

Abstract
The field of immunometabolism investigates and describes the effects of metabolic rewiring in immune cells throughout activation and the fates of these cells. Recently, it has been appreciated that immunometabolism plays an essential role in the progression of viral infections, cancer, and autoimmune diseases. Regarding COVID‐19, the aberrant immune response underlying the progression of diseases establishes two major respiratory pathologies, including acute respiratory distress syndrome (ARDS) or pneumonia‐induced acute lung injury (ALI). Both innate and adaptive immunity (T cell‐based) were impaired in the course of severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) infection. Current findings have deciphered that macrophages (innate immune cells) are involved in the inflammatory response seen in COVID‐19. It has been demonstrated that immune system cells can change metabolic reprogramming in some conditions, including autoimmune diseases, cancer, and infectious disease, including COVID‐19. The growing findings on metabolic reprogramming in COVID‐19 allow an exploration of metabolites with immunomodulatory properties as future therapies to combat this hyperinflammatory response. The elucidation of the exact role and mechanism underlying this metabolic reprograming in immune cells could help apply more precise approaches to initial diagnosis, prognosis, and in‐hospital therapy. This report discusses the latest findings from COVID‐19 on host metabolic reprogramming and immunometabolic responses.
Keywords: COVID‐19, immune reaction, immunometabolism, inflammation, SARS‐CoV2
1. INTRODUCTION
Recently, the field of immunometabolism has emerged as an essential process in the fates of immune response (adaptive and innate) throughout infection, cancer, and autoimmune diseases. 1 , 2 , 3 , 4 Several recent comprehensive investigations have emphasized the essential importance of cellular metabolism in immunity. 3 , 5 , 6 Six important and interrelated metabolic processes mediate the immune response: glycolysis, fatty acid oxidation (FAO), the tricarboxylic acid cycle (TCA), as well recognized as the Krebs cycle, the pentose phosphate pathway (PPP), and the fatty acid synthesis, and amino acid synthesis pathway. 7 , 8 Depending on cell type, development state, stimulation circumstances, and milieu, cells of the immune system predominantly adopt one or more metabolic processes. 9 , 10 , 11 , 12 , 13 For example, activated T cells, M1 macrophages (pro‐inflammatory phenotype), and neutrophils mainly depend on glycolysis. 9 , 10 , 11 , 12 , 13
In addition, M2 macrophages (anti‐inflammatory phenotype) and resting T cells are reliant on oxidative phosphorylation (OXPHOS). 11 Since shifting metabolic processes, also known as metabolic reprogramming, modulate the activity of immune cells activity 14 ; hence, to escape antiviral host defense, many viruses may target cell metabolism in immune cells. It has been found several viral infections, such as the human immunodeficiency virus (HIV), respiratory syncytial virus (RSV), influenza A virus (IVA) (H1N1), Hantaan virus, dengue virus (DENV), cytomegalovirus, and others, can reprogram metabolic processes in immune cells to their benefit. 15 , 16 , 17 , 18
The novel respiratory pathogen that emerged in Wuhan, China, in December 2019 is called SARS‐CoV‐2. 19 , 20 , 21 , 22 , 23 The SARS‐CoV‐2 pandemic had a destructive impact on the national healthcare system, mainly affected the world economy, and imposed much death on humanity to date. 24 , 25 , 26 , 27 , 28 It has been recently found that after infection with SARS‐CoV‐2, this virus may alter the metabolism of immune cells. 29 , 30 , 31 The immune system is influential in predicting coronavirus disease 2019 (COVID‐19) severity and establishing acute lung injury (ALI), or acute respiratory distress syndrome (ARDS). 19 , 26 , 32 The recent evidence revealed that both arms of the immune system, including the innate and adaptive immune response (T cell‐based), were dysregulated upon SARS‐CoV‐2 infection. 19 , 28 , 32 COVID‐19 patients, on the other hand, generate protective antibodies (Abs) against SARS‐CoV‐2. 30 The development of an immune response to a pathogen necessitates the generation of energy production through the immunometabolism procedure. 30 Immunometabolic reprogramming represents immune cells' metabolic state transition from homeostasis to a pro‐inflammatory or infectious milieu.
Viruses may have developed different approaches to modify cellular metabolism for propagation and survival according to their absolute dependence on host cells for replication. 7 , 33 , 34 , 35 Whenever a virus infects a host, it is known that particular host metabolic processes, such as glucose, fatty acid, and nucleotide metabolism, are disrupted. 35 Every viral species is predicted to affect the host cell to undergo a distinct metabolic reprogramming. 35 The transcriptome information collected by the host in reaction to COVID19 contributes to deciphering the alterations in gene expression that occur. 36 In reaction to COVID19, it has already been demonstrated that higher inflammatory cytokine secretion and decreased innate antiviral resistance are present in diverse hosts. 37 , 38 , 39 Particular cell lines have also exhibited autophagic and mitochondrial malfunction, respectively. 38 For COVID19, a thorough perception of metabolic reprogramming in the host is still unknown. Hence, we will thus describe recent findings on the function of host metabolic reprogramming and immunometabolic responses throughout COVID‐19 in the present review.
2. OVERVIEW OF HOST METABOLIC REPROGRAMMING DURING VIRAL INFECTIONS
Many of the host's critical carbon metabolic processes have also been modified by DNA and RNA viruses, including glycolysis, pentose phosphate activities, and the generation of nucleotides, amino acids, and lipids. 40 , 41 While some viruses enhance the requirement for vital nutrient compounds and glutamine and combine relevant metabolic pathways for anabolism, the precise metabolic alterations induced by specific viruses are frequently situational. 40 , 41 They thus can vary slightly even within the same virus family or depend solely on the type of cell infected by the host. 40
Viruses are one of the biological entities that significantly restructure the host cell's metabolic process to survive their life cycle, naming them “metabolic engineers.” 42 Similarities and variations exist in various viruses regarding host cell invasion, proliferation, and spread. 43 Viruses may also attack the host's immune system, potentially directly or indirectly. The latter is particularly visible in persistent viral diseases induced by the herpes viridiae family, which escape the human immune response by triggering the kynurenine pathway. 44 A natural reaction to infections, including the degradation of extracellular tryptophan to restrict virus growth, causes T cell depletion over time and the establishment of regulatory T cells (immunosuppressive). 45 Disruptions in the host metabolic process regulation, or rather the virus's reprogramming, are currently offering insight into the pathogenesis of a viral infection. 45 Beyond infections and severe disease recovery, there is an increasing agreement that the prolonged metabolic expenditures sustained by the host in recovered individuals or postrecovery are significant. 46
Some viral proteins employ host metabolic activities to induce dysregulation in various cellular pathways, increasing cancer risk. 43 For example, the human papillomavirus 16 E6 protein inhibited hypoxia‐inducing factor‐alpha (HIF‐α) degradation and thereby enhanced glycolysis in cells infected with this virus. 47 In Vero cells, it has been found that Enterovirus EV71 can increase glutamine metabolism, whereas pharmaceutical suppression of pyrimidine metabolism enzymes reduced viral proliferation. 48 As indicated by Kespohl et al., 49 in vitro and in vivo infection with Coxsackie B3, host protection strategies cause resistance to the viral invasion, Protein modification with ISG15 (ISGylation) of a 15 kD interferon (IFN)‐stimulated gene resulted in hepatic gluconeogenesis and induction of antiviral state via releasing IFN‐induced proteins. Thaker et al. 40 thoroughly examined the regulation of host cellular metabolism by various viral proteins and emphasized the critical necessity to uncover specific proteins to better comprehend virus‐induced metabolic reprogramming.
Many viruses are well‐acknowledged for their capacity to modify host metabolic processes for their longevity and replication. This is supported by the results of Rayfield et al., 50 who in the early 1970s experimented on a group of healthy men by infecting them with a virus (self‐limiting) that induces mild illness and observes the results. According to observations of viral metabolic regulation, higher glucose absorption resulted in the increased viral proliferation and enhanced redirection of glucose via the hexosamine pathway, respectively. 51 This pathway is well recognized for changing proteins that increase inflammation posttranslationally. For example, the IAV employs the hexosamine pathway to stimulate the expression of pro‐inflammatory genes through the stimulation of IFN regulatory factor 5 (IRF5), which causes the severity of the disease. 52
Moreover, to alter glucose metabolism, viruses also control the production and degradation of lipids, which is essential for their proliferation and survival. 53 Hepatitis C virus (HCV) can cause liver injury; recently, it has been found that dodecenoyl coenzyme A‐delta isomerase, which is a mitochondrial enzyme involved in FAO, is a significant target of this virus, so blocking this enzyme pharmacologically inhibited viral replication in hepatoma cells. 54 The DENV showed that virus‐induced lipid anabolic modulation was crucial for multiplication and virion production in infected cells. 55 The results of host metabolism disruption, particularly lipids perturbation, are recognized in conjunction with immunological responses, such as phagocytes and T cells responding to viral infections, which will be explored in the next section.
Changes in host lipid signaling and metabolism include three stages: viral entrance, complicated replication assembly, and the emergence of additional virions to maintain the infectious disease. 43 , 56 Sterol regulatory element‐binding protein (SREBP)1a, 1c, and 2 are members of the SREBP family, which transcriptionally regulates lipid biosynthesis in response to increased metabolic requirements. 57 SREBPs' downstream targets include genes involved in fatty acid biosynthesis (acetyl CoA carboxylase) and cholesterol production (3‐hydroxy‐3‐methylglutaryl coenzyme A (HMG CoA) reductase) through the mevalonate pathway. 57 Because of the several activities of cholesterol (including immunological control, cholesterol production, and other lipids), it may be a target for viral exploitation. For example, the cholesterol metabolite 25 hydroxycholesterols has been shown to have antiviral activity in macrophages through the signal transducer, and activator of transcription 1 (STAT1) facilitated synthesis of type 1 IFN. 58 , 59 , 60 Substantial evidence suggests viruses influencing host lipid metabolism, including genes' activation, are implicated in lipid synthesis driven by oxidative stress, as shown with HCV‐induced hepatic steatosis. 61
Many efforts have been undertaken to identify important objectives in host lipid metabolism that numerous viruses exploit. 62 Statins, for example, are a prevalent type of medication recommended to decrease blood cholesterol in high‐risk individuals. 63 Cholesterol is widely recognized to have an essential function in the membrane trafficking of the viral protein (including hemagglutinin) and the proliferation of IAV. 64 , 65 In recent years, there has been increased interest in the metabolic profiling of lipids influenced by viruses. A notable example is the nontargeted lipidomic study of plasma and lung tissue from mice infected with RSV, which showed differential management of lipids, including surfactants of the lung tissue, plasmalogens, and acylcarnitine that aggravate inflammation of the airways. 66 In vitro screening of regulators implicated in viral infection and lipid metabolism revealed that cells infected with IAV and Middle Eastern respiratory syndrome (MERS) experience significant alterations in lipid metabolism. 61 Further investigation showed that the retinoic acid receptor‐alpha agonist, 4[(5,6,7,8 tetrahydro 5,5,8,8 tetramethyl 2 naphthalenyl) carboxamido]benzoic acid (AM580), exhibited a high antiviral activity that reduced mouse death rates by blocking SREBP activation, and modifications in lipid metabolism significantly lowered DENV viral load, indicating a potential treatment strategy in viral illnesses by modulating host lipid machinery. 61 , 67
Iron is thought to be involved in the cellular metabolism of all living organisms, such as viruses and their mammalian hosts. 43 A differing class of mediators tightly regulates the host's concentration of iron is circulating, including both unbound and heme‐bound forms, in a way that mitigates oxidative injury (iron‐mediated) and postpones the migration and development of pathogenic organisms both extracellularly (as in blood that harbor's specific parasites and bacteria) and inside cells (as fundamentally observed with viruses). 43 Although most intrinsic iron is recycled through the formation and destruction of erythrocytes, cells also have complicated systems for iron storage. 43 Inflammation is one of the metabolic processes known to modulate cellular iron content in response to tissue damage and infections. Hepatocytes in the liver alter the synthesis of the peptide hormone Hepcidin in response to pathogen infection. 43 In contrast to those infected with hepatitis B or HCV, HIV‐1 demonstrated distinct hepcidin and iron concentration profiles in the plasma of human volunteers in both the phases of illness (acute and chronic) who were taking antiretroviral therapy or other treatments. 68 This emphasizes the importance of understanding viral processes at various phases of infection, resulting in diverse consequences.
Autophagy is the controlled process of “self‐eating in the cell, and its relevance in regulating cellular equilibrium, including its disruption in numerous illnesses, is well documented”. 43 The autophagic pathway is crucial in developing antiviral defenses, although viruses can escape destruction in autophagosomes and continue to infect cells. 69 Recognition is one of the first stages after viral entrance into a vulnerable host cell, proceeded by virus pickup and processing. 43 Pathogen recognition receptors (PRRs), including the toll‐like receptors (TLRs), notably TLR3 and TLR7, and the retinoic acid‐inducible gene 1‐like receptors (RLRs) for processing via autophagy, promote viral uptake. 70 , 71 , 72 Although autophagy is essential in cellular adaptability to chronic metabolic stress, including food deprivation, multiple studies show that autophagy can minimize viral injury and help in host healing. The attachment of the herpes simplex virus type 1 (HSV1) coding the ICP34.5 protein (a neurovirulence protein) to the Beclin1 protein suppressed autophagy, which in turn reduced signaling via the protein kinase R (PKR) axis dedicated to developing antiviral defenses. 73 The modulation of the PKR pathway is a remarkable example of autophagy‐mediated changes in host cellular metabolism in response to pathogens, including viral infection. In cell lines subjected to the HSV1 virus, translational inhibition of the host protein machinery by phosphorylation of the eukaryotic translation initiation factor 2 identifies the involved factors. 74
3. COVID‐19 PATHOGENESIS
Previous research suggests that SARS may be divided into three stages: viral replication, immunological hyperactivity, and respiratory damage. 75 , 76 The viremia episode, acute phase, and recovery period of COVID‐19 have also been suggested as clinical stages. 56 , 77 It is usually assumed that the disease progresses through the subsequent steps: entry and proliferation of viruses, dysregulation of immune responses, multiple organ failure, and healing. 23 , 28 , 78 , 79 The virus initially enters host cells, where it multiplies, assembles, and is transported extracellularly to target cells, triggering direct harm and death of parenchymal cells such as alveolar epithelial cells. 76 Simultaneously, a significant number of the pathogen‐associated molecular pattern (PAMP) and damage‐associated molecular pattern (DAMP) compounds are set to release to boost the innate immunity, elicit inflammatory processes, and secrete considerable amounts of inflammatory mediators, chemokines, free radicals, and proteases, resulting in ARDS, sepsis, and multiple organ dysfunction syndromes (MODS) pathological outcomes of COVID‐19‐induced pneumonia represent those shown in SARS‐CoV and MERS‐CoV infections. 31 , 32 , 80 , 81 Following the first critical phase, the inflammatory reaction progressively resolves, the injured organ steadily recovers, and some of the injured organs progress to the fibrosis and chronic stages, including chronic critical disease, immunosuppression, catabolic syndrome, and chronic inflammation. 76 Figure 1 displays the immunopathogenesis of COVID‐19.
Figure 1.
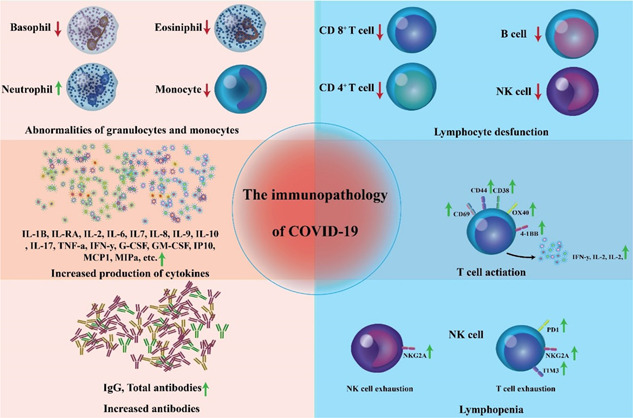
Immunopathogenesis of coronavirus disease (COVID‐19). COVID‐19 immunological characteristics encompass lymphopenia, lymphocyte stimulation and dysregulation, granulocyte and monocyte aberrations, enhanced cytokine secretion, and heightened antibodies. Lymphopenia is a common symptom in COVID‐19 individuals, particularly in severe forms. CD69, CD38, and CD44 are significantly upregulated on CD4+ and CD8+ T cells of these patients. Virus‐specific T cells from severe forms have a central memory profile with elevated amounts of interferon‐gamma (IFN‐γ), tumour necrosis factor α (TNF‐α), and interleukin (IL)‐2. On the other hand, lymphocytes show signs of fatigue due to the high production of PD‐1, TIM‐3, and natural killer group 2A. In serious conditions, neutrophil proportions are substantially higher, while eosinophil, basophil, and monocyte proportions were reduced. Another major feature of severe COVID‐19 is heightened cytokine secretion, particularly of IL‐1, IL‐6, and IL‐10. 82
SARS‐CoV‐2 adheres to the outside of the epithelial barrier of the oral cavity, the mucosal surfaces of the conjunctiva, or the optic canal after viral transmission. 41 , 83 The angiotensin‐converting enzyme 2 (ACE2) is found in a variety of human cells, such as type II alveolar cells (AT2), oral, gastrointestinal (GI), and ileal epithelium, cardiac cells, kidney proximal tubule cells, and bladder urothelial cells, is thought to have a role in the internalization of SARS‐CoV‐2. 84 , 85 The spike (S) protein of SARS‐CoV2 is split at the S1/S2 location by a cellular enzyme called furin, and this degradation is required for viral entry into lung cells. 86 Transmembrane serine protease 2 (TMPRSS2) primes the stimulated S protein before attaching it to ACE2 ligands and entering the host cells. The genetic sequence of SARS‐CoV‐2 is analogous to that of SARS‐CoV, as is the architecture of the S protein of both viruses highly similar.
Data demonstrated that ACE 2/angiotensin (1–7) plays a vital role in inflammation and signaling pathways that contribute to tissue damage. 87 ACE 2's physiological function is to degrade angiotensin II and produce angiotensin (1–7), which counteracts ACE II. 88 Upon viral replication in the host cell, ACE 2 decreased expression reduces angiotensin II degradation into angiotensin (1–7). A disruption in the ACE 2/angiotensin (1–7) axis reflects certain clinical aspects of COVID‐19, including hypokalemia, vasoconstriction, and ARDS establishment. 89 , 90 Notably, the degree of ACE 2 production in the GI, genitourinary (testis) systems, genitourinary, endocrine (pancreas), and cardiovascular is far greater than that in the virus's primary target, the respiratory system. 84 As a result, there is no relationship between viral infection rates and ACE 2 expression. Preliminary data shows that ACE 2 expression is lower in females than in males, which might explain why men have a higher incidence of COVID‐19 cases. 91 , 92 Furthermore, researchers at the Mount Sinai Health System in New York discovered that the production of ACE 2 is age‐dependent. 93
The inflammatory mediators, including interleukin (IL‐)−2, IL‐6, IL‐7, IL‐10, tumor necrosis factor α (TNF‐α), inducible protein (IP‐10), macrophage inflammatory protein‐1 alpha (MIP‐1A), monocyte chemoattractant protein‐1 (MCP‐1), and granulocyte colony‐stimulating factor (G‐CSF) are associated with disease severity in COVID‐19. 83 A substantial lessening in lymphocyte count is seen in COVID‐19. 94 , 95 , 96 A flow cytometric examination of severe COVID‐19 cases reveals a significant decrease in lymphocytic T cells (CD4+ and CD8+) and natural killer (NK) cells. 94 , 95 , 96 Furthermore, in the initial stages of the illness, an elevation in the development of NK group 2A (NKG2A), PD‐1, and T‐cell immunoglobulin (Ig) mucin‐3 (Tim‐3) is related to functional exhaustion of T cells. 94 , 95 , 96 NKG2A is a suppressive NKG2 family member that is present on NK cells, NK T (NKT) cells, and a fraction of CD8+ T cells. The engagement of NKG2A with histocompatibility antigen alpha chain E (HLA‐E) can restrict NK and T cell activation. 97 T lymphocytes and NK cells both express PD‐1. It contributes to suppressing immunological responses and promoting self‐tolerance by reducing T cell activity and boosting the development of regulatory T cells. 98 Tim‐3 is a coinhibitory receptor found on IFN‐producing T cells and innate immune cells, including dendritic cells (DCs) and macrophages. It is crucial in suppressing T helper 1 cell (Th1) reactions and cytokine production, including TNF‐α and IFN‐γ. 99
4. METABOLIC REPROGRAMMING DURING COVID‐19
All viruses lack their metabolic functions and rely on the cells they infect to create the ingredients required to establish new virions. 100 Most viruses significantly alter the metabolism of infected cells, and some eventually shut down all cell metabolism, causing the cell to die. 100 This is the typical fate of SARS‐CoV‐2‐infected cells, and cells like respiratory epithelium must be replaced, or their function, which includes gas exchange and the synthesis of surfactants required for pulmonary function, will end. 101 The data for documenting the metabolic implications of COVID‐19 infection in vivo is being assembled. Some researchers have examined the metabolic profiles of COVID‐19 patients and uninfected people; however, the relevance of these investigations is restricted since they frequently do not specify what phase of disease or severity is being evaluated. 102 Longitudinal studies that repeatedly capture metabolic processes in infected individuals after they become COVID‐19 positive are still needed to determine whether metabolic alterations correspond with the severity and outcome of the illness process.
According to published research on the in vivo impact on metabolism, COVID‐19 infected individuals may have higher blood glucose and fatty acid concentrations and abnormalities in amino acid metabolism. Regarding the latter, genes that encode tryptophan metabolic enzymes, including kynurenine and indoleamine 2, 3‐dioxygenase (IDO), were elevated. 103 , 104 In infected individuals, further intermediates implicated in arginine, aspartate, tyrosine, and lysine metabolism may be altered. 35 , 103 , 104 These case investigations show that numerous components of normal metabolism can be disrupted during the illness process, posing a challenge for any treatment employed to reestablish metabolic balance. The last part explores the importance of targeting metabolic processes to regulate the fate of SARS‐CoV‐2 infection.
Ex vivo experiments on bronchial lavage cells and blood monocytes from COVID‐19 patients, contrasting any metabolic alterations to control subjects using a variety of measurements such as single‐cell RNA‐seq, metabolomics, and transcriptomics, have also been used to record the metabolic consequences of COVID‐19 disease. One such research, which used single‐cell RNA‐seq on bronchial alveolar cells, discovered substantial increases in glycolysis metabolites, including HIF‐1a and genes implicated in the generation of oxidative stress. 105 , 106 This implies that the infected cells require increased cellular energy, which is supplied by glycolysis. Other investigations have revealed alterations in lipid metabolism and modifications in tryptophan metabolism comparable to those previously mentioned in patient examinations. 103
Investigations have been performed utilizing permissive or semipermissive cells infected in vitro, then correlating the metabolic outcomes to those in uninfected cells to establish if the COVID‐19 infection directly impacts the target cell's metabolism. One investigation using peripheral blood monocytes discovered significant metabolic variations. These included enhanced HIF‐1a protein levels and enhanced transcriptional activity of molecules, including glucose transporter 1 (GLUT‐1), 6‐phosphofructo‐2‐kinase/fructose‐2, 6‐biphosphatase 3 (PFKFB3), pyruvate kinase M2 (PKM2), and lactate dehydrogenase A (LDH‐A). 106 However, with this investigation, all cells would not have been infected with the virus. Furthermore, longitudinal research that evaluates metabolic variations in different intervals after synchronized infection in a system where all cells were demonstrated to be infected may yield more potentially valuable data. As a result, it is currently unknown what direct impact COVID‐19 infection has on cellular metabolism and if this knowledge might help reshape events to obtain a more positive outcome once the infection has started.
Another strategy for understanding how COVID‐19 infection affects metabolism is to infect vulnerable cells in vitro and analyze the effect of changing metabolic activity on the consequence of the disease. Several investigations have been conducted to assess the effects of interference with glucose consumption with the medication 2‐deoxy‐d‐glucose (2DG). 106 , 107 This method results in a significant decrease in viral replication, and the infected cells survive. A comparable result was achieved with medications that target either cholesterol metabolism (fenofibrate) or mitochondrial OXPHOS (mitoquinole). 106 , 108 Typically, research was designed to add metabolism modulating drugs earlier or from the time of infection, but processes that can influence the infection results because once given at a later time when propagation occurrences are currently ongoing after infection may be of enhanced therapeutic intrigue. This experimental approach may better represent what might be helpful in patients undergoing active diseases to decrease the severity of their illness.
4.1. Metabolic reprogramming of immune cells against COVID‐19
The progression of SARS‐CoV‐2 illness to a severe form of infection is caused by a dysfunctional immunological reaction, defined as the failure to create a prompt and robust type‐I IFN host defense. 109 This causes a boost in viral load, which is preceded by an inflammatory reaction characterized by inflammatory mediators such as IL‐6, TNF, IL‐1, and IL‐18, and acute phase markers. 110 Because the development of COVID‐19 to severe illness is biphasic, with the initial viral stage being preceded by the inflammatories stage, patients present later in the disease and persistently increased inflammatory markers may benefit more from anti‐inflammatory medication. In the lung infected with SARS‐CoV‐2, the most prevalent inflammatory cells are monocytes and macrophages, which participate in the induction of cytokine storm as seen in severe COVID‐19 infections. 111 SARS‐CoV‐2 infects a wide range of cells, most notably respiratory epithelial cells and macrophages. Macrophages infected with SARS‐CoV‐2 produce metabolic rewiring comparable to that caused by lipopolysaccharides (LPS), with an elevation in aerobic glycolysis, a decrease in the TCA cycle, and a rise in reverse electron transport (RET). 106 This reprogramming in metabolic pathways is facilitated by the manufacturing of mitochondrial reactive oxygen species (ROS) (mtROS), which stabilizes HIF‐1 and glycolytic genes (mediated with HIF‐1) implicated in glucose transfer and glycolysis, such as PFKFB3, PKM2, GLUT‐1, and LDH‐A. 109 LDH, a key enzyme in glycolysis in inflammatory macrophages (M1 phenotype), has also been demonstrated to be a predictive biomarker of disease severity in SARS‐CoV‐2 diseases, highlighting the importance of glycolysis in viral infections such as COVID‐19. 112 Ex vivo investigations with monocytes infected with SARS‐CoV‐2 from cases with COVID‐19 revealed that HIF‐1α protein levels were considerably higher than in uninfected individuals. Also, research on infected monocytes with SARS‐CoV‐2 demonstrates that elevated glucose levels and glycolysis accelerate SARS‐CoV‐2 proliferation, but inhibiting glycolysis reduces viral propagation. Table 1 and Figure 2 show the metabolic reprogramming of immune cells throughout COVID‐19.
Table 1.
Metabolic reprogramming among immune cells during COVID‐19.
| Immune cells | Metabolic reaction | Outcome | Description | References | ||
|---|---|---|---|---|---|---|
| Macrophage | Glycolysis | Pro‐inflammatory cytokine, chemokine generation, virus | The current study found that high glucose levels boost severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) virus replication and release of inflammatory mediators in macrophages taken from the lungs of severe coronavirus disease 2019 (COVID‐19) patients, culminating in a cytokine storm. | [30, 106, 113, 114] | ||
| Replication, activation of hypoxia‐inducible factor 1 alpha (HIF‐1 α) | The extreme inflammatory process and cytokine storm in the respiratory system of COVID‐19‐induced acute lung injury (ALI)/acute respiratory distress syndrome (ARDS) patients creates a sepsis‐like microenvironment, leading to more significant HIF‐1a transcription in infiltrated macrophages, which generate pro‐inflammatory cytokines mediators and chemokines, causing catastrophic COVID‐19‐induced ALI/ARDS. | |||||
| Increased glycolysis in M1 macrophages (pro‐inflammatory phenotype) results in the buildup of Krebs or tricarboxylic acid (TCA) cycle byproducts such as succinate and citrate and TCA cycle‐derived itaconate, which affects inflammatory gene expression. | ||||||
| Periphery CD14+ monocytes in COVID‐19 patients do not create pro‐inflammatory mediators to cause a systemic cytokine storm; therefore, immunometabolic remodeling from oxidative phosphorylation (OXPHOS) to glycolysis and HIF‐1α overexpression does not occur. | ||||||
| Dendritic cell (DC) | Glycolysis, OXPHOS | Reduced mammalian targets of rapamycin (mTOR) signaling, reduced interferon (IFN)‐α production | According to a recent study, COVID‐19 inhibited mTOR signaling and IFN‐a production via toll‐like receptor (TLR) 3 and TLR7/8 signaling, indicating poor immuno‐metabolic reconfiguration in plasmacytoid DCs (pDCs). | [30, 115, 116] | ||
| Because type 1 IFN synthesis is restricted, pDCs infected with SARS‐CoV‐2 do not exhibit the autocrine IFN‐IFNAR signaling generated by type 1 IFN synthesis and adherence to the IFN‐a receptor (IFNAR), which suppresses an increase in OXPHOS and FAO. | ||||||
| Through interferon regulatory transcription factor 3 (IRF3) phosphorylation, SARS‐CoV‐2 infection reduces TANK‐binding kinase‐1 (TBK1) and IKKe–mediated type 1 IFN generation as well as nuclear factor‐κ B (NF‐κB)‐mediated pro‐inflammatory cytokine production in DCs. Due to a deficiency of HK‐II, the metabolism changes to OXPHOS, rendering DCs infected with SARS‐CoV‐2 unable to meet the increased energy requirements required to build protection against viral illness. | ||||||
| As a result, SARS‐CoV‐2 illness may lower the number of DCs in the circulation, lymph nodes, and several organs, including the lungs, by disrupting the immunometabolic remodeling process (OXPHOS to glycolysis). | ||||||
| Natural killer (NK) cell | Glycolysis, OXPHOS | mTORC1 signaling | NK cells depend on both OXPHOS and (at a reduced level) glycolysis for energy during immunological maintenance. | [30] | ||
| A high dose of interleukin‐15 (IL‐15) stimulates mTORC1 signaling, which accelerates glucose absorption through glucose transporter 1 (GLUT‐1) or solute carrier family 2 member 1 (SLC2A1) and, as a result, glycolysis. | ||||||
| T lymphocyte | Glycolysis, glutaminolysis | mTORC1 signaling, induction of HIF‐1a | As seen in the lungs of severe COVID‐19 patients, increased mTORC1 activation in T helper 1 (Th1) cells causes their switch to T helper 17 (Th17) cells. | [30] | ||
| In response to hypoxia, Th17 cells increase glycolysis and glutaminolysis in the lungs of severe COVID‐19 individuals, leading to the activation of HIF‐1 and mTORC1 signaling to promote pro‐inflammatory activity. | ||||||
| B lymphocyte | Glycolysis | mTORC1 signaling generates antibodies (Abs) | As a result, in COVID‐19 instances, B cells create Abs by a glycolysis‐dependent mechanism. In this process, plasma cells develop from B cells to synthesize Immunoglobulin (Ig) IgG, IgM, and IgA, and glycolysis in B cells is not suppressed. | [30, 117] | ||
Figure 2.
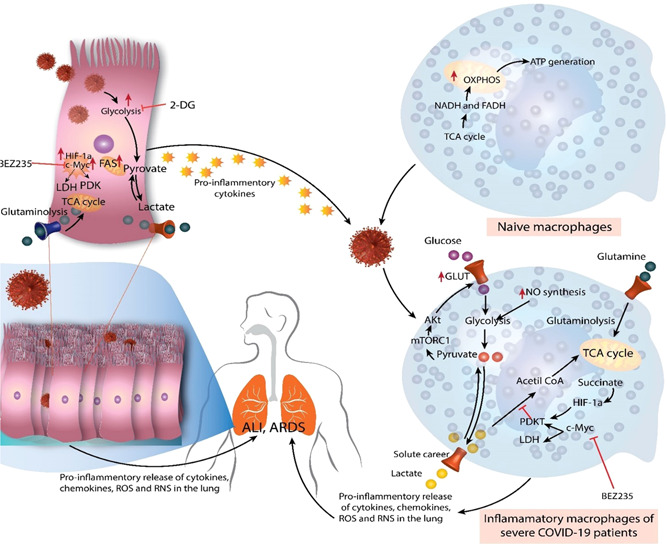
Immunometabolic reactions during coronavirus disease (COVID‐19). During homeostasis, naive macrophages (M0) do not demand substantial energy and rely primarily on oxidative phosphorylation (OXPHOS) and tricarboxylic acid (TCA) or the Krebs cycle for ATP production. Nevertheless, under the effect of the virus and pro‐inflammatory mediators generated by respiratory or pulmonary epithelial cells throughout severe acute respiratory syndrome coronavirus 2 disease, they exhibit a metabolic change. Therefore, these macrophages switch to glycolysis for fuel, which delivers faster energy than OXPHOS. The enhanced glucose absorption by these inflammatory macrophages is caused by excessive glucose transporter 1 (GLUT‐1) production, which is activated by mTORC1 signaling and causes Akt to boost GLUT1 expression. Hypoxia‐inducible factor 1‐alpha (HIF‐1a) and C‐Myc levels also increase, enhancing glycolysis by promoting lactate dehydrogenase (which converts pyruvate to lactate) and PDK1. The deposition of succinate, a TCA cycle byproduct, raises HIF‐1a concentrations. The elevated glutaminolysis contributes to the higher energy consumption of inflammatory macrophages. As a result, augmented cytokine, chemokine, reactive oxygen species (ROS), and reactive nitrogen species (RNS) production by inflammatory macrophages in the lungs leads to the "cytokine storm" that causes acute lung injury (ALI)/acute respiratory distress syndrome (ARDS). Similar to pro‐inflammatory macrophages, respiratory or alveolar epithelial cells infected with SARS‐CoV2 exhibit enhanced glycolysis, glutaminolysis, HIF‐1a, and c‐Myc overexpression. This increases the secretion of pro‐inflammatory mediators, which contributes to the "cytokine storm" and neutrophil and monocyte recruitment in the lungs of severe COVID‐19 individuals. These cells of innate immunity and immunometabolic remodeling processes cause ALI/ARDS in patients with severe COVID‐19. 30
Moreover, enhanced glycolysis elevates IL‐1β synthesis, promoting COVID‐19's inflammatory reaction. HIF‐1α is involved directly in viral proliferation and pro‐inflammatory mediators secretion in these in‐vitro investigations, as SARS‐CoV‐2 proliferation has been demonstrated to be inhibited by HIF‐1α inhibition, and also HIF‐1α stimulation enhanced viral proliferation. Also, HIF‐1α suppression inhibited the release of inflammatory cytokines linked to serious COVID19, such as IFN‐α, IFN‐β, IL1 β, TNF, IL‐6, and ACE2. This provides concrete proof for addressing mtROS‐HIFα‐metabolic pathways reprogramming as a possible therapy for the severe form of COVID‐19 disease by suppressing viral proliferation and inflammatory mediators. 106
Immunometabolism, or metabolic rewiring within and between immune cells, is important in the pathogenesis of inflammatory response and other inflammatory conditions, such as sepsis, in which cytokine storm development performs a critical function in the initiation of ALI/ARDS or multiorgan collapse. 30 , 39 As a result, it is crucial to monitor immunometabolic reprogramming amongst immune effector cells throughout SARS‐CoV2 pathophysiology.
According to previous research, high glucose levels promote SARS‐CoV2 propagation and the production of inflammatory mediators in macrophages derived from the lungs of serious COVID‐19 victims, resulting in cytokine storms. 106 This is caused by the increased expression of many genes implicated in glycolysis in these macrophages after COVID‐19. The infiltration of macrophages derived from monocyte into the lung of COVID‐19 patients following enhanced expression of HIF‐1α demonstrates glycolytic pathway initiation, which supplies common energy as contrasted to OXPHOS, throughout homeostasis where it represents a source of energy for macrophages to transcriptions and translates pro‐inflammatory genes including chemokines and cytokines. 106 In pulmonary macrophages, the metabolic switch from OXPHOS to glycolysis enhances the mammalian target of rapamycin complex 1 (mTORC1), elevating GLUT‐1 in macrophages via Akt to promote glycolysis. 118 Therefore, HIF‐1α supports SARS‐CoV‐2 proliferation in infected macrophages by promoting glycolysis. During COVID‐19, HIF‐1α suppression also reduces cytokine production from SARS‐CoV2‐infected macrophages. 106 Enhanced glycolysis in M1 macrophages (pro‐inflammatory) causes the accumulation of Krebs or succinate and citrate (TCA cycle metabolites) and TCA cycle‐derived itaconate, which regulates the expression of genes (pro‐inflammatory gene). 56 , 113 , 114 The succinate delivered to the cytosol immediately suppresses the action of prolyl hydroxylase (PHD) to reinforce HIF‐1α, so enhancing glycolysis. 106 , 113 , 119 Complex II or succinate dehydrogenase (SDH) stimulates RTE in the mitochondria, enhancing mtROS formation from complex 1, suppressing PHD. 106 , 113 , 119 As a result, succinate oxidation is essential for SARS‐CoV2 proliferation in macrophages infected with SARS‐CoV‐2.
DCs perform an axial function in the Antigen (Ag) presentation and viral infections. 30 The features of DCs as being one of the numerous innate immunity throughout the acute phase of infection, such as sepsis, have already been detailed elsewhere by the author. 119 The specifics of DC immunometabolism under various situations have been addressed previously. 120 , 121 A previous SARS‐CoV study discovered limited viral proliferation in monocyte‐derived DCs (moDCs) following infection, with no increase in virus loads. 122 Even these DCs generate low antiviral type 1 IFNs and cytokines (IL‐12p40) produced in minimal amounts, as are pro‐inflammatory mediators, including IL‐6 and TNF‐α. 122 These DCs, on the other hand, have considerably higher volumes of the chemokines essential to promote neutrophil, monocyte/macrophage, and T cell recruitment. The moDCs infected with SARS‐CoV2 indicate no productive virus proliferation, no antiviral IFN I, II, or III production, and limited inflammatory cytokines secretion. 123
Nevertheless, moDCs infected with SARS‐CoV‐2 do not generate the same chemokines as SARS‐CoV. The virus hijacked a robust antiviral protective immunity by declining to produce numerous antiviral type 1 IFNs and stimulating minimal amounts of pro‐inflammatory mediators. The innate immune cells infected with SARS‐CoV2, for instance, suppress cyclic guanosine monophosphate (GMP)‒adenosine monophosphate (AMP) synthase (cGAS)‒stimulator of IFN genes (STING) signaling‐dependent antiviral type 1 IFN production, RLR, and nuclear factor κ B (NF‐κB)‐dependent pro‐inflammatory mediators. Furthermore, moDCs infected with SARS‐CoV2 suppress IFN signaling in moDCs and phosphorylation in STAT1. 124 It is important to note that DCs infected with SARS‐CoV does not cause maturation or apoptosis. 123 Severe COVID‐19 cases, on the other hand, have a lower amount of conventional DCs (cDCs) and plasmacytoid DCs (pDCs).
Dormant or naive T cells during normal settings for energy sources rely on OXPHOS. 125 Pyruvate is generated by glycolysis, which is required for OXPHOS, FAO, and glutaminolysis due to stimulating TCR signaling and IL‐7‐IL‐7 R coupling to support growth, diversity, longevity, stimulation, and performance. 125 Naive T cells viability is often supported by the interplay of sphingosine 1‐phosphate (S1P) and S1P receptor (S1PR), which keeps OXPHOS and FAO under control. 126 Tonic TCR signaling decreases respiration and glycolysis in periphery CD4+ T cells engaged with peripheral self‐MHC, indicating an inverted link between tonic TCR signaling and basal metabolism. 127 As a result, higher CD4+ T cell engagements with periphery self‐MHC led to decreased basal metabolic rate and vice versa. This reduces the likelihood of autoimmunity and maintains the immune function in balance.
A glycolytic enzyme which is called phosphoglycerate mutase 1 (Pgam1) catalyzes the conversion of 3‐phosphoglycerate (3‐PG) to 2‐phosphoglycerate (2‐PG), which is also crucial in glycolysis. Pgam1 deletion in T cells affects glycolysis, impairing immunological responses from T cells (CD4+ and CD8+ cells). 128 As a result, T cells required the glycolysis pathway for performing immunological reactions, including growth and maturation. Recently, features about T cell metabolism were explored. 125 , 126 Low plasma S1P levels in COVID‐19 subjects alter S1P‐S1PR signaling and mitochondrial mass or frequency in naive T cells, resulting in induction of apoptosis. 129 The lack of DCs following SARS‐CoV2 infection also affects effector CD8+ T cell performance and memory CD8+ T cell production, which are both dependent on them. 30 Due to the lack of DCs following severe form of COVID‐19, decreased IL‐7 signaling and Ag presentation. The proportion of B cells stays unchanged or rises in severe COVID‐19 patients. 30 Plasmablasts cells produce IgG, on the other hand, predominate in severe COVID‐19 cases with the highest levels of NAbs. Exhausted memory B cells, also known as CD21‐CD27‐B cells, have been promoted in both mild and severe COVID‐19 subjects. TLR3 and TLR9 are exclusively expressed by human B cells, although TLR3, TLR7, TLR8, and TLR9 are expressed by human plasma cells. 130
Consequently, activating these TLRs in differentiating plasma cells promotes Ig synthesis. 130 In naïve or noninfected B cells, the STING protein is not detectable. 131 In response to viral infection, they express STING; however, their cGAS‐STING signaling pathway remained defective, blocking them from producing type 1 IFNs. 131 Another research indicates that in the presence of T cell‐dependent antigens, B cells have STING play a critical function in building Abs. 132
Nevertheless, in the existence of alum, cGAS‐STING signaling is inert (adjuvant). 132 Effective Ab response requires RLR‐dependent signaling. 133 As a result, exclusively cytosolic TLRs, RLRs, and cyclic GMP‐AMP synthase (cGAS)‐STING signal transduction pathways are needed for SARS‐CoV‐2 RNA and DNA identification and Ab production. Since they have increased the number of T cells, including such follicular helper T cells (TFHs), Ab manufacturing may be both T cell‐dependent and independent in mild to moderate COVID‐19 instances, but this should be a T cell‐independent process in severe COVID‐19 cases since all types of T cells decrease, and only CD4‐CTLs, Th17 cells, and CD8 T cells boost in the lungs, in which it is a T cell‐dependent procedure. 30 This one should be investigated. For instance, B lymphocytes do not produce ACE2, but only effector, not naïve; B cells exhibit CD147. 134 As a result, this virus neither infects nor manipulates B cells' cytosolic PRR‐based type 1 IFN and cytokine secretion, which is required for increased Ig and Ab synthesis. These data imply that SARS‐CoV‐2 may alter the metabolism of immune cells to advance its pathogenesis; acknowledging the virus's processes for this purpose may, as a result, contribute to the development of novel medicine in treating viral disorders.
Similarly, the biochemical profile of COVID‐19 patients has a significant impact on their survival. 46 To investigate this issue, current findings obtained proteomic, transcriptome, and metabolomic measures from COVID‐19 patients and discovered several connections between specific molecular alterations and disease prognosis. 135 , 136 Nevertheless, there is a paucity of an integrated view of metabolic alterations that may be important in controlling immune function in COVID‐19 as well as other severe acute infections. Lee et al. 137 investigated the metabolic alterations linked with the peripheral immune reaction in 198 people with COVID‐19 using an extensive examination of plasma metabolite and protein concentration and single‐cell multiomics assessment from sequential blood sampling obtained within the first week upon diagnosis. They discovered that rising illness severity coincides with the formation of uncommon but metabolically dominating T cell subgroups and the polarization of monocytes onto two biologically different subsets. We found metabolic rewiring that is extremely precise to distinct immune cell categories, and this switching is linked to alterations in the plasma metabolome and clinical aggravation. They also discovered systemic metabolite classifications of pathogenicity and clinical outcome predictions. This integrative strategy offers the opportunity for researchers to better comprehend the metabolic processes underpinning the immunological response to COVID‐19.
SARS‐CoV‐2 is essentially a pulmonary virus; it has also become apparent that it affects a wide range of organs throughout the body, not only the lungs and airways. 137 As a result, severe COVID‐19 episodes are commonly described as multisystem dysfunction. COVID‐19 induces immediate breathing difficulties in the lungs due to infiltration of inflammatory cells mainly by lymphocytes, extensive pulmonary destruction, and respiratory failure. 138 Furthermore, COVID‐19 individuals have neurological symptoms and renal and hepatic impairment. 139 , 140 , 141 Vascular impairment and thromboembolisms add to the disease's reported multisystem destruction and increased mortality. 142 Schmelter et al. 141 used an experiment to evaluate the metabolic and lipoprotein profiling of hospitalized COVID‐19 cases to many healthy controls and a group of cardiogenic shock cases receiving in the same intensive care unit (ICU) who tested negative for COVID‐19. When COVID‐19 cases were compared to healthy controls, their blood metabolomics signature revealed significant dyslipidemia. Very‐low‐density lipoprotein and intermediate‐density lipoprotein molecules and related apolipoprotein B and intermediate‐density lipoprotein cholesterol were dramatically increased, whereas cholesterol and apolipoprotein A2 were considerably lowered. 141 Furthermore, when 1‐week course was examined, a similar disrupted profile was observed as compared to other patients with cardiogenic shock in the ICU, showing intimate linkages among COVID‐19 and lipid metabolism. 141 This indicates that lipoprotein patterns may be a paradoxical risk factor for COVID‐19, stratifying patients.
Viral sepsis has also been offered as an appropriate terminology to cover all multiorgan impairment and clinical findings in critically ill patients with COVID‐19. 144 Acceptance of this terminology may contribute to the adoption of more precise initial detection, diagnosis, and in‐hospital therapeutic options. Oostdam et al. 144 discovered a comprehensive host‐dependent imbalance of inflammatory markers, neutrophil‐activating chemokines, mitochondrial metabolism, polyamine synthesis glycolysis, lipid metabolism, glycolysis, and amino acid metabolism. In moderate and severely ill cases, dysfunctional metabolites and cytokines/chemokines revealed varied association patterns, showing an interaction among metabolic profile and hyperinflammation. 144
5. METABOLIC DISORDERS AND COVID‐19
Metabolic disorders (including obesity and diabetes) are related to a heightened incidence of viral, parasitic, mycotic, and bacterial infections. 145 Drucker et al. demonstrated that severe respiratory disease is associated with the fast evolution of transitory insulin resistance in other more healthful euglycemic healthy body weight and overweight individuals, showing that pathogens significantly enhance diabetic lethality. 146 A retrospective investigation revealed that the death rate in elderly diabetes individuals was higher. 147 Diabetic condition is implicated considerably in microbial infections and poorer consequences resulting from a combination of impairment of innate immunity and inflammatory pathways. 148 , 149 Also, secondary bacterial infections may aggravate COVID‐19 diseases related to diabetics' reduced epithelial barrier integrity in the lungs and GI system. 146
Chronic inflammation and associated cytokine secretion throughout viral infection cause neutrophilia, coagulation activity, and kidney damage, ultimately contributing to the mortality of SARS‐CoV‐2 victims. 150 , 151 Various investigations have found that in diabetic individuals infected with SARS‐CoV‐2, the overall number of lymphocytes in blood samples is much reduced, whereas the total number of neutrophils is significantly greater. 152 Furthermore, diabetic SARS‐CoV‐2 individuals had elevated blood levels of numerous inflammatory‐related markers than nondiabetic SARS‐CoV‐2 cases. 153 These individuals are distinguished by high blood levels of C reactive protein (CRP) I, TNF‐α, serum ferritin, and IL‐6. In patients with COVID‐19, most investigations indicated that the IL‐6 is a biomarker of disease severity and outcome, and its production period is higher than that of other pro‐inflammatory cytokines, including IL‐1 and TNF‐α. 154 Also, Guo et al. observed evidence of increased ferritin in diabetes patients, demonstrating the stimulation of the monocyte‐macrophage axis, which seems to be a critical component of the cytokine storm. 150 , 154 The scientists determined that diabetic individuals are more likely to experience a cytokine storm, which causes a fast worsening in SARS‐CoV‐2 individuals. 154
Ultimately, hyperglycemia is a significant risk factor for adverse coagulative equilibrium and platelet activation, contributing to the increased thromboembolic abnormalities found in dead COVID‐19 subjects. 155 Multiple pathways have been investigated in diabetic patients that correlate with inflammatory response and coagulative equilibrium. 151 At first, the inflammatory process activates plasmin, which elevates d‐dimer levels. Second, inflammatory conditions and hypoxia stimulate thrombin. The activation of monocyte‐macrophages results in the overexpression of several tissue elements and modulation of the exogenous coagulation pathway, resulting in significant hypercoagulable conditions or even dispersed intravascular coagulation. 150 , 151 Furthermore, higher d‐dimer concentrations have been repeatedly documented, and their steady rise during the disease course is mainly related to the progression of the disease. 156 In sum, longitudinal monitoring of lymphocyte number changes and inflammatory markers such as CRP, ferritin, and IL‐6 during the illness processes may aid in the detection of persons with poor prognoses and early therapy to improve outcomes.
High obesity increases the risk of severe COVID‐19 and lethality through various processes, such as increased inflammatory presses, hypercoagulation, and mechanical impairment. 157 One potential risk factor for serious consequences in those with severe COVID‐19 and obesity is physiological stress on respiration caused by diaphragm excursion restriction. Diabetes and overweight are also implicated in developing chronic obstructive pulmonary disease (pulmonary fibrosis) and a decreased pulmonary rate. 46 The risk of stroke and cardiovascular issues was significantly increased in obesity, obesity, diabetes, and hypertension, and these risk factors are implicated in the induction of severe COVID‐19 presentation. 158 These COVID‐19 cases have an aggravated coagulation reaction to thrombogenic factor upregulation, including coagulation factors (II, VII, VIII, IX, XI, and XII), PAI‐1, and von Willebrand factor, which, particularly combined with pre‐existing disorders, might lead to higher rates of stroke or collapsed lung. 159 , 160
Type 2 diabetes (T2DM) is a chronic condition characterized by insulin resistance, inflammatory processes, and endothelial and ‐cell abnormalities. 161 In situations with severe COVID‐19, the inflammatory response to SARS‐CoV‐2 infection can increase insulin resistance and vascular dysfunction. The interaction between COVID‐19 and T2DM and obesity may intensify the inflammatory reaction and inhibit the activity IFN defenses, resulting in emerging diseases severity in diabetic and obese patients. 162 In addition to stress signaling, Renin–angiotensin–aldosterone system (RAAS) dysfunction and decreased expression of ACE2 can promote insulin resistance. 163 Hence, insulin resistance increases the potential of pulmonary failure and cardiac collapse in diabetic cases by promoting airway hyperreactivity COVID‐19. 163
In addition to their involvement in the immunological reactions to viral diseases, cytokines stimulate the hypothalamc‐pituitary‐adrnal axis, causing the production of adrenal glucocorticoids. 164 Glucocorticoids, in turn, have a negative feedback effect on immune cells, suppressing more cytokine production and secretion. As a result, the host gets a partly protecting signal against the harmful effects of an excessive immune reaction, including tissue injury, septic shock, and autoimmunity. 165 Such adverse effects have also been clinically recorded following dexamethasone therapy, which is the more successful treatment for those with severe COVID‐19 with respiratory complications at the time of writing. 166 Moreover, inhalation of glucocorticoids (e.g., budesonide) has been observed to shorten the time to partial recovery following initial COVID‐19 infection. 167 These data have resulted in a broad prescription of glucocorticoids to treat individuals with moderate‐to‐severe COVID‐19 and, commonly, mild forms.
Upon SARS‐CoV‐2 infection, TLRs 2, 3, and 4 are stimulated, resulting in the production of inflammatory mediators, including IL‐1 and IL‐6. 168 Metaflammation in individuals with metabolic diseases enables a hyperimmune activation that may progress to pathological levels. This inflammatory mediator's secretion promotes fever, inflammation, and sepsis. 168 According to their pathophysiology, infectious disorders, such as viral infections and impaired endocrine and metabolic organs, lead to new‐onset hyperglycemia or insulin resistance in COVID‐19 survivors. SARS‐CoV‐2 can infect and spread in cells of the exocrine and endocrine pancreas, according to studies involving islets taken from human donors infected with SARS‐CoV‐2 and data from postmortem samples from patients who died from COVID‐19. 169 Moreover, in COVID‐19 individuals, inflammatory cells recruitment and necroptosis in the endocrine and exocrine pancreas have been seen. 170 These findings point to COVID‐19 illness of ‐cells may either indirectly or directly impair‐cell performance, resulting in different degrees of metabolic imbalance.
Diabetes, ketoacidosis, hyperglycemia, and severe metabolic comorbidities related to prediabetes are frequently seen in COVID‐19 subjects. 170 Diabetes ketoacidosis and severe ketoacidosis at diabetes diagnosis rose dramatically during the SARS‐CoV‐2 pandemic, especially in children under the age of six, according to data from the German Diabetes Prospective Follow‐up Registry diabetes‐patienten‐verlaufsdokumentation. This countrywide registration covers more than 90% of pediatric patients with T1D. 171 Furthermore, alterations in parental behavior and availability of health care may have had a role in the rise of new‐onset T1D among children in recent decades. 172 Despite this, evidence is mounting that COVID‐19 may be a contributing factor or catalyst to developing new‐onset diabetes. 173 There have also been reports of pancreatitis after the administration of COVID‐19. 174 However, whether COVID‐19 can induce new‐onset diabetes or increase the course of pre‐existing undetected diabetes or prediabetes is still up for debate. 175
6. TARGETING OF HOST METABOLISM FOR THE TREATMENT OF COVID‐19
According to new research, extracellular vesicles, particularly exosomes, contain microRNA (miRNA) targeting immunological and biochemical pathways, such as those affecting the peroxisome proliferator‐activated receptors (PPARs). 4 , 176 , 177 , 178 Exosomal miRNAs, like miR‐155, are increased in obese mice (adipose tissue macrophage) and promote insulin resistance. TNF‐α treatment of human adipocytes resulted in increased miR‐155 synthesis and inflammation, highlighting the relevance of miR‐155. 177 MiR‐155 inhibits PPAR gamma (PPARγ), activating the pro‐inflammatory NF‐kB pathway. 177 , 179 TLR4‐induced NF‐kB signaling pathways were modulated by other miRNAs, including miR‐223, miR‐200b/c, miR‐146a/b, and miR‐203. 180 , 181 , 182 TNF‐α production is also boosted by some miRNAs, including miR‐125b, miR‐221, miR‐579, and miR‐16, resulting in a pro‐inflammatory state. 183 , 184 Since these microRNAs impact the inflammatory reaction on their own, Let‐7 boosts IL‐6 concentrations while also promoting glucose intolerance and decreasing pancreatic insulin production. 185 , 186 Some miRNAs, which have a dual function in immunometabolic processes, might be used as potential therapeutics in individuals with metabolic disorders and COVID‐19.
COVID‐19 is being treated with antibody cocktails that are either obtained directly from infected people or synthesized. 29 Regeneron is sponsoring antibody research targeting COVID‐19, and Eli Lilly is two examples of pharmaceutical companies, both of which have been granted urgent Food and Drug Administration (FDA) approval for the management of COVID‐19. 29 Nanobodies (small antibody‐like molecules) relying on organically released antibodies by camelids are being developed and may be selected due to their cheaper cost and simpler manufacturing procedure. 188 The possible synergistic benefits of combination therapies that reduce inflammation while also controlling metabolism might ameliorate hyperimmune activation in SARS‐CoV‐2 cases with underlying metabolic disorders caused by T2DM or obesity. 29
7. CONCLUSION
Substantial ongoing research into immune cell metabolism provides an overwhelming volume of data. Also, it has been found that immunometabolism has an axial role in the fate of viral infection, including SARS‐CoV‐2. Studies on how SARS‐CoV‐2 changes intracellular metabolism in immune cells such as innate immune cells reveal exciting biochemical aspects such as increased glycolysis and altered OXPHOS. Targeting biochemical (metabolite) alterations in immune cells like macrophages might pave the way for new immunomodulatory strategies such as anti‐inflammatory and antiviral drugs. However, the potential to restrict metabolic rewiring in immunity associated with the viral infection and other infected cells to lessen the raised inflammatory reaction shown throughout the advanced stages of COVID‐19 disease is an intriguing option for treating people who develop from mild to severe COVID‐19 disease. Future investigations should focus on how viruses alter the metabolism of immune cells to advance their pathogenesis since identifying these mechanisms may provide a new therapeutic avenue for better management of viral infections. Innovative treatment methods, such as antagomirs and antibody cocktails, either alone or in conjunction with established anti‐inflammatory medications, including steroids, offer an exciting opportunity to escape the negative consequences of COVID‐19 disease in cases with metabolic abnormalities.
AUTHOR CONTRIBUTIONS
Mohammad Rudiansyah, Saade Abdalkareem Jasim, Rumi Iqbal doewes, Abduladheem Turki Jalil, and Zeinab Gol Mohammad pour have the idea for and planned the study and contributed to the writing of the paper. Sara Sohrabi Athar, D. O. Bokov, Yasser Fakri Mustafa, Ali Salimi Jeda, and Mina Noroozbeygi contributed to the writing of the manuscript. Sajad Karampoor and Rasoul Mirzaei contributed to the critical revision of the report. All authors read and approved the final version.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Rudiansyah M, Jasim SA, Mohammad pour ZG, et al. Coronavirus disease 2019 (COVID‐19) update: from metabolic reprogramming to immunometabolism. J Med Virol. 2022;94:4611‐4627. 10.1002/jmv.27929
Contributor Information
Sajad Karampoor, Email: sajadkarampour1987@gmail.com.
Rasoul Mirzaei, Email: rasul.micro92@gmail.com.
DATA AVAILABILITY STATEMENT
Not applicable.
REFERENCES
- 1. Makowski L, Chaib M, Rathmell JC. Immunometabolism: from basic mechanisms to translation. Immunol Rev. 2020;295(1):5‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ayres JS. Immunometabolism of infections. Nat Rev Immunol. 2020;20(2):79‐80. [DOI] [PubMed] [Google Scholar]
- 3. Mirzaei R, Sholeh M, Jalalifar S, et al. Immunometabolism in human brucellosis: an emerging field of investigation. Microb Pathog. 2021;158:105115. [DOI] [PubMed] [Google Scholar]
- 4. Mirzaei R, Zamani F, Hajibaba M, et al. The pathogenic, therapeutic and diagnostic role of exosomal microrna in the autoimmune diseases. J Neuroimmunol. 2021;358:577640. [DOI] [PubMed] [Google Scholar]
- 5. O'Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. 2016;16(9):553‐565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mirzaei R, Abdi M, Gholami H. The host metabolism following bacterial biofilm: what is the mechanism of action? Rev Med Microbiol. 2020;31(4):175‐182. [Google Scholar]
- 7. Moreno‐Altamirano MMB, Kolstoe SE, Sánchez‐García FJ. Virus control of cell metabolism for replication and evasion of host immune responses. Front Cell Infect Microbiol. 2019;9:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mirzaei R, Sabokroo N, Ahmadyousefi Y, Motamedi H, Karampoor S. Immunometabolism in biofilm infection: lessons from cancer. Mol Med. 2022;28(1):1‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zelante T, Iannitti RG, Cunha C, et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin‐22. Immunity. 2013;39(2):372‐385. [DOI] [PubMed] [Google Scholar]
- 10. Rodríguez‐Prados J‐C, Través PG, Cuenca J, et al. Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J Immunol. 2010;185(1):605‐614. [DOI] [PubMed] [Google Scholar]
- 11. Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity. 2013;38(4):633‐643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mirzaei R, Dehkhodaie E, Bouzari B, et al. Dual role of microbiota‐derived short‐chain fatty acids on host and pathogen. Biomed Pharmacother. 2022;145:112352. [DOI] [PubMed] [Google Scholar]
- 13. Mirzaei R, Ranjbar R. Hijacking host components for bacterial biofilm formation: an advanced mechanism. Int Immunopharmacol. 2022;103:108471. [DOI] [PubMed] [Google Scholar]
- 14. Buck MD, Sowell RT, Kaech SM, Pearce EL. Metabolic instruction of immunity. Cell. 2017;169(4):570‐586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mah AY, Rashidi A, Keppel MP, et al. Glycolytic requirement for NK cell cytotoxicity and cytomegalovirus control. JCI Insight . 2017;2(23). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Raftery MJ, Lalwani P, Krautkrӓmer E, et al. β2 integrin mediates hantavirus‐induced release of neutrophil extracellular traps. J Exp Med. 2014;211(7):1485‐1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Delgado‐Rizo V, Martínez‐Guzmán MA, Iñiguez‐Gutierrez L, García‐Orozco A, Alvarado‐Navarro A, Fafutis‐Morris M. Neutrophil extracellular traps and its implications in inflammation: an overview. Front Immunol. 2017;8:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Martinelli S, Urosevic M, Daryadel A, et al. Induction of genes mediating interferon‐dependent extracellular trap formation during neutrophil differentiation. J Biol Chem. 2004;279(42):44123‐44132. [DOI] [PubMed] [Google Scholar]
- 19. Goodarzi P, Mahdavi F, Mirzaei R, et al. Coronavirus disease 2019 (COVID‐19): immunological approaches and emerging pharmacologic treatments. Int Immunopharmacol. 2020;88:106885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mirzaei R, Mahdavi F, Badrzadeh F, et al. The emerging role of microRNAs in the severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) infection. Int Immunopharmacol. 2021;90:107204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Abdi M, Mirzaei R. Iran without mandatory quarantine and with social distancing strategy against coronavirus disease (COVID‐19). Health Secur. 2020;18(3):257‐259. [DOI] [PubMed] [Google Scholar]
- 22. Hummig W, Cruz MM. Bruxism as a clinical indicator of mental ilness: lessons from the COVID‐19 to the future! Arc Clin Psychiatr. 2021;48:127. [Google Scholar]
- 23. Karampoor S, Hesamizadeh K, Shams Z, et al. The role of lovastatin in the attenuation of COVID‐19. Int Immunopharmacol. 2021;101:108192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mirzaei R, Karampoor S, Sholeh M, Moradi P, Ranjbar R, Ghasemi F. A contemporary review on pathogenesis and immunity of COVID‐19 infection. Mol Biol Rep. 2020;47(7):5365‐5376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li C, Yang Y, Ren L. Genetic evolution analysis of 2019 novel coronavirus and coronavirus from other species. Infect Genet Evol. 2020;82:104285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Karampoor S, Zahednasab H, Farahmand M, et al. A possible pathogenic role of Syndecan‐1 in the pathogenesis of coronavirus disease 2019 (COVID‐19). Int Immunopharmacol. 2021;97:107684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhou J, Li Z, Meng H, Chang Y‐C, Peng N‐H, Wei B. Chinese parental awareness of children's COVID‐19 protective measures. Am J Health Behav. 2021;45(4):657‐664. [DOI] [PubMed] [Google Scholar]
- 28. Karampoor S, Hesamizadeh K, Maleki F, et al. A possible pathogenic correlation between neutrophil elastase (NE) enzyme and inflammation in the pathogenesis of coronavirus disease 2019 (COVID‐19). Int Immunopharmacol. 2021;100:108137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Batabyal R, Freishtat N, Hill E, Rehman M, Freishtat R, Koutroulis I. Metabolic dysfunction and immunometabolism in COVID‐19 pathophysiology and therapeutics. Int J Obes. 2021;45(6):1163‐1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kumar V. How could we forget immunometabolism in SARS‐CoV2 infection or COVID‐19? Int Rev Immunol. 2021;40(1‐2):72‐107. [DOI] [PubMed] [Google Scholar]
- 31. Raheem R, Alsayed R, Yousif E, Hairunisa N. Coronavirus new variants: the mutations cause and the effect on the treatment and vaccination: coronavirus new variants: effect and treatments. Baghdad J Biochem Appl Biol Sci. 2021;2(02):70‐78. [Google Scholar]
- 32. Shafaati M, Saidijam M, Soleimani M, et al. A brief review on DNA vaccines in the era of COVID‐19. Future Virol. 2022;17(1):49‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Thaker SK, Ch'ng J, Christofk HR. Viral hijacking of cellular metabolism. BMC Biol. 2019;17(1):59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mayer KA, Stöckl J, Zlabinger GJ, Gualdoni GA. Hijacking the supplies: metabolism as a novel facet of virus‐host interaction. Front Immunol. 2019;10:1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Moolamalla STR, Balasubramanian R, Chauhan R, Priyakumar UD, Vinod PK. Host metabolic reprogramming in response to SARS‐CoV‐2 infection: a systems biology approach. Microb Pathog. 2021;158:105114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wu M, Chen Y, Xia H, et al. Transcriptional and proteomic insights into the host response in fatal COVID‐19 cases. Proc Natl Acad Sci. 2020;117(45):28336‐28343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Blanco‐Melo D, Nilsson‐Payant BE, Liu WC, et al. Imbalanced host response to SARS‐CoV‐2 drives development of COVID‐19. Cell. 2020;181(5):1036‐1045.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Singh K, Chen YC, Hassanzadeh S, et al. Network analysis and transcriptome profiling identify autophagic and mitochondrial dysfunctions in SARS‐CoV‐2 infection. Front Genet. 2021;12:599261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Karampoor S, Afrashteh F, Laali A. Persistent hiccups after treatment of COVID‐19 with dexamethasone: a case report. Respir Med Case Rep. 2021;34:101515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Thaker SK, Ch'ng J, Christofk HR. Viral hijacking of cellular metabolism. BMC Biol. 2019;17(1):1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Massabeti R, Cipriani MS, Valenti I. Covid‐19: a systemic disease treated with a wide‐ranging approach: a case report. J Popul Ther Clin Pharmacol. 2020;27(S Pt 1):e26‐e30. [DOI] [PubMed] [Google Scholar]
- 42. Maynard ND, Gutschow MV, Birch EW, Covert MW. The virus as metabolic engineer. Biotechnol J. 2010;5(7):686‐694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ganesh GV, Mohanram RK. Metabolic reprogramming and immune regulation in viral diseases. Rev Med Virol. 2022;32(2):e2268. [DOI] [PubMed] [Google Scholar]
- 44. Mehraj V, Routy J‐P. Tryptophan catabolism in chronic viral infections: handling uninvited guests. Int J Tryptophan Res. 2015;8:41‐48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fallarino F, Grohmann U, Hwang KW, et al. Modulation of tryptophan catabolism by regulatory T cells. Nature Immunol. 2003;4(12):1206‐1212. [DOI] [PubMed] [Google Scholar]
- 46. Ayres JS. A metabolic handbook for the COVID‐19 pandemic. Nat Metab. 2020;2(7):572‐585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Guo Y, Meng X, Ma J, et al. Human papillomavirus 16 E6 contributes HIF‐1α induced Warburg effect by attenuating the VHL‐HIF‐1α interaction. Int J Mol Sci. 2014;15(5):7974‐7986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cheng M‐L, Chien K‐Y, Lai C‐H, Li G‐J, Lin J‐F, Ho H‐Y. Metabolic reprogramming of host cells in response to enteroviral infection. Cells. 2020;9(2):473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kespohl M, Bredow C, Klingel K, et al. Protein modification with ISG15 blocks coxsackievirus pathology by antiviral and metabolic reprogramming. Sci Adv. 2020;6(11):eaay1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rayfield EJ, Curnow RT, George DT, Beisel WR. Impaired carbohydrate metabolism during a mild viral illness. N Engl J Med. 1973;289(12):618‐621. [DOI] [PubMed] [Google Scholar]
- 51. Shimano H. Sterol regulatory element‐binding proteins (SREBPs): transcriptional regulators of lipid synthetic genes. Prog Lipid Res. 2001;40(6):439‐452. [DOI] [PubMed] [Google Scholar]
- 52. Blanc M, Hsieh WY, Robertson KA, et al. The transcription factor STAT‐1 couples macrophage synthesis of 25‐hydroxycholesterol to the interferon antiviral response. Immunity. 2013;38(1):106‐118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chang M‐L. Metabolic alterations and hepatitis C: from bench to bedside. World J Gastroenterol. 2016;22(4):1461‐1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Waris G, Felmlee DJ, Negro F, Siddiqui A. Hepatitis C virus induces proteolytic cleavage of sterol regulatory element binding proteins and stimulates their phosphorylation via oxidative stress. J Virol. 2007;81(15):8122‐8130. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 55. Keller P, Simons K. Cholesterol is required for surface transport of influenza virus hemagglutinin. J Cell Biol. 1998;140(6):1357‐1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mirzaei R, Babakhani S, Ajorloo P, et al. The emerging role of exosomal miRNAs as a diagnostic and therapeutic biomarker in mycobacterium tuberculosis infection. Mol Med. 2021;27(1):1‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Episcopio D, Aminov S, Benjamin S, et al. Atorvastatin restricts the ability of influenza virus to generate lipid droplets and severely suppresses the replication of the virus. FASEB J. 2019;33(8):9516‐9525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Shan J, Qian W, Shen C, et al. High‐resolution lipidomics reveals dysregulation of lipid metabolism in respiratory syncytial virus pneumonia mice. RSC Adv. 2018;8(51):29368‐29377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mirzaei R, Afaghi A, Babakhani S, et al. Role of microbiota‐derived short‐chain fatty acids in cancer development and prevention. Biomed Pharmacother. 2021;139:111619. [DOI] [PubMed] [Google Scholar]
- 60. Mirzaei R, Bouzari B, Hosseini‐Fard SR, et al. Role of microbiota‐derived short‐chain fatty acids in nervous system disorders. Biomed Pharmacother. 2021;139:111661. [DOI] [PubMed] [Google Scholar]
- 61. Yuan S, Chu H, Chan JF, et al. SREBP‐dependent lipidomic reprogramming as a broad‐spectrum antiviral target. Nat Commun. 2019;10(1):120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Davies JT, Delfino SF, Feinberg CE, et al. Current and emerging uses of statins in clinical therapeutics: a review. Lipid Insights. 2016;9:13‐29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lardizabal JA, Deedwania PC. Benefits of statin therapy and compliance in high risk cardiovascular patients. Vasc Health Risk Manag. 2010;6:843‐853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Gualdoni GA, Mayer KA, Kapsch AM, et al. Rhinovirus induces an anabolic reprogramming in host cell metabolism essential for viral replication. Proc Natl Acad Sci U S A. 2018;115(30):E7158‐E7165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wang Q, Fang P, He R, et al. O‐GlcNAc transferase promotes influenza A virus‐induced cytokine storm by targeting interferon regulatory factor‐5. Sci Adv. 2020;6(16):eaaz7086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Rasmussen AL, Diamond DL, McDermott JE, et al. Systems virology identifies a mitochondrial fatty acid oxidation enzyme, dodecenoyl coenzyme A delta isomerase, required for hepatitis C virus replication and likely pathogenesis. J Virol. 2011;85(22):11646‐11654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tongluan N, Ramphan S, Wintachai P, et al. Involvement of fatty acid synthase in dengue virus infection. Virol J. 2017;14(1):28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Franchini M, Targher G, Capra F, Montagnana M, Lippi G. The effect of iron depletion on chronic hepatitis C virus infection. Hepatol Int. 2008;2(3):335‐340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Espert L, Codogno P, Biard‐Piechaczyk M. Involvement of autophagy in viral infections: antiviral function and subversion by viruses. J Mol Med (Berl). 2007;85(8):811‐823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lee NR, Ban J, Lee NJ, et al. Activation of RIG‐I‐Mediated antiviral signaling triggers autophagy through the MAVS‐TRAF6‐Beclin‐1 signaling axis. Front Immunol. 2018;9:2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lee HK, Lund JM, Ramanathan B, Mizushima N, Iwasaki A. Autophagy‐dependent viral recognition by plasmacytoid dendritic cells. Science. 2007;315(5817):1398‐1401. [DOI] [PubMed] [Google Scholar]
- 72. Law AH, Lee DC, Yuen KY, Peiris M, Lau AS. Cellular response to influenza virus infection: a potential role for autophagy in CXCL10 and interferon‐alpha induction. Cell Mol Immunol. 2010;7(4):263‐270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Orvedahl A, Alexander D, Tallóczy Z, et al. HSV‐1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe. 2007;1(1):23‐35. [DOI] [PubMed] [Google Scholar]
- 74. He B, Gross M, Roizman B. The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double‐stranded RNA‐activated protein kinase. Proc Natl Acad Sci U S A. 1997;94(3):843‐848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Navas‐Martín SR, Weiss S. Coronavirus replication and pathogenesis: implications for the recent outbreak of severe acute respiratory syndrome (SARS), and the challenge for vaccine development. J Neurovirol. 2004;10(2):75‐85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Li C, He Q, Qian H, Liu J. Overview of the pathogenesis of COVID‐19 (review). Exp Ther Med. 2021;22(3):1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Lin L, Lu L, Cao W, Li T. Hypothesis for potential pathogenesis of SARS‐CoV‐2 infection‐a review of immune changes in patients with viral pneumonia. Emerg Microbes Infect. 2020;9(1):727‐732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Weiss SR, Leibowitz JL. Coronavirus pathogenesis. Adv Virus Res. 2011;81:85‐164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Jimenez‐Guardeño JM, Nieto‐Torres JL, DeDiego ML, et al. The PDZ‐binding motif of severe acute respiratory syndrome coronavirus envelope protein is a determinant of viral pathogenesis. PLoS Pathog. 2014;10(8):e1004320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. von der Thüsen J, van der Eerden M. Histopathology and genetic susceptibility in COVID‐19 pneumonia. Eur J Clin Invest. 2020;50(7):e13259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Hanley B, Lucas SB, Youd E, Swift B, Osborn M. Autopsy in suspected COVID‐19 cases. J Clin Pathol. 2020;73(5):239‐242. [DOI] [PubMed] [Google Scholar]
- 82. Yang L, Liu S, Liu J, et al. COVID‐19: immunopathogenesis and immunotherapeutics. Signal Transduct Target Ther. 2020;5(1):1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kordzadeh‐Kermani E, Khalili H, Karimzadeh I. Pathogenesis, clinical manifestations and complications of coronavirus disease 2019 (COVID‐19). Future Microbiol. 2020;15(13):1287‐1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Zou X, Chen K, Zou J, Han P, Hao J, Han Z. Single‐cell RNA‐seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019‐nCoV infection. Front Med. 2020;14(2):185‐192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Laali A, Kermanshah Z, Keyvani H, Kaveh V, Karampoor S. Idiopathic thrombocytopenic purpura as a hematologic manifestation of COVID‐19 infection: a case report. Respir Med Case Rep. 2021;34:101534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Hoffmann M, Kleine‐Weber H, Pöhlmann S. A multibasic cleavage site in the spike protein of SARS‐CoV‐2 is essential for infection of human lung cells. Mol Cell. 2020;78(4):779‐784.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Rodrigues Prestes TR, Rocha NP, Miranda AS, Teixeira AL, Simoes ESAC. The anti‐inflammatory potential of ACE2/angiotensin‐(1‐7)/Mas receptor axis: evidence from basic and clinical research. l. 2017;18(11):1301‐1313. [DOI] [PubMed] [Google Scholar]
- 88. Tikellis C, Thomas M. Angiotensin‐converting enzyme 2 (ACE2) is a key modulator of the renin angiotensin system in health and disease. Int J Pept. 2012;2012:256294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Pal R, Bhansali A. COVID‐19, diabetes mellitus and ACE2: the conundrum. Diabetes Res Clin Pract. 2020;162:108132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Gheblawi M, Wang K, Viveiros A, et al. Angiotensin‐converting enzyme 2: SARS‐CoV‐2 receptor and regulator of the renin‐angiotensin system: celebrating the 20th anniversary of the discovery of ACE2. Circ Res. 2020;126(10):1456‐1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Zhao Y, Zhao Z, Wang Y, Zhou Y, Ma Y, Zuo W. Single‐cell RNA expression profiling of ACE2, the receptor of SARS‐CoV‐2. Am J Respir Crit Care Med. 2020;202(5):756‐759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Dashraath P, Wong JLJ, Lim MXK, et al. Coronavirus disease 2019 (COVID‐19) pandemic and pregnancy. Am J Obstet Gynecol. 2020;222(6):521‐531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Bunyavanich S, Do A, Vicencio A. Nasal gene expression of angiotensin‐converting enzyme 2 in children and adults. JAMA. 2020;323(23):2427‐2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Zheng M, Gao Y, Wang G, et al. Functional exhaustion of antiviral lymphocytes in COVID‐19 patients. Cell Mol Immunol. 2020;17(5):533‐535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Diao B, Wang C, Tan Y, et al. Reduction and functional exhaustion of T cells in patients with coronavirus disease 2019 (COVID‐19). Front Immunol. 2020;11:827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497‐506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Creelan BC, Antonia SJ. The NKG2A immune checkpoint—a new direction in cancer immunotherapy. Nat Rev Clin Oncol. 2019;16(5):277‐278. [DOI] [PubMed] [Google Scholar]
- 98. Salmaninejad A, Valilou SF, Shabgah AG, et al. PD‐1/PD‐L1 pathway: basic biology and role in cancer immunotherapy. J Cell Physiol. 2019;234(10):16824‐16837. [DOI] [PubMed] [Google Scholar]
- 99. Das M, Zhu C, Kuchroo VK. Tim‐3 and its role in regulating anti‐tumor immunity. Immunol Rev. 2017;276(1):97‐111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Berber E, Sumbria D, Rouse BT. Could targeting immunometabolism be a way to control the burden of COVID‐19 infection? Microb Infect. 2021;23(2‐3):104780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Thomas‐Rüddel D, Winning J, Dickmann P, et al. Coronavirus disease 2019 (COVID‐19): update for anesthesiologists and intensivists March 2020. Anaesthesist. 2021;70(Suppl 1):1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Gardinassi LG, Souza COS, Sales‐Campos H, Fonseca SG. Immune and metabolic signatures of COVID‐19 revealed by transcriptomics data reuse. Front Immunol. 2020;11:1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Thomas T, Stefanoni D, Reisz JA, et al. COVID‐19 infection alters kynurenine and fatty acid metabolism, correlating with IL‐6 levels and renal status. JCI Insight . 2020;5(14). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Blasco H, Bessy C, Plantier L, et al. The specific metabolome profiling of patients infected by SARS‐COV‐2 supports the key role of tryptophan‐nicotinamide pathway and cytosine metabolism. Sci Rep. 2020;10(1):16824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Bost P, Giladi A, Liu Y, et al. Host‐viral infection maps reveal signatures of severe COVID‐19 patients. Cell. 2020;181(7):1475‐1488.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Codo AC, Davanzo GG, Monteiro LB, et al. Elevated glucose levels favor SARS‐CoV‐2 infection and monocyte response through a HIF‐1α/glycolysis‐dependent axis. Cell Metab. 2020;32(3):437‐446.e435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Bojkova D, Klann K. Proteomics of SARS‐CoV‐2‐infected host cells reveals therapy targets. Nature. 2020;583(7816):469‐472. [DOI] [PubMed] [Google Scholar]
- 108. Ehrlich A, Uhl S, Ioannidis K, Hofree M, ten Oever BR, Nahmias Y. The SARS‐CoV‐2 transcriptional metabolic signature in lung epithelium. SSRN Electron J . 2020.
- 109. O'Carroll SM, O'Neill LAJ. Targeting immunometabolism to treat COVID‐19. Immunother Adv. 2021;1(1):ltab013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Hadjadj J, Yatim N. Impaired type I interferon activity and inflammatory responses in severe COVID‐19 patients. Science. 2020;369(6504):718‐724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Merad M, Martin JC. Pathological inflammation in patients with COVID‐19: a key role for monocytes and macrophages. Nat Rev Immunol. 2020;20(6):355‐362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Zhou F, Yu T, Du R, et al. Clinical course and risk factors for mortality of adult inpatients with COVID‐19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395(10229):1054‐1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Ryan DG, O'Neill LAJ. Krebs cycle reborn in macrophage immunometabolism. Annu Rev Immunol. 2020;38:289‐313. [DOI] [PubMed] [Google Scholar]
- 114. Seim GL, Britt EC, John SV, et al. Two‐stage metabolic remodelling in macrophages in response to lipopolysaccharide and interferon‐γ stimulation. Nat Metab. 2019;1(7):731‐742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Arunachalam PS, Wimmers F, Mok CKP, et al. Systems biological assessment of immunity to mild versus severe COVID‐19 infection in humans. Science. 2020;369(6508):1210‐1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Wu D, Sanin DE, Everts B, et al. Type 1 interferons induce changes in core metabolism that are critical for immune function. Immunity. 2016;44(6):1325‐1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Wilson CS, Moore DJ. B cell metabolism: an understudied opportunity to improve immune therapy in autoimmune type 1 diabetes. Immunometabolism. 2020;2(2):e200016. [Google Scholar]
- 118. Kumar V. Targeting macrophage immunometabolism: Dawn in the darkness of sepsis. Int Immunopharmacol. 2018;58:173‐185. [DOI] [PubMed] [Google Scholar]
- 119. Tannahill GM, Curtis AM, Adamik J, et al. Succinate is an inflammatory signal that induces IL‐1β through HIF‐1α. Nature. 2013;496(7444):238‐242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Kumar V. Dendritic cells in sepsis: potential immunoregulatory cells with therapeutic potential. Mol Immunol. 2018;101:615‐626. [DOI] [PubMed] [Google Scholar]
- 121. O'Neill LA, Pearce EJ. Immunometabolism governs dendritic cell and macrophage function. J Exp Med. 2016;213(1):15‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Wculek SK, Khouili SC, Priego E, Heras‐Murillo I, Sancho D. Metabolic control of dendritic cell functions: digesting information. Front Immunol. 2019;10(775):775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Law HK, Cheung CY, Ng HY, et al. Chemokine up‐regulation in SARS‐coronavirus–infected, monocyte‐derived human dendritic cells. Blood. 2005;106(7):2366‐2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Yang D, Chu H, Hou Y, et al. Attenuated interferon and proinflammatory response in SARS‐CoV‐2‐Infected human dendritic cells is associated with viral antagonism of STAT1 phosphorylation. J Infect Dis. 2020;222(5):734‐745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Kumar V. T cells and their immunometabolism: a novel way to understanding sepsis immunopathogenesis and future therapeutics. Eur J Cell Biol. 2018;97(6):379‐392. [DOI] [PubMed] [Google Scholar]
- 126. Geltink RIK, Kyle RL, Pearce EL. Unraveling the complex interplay between T cell metabolism and function. Annu Rev Immunol. 2018;36:461‐488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Milam AAV, Bartleson JM, Buck MD, Tonic TCR. Signaling inversely regulates the basal metabolism of CD4(+) T. Cells. 2020;4(8):485‐497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Toriyama K, Kuwahara M, Kondoh H, et al. T cell‐specific deletion of Pgam1 reveals a critical role for glycolysis in T cell responses. Commun Biol. 2020;3(1):394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Mendoza A, Fang V, Chen C, et al. Lymphatic endothelial S1P promotes mitochondrial function and survival in naive T cells. Nature. 2017;546(7656):158‐161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Dorner M, Brandt S, Tinguely M, et al. Plasma cell toll‐like receptor (TLR) expression differs from that of B cells, and plasma cell TLR triggering enhances immunoglobulin production. Immunology. 2009;128(4):573‐579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Gram AM, Sun C, Landman SL, et al. Human B cells fail to secrete type I interferons upon cytoplasmic DNA exposure. Mol Immunol. 2017;91:225‐237. [DOI] [PubMed] [Google Scholar]
- 132. Walker MM, Crute BWB. Cell‐intrinsic STING signaling triggers cell activation, synergizes with B cell receptor signals, and promotes antibody responses. J Immunol. 2018;201(9):2641‐2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Schenten D, O'Ketch M. Rig‐I‐like receptors (RLRs) regulate humoral immunity to West Nile virus (WNV) infection. J Immunol. 2018;200(1 Suppl):126.115. [Google Scholar]
- 134. Koch C, Staffler G, Hüttinger R, et al. T cell activation‐associated epitopes of CD147 in regulation of the T cell response, and their definition by antibody affinity and antigen density. Int Immunol. 1999;11(5):777‐786. [DOI] [PubMed] [Google Scholar]
- 135. Su Y, Chen D, Yuan D, et al. Multi‐omics resolves a sharp disease‐state shift between mild and moderate COVID‐19. Cell. 2020;183(6):1479‐1495.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Shen B, Yi X, Sun Y, et al. Proteomic and metabolomic characterization of COVID‐19 patient sera. Cell. 2020;182(1):59‐72.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Lee JW, Su Y, Baloni P, et al. Integrated analysis of plasma and single immune cells uncovers metabolic changes in individuals with COVID‐19. Nature Biotechnol. 2022;40(1):110‐120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Zaim S, Chong JH, Sankaranarayanan V, Harky A. COVID‐19 and multiorgan response. Curr Probl Cardiol. 2020;45(8):100618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Xu Z, Shi L, Wang Y, et al. Pathological findings of COVID‐19 associated with acute respiratory distress syndrome. Lancet Respir Med. 2020;8(4):420‐422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Mao L, Jin H, Wang M, et al. Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan, China. JAMA Neurol. 2020;77(6):683‐690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Benedetti C, Waldman M, Zaza G, Riella LV, Cravedi P. COVID‐19 and the kidneys: an update. Front Med (Lausanne). 2020;7:423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Schmelter F, Föh B, Mallagaray A, et al. Metabolic and lipidomic markers differentiate COVID‐19 from non‐hospitalized and other intensive care patients. Front Mol Biosci. 2021;8:737039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Nishiga M, Wang DW, Han Y, Lewis DB, Wu JC. COVID‐19 and cardiovascular disease: from basic mechanisms to clinical perspectives. Nat Rev Cardiol. 2020;17(9):543‐558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Herrera‐Van Oostdam AS, Castañeda‐Delgado JE, Oropeza‐Valdez JJ, et al. Immunometabolic signatures predict risk of progression to sepsis in COVID‐19. PLoS One. 2021;16(8):e0256784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Brufsky A. Hyperglycemia, hydroxychloroquine, and the COVID‐19 pandemic. J Med Virol. 2020;92(7):770‐775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Drucker DJ. Coronavirus infections and type 2 diabetes—shared pathways with therapeutic implications. Endocr Rev. 2020;41(3):457‐470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Seshasai SRK. Diabetes mellitus, fasting glucose, and risk of cause‐specific death. N Engl J Med. 2011;364(13):829‐841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. van Crevel R, van de Vijver S, Moore DA. The global diabetes epidemic: what does it mean for infectious diseases in tropical countries? Lancet Diabetes Endo. 2017;5(6):457‐468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Hodgson K, Morris J, Bridson T, Govan B, Rush C, Ketheesan N. Immunological mechanisms contributing to the double burden of diabetes and intracellular bacterial infections. Immunology. 2015;144(2):171‐185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Guo W, Li M, Dong Y, et al. Diabetes is a risk factor for the progression and prognosis of COVID‐19. Diabetes Metab Res Rev. 2020;36(7):e3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Boccia M, Aronne L, Celia B, et al. COVID‐19 and coagulative axis: review of emerging aspects in a novel disease. Monaldi Arch Chest Dis . 2020;90(2). [DOI] [PubMed] [Google Scholar]
- 152. Shah BR, Hux JE. Quantifying the risk of infectious diseases for people with diabetes. Diabetes Care. 2003;26(2):510‐513. [DOI] [PubMed] [Google Scholar]
- 153. Yang J, Zheng Y, Gou X, et al. Prevalence of comorbidities and its effects in patients infected with SARS‐CoV‐2: a systematic review and meta‐analysis. Int J Infect Dis. 2020;94:91‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Muniyappa R, Gubbi S. COVID‐19 pandemic, coronaviruses, and diabetes mellitus. Am J Physiol Endocrinol Metab. 2020;318(5):E736‐E741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Wichmann D, Sperhake J‐P, Lütgehetmann M, et al. Autopsy findings and venous thromboembolism in patients with COVID‐19: a prospective cohort study. Ann Intern Med. 2020;173(4):268‐277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Terpos E, Ntanasis‐Stathopoulos I. Hematological findings and complications of COVID−19. Am J Hematol. 2020;95(7):834‐847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Seidu S, Gillies C, Zaccardi F, et al. The impact of obesity on severe disease and mortality in people with SARS‐CoV‐2: a systematic review and meta‐analysis. Endocrinol Diabetes Metab. 2020;4(1):e00176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Stefan N. Causes, consequences, and treatment of metabolically unhealthy fat distribution. Lancet Diabetes Endocrinol. 2020;8(7):616‐627. [DOI] [PubMed] [Google Scholar]
- 159. Akhmerov A, Marbán E. COVID‐19 and the heart. Circ Res. 2020;126(10):1443‐1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Avula A, Nalleballe K, Narula N, et al. COVID‐19 presenting as stroke. Brain Behav Immun. 2020;87:115‐119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Schwarz PEH, Timpel P, Harst L, et al. Blood sugar regulation for cardiovascular health promotion and disease prevention: JACC health promotion series. J Am Coll Cardiol. 2018;72(15):1829‐1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Bornstein SR, Rubino F, Ludwig B, et al. Consequences of the COVID‐19 pandemic for patients with metabolic diseases. Nat Metab. 2021;3(3):289‐292. [DOI] [PubMed] [Google Scholar]
- 163. Santos A, Magro DO, Evangelista‐Poderoso R, Saad MJA. Diabetes, obesity, and insulin resistance in COVID‐19: molecular interrelationship and therapeutic implications. Diabetol Metab Syndr. 2021;13(1):23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Marx C, Ehrhart‐Bornstein M, Scherbaum WA, Bornstein SR. Regulation of adrenocortical function by cytokines—relevance for immune‐endocrine interaction. Horm Metab Res. 1998;30(6‐7):416‐420. [DOI] [PubMed] [Google Scholar]
- 165. Steenblock C, Todorov V, Kanczkowski W, et al. Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) and the neuroendocrine stress axis. Mol Psychiatry. 2020;25(8):1611‐1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Horby P, Lim WS, Emberson JR, Mafham M, Bell JL, Linsell L. Dexamethasone in hospitalized patients with Covid‐19. N Engl J Med. 2021;384(8):693‐704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Ramakrishnan S, Nicolau DV Jr., Langford B, et al. Inhaled budesonide in the treatment of early COVID‐19 (STOIC): a phase 2, open‐label, randomised controlled trial. Lancet Respir Med. 2021;9(7):763‐772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Conti P, Ronconi G, Caraffa A, et al. Induction of pro‐inflammatory cytokines (IL‐1 and IL‐6) and lung inflammation by Coronavirus‐19 (COVI‐19 or SARS‐CoV‐2): anti‐inflammatory strategies. J Biol Regul Homeost Agents. 2020;34(2):327‐331. [DOI] [PubMed] [Google Scholar]
- 169. Kusmartseva I, Wu W, Syed F, et al. Expression of SARS‐CoV‐2 entry factors in the pancreas of normal organ donors and individuals with COVID‐19. Cell Metab. 2020;32(6):1041‐1051.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Steenblock C, Richter S, Berger I. Viral infiltration of pancreatic islets in patients with COVID‐19. Nat Commun. 2021;12(1):3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Steenblock C, Schwarz PEH, Ludwig B, et al. COVID‐19 and metabolic disease: mechanisms and clinical management. Lancet Diabetes Endocrinol. 2021;9(11):786‐798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Kamrath C, Mönkemöller K, Biester T, et al. Ketoacidosis in children and adolescents with newly diagnosed type 1 diabetes during the COVID‐19 pandemic in Germany. JAMA. 2020;324(8):801‐804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Salmi H, Heinonen S. New‐onset type 1 diabetes in Finnish children during the COVID‐19 pandemic. Arch Dis Child. 2021;107(2):180‐185. [DOI] [PubMed] [Google Scholar]
- 174. Maestre‐Muñiz MM, Arias Á. Long‐term outcomes of patients with coronavirus disease 2019 at one year after hospital discharge. J Clin Med. 2021;10(13):2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Kumaran NK, Karmakar BK, Taylor OM. Coronavirus disease‐19 (COVID‐19) associated with acute necrotising pancreatitis (ANP). BMJ Case Rep. 2020;13(9):e237903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Accili D. Can COVID‐19 cause diabetes? Nat Metab. 2021;3(2):123‐125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Pan Y, Hui X, Hoo RLC, et al. Adipocyte‐secreted exosomal microRNA‐34a inhibits M2 macrophage polarization to promote obesity‐induced adipose inflammation. J Clin Invest. 2019;129(2):834‐849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Karkeni E, Astier J, Tourniaire F, et al. Obesity‐associated inflammation induces microRNA‐155 expression in adipocytes and adipose tissue: outcome on adipocyte function. J Clin Endocrinol Metab. 2016;101(4):1615‐1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Mirzaei R, Mohammadzadeh R, Mirzaei H, et al. Role of microRNAs in Staphylococcus aureus infectionpotential biomarkers and mechanism. IUBMB Life. 2020;72(9):1856‐1869. [DOI] [PubMed] [Google Scholar]
- 180. Li H, Ooi SQ, Heng CK. The role of NF‐кB in SAA‐induced peroxisome proliferator‐activated receptor γ activation. Atherosclerosis. 2013;227(1):72‐78. [DOI] [PubMed] [Google Scholar]
- 181. Ma X, Becker Buscaglia LE, Barker JR, Li Y. MicroRNAs in NF‐kappaB signaling. J Mol Cell Biol. 2011;3(3):159‐166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Curtale G, Mirolo M, Renzi TA, Rossato M, Bazzoni F, Locati M. Negative regulation of toll‐like receptor 4 signaling by IL‐10–dependent microRNA‐146b. Proc Natl Acad Sci. 2013;110(28):11499‐11504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Wendlandt EB, Graff JW, Gioannini TL, McCaffrey AP, Wilson ME. The role of microRNAs miR‐200b and miR‐200c in TLR4 signaling and NF‐κB activation. Innate Immun. 2012;18(6):846‐855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. El Gazzar M, McCall CE. MicroRNAs distinguish translational from transcriptional silencing during endotoxin tolerance. J Biol Chem. 2010;285(27):20940‐20951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Jing Q, Huang S, Guth S, et al. Involvement of microRNA in AU‐rich element‐mediated mRNA instability. Cell. 2005;120(5):623‐634. [DOI] [PubMed] [Google Scholar]
- 186. Frost RJA, Olson EN. Control of glucose homeostasis and insulin sensitivity by the Let‐7 family of microRNAs. Proc Natl Acad Sci. 2011;108(52):21075‐21080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Iliopoulos D, Hirsch HA, Struhl K. An epigenetic switch involving NF‐kappaB, Lin28, Let‐7 MicroRNA, and IL6 links inflammation to cell transformation. Cell. 2009;139(4):693‐706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Ledford H. The race to make COVID antibody therapies cheaper and more potent. Nature. 2020;587(7832):18. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.