, DOI: 10.3389/fphar.2022.840639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Fire A., Xu S., Montgomery M. K., Kostas S. A., Driver S. E., Mello C. C., Nature 1998, 391, 806–811; [DOI] [PubMed] [Google Scholar]
- 6b. Caplen N. J., Parrish S., Imani F., Fire A., Morgan R. A., Proc. Natl. Acad. Sci. USA 2001, 98, 9742–9747; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6c. Elbashir S., Lendeckel W., Tuschl T., Genes Dev. 2001, 15, 188–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Hu B., Zhong L., Weng Y., Peng L., Huang Y., Zhao Y., Liang X.-J., Signal Transduction Targeted Ther. 2020, 5, 101; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Zhang X., Goel V., Robbie G. J., J. Clin. Pharmacol. 2020, 60, 573–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. V'kovski P., Kratzel A., Steiner S., Stalder H., Thiel V., Nat. Rev. Microbiol. 2021, 19, 155–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Maillard P. V., van der Veen A. G., Poirier E. Z., Reis e Sousa C., EMBO J. 2019, 38, e100941; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9b. Li B.-J., Tang Q., Cheng D., Qin C., Xie F. Y., Wei Q., Xu J., Liu Y., Zheng B.-J., Woodle M. C., Zhong N., Lu P. Y., Nat. Med. 2005, 11, 944–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Selvam C., Mutisya D., Prakash S., Ranganna K., Thilagavathi R., Chem. Biol. Drug Des. 2017, 90, 665–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lan T. C. T., Allan M. F., Malsick L. E., Woo J. Z., Zhu C., Zhang F., Khandwala S., Nyeo S. S. Y., Sun Y., Guo J. U., Bathe M., Näär A., Griffiths A., Rouskin S., Nat. Commun. 2022, 13, 1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wratil P. R., Stern M., Priller A., Willmann A., Almanzar G., Vogel E., Feuerherd M., Cheng C. C., Yazici S., Christa C., Jeske S., Lupoli G., Vogt T., Albanese M., Mejías-Pérez E., Bauernfried S., Graf N., Mijocevic H., Vu M., Tinnefeld K., Wettengel J., Hoffmann D., Muenchhoff M., Daechert C., Mairhofer H., Krebs S., Fingerle V., Graf A., Steininger P., Blum H., Hornung V., Liebl B., Überla K., Prelog M., Knolle P., Keppler O. T., Protzer U., Nat. Med. 2022, 28, 496–503. [DOI] [PubMed] [Google Scholar]
- 13. Pruijssers A. J., George A. S., Schäfer A., Leist S. R., Gralinksi L. E., K. H. Dinnon III , Yount B. L., Agostini M. L., Stevens L. J., Chappell J. D., Lu X., Hughes T. M., Gully K., Martinez D. R., Brown A. J., Graham R. L., Perry J. K., Du Pont V., Pitts J., Ma B., Babusis D., Murakami E., Feng J. Y., Bilello J. P., Porter D. P., Cihlar T., Baric R. S., Denison M. R., Sheahan T. P., Cell Rep. 2020, 32, 107940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Grimson A., Farh K. K.-H., Johnston W. K., Garrett-Engele P., Lim L. P., Bartel D. P., Mol. Cell 2007, 27, 91–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.
- 15a. Setten R. L., Rossi J. J., Han S.-P., Nat. Rev. Drug Discovery 2019, 18, 421–446; [DOI] [PubMed] [Google Scholar]
- 15b. Balwani M., Sardh E., Ventura P., Peiró P. A., Rees D. C., Stölzel U., Bissell D. M., Bonkovsky H. L., Windyga J., Anderson K. E., Parker C., Silver S. M., Keel S. B., Wang J.-D., Stein P. E., Harper P., Vassiliou D., Wang B., Phillips J., Ivanova A., Langendonk J. G., Kauppinen R., Minder E., Horie Y., Penz C., Chen J., Liu S., Ko J. J., Sweetser M. T., Garg P., Vaishnaw A., Kim J. B., Simon A. R., Gouya L., N. Engl. J. Med. 2020, 382, 2289–2301. [DOI] [PubMed] [Google Scholar]
- 16. Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T. S., Herrler G., Wu N.-H., Nitsche A., Müller M. A., Drosten C., Pöhlmann S., Cell 2020, 181, 271–280.e278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.
- 17a. Ballarín-González B., Howard K. A., Adv. Drug Delivery Rev. 2012, 64, 1717–1729; [DOI] [PubMed] [Google Scholar]
- 17b. González B. B., Nielsen E. B., Thomsen T. B., Howard K. A. in RNA Interference from Biology to Therapeutics (Ed.: Howard K. A.), Springer US, Boston, 2013, pp. 97–125. [Google Scholar]
- 18. Cao X., Coyle J. P., Xiong R., Wang Y., Heflich R. H., Ren B., Gwinn W. M., Hayden P., Rojanasakul L., In Vitro Cell. Dev. Biol. Anim. 2021, 57, 104–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.
- 19a.H. Zarkoob, A. Allué-Guardia, Y.-C. Chen, O. Jung, A. Garcia-Vilanova, M. J. Song, J.-G. Park, F. Oladunni, J. Miller, Y.-T. Tung, I. Kosik, D. Schultz, J. Yewdell, J. B. Torrelles, L. Martinez-Sobrido, S. Cherry, M. Ferrer, E. M. Lee, bioRxiv 2021, 2021.05.11.443693;
- 19b.Y. Liu, J. Liu, K. S. Plante, J. A. Plante, X. Xie, X. Zhang, Z. Ku, Z. An, D. Scharton, C. Schindewolf, V. D. Menachery, P.-Y. Shi, S. C. Weaver, bioRxiv 2021, 2021.03.08.434499;
- 19c. Hattori S.-I., Higashi-Kuwata N., Hayashi H., Allu S. R., Raghavaiah J., Bulut H., Das D., Anson B. J., Lendy E. K., Takamatsu Y., Takamune N., Kishimoto N., Murayama K., Hasegawa K., Li M., Davis D. A., Kodama E. N., Yarchoan R., Wlodawer A., Misumi S., Mesecar A. D., Ghosh A. K., Mitsuya H., Nat. Commun. 2021, 12, 668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gramlich P. M. E., Warncke S., Gierlich J., Carell T., Angew. Chem. Int. Ed. 2008, 47, 3442–3444; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 3491–3493. [Google Scholar]
- 21. Struck A.-W., Axmann M., Pfefferle S., Drosten C., Meyer B., Antiviral Res. 2012, 94, 288–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.
- 22a. Brunner K., Harder J., Halbach T., Willibald J., Spada F., Gnerlich F., Sparrer K., Beil A., Möckl L., Bräuchle C., Conzelmann K.-K., Carell T., Angew. Chem. Int. Ed. 2015, 54, 1946–1949; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 1968–1971; [Google Scholar]
- 22b. Willibald J., Harder J., Sparrer K., Conzelmann K.-K., Carell T., J. Am. Chem. Soc. 2012, 134, 12330–12333. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.