

To the Editor,
I found the article published by Magalis et al. 1 Magalis et al. 1 particularly interesting as the information presented in this article covers two significant aspects that should be considered as a part of the control of the coronavirus disease 2019 (COVID‐19) pandemic. The first of these aspects is the potential role of the low‐frequency variants in the epidemiology of COVID‐19, an issue directly linked with the evolutionary profile of severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2), and the second aspect is the presentation of breakthrough infections among vaccinated individuals, a situation associated with the effectiveness of the current vaccines used during this pandemic.
To date (June 27, 2022), official data by the World Health Organization indicates that the COVID‐19 pandemic has produced a total of 539 893 858 cases of COVID‐19 resulting in a total of 6 324 112 deaths worldwide (https://covid19.who.int/). These numbers reflect the complexity of this situation in terms of the control of this pandemic and highlights the importance of increasing the knowledge in multiple aspects of the epidemiological triad of SARS‐CoV‐2 2 , 3
As mentioned above, I believe that one of the main complications in controlling this pandemic is due to the extraordinary and unexpected genome plasticity shown by SARS‐CoV‐2, a condition reported since the initial stages of this pandemic. 4 As a consequence, more than 1300 lineages have been identified based on the Pango lineage designation. 5 The genetic relationship among these lineages can be phylogenetically represented by the existence of 14 divergent clades (Figure 1A). 6 Furthermore, the rapid evolutionary divergence shown by SARS‐CoV‐2 during this pandemic, is a phenomenon that may be highly explained by the role of natural selection. In this sense, based on the analysis reported by the adaptive evolution server Datamonkey, 7 at least 862 sites in the genome of SARS‐CoV‐2 have been predicted to be under positive selection. The lack of correlation between the number of sites under positive selection and the length of different genes, indicates that the detection of these sites is not just a mere artifact influenced by the length of different genes (R 2 = 0.073, p = 0.221; Figure 1B). Similar results were obtained when validated/predicted cytotoxic T lymphocytes (CTL) epitopes were evaluated 8 (Figure 1C).
Figure 1.
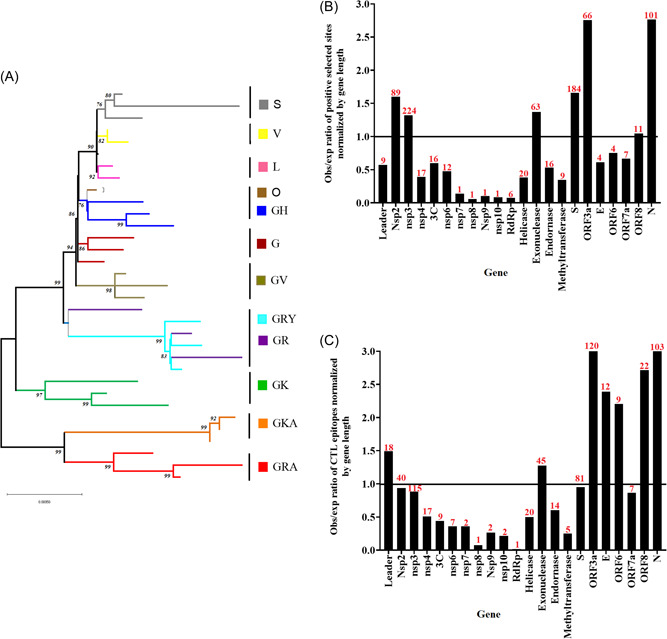
Evolutionary profile of SARS‐CoV‐2 during the pandemic of COVID‐19. (A) Phylogenetic tree reconstructed by Maximum likelihood and the general time reversible model, illustrating the 14 clades (based on GISAID classification) predicted during the evolution of SARS‐CoV‐2. This tree was constructed using a total of 32 full‐length sequences representing different clades. Representative sequences were obtained from GISAID database (https://www.gisaid.org/). (B) Observed/expected ratio (obs/exp) of sites under positive selection at different genes sections of SARS‐CoV‐2 normalized by gene length. (C) obs/exp ratio of CTL epitopes at different gene sections of SARS‐CoV‐2 normalized by gene length. (Obs/exp = proportion of the total number of predicted positive selected sites or CTL epitopes found at specific genome sections/proportion of length at the overall coding region of SARS‐CoV‐2 represented by different gene sections). Red numbers on the bars represent the raw number of either sites under positive selection or CTL epitopes (predicted/validated) at different gene sections of SARS‐CoV‐2. Genes of SARS‐CoV‐2 where no information was available about sites under positive selection or epitopes were excluded from the graphics. The horizontal line at 1.0 indicate obs/exp values under the expected gene size. Information for (B) and (C) were obtained from the adaptive evolution server Datamonkey (https://observablehq.com/@spond/revised‐sars‐cov‐2‐analytics‐page). COVID‐19, coronavirus disease 2019; CTL, cytotoxic T lymphocyte; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2.
Interestingly, the correlation between the prediction of some positively selected sites and the presence of distinct CTL epitopes (R 2 = 0.630, p ≤ 0.0001) suggests that the host immune response may be one of the main drivers in the evolution of SARS‐CoV2. In this context, the presence of positively selected sites associated with CTL‐escape mutations must be influenced by differences in human leukocyte antigen allele distribution worldwide.
The above data may help to explain the circulation dynamics of different lineages, a condition that can be exemplified by the fluctuation in the proportion of different divergent clades during the pandemic (Figure 2A). Based on the mutation profile in the spike protein, the World Health Organization has classified specific groups of variant lineages as variant being monitored, variant of concern (VOC), or variant of interest. The dominance of these variants has been fluctuating during the pandemic, with the omicron VOC (B.11.529) being the one dominating the infections worldwide during 2022 9 (Figure 2B).
Figure 2.
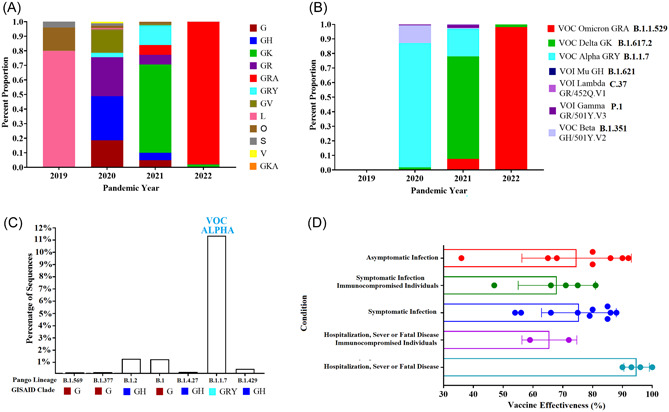
Lineage circulation dynamics of SARS‐CoV‐2 during the pandemic, and vaccine effectiveness of RNA vaccines against COVID‐19. (A) Relevance of different divergent clades of SARS‐CoV‐2 during the pandemic (based on GISAID classification). (B) Circulation dynamic of different variants during the pandemic. (C) Percentage of available sequences at the GISAID database associated with the lineages described by Magalis et al., 1 representing the low circulation of these lineages during the pandemic around the world. However, it is important to consider the huge temporal and geographical biases in sequencing and the presence of locally dominant variants that might have predominated in a single region/or country for this reason the concept of low‐circulating lineage must be taken with caution. Information about the number of sequences associated with different clades and lineages was obtained from the GISAID database. (D) Summary of the effectiveness at different clinical conditions of Moderna and Pfizer BioNTech vaccines against SARS‐COV‐2. Information was obtained from the Centers of Disease Control and Prevention (https://www.cdc.gov/coronavirus/2019-ncov/science/science-briefs/fully-vaccinated-people.html. COVID‐19, coronavirus disease 2019; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2.
Notably, the work published by Magalis et al. 1 presents an interesting perspective that the VOC where not the only variants producing breakthrough infections in vaccinated individuals, but in addition variants of low circulation also played a part in this equation during the pandemic (Figure 2C). The authors report a new set of mutations in the spike protein of lineage B.1.1.7 (L18F and K1191N), which have also come under positive selection during the course of the pandemic (https://observablehq.com/@spond/revised‐sars‐cov‐2‐analytics‐page). These residues as mentioned previously are predicted/validated to be associated with CTL epitopes. 8 , 10 Similarly, Magalis et al. 1 describes T572I mutation in the spike protein of lineage B.1.2, which also appear to be under positive selection. Future research is needed to understand the role of this mutation.
Finally, this study indirectly covers a relevant issue associated with the effectiveness of RNA vaccines and the presentation of breakthrough infections, a situation that must be considered as a potential issue for the generation of new variants especially in immunocompromised people (Figure 2D).
In conclusion, the work published by Magalis et al. 1 reflects the importance of the continuous research on the evolution of SARS‐CoV‐2, and the necessity to improve current vaccines to promote the control of the COVID 19 pandemic.
CONFLICT OF INTEREST
The author declares no conflict of interest
ACKNOWLEDGMENTS
The author thanks Dr. Lauren Holinka for her critical review of this letter. This study was conceived in absence of funding.
DATA AVAILABILITY STATEMENT
No datasets were generated during this study.
REFERENCES
- 1. Magalis BR, Mavian C, Tagliamonte M, et al. Low‐frequency variants in mildly symptomatic vaccine breakthrough infections presents a doubled‐edged sword. J Med Virol. 2022;94:3192‐3202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schafer EA, Eberl S, Velazquez‐Salinas L. Beyond the biology: evaluating the role of political, economic, and social factors associated with the incidence and mortality of SARS‐CoV‐2 during the first seven months of the COVID‐19 pandemic. arXiv. 2022;2204:13995. [Google Scholar]
- 3. Schafer EA, Velazquez‐Salinas L. Controlling the COVID‐19 pandemic: the complex epidemiological triad of SARS‐CoV‐2. Int J Infect Dis Epidemiol. 2021;2:41‐42. [Google Scholar]
- 4. Velazquez‐Salinas L, Zarate S, Eberl S, Gladue DP, Novella I, Borca MV. Positive selection of ORF1ab, ORF3a, and ORF8 genes drives the early evolutionary trends of SARS‐CoV‐2 during the 2020 COVID‐19 pandemic. Front Microbiol. 2020;11:550674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. O'Toole A, Pybus OG, Abram ME, Kelly EJ, Rambaut A. Pango lineage designation and assignment using SARS‐CoV‐2 spike gene nucleotide sequences. BMC Genomics. 2022;23:121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Khare S, Gurry C, Freitas L, et al. GISAID's role in pandemic response. China CDC Wkly. 2021;3:1049‐1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Weaver S, Shank SD, Spielman SJ, Li M, Muse SV, Kosakovsky Pond SL. Datamonkey 2.0: a modern web application for characterizing selective and other evolutionary processes. Mol Biol Evol. 2018;35:773‐777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nelde A, Bilich T, Heitmann JS, et al. SARS‐CoV‐2‐derived peptides define heterologous and COVID‐19‐induced T cell recognition. Nat Immunol. 2021;22:74‐85. [DOI] [PubMed] [Google Scholar]
- 9. Harvey WT, Carabelli AM, Jackson B, et al. SARS‐CoV‐2 variants, spike mutations and immune escape. Nat Rev Microbiol. 2021;19:409‐424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Campbell KM, Steiner G, Wells DK, Ribas A, Kalbasi A. Prioritization of SARS‐CoV‐2 epitopes using a pan‐HLA and global population inference approach. bioRxiv . [Preprint]. 2020. 10.1101/2020.03.30.016931 [DOI]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No datasets were generated during this study.