

Abstract
The precise interaction between the immune system and severe acute respiratory syndrome coronavirus 2 (SARS‑CoV‑2) is critical in deciphering the pathogenesis of coronavirus disease 2019 (COVID‐19) and is also vital for developing novel therapeutic tools, including monoclonal antibodies, antivirals drugs, and vaccines. Viral infections need innate and adaptive immune reactions since the various immune components, such as neutrophils, macrophages, CD4+ T, CD8+ T, and B lymphocytes, play different roles in various infections. Consequently, the characterization of innate and adaptive immune reactions toward SARS‐CoV‐2 is crucial for defining the pathogenicity of COVID‐19. In this study, we explain what is currently understood concerning the conventional immune reactions to SARS‐CoV‐2 infection to shed light on the protective and pathogenic role of immune response in this case. Also, in particular, we investigate the in‐depth roles of other immune mediators, including neutrophil elastase, serum amyloid A, and syndecan, in the immunopathogenesis of COVID‐19.
Keywords: adaptive immunity, COVID‐19, innate immunity, neutrophil elastase, pathogenesis, syndecan
1. INTRODUCTION
As the 2019 coronavirus disease (COVID‐19) etiologic agent, the novel emerging coronavirus known as severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2), its gaps in deciphering the pathogenesis are just completing. 1 , 2 , 3 , 4 , 5 , 6 Early and moderate COVID‐19 stays in the upper respiratory tract, eliciting a minimal innate immune response. 7 Immune cells and molecules, including neutralizing antibodies (NAbs), are present in sufficient numbers in recovered individuals at this stage to assist in combat infection, and this is evidenced by their effective elimination of the virus. 8 , 9
The function of cellular immunity in a protective immune response to COVID‐19 is becoming evident. Recent research has shown that SARS‐CoV‐2 elicits a strong and highly potent T‐cell‐mediated immunity (even in antibody‐seronegative individuals), which provides long‐term immunity. 10 , 11 Many factors involved in immunity toward SARS‐CoV‐2 have been discovered, varying from innate to adaptive immunity. 12 , 13 More T‐cell activation is associated with less disease and mortality, according to research. 14 Rehabilitation following COVID‐19 requires a robust Th1 response, even though non‐Th1 mediators have been associated with respiratory failure. 15 , 16 Several studies have demonstrated the effectiveness of the antibody response, notably the immunoglobulin G (IgG) reaction in predicting patient survival. 15 , 16 Furthermore, a synchronized T‐ and B‐cell reaction, particularly anti‐spike (S) IgG in combination with interleukin‐2 (IL−2)/interferon‐γ (IFN‐γ) producing activated CD4+ and CD8+ T cells, is involved in viral elimination. 17 , 18 Although an ordered adaptive immunity is required for mild illness, the significance of innate cytokines in establishing a significant T‐cell reaction in COVID‐19 remains unclear. 19
Further information and insight into pathophysiological mechanisms underlying SARS‐CoV‐2 disease and COVID‐19 development are starting to emerge, emphasizing the essential importance of immunological hyper‐response, defined by widely spread endothelial dysfunction, systemic microangiopathy, and complement‐induced blood coagulation in illness aggravation. 20 As previously discussed, the host's substantial proinflammatory reaction may promote endothelial dysfunction in COVID‐19, notably through the activity of IL‐6 as well as the concentration of tumor necrosis factor‐α (TNF‐α), which are significantly elevated in severe forms of the disease. 20 , 21 Besides, COVID‐19 appears to be more than simply a respiratory illness; it may be the result of a systemic malfunction brought on by a bradykinin storm that begins in the lungs. 22 , 23 In this review, we summarize what is known about the innate and adaptive immunity to SARS‐CoV‐2, as well as the function of additional immunological mediators in COVID‐19, such as neutrophil elastase (NE), serum amyloid A (SAA), and syndecan (SDC).
2. COVID‐19 IMMUNOPATHOGENESIS
The precise mechanism of the pathogenicity of COVID‐19 is not entirely understood; however, many findings show that it is similar to another coronavirus, SARS‐CoV‐1. 24 The mode of transmission from person to person has a significant impact on pathophysiology (Figure 1). 25 , 26 A receptor for SARS‐CoV‐2 entrance is angiotensin‐converting enzyme 2 (ACE2), whereas an activator for virus entry is proteases, and SARS‐CoV‐2 can also bind via CD147, making it easier for the virus to spread. 27 , 28 , 29
Figure 1.
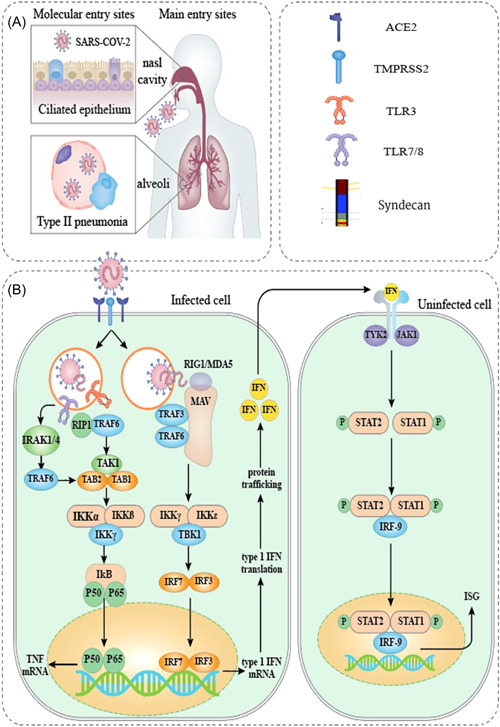
. The schematic representation of SARS‐CoV‐2 pathophysiology. (A) SARS‐CoV‐2 enters the body primarily through cells in the nasal cavity and the upper and lower respiratory tracts. (B) Several PRRs that identify foreign RNA, such as endosomal TLR3 and TLR7, and cytoplasmic RIG‐I and MDA5, are thought to be involved in recognizing SARS‐CoV‐2. Results from genetic research, functional and clinical findings, interaction modeling, and CRISPR screens are used to estimate downstream signaling occurrences. Direct communication among viral or host proteins and interplay among SARS‐CoV‐2‐derived proteins and cellular mechanisms as defined by interaction mapping derived information. ORF3b was found to be functionally active in the suppression of type I IFN, but no specific target was recognized. 12 CRISPR, clustered regularly interspaced short palindromic repeat; IFN, interferon; MDA5, melanoma differentiation‐associated protein 5; ORF, open reading frame; PRR, pattern recognition receptor; TLR, Toll‐like receptor; RIG‐I, retinoic acid‐inducible gene I; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2.
The essential protease in activating SARS‐CoV‐2 is transmembrane serine protease 2/CD147. 24 In addition to ACE2, pattern recognition receptors (PRRs) play a role in the pathophysiology of virus‐mediated immunopathology as well. 24 PRRs contribute to identifying pathogens, such as viruses. 30 Upon direct interaction with the viral receptors on the surface proteins, viruses trigger a variety of host immunity reactions, including the induction and enhancement of inflammatory mediators, the development and increased activity of dendritic cells (DCs), and the elevated expression of IFNs to block virus dissemination and replication. 30
An increasing body of research has improved our knowledge of how dysfunctional immune cells contribute to the inflammatory response in COVID‐19 patients. Some groups have used RNA‐sequencing (RNA‐seq) as a method for investigating the functionality of various immune reactions. Yao et al. 31 investigated the transcriptome of healthy and COVID‐19 patient peripheral blood mononuclear cells (PBMCs) using RNA‐seq. Although most immune cell compartments exhibited the predicted hyperinflammatory response in very unwell patients, they discovered that numerous essential pathways were malfunctioning, which may have contributed to their inability to manage the viral infection. In fact, PBMCs from the severe group exhibited a transcriptomic signal indicating deficits in virus‐clearing processes, including cytotoxic killing in natural killer (NK) and CD8 T cells, B‐cell activation, and reduced antigen presentation by monocytes. 31
SARS‐CoV‐2 elicited various immune reactions in infected individuals. 24 Upon being activated by the virus, CD4+ T cells generate some mediators and cytokines that stimulate the production of B cells as well as cytotoxic T lymphocytes. 24 Activated B cells subsequently generate antibodies (IgG and immunoglobulin M (IgM)) specific to the virus. 24 The cytotoxicity is mediated by activated CD8+ T lymphocytes, engulfing and destroying the virus‐infected cells. It is crucial to highlight that, even though the existence of complement factors (C3a and C5a) and antibodies are required to combat viral attacks, SARS‐CoV‐2 can evade host immunity by T‐cell function via the induction of apoptosis in T cells. 32 Serological examinations of rescued symptomatic individuals have shown elevated levels of virus‐specific nAbs and enhanced synthesis of antibodies secreting B cells by the immune system. 9 , 24 Moreover, several clinical investigations have demonstrated that recovered patients had a rise in T cells such as CD8+ and CD4+ cells and T follicular helper (TFH) cells. 33 , 34 However, emerging reports have revealed that an excessive and disrupted immune reaction in severe COVID‐19 patients with higher inflammatory mediators is assumed to be a starting point for pathophysiology and results in severe abnormalities and pulmonary worsening. 35
According to increasing data, COVID‐19 pneumonia may be caused by the T helper type 17 (Th17) inflammatory response. The release of cytokines such as IL‐17 and granulocyte–macrophage colony‐stimulating factor (GM‐CSF), stimulation of neutrophil migration, and a decrease in the regulatory T‐cell (Treg) response all contribute to the immune response's exaggeration. Treg cells, unlike Th17 cells, produce anti‐inflammatory mediators (IL‐4, IL‐10, and transforming growth factor‐β [TGF‐β]) and play a critical role in reducing hyperactive immune responses. 36 Patients with severe COVID‐19 have a lower Treg/Th17 cell ratio, indicating that proinflammatory responses are not adequately controlled. 37 COVID‐19 patients may have a disproportionately high proportion of Tregs, rather than Th17 cells, in their immune system, which may contribute to the unregulated release of cytokines and chemokines, resulting in tissue damage. 36 Th17 cells in bronchoalveolar lavage fluid (BALF) from individuals with COVID‐19 were shown to be more prevalent than in healthy subjects in several investigations. 37 , 38
Acute and chronic pulmonary repercussions in SARS‐CoV‐2 infected patients, including pneumonia, acute respiratory distress syndrome (ARDS), and lung fibrosis, continue to be key concerns despite rapid advancements in early detection, illness treatment, and vaccine development. In this regard, Wang et al. 39 studied the circulating soluble factors and single‐cell RNA seq (scRNA‐seq) of PBMCs in individuals with severe COVID‐19. In pulmonary fibrosis‐high patients, the expression of genes enriched in IFN signaling, innate immune response, and adaptive immune response were lower in T cells, NK cells, and monocytes than in pulmonary fibrosis‐low patients. In conclusion, their findings suggested that reduced IFN‐responsive genes and their associated signaling pathways may be crucial for the advancement of pulmonary fibrosis in COVID‐19 patients. A multiomic single‐cell immune profiling was carried out by Wilk et al. 40 in COVID‐19 patients with varying degrees of disease severity, ranging from mild outpatient cases to fatal ones. They discovered the significant failure of innate immunity, including strong hyperactivation signals in neutrophils and NK cells, in severe and lethal COVID‐19. They also discovered alterations in chromatin accessibility at nuclear factor‐κB (NF‐κB) binding sites within cytokine gene loci as a potential reason for the dramatic absence of proinflammatory cytokine production reported in monocytes with severe and fatal COVID‐19. Wilk et al. 40 found further that emergency myelopoiesis is a key characteristic of COVID‐19. These new findings show immunological phenotypes linked with disease severity in COVID‐19 and suggest pathogenesis‐related pathways that are possible treatment targets. Readers refer to other comprehensive reviews for more details on COVID‐19 immunopathogenesis. 37 , 41 , 42
3. IMMUNE REACTIONS AND COVID‐19
Several molecular mechanisms have been described to better explain the complicated molecular mechanisms underlying the cytokine storm reaction in COVID‐19 cases. 43 Since it promotes lymphopenia and lymphocyte malfunction, understanding the cytokine storm mechanism would be critical, and defects in cytotoxicity of NK cells from the innate immunity and cytolytic T cells from the adaptive immunity are cited as reasons for the cytokine storm's progression. 44 , 45 Nevertheless, this dysfunctional state, whether hereditary or acquired, prevents cytolytic cells from inducing apoptosis in infected and activated antigen‐presenting cells (APCs). Many proinflammatory mediators are produced as a result of the prolonged and excessive interplay among innate and adaptive immunity, and the analysis revealed that the amounts of immune cells, including NK, B, CD4+ T, and CD8+ T cells are significantly altered in COVID‐19 cases. 44 However, little is known about the immunological response in asymptomatic and redetectable‐positive individuals. PBMC samples from individuals with various COVID‐19 presentations were examined by Vigón et al. 46 for some characteristics associated with the cellular immune response. They discovered that the severely cytotoxic CD8+ T‐cell subset was present in low levels in individuals with serious COVID‐19. In contrast, high Treg levels, low plasma IL‐2 levels, and poor Th1 differentiation were associated with a significantly lower CD4 count. In this section, we look at how far we have come in decoding the immune reactions to COVID‐19 (Figure 2).
Figure 2.
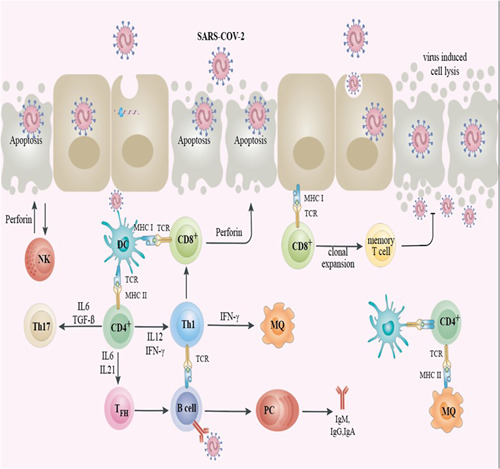
The schematic representation of the immune reaction against SARS‐CoV‐2. When SARS‐CoV‐2 infects the epithelium, cells may undergo lysis and significant injury to the epithelial cell during virus replication. The viral antigens were presented to CD8+ T cells by the epithelial cell. CD8+ T cells and NK cells could cytolyze the endothelial cells infected by SARS‐CoV‐2 with their perforin and granzymes, causing programmed cell death (apoptosis). DC in subepithelial recognize SARS‐CoV‐2 antigens and then the processed antigens presented to the T CD4+, causing these T cells to differentiate toward memory Th1, Th17, and memory TFH. TFH supports the development of B cells into PC and the development of specific antibodies against SARS‐CoV‐2 (IgA, IgM, and IgG). Moreover, SARS‐CoV‐2 antigens were presented to the T CD4+ cells by DCs and tissue MΦ. 33 DC, dendritic cell; Ig, immunoglobulin; MΦ, macrophage; NK, natural killer; PC, plasma cells; TFH, T follicular helper cells; Th, helper T cell; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2.
3.1. Innate immunity and COVID‐19 pathogenesis
Effective immunity to pathogenic organisms prompts the initial stimulation of innate immune responses, as well as the maintenance of specific adaptive immune reactions, which significantly contribute to infection clearance and prevention of reinfection by the same infectious agent. 47 , 48 , 49 When tissue‐resident cells identify SARS‐CoV‐2, local immunological responses occur, resulting in the recruitment of several innate mediators from the bloodstream circulation. 1 , 47
SARS‐CoV‐2 can escape innate immunity identification, signaling, IFN production, and IFN‐stimulated genes (ISGs) by expressing a plethora of viral proteins that disrupt these processes. 50 As a result, SARS‐CoV‐2‐infected patients have lower rates of IFN‐I or IFN‐III in their lungs or peripheral blood compared to other respiratory pathogens. 51 , 52 The generation of IFN‐1 and IFN‐III and ISGs in the upper airways is linked with lower disease severity, while the production of IFN‐II and type I IFNs (but not ISGs) is associated with the severity of COVID‐19. 53 COVID‐19 would be life‐threatening in those who have genetic abnormalities or autoantibodies that impair IFN systems, as detailed below. Persistent IFN secretion is linked with poorer clinical outcomes in advanced stages, probably via the production of chemokines that attract inflammatory cellular infiltrates. 54 , 55 , 56 Furthermore, COVID‐19 is concomitant with a considerable decrease in the number of immunological sensor cells in the blood and lungs, both plasmacytoid DCs (pDCs) and conventional DCs (cDCs). 54 In this part, we describe the most recent research on innate immune cells and COVID‐19 (Table 1).
Table 1.
Overview of innate immune reactions to SARS‐CoV‐2
| Innate immune response | Reaction | Outcome | References |
|---|---|---|---|
| Macrophage |
After SARS‐CoV‐2 infection, the renal, splenic, and alveolar macrophages are stimulated and then heightened the formation of proinflammatory cytokines such as IL‐6, IL‐10, and TNF‐α. In sum, the accumulating evidence indicated that in severe cases with COVID‐19, alveolar macrophages are likely to generate chemokines that select further neutrophils and monocytes to the lung, which contribute to the excessive formation of proinflammatory agents. |
Induction of highly inflammatory response and potent chemokines, ARDS |
47 , 57 , 58 |
| Neutrophil |
Neutrophils serve as hyperinflammation operators using increased cell degranulation and cytokine production in patients with COVID‐19. Notably, investigations explained that the exhibition of neutrophils from healthy subjects to cases infected with SARS‐CoV‐2 sera supports the NET activity, suggesting that NETs might act as a possible target in severe cases with COVID‐19. |
Tissue injury due to potent inflammatory reactions | 59 , 60 , 61 |
| NK cell |
The rate of CD56dimCD16+KIR+ NK cells was significantly decreased in the blood sample of COVID‐19 patients, implying either disrupted maturation or expanded recruitment of NK toward tissues infected with SARS‐CoV‐2. The recent finding demonstrated that COVID‐19 could modulate the cytotoxic activity of NK cells by provoking the upregulation of the NKG2A. The impaired cytotoxic activity and decreased number of NK cells in circulation were noticed in severe cases with COVID‐19, in mild patients, and in dead versus survivor cases, proposing that the functional impairment of NK cells activity points to enhanced cell activation innate immunity with an extensive production of proinflammatory cytokine. |
The induction of massive production of proinflammatory cytokine due to increased activation of innate immunity cells | 47 , 62 , 63 , 64 |
| MDSC |
Current reports have indicated a dysregulation in the myeloid cells in COVID‐19 severe cases, with heightened levels and activity of MDSC relating to disease severity. The enhanced ratio of MDSC to T CD8+ effector cells (memory) was found in severe COVID‐19 cases with ARDS compared to moderate pneumonia cases with COVID‐19; this finding showed that MDSC related to COVID‐19 augmentation is directly associated with lymphopenia and heightened arginase activity. The accumulating data proposed that G‐MDSCs and other myeloid cells signify unlimited negative feedback, eventually establishing pan‐immunosuppression and following dysregulation in adaptive immune responses. |
Modulating immunity against SARS‐CoV‐2 (immunosuppressive properties) increases cytokine levels and other proinflammatory markers | 65 , 66 , 67 , 68 , 69 |
| Eosinophil |
Comprehensive examination showed that COVID‐19 severity is correlated with intensified eosinophil‐mediated pulmonary inflammation. The recent finding showed that SARS‐CoV‐2 infection distinct innate immune responses, including inflammatory conditions related to eosinophil and following Th2 reactions, contributing to severe pneumonia associated with COVID‐19. |
Pulmonary inflammation | 57 , 70 , 71 , 72 |
| DCs |
The investigation revealed that isolated pDCs are stimulated through diversification into P1 and P2, as well as P3 subpopulations. It has been shown that BALFs from severe and critical COVID‐19 cases comprise fewer pDCs than moderate cases. The pDCs stimulated in COVID‐19 generate high concentrations of IFNs by the TLR‐7 pathway. |
Impaired IFN‐α production | 73 , 74 , 75 |
Abbreviations: ARDS, acute respiratory distress syndrome; BALF, bronchoalveolar lavage fluid; COVID‐19, coronavirus disease 2019; DC, dendritic cell; IFN, interferon; IL, interleukin; G‐MDSC, granulocyte‐myeloid‐derived suppressor cell; NET, neutrophil extracellular trap; NK, natural killer; NKG2A, NK group 2 member A; pDCs, plasmacytoid dendritic cells; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2; TLR, Toll‐like receptor 8; TNF‐α, tumor necrosis factor‐α.
3.1.1. Macrophages
Among the principal determinants of innate immunity in reaction to COVID‐19, macrophage activity is responsible for both inflammatory reactions and a benefit for the pathophysiology of COVID‐19 in patients. 76 These macrophages can inhibit initial viral replication by triggering IFN‐I activity and the inflammatory reaction that recruits further numbers of leukocytes. 77 Even though cytokine storms are necessary to activate the immune reactions toward SARS‐CoV‐2, excessive inflammation leads to COVID‐19‐associated mortality. 18 , 78 Macrophages in the lung include interstitial macrophages, which are found in the interstitial space, as well as alveolar macrophages (AMs), which are found in the alveolar space. They could be critical for limiting inflammatory responses in reactions to corona infection. 79 AMs have antiviral and proinflammatory functions, according to these observations while nerve‐ and airway‐associated macrophages (NAMs) eliminate unnecessary and detrimental inflammatory reactions. Notably, NAMs decrease IL‐6 secretion during influenza, suggesting that NAMs are a significant control in regulating IL‐6 concentrations and, as a result, can govern the COVID‐19 cytokine storm. 80 According to these findings, different macrophage communities were recognized in the lung of COVID‐19 individuals. Even though these researchers did not examine NAMs, they did find a richness of anti‐inflammatory macrophages in cases with moderate illness, while high numbers of inflammatory AM communities predominated in COVID‐19 severe cases. 81 These results suggest that polarization of macrophages and the relative percentage of their subgroups are essential determinants in COVID‐19 pathogenesis.
Because SARS‐CoV‐2 can infect macrophages, this suggests that the virus actively tries to manipulate macrophages to escape immune response. 76 However, it is not clear how SARS‐CoV‐2 infection affects macrophage activity; other coronaviruses have been shown to influence macrophage activity. 82 The Middle East respiratory syndrome coronavirus may be interfering with major histocompatibility complex II (MHCII)'s presentation because MHC I, CD80, and CD86 are all strongly expressed in infected macrophages, but not MHC II. 83 , 84 In monocytes and B cells of COVID‐19 patients, MHC II was shown to be reduced. 85 Aside from that, human leukocyte antigen‐DR isotype (HLA‐DR) expressed on monocyte is significantly reduced in patients with severe COVID‐19, whereas its expression could be partly reversed by an IL‐6 antagonist. 86 MHC II downregulation is not fully recognized, although it is thought to be caused by alterations in the epigenetic landscape of infected cells, partially responsible for this phenomenon. 87 The epigenetic event can decrease the expression of MHC II, which is a process conserved by other coronaviruses, such as the human coronavirus‐EMC, which reprograms MHC II epigenetically. 88 Nevertheless, epigenetic reprogramming of antigen presentation is not typical; for instance, since SARS‐CoV limits antigen presentation on MHC II, it is not a characteristic of this virus. 88
The recently discovered interaction of SARS‐CoV‐2 with the host provides some insights into how this virus interacts with the function of macrophages. 89 Specifically, nonstructural protein 5 (Nsp 5), a protein belonging to SARS‐CoV‐2, can interface with the histone deacetylase 2 (HDAC2) and can control MHC II generation and cytokine secretion. 89 , 90 , 91 Since it is not determined whether SARS‐CoV‐2 suppresses or promotes HDAC2 function, this interplay suggests that the virus may directly influence the cytokine storm as well as antigen presentation. SARS‐CoV‐2 genes, namely, Nsp13 and open‐reading frame 8 (ORF8), communicate with various parts of the Golgi trafficking network, which can be used to prevent MHC from exporting into the cell. According to current work, ORF8 bind to MHC I in the endoplasmic reticulum and direct it to autolysosomes, where it can degrade. 92 To restrict the presentation of antigen via MHC I, viruses often redirect MHC transport to the Golgi, as the human immunodeficiency virus (HIV) Nef protein does. 93 , 94 Nsp10 also interacts with Adaptor protein complex 2, which has a central role in regulating MHC II transportation to antigen‐processing compartments. 89 , 95 , 96
3.1.2. Neutrophils
Neutrophils are the main cells to be drawn to the inflamed tissue following the activation by chemotactic factors produced by infected cells, and they seem to play a substantial role in generating ARDS as well as acute lung injury, and as the disease progresses, the activation and recruitment of neutrophils is a common symptom. 97 , 98 , 99 In this regard, the relationship between neutrophil infiltration and pathogenic evens in COVID‐19 has been noted. 100 Besides, the high number of peripheral neutrophils has been reported that can be considered a predictor of poor outcomes in COVID‐19 patients with ARDS. As a result, the research found that individuals with more severe symptoms had greater peripheral neutrophil count. 101 , 102 Neutrophil chemoattractant chemokine (C–X–C motif) ligands 2 and 8 (CXCL 2 and 8) in COVID‐19 patients' BALF and PBMCs can support the link between enhanced neutrophil recruitment and COVID‐19 patients' disease severity. 103 Another important metric for predicting COVID pathogenesis is the neutrophil‐to‐lymphocyte ratio. 101
Neutrophil pathology in COVID‐19 patients may be caused by more than only infiltration. 104 As a result of neutrophil activation proteases, neutrophil extracellular traps (NETs), and reactive oxygen species (ROS) are potentially pathogenic substances. 104 Infection with SARS‐CoV‐2 generates redox imbalance and ROS, which leads to thrombosis, tissue damage, and red blood cell imbalance, all of which contribute to the severity of COVID‐19. 105 , 106 NETs generated by neutrophils are implicated in organ damage and death in COVID‐19 cases. 59 , 107 Finally, a cytokine storm can be caused by NETs. 107 Strategies that decrease NET synthesis or encourage fragmentation are proposed to treat COVID‐19. Clinical studies have used inhibitors of NE, peptidyl arginine deiminase type 4, and gasdermin D to treat COVID‐19. 107 , 108 Takan together, targeting proinflammatory cytokines and neutrophil‐produced compounds may be a viable way to treat COVID‐19.
3.1.3. NK cells
Both perforin‐mediated and Ab cell‐mediated cytotoxicity are necessary for the NK cell to detect and kill virus‐infected cells to control viral infections. 109 NK cells, plus destroying, have immunoregulatory capabilities, as they may reduce the inflammatory response induced by a viral infection, limiting host injury and disease development. 110 A considerable reduction in the proportion of CD56dimCD16+KIR+ NK cells has been reported in whole blood samples from cases who were infected with SARS‐CoV‐2, indicating either delayed development or increased recruiting and selection of circulatory NK cells towards damaged tissue, respectively. 62 , 63 Besides, NK group 2 member A (NKG2A), an inhibitory receptor, was shown to be upregulated by SARS‐CoV‐2, which has been linked to NK‐mediated cytotoxicity. 111
Similarly, the NKG2A receptor is upregulated in NK cells of infected individuals with SARS‐CoV‐2 than in healthy controls, although the production of stimulatory markers, namely, IFN‐γ, IL‐2, CD107a, and TNF‐α, decreased. 47 The adverse impact of SARS‐CoV‐2 on the activation condition of NK cells and cytolytic capability is also associated with the upregulation of lymphocyte‐activation gene 3 and T‐cell immunoglobulin and mucin protein 3, which are found in NK cells. 52 , 102 NKG2A and NK cell numbers were restored in COVID‐19 individuals after they recovered from their diseases. 55 , 111 A significant correlation exists between the rise in IL‐6 levels in individuals who died from SARS‐CoV‐2 infection as well as decreased NK cell counts and decreased anti‐inflammatory function in the most severe instances and in those who died versus those who survived. These findings suggest that the decreased NK cell activity results in an increased release of cytokines by innate immunity. 47 , 55 , 64 In the initial stage of SARS‐CoV‐2 infection, the NK cells' function and immunoregulatory activity were exhausted, and this phenotype is correlated with disease development. A study performed on the alveolar compartment of COVID‐19 subjects found that although resting NK cells were significantly reduced, no significant changes were detected in stimulated NK cells. However, another report indicates that individuals with severe COVID‐19 had more numbers of NK cells in their alveolar compartment than those with moderately infected or healthy people. 57 , 60 These findings on the presence of NK cells in the tissue of infected individuals are not fully completed and conflicting, most likely due to variations in specimen collection timing or sickness severity.
In a study by Leem et al., 112 NK cells in the RNA‐seq investigation had unique characteristics in comparison to healthy donors, including a notable enrichment of proinflammatory cytokine‐mediated signaling pathways. Intriguingly, they discovered that NK‐cell cytotoxicity reduced and the unusual CD56dim CD16neg NK‐cell population of PBMCs from COVID‐19 patients independent of the severity of the illness. 112 In patients with moderate COVID‐19, the NK‐cell population quickly returned to normal along with the elimination of unusual CD56dim CD16neg NK cells and the restoration of NK‐cell cytotoxicity, but this process took much longer in patients with severe COVID‐19. Finally, using scRNA‐seq on PBMCs and isolated NK cells, Guo et al. 113 identified a memory‐like NK subpopulation (NK1) that increases with age and correlates with disease severity in COVID‐19. Their findings suggested that memory‐like NK2.1 cells may be used to create immunotherapies for COVID‐19 to treat age‐related immunological dysfunctions.
3.1.4. Myeloid suppressor cells
Myeloid‐derived suppressor cells (MDSCs) are innate immune cells that regulate adaptive immunological responses. 65 Various infectious diseases have been shown to increase the activity of MDSCs. 65 , 114 Studies revealed that individuals with severe COVID‐19 have dysregulated myeloid cell components with higher MDSCs and activity related to the severity of COVID‐19. 65 Inflammatory monocytes (HLA‐DRhiCD11chi) with ISG signatures, indicating terminally differentiated monocytes, have been observed in mild COVID‐19 cases. A deficiency in type I IFNs, classical monocytes (HLA‐DRlow), and neutrophils (CD10lowCD101−CXCR4+/−) with immunosuppressive properties in the circulation and lungs of patients with severe COVID‐19 are frequently reported in severe cases indicating the urgent myelopoiesis. 115 , 116 , 117
According to the research, severe COVID‐19 pneumonia patients had a greater MDSC to T‐cell (CD8 effector memory) ratio compared to those with moderate COVID‐19 pneumonia, and the formation of MDSC is directly linked to lymphopenia and enhanced arginase activity in patients. 66 Up to 90% of total blood mononuclear cells were found to be MDSCs in cases with severe conditions, while such a percentage would be 25% in patients with mild conditions, and this proportion is decreased when the disease condition is improved. 118 In COVID‐19, granulocytic markers are elevated and applied to distinguish between individuals with mild and severe forms of diseases, suggesting the contribution of polymorphonuclear leukocyte‐MDSCs in the COVID‐19 pathogenesis. 119 Enhanced CD15+CD16+ neutrophil numbers, reduced integrin CD11b granulocytic expression, and decreased expression of chemoattractant receptor‐homologous molecule expressed on Th2 cells associated with Th2 in eosinophils and basophils, effectively involved in the development of COVID‐19 hallmarks. Also, regarding the basophils and eosinophils, the emergence of the expression of the programmed death‐ligand 1 (PD‐L1) checkpoint was linked to the severity of symptoms. 119 Because myeloid cells are the predominant immune cell subgroups linked with COVID‐19 severity, identifying their inflammatory and chemotactic profiles might have diagnostic and therapeutic implications. 120
3.1.5. Eosinophil
Severe COVID‐19 has been shown to have self‐perpetuating pathological hyperinflammation situations like cytokine storm. 121 , 122 , 123 Cellular responses such as margination and apoptosis can be modulated by cytokines acting alone or in combination with one another under certain circumstances. Significantly, moderate‐to‐severe stress hinders cortisol responses, contributing to eosinopenia in other situations. 124 , 125 Besides, systematic investigations of leukocyte subsets and plasma cytokines in COVID‐19 patients have shown an array of intriguing results. Patients with COVID‐19 who required hospitalization had a longitudinal profile of plasma cytokines and peripheral blood leukocytes, according to Lucas et al. 54 results. According to their results, increased aggravation was related to abnormal Th2 and eosinophil responses, comprising raised levels of IL‐5, IL‐13, immunoglobulin E, and eotaxin‐2, as well as a rise in the eosinophil counts in the circulation. Rodriguez et al. 126 evaluated the circulatory immune cells of individuals who recovered from a severe form of COVID‐19. They discovered a distinct subgroup of IFN‐induced CD62L+ eosinophils that were promptly increased before worsening in COVID‐19 patients. The above findings are rather surprising since proinflammatory stimulation generally leads to decreased expression of CD62L in eosinophils; consequently, the therapeutic implications of this immunoregulatory response have yet to be determined. 127 Accordingly, Vitte et al. 119 conducted an unbiased mapping investigation focusing on important surface indicators of circulatory leukocytes in COVID‐19 patients. Eosinophil‐mediated overexpression of PD‐L1 is directly associated with clinical outcomes in these patients. Also, Onodi et al. 73 recently explored that IFN promotes PD‐L1 upregulation in eosinophils. Numerous studies have indicated that IFN‐γ acts as a critical element of cytokine storm in COVID‐19. 121 Eosinophils and their responses to COVID‐19 can be better understood by studying the kinetics and dynamics of IFN production and signaling. Surprisingly, although the modulation of peripheral eosinophils occurred during the progression of this condition, few eosinophils have already been found in bronchoscopy samples and very rarely in lung tissue in postmortem specimens. 128 , 129 Furthermore, Zein et al. 130 discovered that eosinophils have antiviral properties in addition to their involvement in inflammation. COVID‐19 individuals who were given inhaled corticosteroids had a reduction in coronavirus proliferation, which was connected to better outcomes. 130 Nevertheless, the interaction of SARS‐CoV‐2 and eosinophil and its effect on COVID‐19 require additional investigations. The relationship between eosinophilia and improved outcomes of COVID‐19 is dependent on the inhaled corticosteroids. Prospective randomized controlled investigations are required to assess the function of inhaled corticosteroids in COVID‐19 treatment and their interplay with eosinophilia.
3.1.6. Other innate immune cells
APCs include DCs that effectively process and deliver antigens to T cells to prime T‐cell activation to particular antigens. 131 cDCs and pDCs are the two main types of DCs. There are two types of cDCs: type 1 cDCs (CD103 or CD8 expressing) and type 2 cDCs (CD11b+), which include cross‐presentation and CD4+ T‐cell responses. 132 Many respiratory infections, especially COVID‐19, might be caused by DC dysfunction because of their crucial role in protecting the body from respiratory infections. 102 , 133 In severe forms of COVID‐19, the rate of DCs is decreased in PBMCs from COVID‐19 cases. In contrast to healthy donors, they did not increase the formation of costimulatory molecules like CD80 following maturation stimulation. 131 Additionally, unlike cDCs obtained from healthy donors, cDCs obtained from severe cases are not capable of stimulating T‐cell activation or the synthesis of antiviral compounds, implying that cDCs of severe patients are deficient for activation, and development of T cells. 133 This suggests that they are ineffective in eliciting an effective immune reaction following COVID‐19. In addition to cDCs, pDCs, which are leading suppliers of type I IFNs, are reduced in the blood, and functionally impaired after COVID‐19. 117 Therefore, additional research is needed to determine if these abnormal DCs are linked to condition severity in COVID‐19 patients.
Finally, basophils are decreased in COVID‐19 patients, demonstrating higher recruitment of these types of cells to injured lungs. 134 Because basophil can play a significant activity in tissue healing and create coagulants, its reduction sometimes causes long‐term lung inflammation and thrombosis. Basophil depletion usually occurs before the onset of the disease. 135
3.1.7. Innate lymphoid cells and COVID‐19
To date, little is known about the role of innate lymphoid cells (ILCs) in COVID‐19 pathophysiology. In reaction to an infectious agent or microenvironmental alterations, ILCs residing in pulmonary epithelial tissue play an important role in host defense and make a significant contribution to lung protection, pathophysiology, and diseases. 136 Because ILCs lack antigen‐specific receptors, it is hypothesized that they trigger by proinflammatory cytokines and unknown receptors. ILC may be indirectly or directly stimulated by the combinations of pathogen‐associated molecular patterns with PRRs. 137 Although helper ILC subtypes are mostly found in tissues, they could be present in the blood as well. 138 , 139 As a result, our knowledge of COVID‐19‐derived ILC cells is primarily restricted to changes in the peripheral blood. The ILC2 subgroup in the lungs inhibits allergen and viral‐induced type 2 reactions, eosinophil migration, inflammatory reaction cessation, and tissue healing. 140 Concerning the NK subset, overall amounts of the ILCs subgroups and ILC progenitors (ILCp) subsets are reduced in the peripheral blood of patients with mild and severe COVID19; nevertheless, when estimated as a proportion of ILCs, only the ILC2 keeps increasing in the peripheral circulation of mild COVID‐19 compared with normal individuals. 141 , 142 The proportions of helper ILC in specimens from recovered cases are comparable to those found in healthy subjects. 141 , 142 The SARS‐CoV‐2 is dependent on the papain‐like proteases to produce functioning replicase complexes that control viral propagation and the innate immune system. 143 Throughout allergic inflammation and asthma, papain has been found to increase the respiratory capacity of ILC2 cells in the lungs. 144 It has been found that injection of SARS‐CoV‐2 papain‐like proteases in the lungs of mice rises the levels of IL‐5‐producing ILC2 in pulmonary tissue. 145 Moderate COVID‐19 cases had higher levels of IL‐13, ILC2, and IL‐5, as well as IL‐33. 142 Not only is there a general decline in the overall ILC number, but there are also changes in the expression of stimulation, migratory, and differentiating characteristics associated with disease severity in COVID‐19 individuals. 146 ILC2 and ILCp show a greater degree of CD69 expression while exhibiting lower rates of CXCR3 and C–C motif chemokine receptor 4 expression. 141 , 142 There seems to be an enhancement in the stimulating receptors NKG2D+ in the ILC2 subgroup and a substantial reduction in the inhibitory receptors CD25 and KLRG1 in severe COVID‐19 cases. 141 , 142 These findings imply that COVID‐19 alters the rate of the whole ILC population in the peripheral circulation, and the ILC2 subgroup undergoes major modifications. These alterations in the ILC subtype in peripheral circulation are characterized by the formation of cytokines like IL‐5 and IL‐13, which are released by the ILC2. 142 Furthermore, severe COVID‐19 cases need hospitalization, and the length of hospital stay is associated with a decrease in the number of ILCs, showing the critical involvement of ILCs in COVID‐19. 146
3.2. Adaptive immunity and COVID‐19 pathogenesis
Adaptive immunity is essential for the elimination and control of the majority of viral diseases. 147 , 148 B lymphocytes (the producer of antibodies), CD4+, and CD8+ T cells constitute an essential part of adaptive immunity. 5 , 149 There are still many unknowns about the role of CD4+, CD8+ T cells, and nAbs in controlling SARS‐CoV‐2 in COVID‐19 cases. 147 SARS‐CoV‐2‐induced CD4+ and CD8+ T cells are targeted against a variety of antigens comprising structural and Nsps and are strongly related to milder forms of COVID‐19. 17 Antibody‐mediated reduction of CD8+ T cells in convalescent macaques reduces immunity toward SARS‐CoV‐2 rechallenge, implying a function for CD8+ T cells in the context of diminishing antibody reactions (Table 2). 153 The response of CD4 + T cells to protein S has been investigated for its importance in the production of nAbs using prediction models, peptide or protein priming, and T‐cell isolation to protein S at significant depths in recovering and vaccinated individuals. 150 , 165
Table 2.
Overview of adaptive immune reactions to SARS‐CoV‐2
| Adaptive immune response | Reaction | Outcome | Reference |
|---|---|---|---|
| T CD4 lymphocyte |
According to the research findings that examined CD4+ T cell reaction to proteins of SARS‐CoV‐2 in recovered COVID‐19 patients, reactions were identified toward approximately all SARS‐CoV‐2 proteins, with CD4+ T‐cell responses being unrecognizable only for one of the smallest proteins. Remarkably, CD4+ T cells special for SARS‐CoV‐2 were reported to significantly correlate with reduced COVID‐19 disease severity. IFNγ is the prevailing cytokine generated by SARS‐CoV‐2‐specific CD4+ T cells from cases with COVID‐19, with a distinguishable IFNγ, TNF, and IL‐2 protein signature of classical Th1 cells. A subset of T CD4+ expressed CCR6 specific to SARS‐CoV‐2 indicates underlying Th17 characteristics of those cells, but the reports have suggested the low or undetectable levels of IL‐17α protein expression in COVID‐19 patients. T CD4+ cells (SARS‐CoV‐2‐specific) can express a high level of IL‐22. |
B‐cell affinity maturation and antibody production, Initiation of CD8 T‐cell proliferation and differentiation, direct cytotoxic activity, regulation of primary SARS‐CoV‐2 disease, reduction in COVID‐19 pathogenicity, and increased viral removal |
17 , 34 , 147 , 150 , 151 , 152 |
| T CD8 lymphocyte |
The existence of virus‐specific CD8+ T cells has now been linked to improved COVID‐19 consequences. T CD8+ cells recognize various SARS‐CoV‐2 antigens, including spike, nucleocapsid, M, and ORF3a. Specific CD8+ T for SARS‐CoV‐2 express many molecules related to potent cytotoxic activity, including IFNγ, perforin, CD107, and granzyme B. Furthermore, depending on the increased expression of inhibitory receptors, several researchers have described exhaustion phenotypes of CD8+ T cells in severe COVID‐19 cases. |
Protection against the expansion of severe COVID‐19, the killing of virus‐infected cells, the production of effector cytokines, and the impairment of host defense mechanisms | 17 , 150 , 153 , 154 , 155 , 156 , 157 , 158 , 159 |
| B lymphocyte |
Upon infection with SARS‐CoV‐2, the naive B cells, or possibly pre‐existing memory B cells from previous HCoVs illnesses, are stimulated by antigen identification, and CD4+ T cells support. Definitions of circulatory B cells in the early weeks of an acute SARS‐CoV‐2 disease have revealed moderate relative B cell lymphopenia and changeable enhancement in plasmablasts frequencies, which in some cases exceeded 30% of total B cells. Plasma cells and memory B cells that secrete antibodies can access the blood and (presumably) the mucosa. They assisted in the battle against viral illness and defended against reinfection. Indeed, severe COVID‐19 cases exhibited higher rates of the DN2 B cells as opposed to those with mild cases and additionally had higher plasmablast numbers. |
Affinity maturation, resulting in long‐lived plasma cells and memory B cells, particular antibody generation, rise in secondary reactions | 160 , 161 , 162 , 163 , 164 |
Abbreviations: COVID‐19, coronavirus disease 2019; DN2 B cells, double‐negative (DN) B cells; HcoVs, human coronaviruses; IL, interleukin; Th17, T helper 17 cells; TNF, tumor necrosis factor; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2.
3.2.1. B cells and COVID‐19
The neutralization by specific antibodies is a crucial stage in viral eradication, although the specificity of the NAbs remained unclear. 166 The S protein of SARS‐CoV‐2 contains a 193 amino acid region called the receptor‐binding domain (RBD). This area binds to the ACE2 receptor, and RBD is a primary target for NAbs. 166 In addition, earlier reported monoclonal antibodies toward other coronaviruses could also attach to SARS‐CoV‐2, although their epitope specificity might not even match with the ACE2‐binding domain. 167
The activation of B cells and accelerated generation of antigen‐specific antibodies by antibody‐secreting cells (ASCs) are vital to managing viral diseases. 166 Several studies indicated decreased CD5+ B‐cell counts, increased plasmablasts, and SARS‐CoV‐2‐specific antibody production. 14 , 166 Despite reports of lymphopenia, COVID‐19 patients had a higher number of PBMCs and CD19+ B cells than control participants. 168 B cells contain five key communities: transitional, naïve, double‐negative, memory, and ASCs, and these five basic populations were further classified into 14 subpopulations depending on their features. 169 Individuals with moderate diseases, individuals in the intensive care unit (ICU), and healthy individuals all had unique B‐cell profiles, which were particularly notable. 168 The developments of ASCs and double‐negative lymphocyte cells were found in B cells isolated from ICU cases. 168 In contrast, transitional cells were found in those patients with mild conditions. 166 Extrafollicular responses are known to involve these cell types, and interestingly, comparable B cells have been discovered in animal models of autoimmune and viral clearance. 170 , 171 , 172 As a result, individuals with a background of SARS‐CoV‐2 disease, especially those diagnosed with severe infection, should be closely examined for manifestations of autoimmunity.
Furthermore, as compared to patients with moderate illness, individuals in the ICU had greater levels of ASCs, suggesting that circulating ASCs have an immunopathologic function in severe COVID‐19. The plasma cell maturation marker, or CD138, expressed on multiple ASCs, indicates that ASCs are exposed to a highly inflammatory environment in ICU cases. 168 Those in the ICU had higher levels of CD21lo transitional B cells than those in the general population, as well, in which cells accounted for less than 25% of the B‐cell population. 168 Next research must focus on elucidating the pathways through which CD21lo transitional cells exert their protective effects. 168
Yao et al. 31 investigated PBMCs of COVID‐19 patients using RNA‐seq, despite the reduced number of CD4+ T cells found in COVID‐19 patients, their activity was normal. In contrast, pathways involved in B‐cell activation were downregulated in the severe group, indicating a B‐lymphocyte compartment malfunction that restricts their activity.
Interestingly, Kang et al. 173 assessed titers of various isotypes of Abs against SARS‐CoV‐2 antigens, phagocytic capacity, and memory B responses in PBMCs and plasma samples obtained from individuals who suffered asymptomatic, moderate, and severe disease 1 year after COVID‐19. They demonstrated that the phagocytic capability of Abs and memory B‐cell responses, which are key factors in guarding against reinfection with SARS‐CoV‐2, are linked with disease severity at 1 year post‐COVID‐19. To better our understanding of B cells and their Ab expression in COVID‐19 and ultimately improve vaccination approaches, more research on these immune responses to SARS‐CoV‐2 is necessary.
3.2.2. T cells and COVID‐19
T cells are critical against viral infections and the fate of disease. CD4+ T cells assist B cells in synthesizing antibodies and stimulating the CD8+ T cells' response and other immune cells in the face of infection. 174 T‐cell reactions emerge early and associate with survival, but they are significantly reduced in severe COVID‐19 and are linked with high stimulation and lymphopenia. A fraction of seasonal coronavirus‐sensitive T lymphocytes interact with SARS‐CoV‐2 and even contribute to clinical prevention, especially in the early stages of infection. 175 T‐cell memory includes wide recognition of viral proteins, thought to be approximately 30 epitopes per individual, and appears to be successfully maintained thus far. 175 This diversity of identification has the potential to restrict the effect of particular viral alterations and is likely to support the defense against severe illnesses caused by viral variations such as Omicron. 176
CD4+ and CD8+ T‐cell frequencies are significantly reduced in COVID‐19 patients, while T‐cell activity is increased. 54 Patients with severe COVID‐19 had a deposit of mononuclear cells in the lungs and lower rates of hyperactive T cells in the circulation, according to postmortem data. 177 These results imply that T cells from the blood are transferred into infected lung tissues to inhibit viral infection. 174 Furthermore, the immune reaction demonstrates some components of therapeutic antiviral protection and subtle aspects of sepsis. 135 , 178 , 179 Gamma delta T cells (γδ T cells) have been proven to possess a protecting antiviral effect in influenza pneumonia and are hence expected to be beneficial in COVID‐19. 179 Furthermore, COVID‐19 severity is mediated by selective T‐cell expansion, exhaustion, and depletion. Cytolytic memory CD8+ T effector cells in patients versus healthy controls show that activated T cells can be used as a therapeutic tool for treating SARS‐CoV‐2 infection. 135 As a result, immunological linkages and other assays (including C‐reactive protein (CRP) and d‐dimer) may help identify individuals at high risk of severe illness. 135
After acquiring SARS‐CoV disease and recovering, individuals who had a severe infection but subsequently survived produced specific memory T cells that remained active for 2 years after the illness. 180 , 181 IL‐2, IFN‐γ, and TNF are produced by CD4+ T cells from SARS‐CoV patients, indicating that cellular immunity is crucial for managing the disease and preventing its spread. 182 , 183 The inflammatory mediators produced by these cells contribute to pathophysiology, but the viral elimination depends on this reaction since the loss of these cells causes significant lung inflammation in mice. 174 , 184 Another benefit is that the growth of CD4+ and CD8+ T lymphocytes in the lung is increased by immunization with DCs expressing SARS CoV antigens. 185 , 186 T cells have a critical role in infection management, as shown by transplanting these cells into immunodeficient animals improved resistance toward SARS‐CoV disease. 186
Compared with healthy subjects, CD8+ T cells from individuals with COVID‐19 had lower levels of inhibitory receptor expression. 102 CD8+ T cells from patients with severe diseases have released fewer cytokines once stimulated. 111 On the other hand, other research found an overactive CD8+ T‐cell reactivity, upregulation of NK‐associated markers, and enhanced cytotoxicity. 111 , 187 , 188 Moreover, increasing proportions of CD38+HLA‐DR+ functional CD8+ T cells or propagating CD8+ T cells were detected in the majority of patients with COVID‐19. 160 , 189 It should be noted that this scenario is not true in all patients, indicating that CD8+ T‐cell reactions in COVID‐19 might manifest themselves in a variety of ways. 160 As a result, there appears to be variability in the immune reaction to SARS‐CoV‐2, and various immunotypes may be correlated with different clinical characteristics. 86 , 160 Predictably, a study verified the relevance of respiratory CD8+ T cell reactions, which entail connections (especially the IFN axis) among CD8+ T cells and upper respiratory epithelial cells. 57 , 190 However, mild COVID‐19 may be linked with more powerful clonal proliferation of CD8+ T cells. 191 Also, specific CD8+ T cells have been found in recovered cases, confirming the development of SARS‐CoV‐2‐specific CD8+ T‐cell reactions and the presence of CD8+ T‐cell memory. 150 , 192 The precise involvement of CD8+ T cells specific for SARS‐CoV‐2 in regulating the initial acute infection and providing protection against subsequent infections remains unknown. 193
In individuals with COVID‐19, CD4+ T cells demonstrate impairments, as well as the upregulation, activation, and/or exhaustion markers. 194 , 195 According to research, individuals with a moderate form of COVID‐19 had a more significant percentage of IFN‐γ‐producing Th1‐like cells than in severe cases. 196 There have been reports of SARS‐CoV‐2‐specific CD4+ T cells throughout acute infection, and the inflammatory profile of these cells is consistent with that of a Th1. 197 A normal Th2 response is seen in moderate instances of COVID‐19, but the relevance of these cells in severe cases is not yet clear. 198 The additional data reported the involvement of pathological Th17 cell reactions in severe COVID‐19 patients, including a significant reaction by CD4+ T cells that coexpress CCR6. 177 , 199 The presence of enhanced CD4+ T‐cell reactions that produce TGF‐β, as well as an elevated subset of CD4+ T‐cell reactions that produce IL‐6 and GM‐CSF, has been found in COVID‐19 cases. 200 , 201 The presence of virus‐specific memory CD4+ T cells in individuals after recovery from COVID‐19 is significant because it suggests the formation of the protective immune response. 150 , 192 , 202 Likewise, individuals who survived mild COVID‐19 established memory CD4+ T cells that indicated high numbers of the IL‐7 receptors (IL‐7R) throughout their recovery. 202
Additionally, in a study, Sekine et al. 154 stimulated PBMCs with nucleocapsid, S, and membrane peptides to assess the functional capacities of memory CD8+ and CD4+ T cells in recovering COVID‐19. They demonstrated that whereas CD8+ T cells are characterized by IFN production and mobilization CD107a expression, CD4+ T cells specific for SARS‐CoV‐2 produce IFN, IL‐2, and TNF. Notably, membrane‐ and nucleocapsid‐specific CD4+ T cells were developed into Th1 or Th1/Th17 cells, but S‐specific CD4+ T cells were biassed toward circulating TFH cells. In addition, Yao et al. 31 discovered that CD8+ T lymphocytes cannot destroy cells, which may contribute to the pathobiology of ARDS in COVID‐19 patients.
Furthermore, there are very few studies on COVID‐19 subjects with double negative (CD3+CD4− CD8−) T cells. In Zahran et al. 203 study, they showed that double‐negative T cells were higher in COVID‐19 patients than in other lymphocyte subgroups. Besides, in some other studies, T‐lymphocyte subset absolute counts (overall CD3+, CD3+CD4+, CD3+CD8+, CD3+CD4+CD8+ double positive, and CD3+CD4−CD8− double negative) were lower in nonsurvivors and patients with severe illness compared with individuals who survived and nonsevere cases. 204 Therefore, more investigation is required to determine the involvement of double‐negative cells in the development and regulation of COVID‐19.
4. CYTOKINE STORM AND COVID‐19
Cytokine release is excessive in severe COVID‐19 patients named a cytokine storm and impacts the human body. 205 The processes through which SARS‐CoV‐2 disease causes cytokine overproduction remain unknown. The ACE2 protein is present in the highest concentrations in lung respiratory epithelial cells and small intestine enterocytes. 206 In addition, ACE2 is abundantly expressed in smooth muscle and endothelial cells of arteries and veins in all organs investigated. 206 , 207 The evaluation of autopsy specimens obtained from SARS patients revealed that SARS‐CoV predominantly infected pulmonary epithelium, which was consistent with ACE2 expression levels. 207 Given the presence of ACE2 expression, SARS‐CoV can infect and cause damage to immune cells, such as T lymphocytes, monocytes, and macrophages. 206 , 207 Surprisingly, despite the presence of SARS‐CoV virus particles in other cell types, such as gastrointestinal system epithelial cells, brain neurons, and renal cells, numerous organs with ACE2 activity maintained uninfected. 206 , 207 These inconsistencies support the idea that SARS‐CoV cell entrance is not completely dependent on ACE2.
After infection with SARS‐CoV‐2, CD4+ T cells are rapidly stimulated by pathogenic Th1 cells, which secrete GM‐CSF, and produce CD14+CD16+ monocytes with high‐speed IL‐6, which intensifies the inflammatory response. 205 According to single‐cell research, in COVID‐19 patients, immune cell interplay is defined by a rise in a subgroup of CD14+IL‐1+ monocytes, which may support enhanced IL‐1β release. 191 Th17 cells produce some proinfla mediators, such as IL‐17, which recruits monocytes/macrophages and neutrophils to the area of inflammation and stimulates other inflammatory cascades, including IL‐1 and IL‐6, among others. 208 Among these mediators, IL‐6 plays a vital role in developing cytokine storm in COVID‐19 patients. 205
The cytokine storm in COVID‐19 is caused by many activated cells, including neutrophils, B, T, DC, NK, macrophage, and tissue‐resident cells. 47 COVID‐19 patients have a higher rate of proinflammatory cytokines and chemokines than healthy controls, including IL‐1β, IL‐1 receptor antagonist, IL‐2, IL‐7, IL‐6, TNF‐α, IL‐10, IFN‐γ, GM‐CSF, granulocyte‐GCF (G‐CSF), fibroblast growth factor, platelet‐derived growth factor, vascular endothelial growth factor, chemokine (C–C motif) ligand (CCL) 2 (CCL2), CCL3, CCL4, CCL8, CXCL2, CXCL8, CXCL9, CXCL10, and CXCL16. 47 Furthermore, a statistically significant relationship has been found between the severity of COVID‐19 and the blood levels of TNF, CXCL8, CXCL10, CCL2, CCL3, IL‐1, IL‐2, IL‐6, IL‐7, IL‐10, and G‐CSF, among others. 47 , 51 , 199 Another supplementary result confirms that severe COVID‐19 patients have considerably higher plasma concentrations of IL‐6, IL‐10, and TNF‐α than those with moderate forms of COVID‐19. 209 , 210 In this regard, elevated concentrations of IL‐6, TNF, and IL‐10 found in severe COVID‐19 cases are significantly correlated with lower levels of inflammatory T cells. 194 Longitudinal studies in individuals with mild to severe COVID‐19 who had the same expression profiles of inflammatory markers for up to 10 days after the onset of infection showed that this is an important component in the COVID‐19 pathogenesis. Meanwhile, in subsequent periods, levels of TNF‐α, IL‐6, and IL‐10 decreased rapidly in patients with mild disease, but remained high in patients with severe COVID‐19. 54 In this context, IL‐6 is a useful biomarker since its plasma level correlates with both viral load and lung damage in critically ill patients. 211
5. OTHER MARKERS AND COVID‐19 SEVERITY
Individuals with COVID‐19 showed a variety of clinical characteristics, including mild, moderate, severe, and critical forms. Although the majority of COVID‐19 individuals have mild to moderate signs and indications, a Chinese study found that around 14% of patients had severe symptoms and signs, and 5% had critical signs and indicators. 212 Earlier research and clinical experience demonstrated that the degree of severity was related to clinical therapy and illness prognosis. 25 , 212 The overall case‐fatality rate of verified COVID‐19 individuals was 2.3% on average, but this increased to 49% in critical patients. 212 Misdiagnosis delays appropriate treatment and increases the likelihood of a poor outcome. The treatment for severe or critical COVID‐19 patients, on the other hand, necessitates comprehensive medical resources, and multiple misdiagnoses will exhaust those resources and exacerbate the medical burden. As a result, early diagnosis of individuals who are at risk of developing severe or critical COVID‐19 is vital for clinical management and epidemic management. The severity of COVID‐19 is classified into four stages, namely, mild, moderate, severe, and critical. 213 This categorization is mostly made based on signs, oxygen saturation (SaO2), and computed tomography imaging data. However, there is no evidence of laboratory indicators to diagnose COVID‐19. Previous research has linked lymphopenia, organ failure, coagulopathy, and high d‐dimer concentrations to the severity of the disease. 25 , 212 , 214
Besides, the expression of numerous inflammation‐related genes, such as arginase 1 and IL‐1 receptor 2, was found to be highly increased in the PBMCs of COVID‐19 patients, despite individual variances in Yang et al. 215 study. Patients with COVID‐19 have abnormal levels of the coagulation‐related genes Von Willebrand factor and protein S. Certain gene expression patterns, such as IL‐1 receptor, were associated with their histone methylation marks. In the TGF‐β, IL‐1β, IL‐6, and IL‐17 pathways, the majority of the dysregulated genes were found. Also, in Yang et al. 215 study the expression of bone marrow kinase X, which is part of the TEC family, was enhanced in the PBMCs of COVID‐19 patients. We tried to summarize some of those factors linked with COVID‐19 severity in this section.
6. SERUM AMYLOID‐A AND COVID‐19
The acute‐phase reaction, which includes various phenomena, indicates the existence of infection and inflammation, such as elevated temperature and hormonal and metabolic changes, and dramatically activates SAA. 216 Peripheral SAA levels, which are generally moderate under normal conditions (20–50 mg/l), may increase 1000‐fold during the first 24 to 48 h of the acute‐phase response. This is due to higher production in the liver, which is activated by a variety of factors like TNF‐α, IFN‐γ, IL‐1β, and IL‐6. 217 , 218 SAA, in turn, may stimulate the complement system activation and the nucleotide‐binding domain leucine‐rich repeat‐containing family pyrin‐domain containing 3 inflammasome, increasing IL‐1β, TNF‐α, and IL‐6 production and activating additional proinflammatory mediators, including IL‐1α and IL‐23. 219 , 220 Significantly, these agents have been demonstrated to display an essential activity in initiating the cytokine storm and its adverse clinical effects on COVID‐19. 221 As a result, it is possible that the immediate rise in SAA levels in COVID‐19 patients reflects the existence of acute phase response and anticipates the onset of the cytokine storm and, as an outcome, multiorgan collapse and an elevated chance of detrimental consequences.
Besides its possible function in the pathophysiology of the cytokine storm, it has recently been shown that SAA may also have procoagulant properties, facilitated by an elevation in fibrinogen and concurrent platelet aggregation and prothrombotic conditions. 222 In summary, pending further investigation, an acute increase in SAA levels may indicate an important component linking inflammatory processes and prothrombotic cascades. The interaction between inflammatory response and thrombosis has also been found in COVID‐19, a disorder frequently characterized by significant coagulation abnormalities and a prothrombotic situation, especially in individuals with severe forms of COVID‐19. 223
The blood levels of SAA in severe COVID‐19 subjects are more than a thousand times greater than those reported in individuals with other malignancies or inflammatory disorders when SAA upregulation is correlated with systemic amyloidosis as a secondary disorder. 224 , 225 SAA amyloidosis is defined by the production and accumulation of SAA amyloids in blood vasculature, resulting in thrombosis, inflammation, and, ultimately, organ failure. 226 The prevalent consequence of SAA amyloidosis, such as renal dysfunction or elevated thrombosis rates, is observed in COVID‐19 cases. 226 , 227 The pattern of signs implies that SAA amyloidosis could increase COVID‐19 symptoms. 228
7. NE AND COVID‐19
Neutrophils perform a significant role in the development of ARDS by releasing toxic molecules, such as ROS and proteases, particularly elastase. 229 , 230 Neutrophils may also release IL‐6 in reaction to viral diseases, particularly single‐stranded RNA viruses, including SARS‐CoV‐2, via a process involving the Toll‐like receptor 8 (TLR8). 231 The lungs depend on these cells to produce soluble IL‐6 receptors (IL‐6Rs), which may play a role in developing chronic respiratory disorders characterized by pathogenic IL‐6R trans‐signaling. 232 The significance of this type of communication in cytokine release syndrome (CRS) establishment has been established in lymphoma patients who received chimeric antigen receptor T‐cell therapy. 233 According to this study, an elevated neutrophil count in individuals with ARDS may contribute to CRS and lung damage. The elastase enzyme produced by these cells has also been demonstrated to be one of the crucial proteolytic enzymes required to activate coronaviruses' S protein and alter the virus's entrance path to a low pH‐independent pathway. 234
Although it serves a physiological purpose as a potent host defense, NE is also recognized as one of the most detrimental enzymes in the human body. 235 An excessive release of enzymatically active NE from the neutrophils might damage local tissue. 235 In addition, it has been observed that NE may stimulate the COV protein S and cause the virus to enter the cell through a low pH‐independent pathway. 234 As a result, high NE levels were detected in patients with SARS‐CoV‐2 by Akgun et al. 235 validates these results.
8. SDC AND COVID‐19
SDCs (SDC‐1, SDC‐2, SDC‐3, and SDC‐4) are type I transmembrane heparan sulfate proteoglycans that may interact with inflammatory mediators, adhesion molecules, proteolytic enzymes, and cytokines. 236 SDCs and their ligands interact to initiate biological signaling events related to inflammation, angiogenesis, cell attachment, and tissue repair. 237 SDCs help maintains cellular homeostasis in normal conditions while also controlling inflammatory responses after trauma and diseases. 238 SDC‐1 has recently been shown to have a critical key role in developing inflammatory disorders, malignancies, and infectious diseases, according to research conducted on animal models of different conditions. 237 In vitro and in vivo investigations have shown that these SDC‐1 activities are crucial for understanding the pathogenesis of infectious diseases. 239 , 240 , 241 SDC‐1 depletion or deletion confers considerable resistance to infection by various viral and bacterial pathogens. 239 , 240 , 241
A recent study has recently confirmed the role of SDC‐1 in viral infection pathobiology. Bermejo‐Jambrina et al. 242 found that the cell surface of heparan sulfate proteoglycan (HSPG), including SDC‐1 and SDC‐4, is required for SARS‐CoV‐2 infection in permissive cells, and SARS‐CoV‐2 infection in AMs was effectively impeded by low‐molecular‐weight heparins. Regarding the intriguing function of SDC‐1 in the course of inflammatory conditions, such as respiratory viral disease, Karampoor et al. 237 discovered dynamic changes in SDC‐1 levels along with certain indicators, such as IL‐6, IL‐10, IL‐18, CRP, and vitamin D in COVID‐19 patients.
The primary receptor for SARS‐CoV‐2 cellular entrance has been identified to be ACE2. However, new research reveals that other membrane proteins, including HSPGs, have a role in SARS‐CoV‐2 internalization. 243 Hudák et al. 243 discovered that SDCs enable SARS‐CoV‐2 cellular entrance. Among SDCs, SDC‐4 was the most effective in facilitating SARS‐CoV‐2 uptake in their investigation, although the upregulation of other isoforms, especially neuronal SDC‐3, also boosted SARS‐CoV‐2 internalization. According to the literature, the S1 component of the SARS‐CoV‐2 S protein is crucial in the virus's interaction with SDCs. 243 Other elements of the SDC ectodomain like the cell‐attachment domain participate in the interface with SARS‐CoV‐2 in addition to the polyanionic heparan sulfates, which are binding sites for various viruses. 243 SDCs colocalize with ACE2 during viral internalization, indicating that the two proteins are involved in the same internalization pathway. 243 Both ACE2 and SDCs inhibitors were shown to be effective in inhibiting the cellular entrance of SARS‐CoV‐2, indicating that internalization is a multifaceted process. 243
9. TARGETING IMMUNE RESPONSES TOWARD COVID‐19
Therapeutic approaches widely used for the treatment of SARS‐CoV‐2 are categorized as (1) treatments focused on IFNs, (2) therapies addressing pathological inflammatory reactions, and (3) therapies targeting noncanonical pathways. 244 In addition, monoclonal antibodies that target the virus are effective in minimizing the pathophysiology and critical illnesses, but have not been further explored here. 245 When delivered early, SARS‐CoV‐2 is particularly susceptible to IFN therapy in vitro and in vivo. 246 , 247 Similarly, a retinoic acid‐inducible gene I (RIG‐I) PRR antagonists and stimulators of IFN genes, both prophylactically and timely after infection, effectively limit the release of SARS‐CoV‐2 in vivo in an IFN‐type I‐dependent manner. 248 , 249 Human IFN therapy is currently under investigation. A retrospective observational research examining the efficacy of intranasal aerosolized IFNa (IFNa2B) therapy in COVID‐19 participants revealed that prompt IFNa2b delivery was linked with lower in‐hospital death, but late treatments were correlated with greater death rates and delayed rehabilitation. 250 Different investigations failed to discover the therapeutic benefits of subcutaneous pegylated IFN1a and IFN on mild and moderate forms of COVID‐19. 251 , 252 In these circumstances, the pharmacological use of IFN‐β based on its physiological effect and the absence of IFN‐β‐neutralizing autoantibodies in cases has been suggested in a large proportion of patients compared to the presence of such autoantibodies to IFN‐α and IFN‐ω. 253 , 254 Significant worsening of symptoms reported after the late initiation of IFNa2b is likely due to some proinflammatory action of IFNs, especially when associated with a preinflamed respiratory tract. For example, type I IFN substantially promotes ZBP1, which stimulates NF‐κB‐driven proinflammatory cytokine production in response to SARS‐CoV‐2 infection. 250 , 255 Surprisingly, the SARS‐CoV‐2 protein cleaves NSP3 ISG15 and alters the physiological role of ISG15 from supporting ISG production to increasing NF‐κB‐driven proinflammatory mediators. 256 In addition, IFN type I in invading proinflammatory monocytes destroys the pulmonary epithelium via TNF‐associated apoptosis‐inducing ligand. 257 This, together with the effect of inhibiting IFN type I on alveolar epithelial cell growth, disrupts lung regeneration. 258 The ectopic inflammatory process is the second treatment strategy for COVID‐19. The clinical efficacy of dexamethasone provides conceptual evidence for this COVID‐19 treatment. 259 Furthermore, the findings of a pilot trial revealed that while anti‐IL‐17 antibody may lower the inflammatory reaction and improve oxygenation, it does not reduce the risk of death. 260 Regarding TNF‐directed medicines, descriptive clinical evidence and case series suggest the feasibility and promise of anti‐TNF medications as a COVID‐19 therapy, but broader clinical studies are required. 261 Finally, in severe COVID‐19, the complement system remains a potential therapeutic target. In patients with COVID‐19, for instance, currently, underway clinical trials are assessing the possibility of a humanized monoclonal C1 esterase antagonist as a multitarget suppressor of inflammatory feedback loops such as kinin–kallikrein, the contact activation system, and complement in attempting to reduce lung inflammation and pathogenesis. 262
10. CONCLUSION
SARS‐CoV‐2 infection overproduces proinflammatory cytokines, leading to cytokine syndrome. This condition leads to uncontrollable inflammation, which mostly leads to multiple organ failures. SARS‐CoV‐2 triggers both innate and adaptive immune reactions too much, resulting in tissue damage. Therefore, understanding the most important features and evolving intrinsic and adaptive immunity against this virus is crucial in predicting the consequences of COVID‐19 and in the administration of useful approaches for controlling the disease. The management of the inflammatory response is critical for targeting viral infection in this regard, thus it is critical to understand the processes driving hyperinflammation to develop a better treatment strategy to limit viral proliferation. COVID‐19 prevention and treatment might benefit from ongoing clinical trials evaluating the immunogenicity of COVID‐19‐targeting medicines. We should look at all possible paths of study to find out how SARS‐CoV‐2 infection is affecting people's immune systems to properly implement a multifaceted strategy.
AUTHOR CONTRIBUTIONS
All authors equally contributed to this study. Saade Abdalkareem Jasim, Roaa Salih Mahdi, Dmitry Olegovich Bokov Mazin A. A. Najm, Sajad Karampoor, and Rasoul Mirzaei participated in the study design, drafting of the manuscript, and collecting of the documentation materials. Guzal N. Sobirova, Zarnigor O. Bafoyeva, Ahmed Taifi, Ola Kamal A. Alkadir, and Yasser Fakri Mustafa participated in writing the manuscript and drawing figures. All authors read and approved the final version of the manuscript.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Jasim SA, Mahdi RS, Bokov DO, et al. The deciphering of the immune cells and marker signature in COVID‐19 pathogenesis: an update. J Med Virol. 2022;94:5128‐5148. 10.1002/jmv.28000
Contributor Information
Rasoul Mirzaei, Email: rasul.micro92@gmail.com.
Sajad Karampoor, Email: karampour.s@iums.ac.ir, Email: sajadkarampour1987@gmail.com.
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- 1. Karampoor S, Hesamizadeh K, Shams Z, et al. The role of lovastatin in the attenuation of COVID‐19. Int Immunopharmacol. 2021;101:108192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Karampoor S, Hesamizadeh K, Maleki F, et al. A possible pathogenic correlation between neutrophil elastase (NE) enzyme and inflammation in the pathogenesis of coronavirus disease 2019 (COVID‐19). Int Immunopharmacol. 2021;100:108137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mirzaei R, Mahdavi F, Badrzadeh F, et al. The emerging role of microRNAs in the severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) infection. Int Immunopharmacol. 2021;90:107204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mirzaei R, Mohammadzadeh R, Mahdavi F, et al. Overview of the current promising approaches for the development of an effective severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) vaccine. Int Immunopharmacol. 2020;88:106928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Goodarzi P, Mahdavi F, Mirzaei R, et al. Coronavirus disease 2019 (COVID‐19): immunological approaches and emerging pharmacologic treatments. Int Immunopharmacol. 2020;88:106885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mirzaei R, Goodarzi P, Asadi M, et al. Bacterial co‐infections with SARS‐CoV‐2. IUBMB Life. 2020;72(10):2097‐2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pan Y, Jiang X, Yang L, et al. SARS‐CoV‐2‐specific immune response in COVID‐19 convalescent individuals. Signal Transduct Target Ther. 2021;6(1):256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ko J‐H, Joo E‐J, Park S‐J, et al. Neutralizing antibody production in asymptomatic and mild COVID‐19 patients, in comparison with pneumonic COVID‐19 patients. J Clin Med. 2020;9(7):2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Goel RR, Apostolidis SA, Painter MM, et al. Distinct antibody and memory B cell responses in SARS‐CoV‐2 naïve and recovered individuals after mRNA vaccination. Science immunology. 2021;6(58):eabi6950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. PriyankaChoudhary OP, Singh I. Protective immunity against COVID‐19: unravelling the evidences for humoral vs. cellular components. Travel Med Infect Dis. 2021;39:101911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kojima N, Klausner JD. Protective immunity after recovery from SARS‐CoV‐2 infection. Lancet Infect Dis. 2022;22(1):12‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schultze JL, Aschenbrenner AC. COVID‐19 and the human innate immune system. Cell. 2021;184(7):1671‐1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Karampoor S, Afrashteh F, Laali A. Persistent hiccups after treatment of COVID‐19 with dexamethasone: a case report. Respir Med Case Rep. 2021;34:101515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mathew D, Giles JR. Deep immune profiling of COVID‐19 patients reveals distinct immunotypes with therapeutic implications. Science. 2020;369(6508):eabc8511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bülow Anderberg S, Luther T, Berglund M, et al. Increased levels of plasma cytokines and correlations to organ failure and 30‐day mortality in critically ill Covid‐19 patients. Cytokine. 2021;138:155389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Secchi M, Bazzigaluppi E, Brigatti C, et al. COVID‐19 survival associates with the immunoglobulin response to the SARS‐CoV‐2 spike receptor binding domain. J Clin Invest. 2020;130(12):6366‐6378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rydyznski Moderbacher C, Ramirez SI, Dan JM, et al. Antigen‐specific adaptive immunity to SARS‐CoV‐2 in acute COVID‐19 and associations with age and disease severity. Cell. 2020;183(4):996‐1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shafaati M, Saidijam M, Soleimani M, et al. A brief review on DNA vaccines in the era of COVID‐19. Future Virol. 2022;17(1):49‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jordan SC. Innate and adaptive immune responses to SARS‐CoV‐2 in humans: relevance to acquired immunity and vaccine responses. Clin Exp Immunol. 2021;204(3):310‐320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Perico L, Benigni A, Casiraghi F, Ng LFP. Immunity, endothelial injury and complement‐induced coagulopathy in COVID‐19. Nat Rev Nephrol. 2021;17(1):46‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Perico L, Benigni A, Casiraghi F, Ng LFP, Renia L, Remuzzi G. Immunity, endothelial injury and complement‐induced coagulopathy in COVID‐19. Nat Rev Nephrol. 2021;17(1):46‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mortaz E, Tabarsi P, Varahram M, Folkerts G, Adcock IM. The immune response and immunopathology of COVID‐19. Front Immunol. 2020;11:2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Channappanavar R, Perlman S. Pathogenic human coronavirus infections: causes and consequences of cytokine storm and immunopathology. Paper presented at: Seminars in immunopathology 2017. [DOI] [PMC free article] [PubMed]
- 24. Rabaan AA, Al‐Ahmed SH, Al Mutair A, et al. Immunopathogenesis and immunobiology of SARS‐CoV‐2. Infez Med. 2021;29(2):167‐180. [PubMed] [Google Scholar]
- 25. Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497‐506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhou P, Yang X‐L, Wang X‐G, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Seyedpour S, Khodaei B, Loghman AH, et al. Targeted therapy strategies against SARS‐CoV‐2 cell entry mechanisms: a systematic review of in vitro and in vivo studies. J Cell Physiol. 2021;236(4):2364‐2392. [DOI] [PubMed] [Google Scholar]
- 28. Wang K, Chen W, Zhang Z, et al. CD147‐spike protein is a novel route for SARS‐CoV‐2 infection to host cells. Signal Transduct Target Ther. 2020;5(1):283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu C, von Brunn A, Zhu D. Cyclophilin A and CD147: novel therapeutic targets for the treatment of COVID‐19. Med Drug Discov. 2020;7:100056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Addi AB, Lefort A, Hua X, et al. Modulation of murine dendritic cell function by adenine nucleotides and adenosine: involvement of the A2B receptor. Eur J Immunol. 2008;38(6):1610‐1620. [DOI] [PubMed] [Google Scholar]
- 31. Yao C, Bora SA, Parimon T, et al. Cell‐type‐specific immune dysregulation in severely ill COVID‐19 patients. Cell Rep. 2021;34(1):108590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Niu P, Zhang S, Zhou P, et al. Ultrapotent human neutralizing antibody repertoires against Middle East respiratory syndrome coronavirus from a recovered patient. J Infect Dis. 2018;218(8):1249‐1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Azkur AK, Akdis M, Azkur D, et al. Immune response to SARS‐CoV‐2 and mechanisms of immunopathological changes in COVID‐19. Allergy. 2020;75(7):1564‐1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Weiskopf D, Schmitz KS, Raadsen MP, et al. Phenotype and kinetics of SARS‐CoV‐2‐specific T cells in COVID‐19 patients with acute respiratory distress syndrome. Sci Immunol. 2020;5(48):eabd2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zafer MM, El‐Mahallawy HA, Ashour HM. Severe COVID‐19 and sepsis: immune pathogenesis and laboratory markers. Microorganisms. 2021;9(1):159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Muyayalo KP, Huang DH, Zhao SJ, Xie T, Mor G, Liao AH. COVID‐19 and Treg/Th17 imbalance: potential relationship to pregnancy outcomes. Am J Reprod Immunol. 2020;84(5):e13304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Martonik D, Parfieniuk‐Kowerda A, Rogalska M, Flisiak R. The role of Th17 response in COVID‐19. Cells. 2021;10(6):1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. De Biasi S, Meschiari M, Gibellini L, et al. Marked T cell activation, senescence, exhaustion and skewing towards TH17 in patients with COVID‐19. Pneumonia. 2020;11(1):3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang Z, Zhang Y, Yang R, et al. Landscape of peripheral blood mononuclear cells and soluble factors in severe COVID‐19 patients with pulmonary fibrosis development. Front Immunol. 2022;13:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wilk AJ, Lee MJ, Wei B, et al. Multi‐omic profiling reveals widespread dysregulation of innate immunity and hematopoiesis in COVID‐19. J Exp Med. 2021;218(8):e20210582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bhardwaj A, Sapra L, Saini C, et al. COVID‐19: immunology, immunopathogenesis and potential therapies. Int Rev Immunol. 2022;41(2):171‐206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Merad M, Blish CA, Sallusto F, Iwasaki A. The immunology and immunopathology of COVID‐19. Science. 2022;375(6585):1122‐1127. [DOI] [PubMed] [Google Scholar]
- 43. Zavvar M, Yahyapoor A, Baghdadi H, et al. COVID‐19 immunotherapy: treatment based on the immune cell‐mediated approaches. Int Immunopharmacol. 2022;107:108655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Delshad M, Tavakolinia N, Pourbagheri‐Sigaroodi A, Safaroghli‐Azar A, Bagheri N, Bashash D. The contributory role of lymphocyte subsets, pathophysiology of lymphopenia and its implication as prognostic and therapeutic opportunity in COVID‐19. Int Immunopharmacol. 2021;95:107586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Soy M, Keser G, Atagündüz P, Tabak F, Atagündüz I, Kayhan S. Cytokine storm in COVID‐19: pathogenesis and overview of anti‐inflammatory agents used in treatment. Clin Rheumatol. 2020;39(7):2085‐2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Vigón L, Fuertes D, García‐Pérez J, et al. Impaired cytotoxic response in PBMCs from patients with COVID‐19 admitted to the ICU: biomarkers to predict disease severity. Front Immunol. 2021;12:665329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ricci D, Etna MP, Rizzo F, Sandini S, Severa M, Coccia EM. Innate immune response to SARS‐CoV‐2 infection: from cells to soluble mediators. Int J Mol Sci. 2021;22(13):7017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mirzaei R, Afaghi A, Babakhani S, et al. Role of microbiota‐derived short‐chain fatty acids in cancer development and prevention. Biomed Pharmacother. 2021;139:111619. [DOI] [PubMed] [Google Scholar]
- 49. Mirzaei R, Bouzari B, Hosseini‐Fard SR, et al. Role of microbiota‐derived short‐chain fatty acids in nervous system disorders. Biomed Pharmacother. 2021;139:111661. [DOI] [PubMed] [Google Scholar]
- 50. Kasuga Y, Zhu B. Innate immune sensing of coronavirus and viral evasion strategies. Exp Mol Med. 2021;53(5):723‐736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Blanco‐Melo D, Nilsson‐Payant BE, Liu WC, et al. Imbalanced host response to SARS‐CoV‐2 drives development of COVID‐19. Cell. 2020;181(5):1036‐1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hadjadj J, Yatim N. Impaired type I interferon activity and inflammatory responses in severe COVID‐19 patients. Science. 2020;369(6504):718‐724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sposito B, Broggi A, Pandolfi L, et al. The interferon landscape along the respiratory tract impacts the severity of COVID‐19. Cell. 2021;184(19):4953‐4968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lucas C, Wong P, Klein J. Longitudinal analyses reveal immunological misfiring in severe COVID‐19. Nature. 2020;584(7821):463‐469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lee JS, Park S. Immunophenotyping of COVID‐19 and influenza highlights the role of type I interferons in development of severe COVID‐19. Sci Immunol. 2020;5(49):eabd1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Israelow B, Song E, Mao T, et al. Mouse model of SARS‐CoV‐2 reveals inflammatory role of type I interferon signaling. J Exp Med. 2020;217:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Liao M, Liu Y, Yuan J, et al. Single‐cell landscape of bronchoalveolar immune cells in patients with COVID‐19. Nat Med. 2020;26(6):842‐844. [DOI] [PubMed] [Google Scholar]
- 58. Konig MF, Powell M, Staedtke V, et al. Preventing cytokine storm syndrome in COVID‐19 using α‐1 adrenergic receptor antagonists. J Clin Invest. 2020;130(7):3345‐3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zuo Y, Yalavarthi S, Shi H, et al. Neutrophil extracellular traps in COVID‐19. JCI Insight. 2020;5(11):e138999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhou Z, Ren L, Zhang L, et al. Heightened innate immune responses in the respiratory tract of COVID‐19 patients. Cell Host Microbe. 2020;27(6):883‐890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Parackova Z, Zentsova I. Disharmonic inflammatory signatures in COVID‐19: augmented neutrophils' but impaired monocytes' and dendritic cells' responsiveness. Cells. 2020;9(10):2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. The involvement of natural killer cells in the pathogenesis of severe acute respiratory syndrome. Am J Clin Pathol. 2004;121(4):507‐511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wang F, Nie J, Wang H, et al. Characteristics of peripheral lymphocyte subset alteration in COVID‐19 pneumonia. J Infect Dis. 2020;221(11):1762‐1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Mu J, Fang Y, Yang Q, et al. SARS‐CoV‐2 N protein antagonizes type I interferon signaling by suppressing phosphorylation and nuclear translocation of STAT1 and STAT2. Cell Discov. 2020;6:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Rowlands M, Segal F, Hartl D. Myeloid‐Derived suppressor cells as a potential biomarker and therapeutic target in COVID‐19. Front Immunol. 2021;12:2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Reizine F, Lesouhaitier M, Gregoire M, et al. SARS‐CoV‐2‐induced ARDS associates with MDSC expansion, lymphocyte dysfunction, and arginine shortage. J Clin Immunol. 2021;41(3):515‐525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bordoni V, Sacchi A, Cimini E, et al. An inflammatory profile correlates with decreased frequency of cytotoxic cells in coronavirus disease 2019. Clin Infect Dis. 2020;71(16):2272‐2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Xue G, Jiang M, Zhao R, Le A, Li J. Elevated frequencies of CD14+ HLA‐DRlo/neg MDSCs in COVID‐19 patients. Aging. 2021;13(5):6236‐6246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Koushki K, Salemi M, Miri SM, Arjeini Y, Keshavarz M, Ghaemi A. Role of myeloid‐derived suppressor cells in viral respiratory infections; hints for discovering therapeutic targets for COVID‐19. Biomed Pharmacother. 2021;144:112346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Chua RL, Lukassen S, Trump S, et al. COVID‐19 severity correlates with airway epithelium–immune cell interactions identified by single‐cell analysis. Nat. Biotechnol. 2020;38(8):970‐979. [DOI] [PubMed] [Google Scholar]
- 71. Kim D‐M, Kim Y, Seo J‐W, et al. Enhanced eosinophil‐mediated inflammation associated with antibody and complement‐dependent pneumonic insults in critical COVID‐19. Cell Rep. 2021;37(1):109798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Gebremeskel S, Schanin J, Coyle KM, et al. Mast cell and eosinophil activation are associated with COVID‐19 and TLR‐mediated viral inflammation: implications for an anti‐Siglec‐8 antibody. Front Immunol. 2021;12(641):650331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Onodi F, Bonnet‐Madin L, Meertens L, et al. SARS‐CoV‐2 induces human plasmacytoid predendritic cell diversification via UNC93B and IRAK4. J Exp Med. 2021;218(4):e20201387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Severa M, Diotti RA, Etna MP, et al. Differential plasmacytoid dendritic cell phenotype and type I interferon response in asymptomatic and severe COVID‐19 infection. PLoS Pathog. 2021;17:e1009878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Arunachalam PS, Wimmers F. Systems biological assessment of immunity to mild versus severe COVID‐19 infection in humans. Science. 2020;369(6508):1210‐1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Feng Z, Diao B, Wang R, et al. The novel severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) directly decimates human spleens and lymph nodes. medRxiv . 2020. 10.1101/2020.03.27.20045427 [DOI]
- 77. Mirzaei R, Babakhani S, Ajorloo P, et al. The emerging role of exosomal miRNAs as a diagnostic and therapeutic biomarker in Mycobacterium tuberculosis infection. Mol Med. 2021;27(1):1‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Merad M, Martin JC. Pathological inflammation in patients with COVID‐19: a key role for monocytes and macrophages. Nat Rev Immunol. 2020;20(6):355‐362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. de Groen RA, Boltjes A, Hou J, et al. IFN‐λ‐mediated IL‐12 production in macrophages induces IFN‐γ production in human NK cells. Eur J Immunol. 2015;45(1):250‐259. [DOI] [PubMed] [Google Scholar]
- 80. Ural BB, Yeung ST. Identification of a nerve‐associated, lung‐resident interstitial macrophage subset with distinct localization and immunoregulatory properties. Sci Immunol. 2020;5(45):eaax8756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Liao H, Marley G, Si Y, et al. The landscape of lung bronchoalveolar immune cells in COVID‐19 revealed by single‐cell RNA sequencing. medRxiv . 2020. 10.1101/2020.02.23.20026690 [DOI]
- 82. Taefehshokr N, Taefehshokr S, Hemmat N, Heit B. Covid‐19: perspectives on innate immune evasion. Front Immunol. 2020;11:580641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Channappanavar R, Fehr AR, Zheng J, et al. IFN‐I response timing relative to virus replication determines MERS coronavirus infection outcomes. J Clin Invest. 2019;129(9):3625‐3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Zhou J, Chu H, Li C, et al. Active replication of Middle East respiratory syndrome coronavirus and aberrant induction of inflammatory cytokines and chemokines in human macrophages: implications for pathogenesis. J Infect Dis. 2014;209(9):1331‐1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Wilk AJ, Rustagi A, Zhao NQ, et al. A single‐cell atlas of the peripheral immune response in patients with severe COVID‐19. Nat Med. 2020;26(7):1070‐1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Giamarellos‐Bourboulis EJ, Netea MG, Rovina N, et al. Complex immune dysregulation in COVID‐19 patients with severe respiratory failure. Cell Host Microbe. 2020;27(6):992‐1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Menachery VD, Schäfer A, Burnum‐Johnson KE. MERS‐CoV and H5N1 influenza virus antagonize antigen presentation by altering the epigenetic landscape. Proc Natl Acad Sci USA. 2018;115(5):E1012‐E1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Josset L, Menachery VD, Gralinski LE, et al. Cell host response to infection with novel human coronavirus EMC predicts potential antivirals and important differences with SARS coronavirus. mBio. 2013;4(3):e00165‐e00113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K. A SARS‐CoV‐2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583(7816):459‐468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Fang WF, Chen YM, Lin CY, et al. Histone deacetylase 2 (HDAC2) attenuates lipopolysaccharide (LPS)‐induced inflammation by regulating PAI‐1 expression. J Inflamm. 2018;15:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Kong X, Fang M, Li P, Fang F, Xu Y. HDAC2 deacetylates class II transactivator and suppresses its activity in macrophages and smooth muscle cells. J Mol Cell Cardiol. 2009;46(3):292‐299. [DOI] [PubMed] [Google Scholar]
- 92. Zhang Y, Zhang J, Chen Y, et al. The ORF8 protein of SARS‐CoV‐2 mediates immune evasion through potently downregulating MHC‐I. bioRxiv . 2020. 10.1101/2020.05.24.111823 [DOI]
- 93. Dirk BS, Pawlak EN, Johnson AL, et al. HIV‐1 Nef sequesters MHC‐I intracellularly by targeting early stages of endocytosis and recycling. Sci Rep. 2016;6:37021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Dirk BS, Heit B, Dikeakos JD. Visualizing interactions between HIV‐1 nef and host cellular proteins using ground‐state depletion microscopy. AIDS Res Hum Retroviruses. 2015;31(7):671‐672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Cendrowski J, Mamińska A, Miaczynska M. Endocytic regulation of cytokine receptor signaling. Cytokine Growth Factor Rev. 2016;32:63‐73. [DOI] [PubMed] [Google Scholar]
- 96. McCormick PJ, Martina JA, Bonifacino JS. Involvement of clathrin and AP‐2 in the trafficking of MHC class II molecules to antigen‐processing compartments. Proc Natl Acad Sci USA. 2005;102(22):7910‐7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Williams AE, José RJ, Mercer PF, et al. Evidence for chemokine synergy during neutrophil migration in ARDS. Thorax. 2017;72(1):66‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Grommes J, Soehnlein O. Contribution of neutrophils to acute lung injury. Mol Med. 2011;17(3‐4):293‐307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. ARDS Definition Task Force, Ranieri VM, Rubenfeld GD, et al. Acute respiratory distress syndrome: the Berlin definition. JAMA. 2012;307(23):2526‐2533. [DOI] [PubMed] [Google Scholar]
- 100. Fox SE, Akmatbekov A, Harbert JL, et al. Pulmonary and cardiac pathology in African American patients with COVID‐19: an autopsy series from New Orleans. Lancet Respir Med. 2020;8(7):681‐686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Liu J, Liu Y, Xiang P, et al. Neutrophil‐to‐lymphocyte ratio predicts critical illness patients with 2019 coronavirus disease in the early stage. J Transl Med. 2020;18(1):206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Wilk AJ, Rustagi A. A single‐cell atlas of the peripheral immune response in patients with severe COVID‐19. Nat Med. 2020;26(7):1070‐1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Xiong Y, Liu Y, Cao L, et al. Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID‐19 patients. Emerging microbes & infections. 2020;9(1):761‐770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Kang Y‐W, Park S, Lee K‐J, Moon D, Kim Y‐M, Lee S‐W. Understanding the host innate immune responses against SARS‐CoV‐2 infection and COVID‐19 pathogenesis. Immune Netw. 2021;21(1):e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Laforge M, Elbim C, Frère C. Tissue damage from neutrophil‐induced oxidative stress in COVID‐19. Nat Rev Immunol. 2020;20(9):515‐516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Klok FA, Kruip M, van der Meer NJM, et al. Confirmation of the high cumulative incidence of thrombotic complications in critically ill ICU patients with COVID‐19: an updated analysis. Thromb Res. 2020;191:148‐150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Barnes BJ, Adrover JM, Baxter‐Stoltzfus A, et al. Targeting potential drivers of COVID‐19: neutrophil extracellular traps. J Exp Med. 2020;217(6):e20200652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Veras FP, Pontelli MC, Silva CM, et al. SARS‐CoV‐2–triggered neutrophil extracellular traps mediate COVID‐19 pathology. J Exp Med. 2020;217(12):e20201129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Ricci D, Etna MP, Rizzo F. Innate immune response to SARS‐CoV‐2 infection: from cells to soluble mediators. Int J Mol Sci. 2021;22(13):7017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Sivori S, Vacca P, Del Zotto G. Human NK cells: surface receptors, inhibitory checkpoints, and translational applications. Cell Mol Immunol. 2019;16(5):430‐441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Zheng M, Gao Y, Wang G, et al. Functional exhaustion of antiviral lymphocytes in COVID‐19 patients. Cell Mol Immunol. 2020;17(5):533‐535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Leem G, Cheon S, Lee H, et al. Abnormality in the NK‐cell population is prolonged in severe COVID‐19 patients. J Allergy Clin Immunol. 2021;148(4):996‐1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Guo C, Wu M, Huang B, et al. Single‐cell transcriptomics reveal a unique memory‐like NK cell subset that accumulates with ageing and correlates with disease severity in COVID‐19. Genome Med. 2022;14(1):46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Mahdiun F, Mansouri S, Khazaeli P, Mirzaei R. The effect of tobramycin incorporated with bismuth‐ethanedithiol loaded on niosomes on the quorum sensing and biofilm formation of Pseudomonas aeruginosa . Microb Pathog. 2017;107:129‐135. [DOI] [PubMed] [Google Scholar]
- 115. Schulte‐Schrepping J, Reusch N, Paclik D, et al. Severe COVID‐19 is marked by a dysregulated myeloid cell compartment. Cell. 2020;182(6):1419‐1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Silvin A, Chapuis N, Dunsmore G, et al. Elevated calprotectin and abnormal myeloid cell subsets discriminate severe from mild COVID‐19. Cell. 2020;182(6):1401‐1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Arunachalam PS, Wimmers F, Mok CKP, et al. Systems biological assessment of immunity to mild versus severe COVID‐19 infection in humans. Science. 2020;369(6508):1210‐1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Agrati C, Sacchi A, Bordoni V, et al. Expansion of myeloid‐derived suppressor cells in patients with severe coronavirus disease (COVID‐19). Cell Death Differ. 2020;27(11):3196‐3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Vitte J, Diallo AB, Boumaza A, et al. A granulocytic signature identifies COVID‐19 and its severity. J Infect Dis. 2020;222(12):1985‐1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Shaath H, Vishnubalaji R, Elkord E, Alajez NM. Single‐cell transcriptome analysis highlights a role for neutrophils and inflammatory macrophages in the pathogenesis of severe COVID‐19. Cells. 2020;9(11):2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Rosenberg HF, Foster PS. Eosinophils and COVID‐19: diagnosis, prognosis, and vaccination strategies. Semin Immunopathol. 2021;43(3):383‐392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Pum A, Ennemoser M, Adage T, Kungl AJ. Cytokines and chemokines in SARS‐CoV‐2 Infections‐Therapeutic strategies targeting cytokine storm. Biomolecules. 2021;11(1):91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Rokni M, Hamblin MR, Rezaei N. Cytokines and COVID‐19: friends or foes? Hum Vaccin Immunother. 2020;16(10):2363‐2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Hashim M, Athar S, Gaba WH. New onset adrenal insufficiency in a patient with COVID‐19. BMJ Case Rep. 2021;14(1):e237690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Freire Santana M, Borba MGS, Baía‐da‐Silva DC, et al. Case report: adrenal pathology findings in severe COVID‐19: an autopsy study. Am J Trop Med Hyg. 2020;103(4):1604‐1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Rodriguez L, Pekkarinen PT, Lakshmikanth T, et al. Systems‐Level immunomonitoring from acute to recovery phase of severe COVID‐19. Cell Rep Med. 2020;1(5):100078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Johansson MW. Activation states of blood eosinophils in asthma. Clin Exp Allergy. 2014;44(4):482‐498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Barton LM, Duval EJ, Stroberg E, Ghosh S, Mukhopadhyay S. COVID‐19 autopsies, Oklahoma, USA. Am J Clin Pathol. 2020;153(6):725‐733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Damiani S, Fiorentino M. Pathological post‐mortem findings in lungs infected with SARS‐CoV‐2. J Pathol. 2021;253(1):31‐40. [DOI] [PubMed] [Google Scholar]
- 130. Zein JG, Strauss R, Attaway AH, et al. Eosinophilia is associated with improved COVID‐19 outcomes in inhaled corticosteroid‐treated patients. J Allergy Clin Immunol. 2022;10(3):742‐750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Kang YW, Park S. Understanding the host innate immune responses against SARS‐CoV‐2 infection and COVID‐19 pathogenesis. Immune Netw. 2021;21(1):e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Eisenbarth SC. Dendritic cell subsets in T cell programming: location dictates function. Nat Rev Immunol. 2019;19(2):89‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Zhou R, To KK, Wong YC, et al. Acute SARS‐CoV‐2 infection impairs dendritic cell and T cell responses. Immunity. 2020;53(4):864‐877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Hasan A, Al‐Ozairi E, Al‐Baqsumi Z, Ahmad R. Cellular and humoral immune responses in Covid‐19 and immunotherapeutic approaches. Immunotargets Ther. 2021;10:63‐85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Laing AG, Lorenc A. A dynamic COVID‐19 immune signature includes associations with poor prognosis. Nat Med. 2020;26(10):1623‐1635. [DOI] [PubMed] [Google Scholar]
- 136. Stehle C, Hernández DC. Innate lymphoid cells in lung infection and immunity. Immunol Rev. 2018;286(1):102‐119. [DOI] [PubMed] [Google Scholar]
- 137. Ranjan P, Bowzard JB, Schwerzmann JW, Jeisy‐Scott V, Fujita T, Sambhara S. Cytoplasmic nucleic acid sensors in antiviral immunity. Trends Mol Med. 2009;15(8):359‐368. [DOI] [PubMed] [Google Scholar]
- 138. Wang Y, Lifshitz L, Silverstein N, et al. Clarification of human blood ILC subtype interrelatedness and discovery of amphiregulin production by human NK cells shed light on HIV‐1 pathogenesis. bioRxiv . 2021. 10.1101/2021.04.20.440368 [DOI]
- 139. Loyon R, Jary M, Salomé B, et al. Peripheral innate lymphoid cells are increased in first line metastatic colorectal carcinoma patients: a negative correlation with Th1 immune responses. Front Immunol. 2019;10:2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. van der Ploeg EK, Carreras Mascaro A, Huylebroeck D, Hendriks RW, Stadhouders R. Group 2 innate lymphoid cells in human respiratory disorders. J Innate Immun. 2020;12(1):47‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. García M, Kokkinou E. Innate lymphoid cell composition associates with COVID‐19 disease severity. Clin Transl immunol. 2020;9(12):e1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Gomez‐Cadena A, Spehner L, Kroemer M, et al. Severe COVID‐19 patients exhibit an ILC2 NKG2D(+) population in their impaired ILC compartment. Cell Mol Immunol. 2021;18(2):484‐486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Shin D, Mukherjee R. Papain‐like protease regulates SARS‐CoV‐2 viral spread and innate immunity. Nature. 2020;587(7835):657‐662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Halim TY, Steer CA, Mathä L, et al. Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell‐mediated allergic lung inflammation. Immunity. 2014;40(3):425‐435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. García M, Kokkinou E, Carrasco García A, et al. Innate lymphoid cell composition associates with COVID‐19 disease severity. Clin Transl Immunol. 2020;9(12):e1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Silverstein NJ, Wang Y, Manickas‐Hill Z, et al. Innate lymphoid cells and disease tolerance in SARS‐CoV‐2 infection. medRxiv . 2021. 10.1101/2021.01.14.21249839 [DOI]
- 147. Sette A, Crotty S. Adaptive immunity to SARS‐CoV‐2 and COVID‐19. Cell. 2021;184(4):861‐880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Rasoul M, Rokhsareh M, Mohammad SM, Sajad K, Ahmadreza M. The human immune system against Staphylococcus epidermidis . Crit Rev Immunol. 2019;39:3. [DOI] [PubMed] [Google Scholar]
- 149. Mirzaei R, Attar A, Papizadeh S, et al. The emerging role of probiotics as a mitigation strategy against coronavirus disease 2019 (COVID‐19). Arch Virol. 2021;166:1‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Grifoni A, Weiskopf D, Ramirez SI, et al. Targets of T cell responses to SARS‐CoV‐2 coronavirus in humans with COVID‐19 disease and unexposed individuals. Cell. 2020;181(7):1489‐1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Tan AT, Linster M, Tan CW, et al. Early induction of SARS‐CoV‐2 specific T cells associates with rapid viral clearance and mild disease in COVID‐19 patients. Cell Rep. 2020;34(6):108728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Braun J, Loyal L, Frentsch M, et al. SARS‐CoV‐2‐reactive T cells in healthy donors and patients with COVID‐19. Nature. 2020;587(7833):270‐274. [DOI] [PubMed] [Google Scholar]
- 153. McMahan K, Yu J, Mercado NB. Correlates of protection against SARS‐CoV‐2 in rhesus macaques. Nature. 2021;590(7847):630‐634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Sekine T, Perez‐Potti A, Rivera‐Ballesteros O, et al. Robust T cell immunity in convalescent individuals with asymptomatic or mild COVID‐19. Cell. 2020;183(1):158‐168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Peng Y, Mentzer AJ, Liu G, et al. Broad and strong memory CD4+ and CD8+ T cells induced by SARS‐CoV‐2 in UK convalescent individuals following COVID‐19. Nat Immunol. 2020;21(11):1336‐1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Le Bert N, Tan AT, Kunasegaran K, et al. SARS‐CoV‐2‐specific T cell immunity in cases of COVID‐19 and SARS, and uninfected controls. Nature. 2020;584(7821):457‐462. [DOI] [PubMed] [Google Scholar]
- 157. Nelde A, Bilich T, Heitmann JS, et al. SARS‐CoV‐2‐derived peptides define heterologous and COVID‐19‐induced T cell recognition. Nat Immunol. 2021;22(1):74‐85. [DOI] [PubMed] [Google Scholar]
- 158. Schulien I, Kemming J, Oberhardt V, et al. Characterization of pre‐existing and induced SARS‐CoV‐2‐specific CD8+ T cells. Nat Med. 2021;27(1):78‐85. [DOI] [PubMed] [Google Scholar]
- 159. Rha M‐S, Shin E‐C. Activation or exhaustion of CD8+ T cells in patients with COVID‐19. Cell Mol Immunol. 2021;18(10):2325‐2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Mathew D, Giles JR, Baxter AE, et al. Deep immune profiling of COVID‐19 patients reveals distinct immunotypes with therapeutic implications. Science. 2020;369(6508):eabc8511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Röltgen K, Boyd SD. Antibody and B cell responses to SARS‐CoV‐2 infection and vaccination. Cell Host Microbe. 2021;29(7):1063‐1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Iyer AS, Jones FK, Nodoushani A, et al. Persistence and decay of human antibody responses to the receptor binding domain of SARS‐CoV‐2 spike protein in COVID‐19 patients. Sci Immunol. 2020;5(52):eabe0367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Long Q‐X, Liu B‐Z, Deng H‐J, et al. Antibody responses to SARS‐CoV‐2 in patients with COVID‐19. Nat Med. 2020;26(6):845‐848. [DOI] [PubMed] [Google Scholar]
- 164. Kaneko N, Kuo H‐H, Boucau J, et al. Loss of Bcl‐6‐expressing T follicular helper cells and germinal centers in COVID‐19. Cell. 2020;183(1):143‐157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Low JS, Vaqueirinho D. Clonal analysis of immunodominance and cross‐reactivity of the CD4 T cell response to SARS‐CoV‐2. Science (New York, N.Y.). 2021;372(6548):1336‐1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Hasan A, Al‐Ozairi E, Al‐Baqsumi Z, Ahmad R, Al‐Mulla F. Cellular and humoral immune responses in Covid‐19 and immunotherapeutic approaches. Immunotargets Ther. 2021;10:63‐85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Wang C, Li W. Publisher correction: A human monoclonal antibody blocking SARS‐CoV‐2. Infection. 2020;11(1):2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Woodruff MC, Ramonell RP. Extrafollicular B cell responses correlate with neutralizing antibodies and morbidity in COVID‐19. Nat Immunol. 2020;21(12):1506‐1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Woodruff MC, Ramonell RP, Nguyen DC, et al. Extrafollicular B cell responses correlate with neutralizing antibodies and morbidity in COVID‐19. Nat Immunol. 2020;21(12):1506‐1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Zhang W, Zhang H, Liu S, et al. Excessive CD11c(+)Tbet(+) B cells promote aberrant T(FH) differentiation and affinity‐based germinal center selection in lupus. Proc Natl Acad Sci USA. 2019;116(37):18550‐18560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Mirzaei R, Zamani F, Hajibaba M, et al. The pathogenic, therapeutic and diagnostic role of exosomal microRNA in the autoimmune diseases. J Neuroimmunol. 2021;358:577640. [DOI] [PubMed] [Google Scholar]
- 172. Ranjbar R, Karampoor S, Jalilian FA. The protective effect of Helicobacter pylori infection on the susceptibility of multiple sclerosis. J Neuroimmunol. 2019;337:577069. [DOI] [PubMed] [Google Scholar]
- 173. Kang CK, Kim M, Hong J, et al. Distinct immune response at 1 year post‐COVID‐19 according to disease severity. Front Immunol. 2022;13:830433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Tay MZ, Poh CM, Rénia L. The trinity of COVID‐19: immunity, inflammation and intervention. Nat Rev Immunol. 2020;20(6):363‐374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Moss P. The T cell immune response against SARS‐CoV‐2. Nat Immunol. 2022;23(2):186‐193. [DOI] [PubMed] [Google Scholar]
- 176. Jung MK, Jeong SD, Noh JY, et al. BNT162b2‐induced memory T cells respond to the Omicron variant with preserved polyfunctionality. Nat Microbiol. 2022;7(6):909‐917. [DOI] [PubMed] [Google Scholar]
- 177. Xu Z, Shi L, Wang Y, et al. Pathological findings of COVID‐19 associated with acute respiratory distress syndrome. Lancet Respir Med. 2020;8(4):420‐422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Guo C, Li B, Ma H, et al. Single‐cell analysis of two severe COVID‐19 patients reveals a monocyte‐associated and tocilizumab‐responding cytokine storm. Nat Commun. 2020;11(1):3924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Zheng J, Liu Y, Lau YL, Tu W. γδ‐T cells: an unpolished sword in human anti‐infection immunity. Cell Mol Immunol. 2013;10(1):50‐57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Libraty DH, O'Neil KM, Baker LM, Acosta LP, Olveda RM. Human CD4(+) memory t‐lymphocyte responses to SARS coronavirus infection. Virology. 2007;368(2):317‐321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Yang LT, Peng H, Zhu ZL, et al. Long‐lived effector/central memory t‐cell responses to severe acute respiratory syndrome coronavirus (SARS‐CoV) S antigen in recovered SARS patients. Clin Immunol. 2006;120(2):171‐178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Shin HS, Kim Y, Kim G, et al. Immune responses to Middle East respiratory syndrome coronavirus during the acute and convalescent phases of human infection. Clin Infect Dis. 2019;68(6):984‐992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Janice Oh HL, Ken‐En Gan S, Bertoletti A, Tan YJ. Understanding the T cell immune response in SARS coronavirus infection. Emerg Microbes Infect. 2012;1(9):e23‐e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Chen J, Lau YF, Lamirande EW, et al. Cellular immune responses to severe acute respiratory syndrome coronavirus (SARS‐CoV) infection in senescent BALB/c mice: CD4+ T cells are important in control of SARS‐CoV infection. J Virol. 2010;84(3):1289‐1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Roberts A, Deming D, Paddock CD, et al. A mouse‐adapted SARS‐coronavirus causes disease and mortality in BALB/c mice. PLoS Pathog. 2007;3(1):e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Zhao J, Zhao J, Perlman S. T cell responses are required for protection from clinical disease and for virus clearance in severe acute respiratory syndrome coronavirus‐infected mice. J Virol. 2010;84(18):9318‐9325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Kuri‐Cervantes L, Pampena MB. Comprehensive mapping of immune perturbations associated with severe COVID‐19. Sci Immunol. 2020;5(49):eabd7114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Yu K, He J, Wu Y, et al. Dysregulated adaptive immune response contributes to severe COVID‐19. Cell Res. 2020;30(9):814‐816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Kuri‐Cervantes L, Pampena MB, Meng W, et al. Comprehensive mapping of immune perturbations associated with severe COVID‐19. Sci Immunol. 2020;5(49):eabd7114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Chua RL, Lukassen S. COVID‐19 severity correlates with airway epithelium‐immune cell interactions identified by single‐cell analysis. Nat Biotechnol. 2020;38(8):970‐979. [DOI] [PubMed] [Google Scholar]
- 191. Wen W, Su W, Tang H, et al. Immune cell profiling of COVID‐19 patients in the recovery stageby single‐cell sequencing. Cell Discov. 2020;6(1):31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Ni L, Ye F, Cheng M‐L, et al. Detection of SARS‐CoV‐2‐Specific humoral and cellular immunity in COVID‐19 convalescent individuals. Immunity. 2020;52(6):971‐977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Chen Z, John, Wherry E. T cell responses in patients with COVID‐19. Nat Rev Immunol. 2020;20(9):529‐536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Diao B, Wang C, Tan Y, et al. Reduction and functional exhaustion of T cells in patients with coronavirus disease 2019 (COVID‐19). Front Immunol. 2020;11:827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Mazzoni A, Salvati L, Maggi L, et al. Impaired immune cell cytotoxicity in severe COVID‐19 is IL‐6 dependent. J Clin Invest. 2020;130(9):4694‐4703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Chen G, Wu D, Guo W, et al. Clinical and immunologic features in severe and moderate forms of coronavirus disease 2019. medRxiv. 2020;130(5):2620‐2629. 10.1101/2020.02.16.20023903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Weiskopf D, Schmitz KS, Raadsen MP, et al. Phenotype and kinetics of SARS‐CoV‐2‐specific T cells in COVID‐19 patients with acute respiratory distress syndrome. Sci Immunol. 2020;5(48):eabd2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Gil‐Etayo FJ, Suàrez‐Fernández P, Cabrera‐Marante O, et al. T‐helper cell subset response is a determining factor in COVID‐19 progression. Front Cell Infect Microbiol. 2021;11:624483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Qin C, Zhou L, Hu Z, et al. Dysregulation of immune response in patients with coronavirus 2019 (COVID‐19) in Wuhan, China. Clin Infect Dis. 2020;71(15):762‐768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Wang W, Su B, Pang L, et al. High‐dimensional immune profiling by mass cytometry revealed immunosuppression and dysfunction of immunity in COVID‐19 patients. Cell Mol Immunol. 2020;17(6):650‐652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Zhou Y, Fu B, Zheng X, et al. Pathogenic T cells and inflammatory monocytes incite inflammatory storm in severe COVID‐19 patients. Natl Sci Rev. 2020;7:nwaa041‐nwaa1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Neidleman J, Luo X, Frouard J, et al. SARS‐CoV‐2‐Specific T cells exhibit phenotypic features of helper function, lack of terminal differentiation, and high proliferation potential. Cell Reports Medicine. 2020;1(6):100081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Zahran AM, Zahran ZAM, Mady YH, et al. Differential alterations in peripheral lymphocyte subsets in COVID‐19 patients: upregulation of double‐positive and double‐negative T cells. Multidiscip Respir Med. 2021;16(2):758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Iannetta M, Buccisano F, Fraboni D, et al. Baseline T‐lymphocyte subset absolute counts can predict both outcome and severity in SARS‐CoV‐2 infected patients: a single centre study. Sci Rep. 2021;11(1):12762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Yang L, Liu S, Liu J, et al. COVID‐19: immunopathogenesis and immunotherapeutics. Signal Transduct Target Ther. 2020;5(1):128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. 2004;203(2):631‐637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Gu J, Gong E, Zhang B, et al. Multiple organ infection and the pathogenesis of SARS. J Exp Med. 2005;202(3):415‐424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Wu D, Yang XO. TH17 responses in cytokine storm of COVID‐19: an emerging target of JAK2 inhibitor fedratinib. J Microbiol Immunol Infect. 2020;53(3):368‐370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Han H, Ma Q, Li C, et al. Profiling serum cytokines in COVID‐19 patients reveals IL‐6 and IL‐10 are disease severity predictors. Emerg Microbes Infect. 2020;9(1):1123‐1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Halim C, Mirza AF, Sari MI. The association between TNF‐α, IL‐6, and vitamin D levels and COVID‐19 severity and mortality: a systematic review and meta‐analysis. Pathogens. 2022;11(2):195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Liu Y, Zhang C, Huang F, et al. Elevated plasma levels of selective cytokines in COVID‐19 patients reflect viral load and lung injury. Natl Sci Rev. 2020;7(6):1003‐1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Wu Z, McGoogan JM. Characteristics of and important lessons from the coronavirus disease 2019 (COVID‐19) outbreak in China: summary of a report of 72 314 cases from the Chinese Center for Disease Control and Prevention. JAMA. 2020;323(13):1239‐1242. [DOI] [PubMed] [Google Scholar]
- 213. Wang D, Hu B, Hu C, et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus‐infected pneumonia in Wuhan, China. JAMA. 2020;323(11):1061‐1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Chen N, Zhou M, Dong X, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in wuhan, China: a descriptive study. Lancet. 2020;395(10223):507‐513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Yang X, Rutkovsky AC, Zhou J, et al. Characterization of altered gene expression and histone methylation in peripheral blood mononuclear cells regulating inflammation in COVID‐19 patients. J Immunol. 2022;208(8):1968‐1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Yoo JY, Desiderio S. Innate and acquired immunity intersect in a global view of the acute‐phase response. Proc Natl Acad Sci USA. 2003;100(3):1157‐1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Uhlar CM, Whitehead AS. The kinetics and magnitude of the synergistic activation of the serum amyloid A promoter by IL‐1 beta and IL‐6 is determined by the order of cytokine addition. Scand J Immunol. 1999;49(4):399‐404. [DOI] [PubMed] [Google Scholar]
- 218. Zinellu A, Paliogiannis P, Carru C, Mangoni AA. Serum amyloid A concentrations, COVID‐19 severity and mortality: an updated systematic review and meta‐analysis. Int J Infect Dis. 2021;105:668‐674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Ather JL, Ckless K, Martin R, et al. Serum amyloid A activates the NLRP3 inflammasome and promotes Th17 allergic asthma in mice. J Immunol. 2011;187:64‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. De Buck M, Gouwy M, Wang JM, et al. The cytokine‐serum amyloid A—chemokine network. Cytokine Growth Factor Rev. 2016;30:55‐69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Fajgenbaum DC, June CH. Cytokine storm. N Engl J Med. 2020;383(23):2255‐2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Page MJ, Thomson GJA, Nunes JM, Engelbrecht AM, Nell TA. Serum amyloid A binds to fibrin(ogen), promoting fibrin amyloid formation. Sci Rep. 2019;9(1):3102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Al‐Samkari H, Karp Leaf RS, Dzik WH, et al. COVID‐19 and coagulation: bleeding and thrombotic manifestations of SARS‐CoV‐2 infection. Blood. 2020;136(4):489‐500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Liu J, Tu C, Zhu M, et al. The clinical course and prognostic factors of severe COVID‐19 in Wuhan, China: a retrospective case–control study. Medicine. 2021;100(8):e23996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Li H, Xiang X, Ren H, et al. Serum amyloid A is a biomarker of severe coronavirus disease and poor prognosis. J Infect. 2020;80(6):646‐655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Jana AK, Greenwood AB, Hansmann UHE. Presence of a SARS‐COV‐2 protein enhances amyloid formation of serum amyloid A. J Phys Chem B. 2021;125(32):9155‐9167. 10.1101/2021.05.18.444723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Fabrizi F, Alfieri CM, Cerutti R, Lunghi G, Messa P. COVID‐19 and acute kidney injury: a systematic review and Meta‐Analysis. Pathogens. 2020;9(12):1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Galkin AP. Hypothesis: AA amyloidosis is a factor causing systemic complications after coronavirus disease. Prion. 2021;15(1):53‐55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Zheutlin LM, Thonar EJMA, Jacobs ER, Hanley ME, Balk RA, Bone RC. Plasma elastase levels in the adult respiratory distress syndrome. J Crit Care. 1986;1(1):39‐44. [Google Scholar]
- 230. Mohamed MMA, El‐Shimy IA, Hadi MA. Neutrophil elastase inhibitors: A potential prophylactic treatment option for SARS‐CoV‐2‐induced respiratory complications? Crit Care. 2020;24(1):311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Zimmermann M, Arruda‐Silva F, Bianchetto‐Aguilera F, et al. IFNα enhances the production of IL‐6 by human neutrophils activated via TLR8. Sci Rep. 2016;6:19674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Farahi N, Paige E, Balla J, et al. Neutrophil‐mediated IL‐6 receptor trans‐signaling and the risk of chronic obstructive pulmonary disease and asthma. Hum Mol Genet. 2017;26(8):1584‐1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Li C, Zhang C, Chen X, et al. Relative depletion of soluble interleukin 6 receptors abolished the development of cytokine release syndrome after CART19/22 and lenalidomide treatment for lymphoma. Blood. 2019;134:5313. [Google Scholar]
- 234. Belouzard S, Madu I, Whittaker GR. Elastase‐mediated activation of the severe acute respiratory syndrome coronavirus spike protein at discrete sites within the S2 domain. J Biol Chem. 2010;285(30):22758‐22763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Akgun E, Tuzuner MB, Sahin B, et al. Proteins associated with neutrophil degranulation are upregulated in nasopharyngeal swabs from SARS‐CoV‐2 patients. PLoS One. 2020;15(10):e0240012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Xian X, Gopal S, Couchman JR. Syndecans as receptors and organizers of the extracellular matrix. Cell Tissue Res. 2010;339(1):31‐46. [DOI] [PubMed] [Google Scholar]
- 237. Karampoor S, Zahednasab H, Farahmand M, et al. A possible pathogenic role of syndecan‐1 in the pathogenesis of coronavirus disease 2019 (COVID‐19). Int Immunopharmacol. 2021;97:107684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Gopal S. Syndecans in inflammation at a glance. Front Immunol. 2020;11:227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Kalia M, Chandra V, Rahman SA, Sehgal D, Jameel S. Heparan sulfate proteoglycans are required for cellular binding of the hepatitis E virus ORF2 capsid protein and for viral infection. J Virol. 2009;83(24):12714‐12724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Bacsa S, Karasneh G, Dosa S, Liu J, Valyi‐Nagy T, Shukla D. Syndecan‐1 and syndecan‐2 play key roles in herpes simplex virus type‐1 infection. J Gen Virol. 2011;92(pt 4):733‐743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Hayashida A, Amano S, Park PW. Syndecan‐1 promotes Staphylococcus aureus corneal infection by counteracting neutrophil‐mediated host defense. J Biol Chem. 2011;286(5):3288‐3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Bermejo‐Jambrina M, Eder J, Kaptein TM, et al. SARS‐CoV‐2 infection and transmission depends on heparan sulfates and is blocked by low molecular weight heparins. bioRxiv . 2020. 10.1101/2020.08.18.255810 [DOI]
- 243. Hudák A, Letoha A, Szilák L, Letoha T. Contribution of syndecans to the cellular entry of SARS‐CoV‐2. Int J Mol Sci. 2021;22(10):5336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Pandey A, Nikam AN, Shreya AB, et al. Potential therapeutic targets for combating SARS‐CoV‐2: drug repurposing, clinical trials and recent advancements. Life Sci. 2020;256:117883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. O'Brien MP, Forleo‐Neto E, Musser BJ, et al. Subcutaneous REGEN‐COV antibody combination to prevent Covid‐19. N Engl J Med. 2021;385(13):1184‐1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Felgenhauer U, Schoen A, Gad HH, et al. Inhibition of SARS‐CoV‐2 by type I and type III interferons. J Biol Chem. 2020;295(41):13958‐13964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Bessière P, Wasniewski M. Intranasal type I interferon treatment is beneficial only when administered before clinical signs onset in the SARS‐CoV‐2 hamster model. PLoS Pathog. 2021;17(8):e1009427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Mao T, Israelow B. A stem‐loop RNA RIG‐I agonist protects against acute and chronic SARS‐CoV‐2 infection in mice. J Exp Med. 2022;219(1):e20211818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Li M, Ferretti M. Pharmacological activation of STING blocks SARS‐CoV‐2. Sci Immunol. 2021;6(59):eabi9007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Wang N, Zhan Y, Zhu L, et al. Retrospective multicenter cohort study shows early interferon therapy is associated with favorable clinical responses in COVID‐19 patients. Cell Host Microbe. 2020;28(3):455‐464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Jagannathan P, Andrews JR, Bonilla H, Hedlin H. Peginterferon Lambda‐1a for treatment of outpatients with uncomplicated COVID‐19: a randomized placebo‐controlled trial. Nat Commun. 2021;12(1):1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Alavi Darazam I, Shokouhi S, Pourhoseingholi MA, et al. Role of interferon therapy in severe COVID‐19: the COVIFERON randomized controlled trial. Sci Rep. 2021;11(1):8059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Bastard P, Gervais A, Le Voyer T, et al. Autoantibodies neutralizing type I IFNs are present in ~4% of uninfected individuals over 70 years old and account for ~20% of COVID‐19 deaths. Sci Immunol. 2021;6(62):eabl4340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Vinh DC, Abel L, Bastard P, et al. Harnessing Type I IFN immunity against SARS‐CoV‐2 with early administration of IFN‐β. J Clin Immunol. 2021;41(7):1425‐1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. McNab F, Mayer‐Barber K, Sher A, Wack A, O'Garra A. Type I interferons in infectious disease. Nat Rev Immunol. 2015;15(2):87‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Munnur D, Teo Q, Eggermont D. Altered ISGylation drives aberrant macrophage‐dependent immune responses during SARS‐CoV‐2 infection. Nat Immunol. 2021;22(11):1416‐1427. [DOI] [PubMed] [Google Scholar]
- 257. Davidson S, Crotta S, McCabe TM, Wack A. Pathogenic potential of interferon αβ in acute influenza infection. Nat Commun. 2014;5:3864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Major J, Crotta S. Type I and III interferons disrupt lung epithelial repair during recovery from viral infection. Science. 2020;369(6504):712‐717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Horby P, Lim WS, Emberson JR, Mafham M, Bell JL, Linsell L. Dexamethasone in hospitalized patients with Covid‐19. N Engl J Med. 2021;384(8):693‐704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Avdeev SN, Trushenko NV, Tsareva NA, et al. Anti‐IL‐17 monoclonal antibodies in hospitalized patients with severe COVID‐19: a pilot study. Cytokine. 2021;146:155627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Robinson PC, Richards D, Tanner HL, Feldmann M. Accumulating evidence suggests anti‐TNF therapy needs to be given trial priority in COVID‐19 treatment. Lancet Rheumatol. 2020;2(11):e653‐e655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Urwyler P, Charitos P, Moser S, et al. Recombinant human C1 esterase inhibitor (conestat alfa) in the prevention of severe SARS‐CoV‐2 infection in hospitalized patients with COVID‐19: a structured summary of a study protocol for a randomized, parallel‐group, open‐label, multi‐center pilot trial (PROTECT‐COVID‐19). Trials. 2021;22(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.