

Abstract
Among numerous severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) variants of concerns, Omicron is more infectious and immune‐escaping, while Delta is more pathogenic. Here, we provide evidence for both intervariant and intravariant recombination of the rapidly evolving new SARS‐CoV‐2 genomes, including XD/XE/XF and BA.3, raising concerns of potential more infectious, immune‐escaping, and disease‐causing Omicron and Delta–Omicron variants.
Keywords: Delta‐Omicron XD, Omicron, recombination, SARS‐CoV‐2
1. INTRODUCTION
The global coronavirus disease‐2019 (COVID‐19 pandemic caused by severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) has incurred over half‐billion confirmed cases and more than 6 million fatalities worldwide (https://covid19.who.int). The virus continuously evolves into new variants of concerns (VOCs) and rapidly spreads into different parts of the world. The Omicron variant is believed to be more infectious and capable of immune escape, whereas the Delta variant may cause more severe COVID‐19 diseases. 1 The significantly increased infectivity of Omicron VOCs dominates them over the Delta variants worldwide in only 3 months, providing grounds for multiple lineages circulations, and aiding potential recombination events. The Omicron variant carries over 30 mutations in its spike protein compared with the ancestral Wuhan‐1 reference strain. 2 , 3 Although the Omicron variant has caused comparatively milder symptoms since first identified in Johannesburg, South Africa, in November 2021, its explosive spread over the past few months, including vaccinated individuals, suggests that these mutations could effectively evade prior immunization. The ability of the Omicron variant to circumvent antibody protection was confirmed recently. 4
The cocirculation of multiple variants can introduce co‐infections and result in recombination events, 5 especially in immunocompromised or unvaccinated populations, where different virus variants may co‐exist for a more extended period. The earliest report of possible recombination of the Delta and Omicron variant was in late 2021 but was later considered an error due to sample contamination. In the meantime, the possibility of recombination between Delta and Omicron has been discussed by researchers on Twitter (https://twitter.com/PeacockFlu/) and GitHub (https://github.com/cov-lineages/pango-designation/issues), and much molecular evidence was detected to support the potential existence of Delta–Omicron recombinant. On January 17, 2022, the Institut Pasteur of France reported the isolation of Delta–Omicron (XD) recombinant genomes. 6 The raw reads from the sequences have overall high sequence coverage and no minor variant populations to rule‐out potential contamination or co‐infection. The United States has also identified similar variants. 7 The World Health Organization (WHO) confirmed the occurrence of Delta–Omicron recombination later and classified this variant as Variants under monitoring (VUMs) on March 9, 2022. Besides the XD variant, additional Delta–Omicron or Omicron subvariants including XE, XF, BA.1, BA.2, BA.3, BA.4, and BA.5 were also identified. However, the molecular mechanisms responsible for the rapid revolution of these variants remain to be elucidated. In this study, we have identified multiple recombination events between Delta and Omicron BA.1 resulted in the formation of Delta–Omicron XD. Systematic sequence scanning of the ancestral genome of BA.1‐3 revealed that Omicron BA.3 might have been aided by multiple recombination events from ancestral genomes of BA.1 and BA.2. At the same time, results from the molecular clock implied that BA.4 and BA.5 are more likely to evolve independently from the common ancestry of BA.2 subvariant.
2. MATERIALS AND METHODS
2.1. Data acquisition
All consensus sequences belong to the genomic sequences shared via the Global Initiative on Sharing All Influenza Data (GISAID) 8 database, the global data science intiative and were aligned to the Wuhan Hu‐1 reference sequence (GenBank: MN908947.3, GISAID accession: EPI_ISL_402125) using MAFFT 9 v7.487, with the auto option. Then, those genomes with less than 1% ambiguous nucleotides and complete collection dates were retained for subsequential analyses. Besides, the global proportion of amino acid mutations for Omicron sublineages was obtained from the CoV‐Spectrum website 10 on May 03, 2022. These finds are based on analysis of approximately 45 sequences, accessible at https://doi.org/10.55876/gis8.220607zp.
2.2. Phylogenetic analysis
The phylogenetic relationships of aligned genomes were reconstructed with IQTREE 11 v2.1.4 under the Hasegawa–Kishino–Yano (HKY) nucleotide substitution model. Tree node confidence was measured using 1000 ultrafast bootstrap replicates, and trees were rooted on the reference sequence. All phylogenetic tree files were visualized in the R package ggtree 12 v.3.0.4.
2.3. Recombinant analysis
For each putative recombinant and the corresponding parental sequences, we first explored and visualized the single nucleotide differences using the snipit software (https://github.com/aineniamh/snipit). Second, we constructed the maximum likelihood (ML) trees for the whole genome or the partial genome regions fragmented by breakpoints according to the possible breakpoints inferred from the visualized mosaic structures. Third, the recombinant detection and sequence comparison analyses were performed using Recombinant Analysis Tool (RAT) 13 with a sliding window of 200nt and a step size of 20nt.
2.4. Represented genomes identification
Representative sequences of BA.1, BA.2, and BA.3 lineages were subsampled with covSampler (http://github.com/wuaipinglab/covSampler). The sampled location was fixed in South Africa, and the collection date range was from the SARS‐CoV‐2 outbreak to February 11, 2022. Then, after the ML trees were constructed, we identified the high‐quality sequence closest to the root node as the represented genome for each lineage.
2.5. Omicron spike protein structure and linear mapping
The amino acid residues in the SARS‐CoV Spike trimer or monomer structure (PDB: 7TB4) were labeled according to the site of mutations from the Omicron variant. The structural illustration was generated using PDB: Mol Viewer. The linear mapping of amino acid mutations and labeling of SARS‐CoV‐2 susceptibility were obtained from Coronavirus Resistance Database (CoV‐RDB). 14
2.6. Molecular clock inference
The representative sequences were selected through prephylogenetic analyses. In brief, representative sequences of each main subvariant or minor subvariants in the GISAID database were used for phylogenetic analysis. The genome with the earliest isolated date and nearest distance to the root was selected as the representative sequences. Bayesian coalescent analyses were performed in BEAST 15 v1·10.4, using a Markov chain Monte Carlo (MCMC) length of 50 million steps and sampling every 1000 steps. We used an HKY substitution model with Γ distributed rate heterogeneity with invariant sites and the uncorrelated relaxed clock with an exponential distribution using default parameters. Runs were assessed in Tracer v1.7.1 for sufficient convergence (Effective sample size [ESS] > 200), and maximum clade credibility trees were generated in TreeAnnotator v1.10.4 after discarding 10% of runs as burn‐in.
3. RESULTS
3.1. Potential multiple recombination events at the spike protein of the Delta–Omicron XD
To study the evolution of XD, we performed a phylogenetic analysis of a subsampling of all VOC, variants of interest (VOI), and Omicron sub‐variants recommended by the WHO (Figure 1A). Our data confirm that XD shares the same lineage with Delta with additional mutations. We performed recombination analysis using the Recombination Analysis Tool (RAT) with representative recombination sequences EPI_ISL_10819657 (XD) from France and the sequence of related regional dominant Delta sub‐variant AY.4 (EPI_ISL_9315567) as well as that of Omicron sub‐variant BA.1 (EPI_ISL_9402095) (Figure 1B). We found that XD had the Delta backbone except for the region in the Spike protein.
Figure 1.
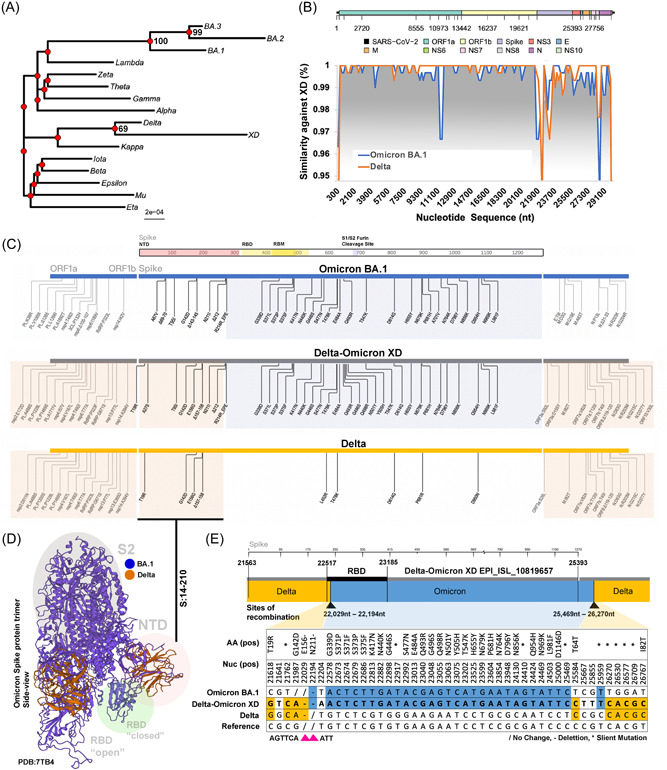
Multiple recombination events of the Delta–Omicron XD. (A) Phylogenetic analysis of the first reported sequence of Delta–Omicron XD by Institut Pasteur has been compared with VOCs, VOIs, and Omicron major subvariants using ML with 1000 bootstrap replicate. (B) Recombination analysis using Delta–Omicron XD as the background template. Recombination events against Omicron BA.1 are highlighted in light blue, and recombination events against Delta are highlighted in light orange. (C) Graphical illustration of amino acid comparison between Delta–Omicron XD, Delta, and Omicron BA.1, emphasizing Spike gene. Similar shared amino acid mutations were grouped and highlighted in orange (Delta–Omicron XD and Delta) and blue (Delta‐Omicron XD and Omicron BA.1). (D) Shared mutations were illustrated using Omicron BA.1 Spike structure (PDB: 7TB4). Regions in the Spike protein were highlighted for NTD (red), RBD (green), and S2 (grey). (E) Pairwise amino acid and nucleotide comparisons of shared mutations were illustrated between Delta–Omicron XD, Delta, and Omicron BA.1. Potential sites or ranges of recombination were depicted using black arrows. ML, maximum likelihood; VOCs, variants of concerns; VOIs, variants of interest.
Further examining the mutations in the Spike protein (PDB: 7TB4) showed potential recombinations in the RBD‐S2 (aa. 210‐1273) region (Figure 1C,D). Finally, comparing the nucleotide mutations of the sequences showed one or more potential recombination events: The first event between 22 029nt–22 194nt and the second event between 25 469nt–26 270nt. The total Omicron BA.1 recombinant size is between 3275 and 4241 bp (Figure 1E).
Furthermore, the phylogenetic analyses for partial regions segmented by pairwise nucleotide comparison suggested that the Omicron BA.1 or Delta AY.4 recombined region that matches in the XD variant showed the highest phylogenetic similarity to the corresponding genomes (Figure 2A,B). Our data implied potential one or more recombination events between Delta and Omicron variants leading to the formation of the Delta–Omicron XD.
Figure 2.
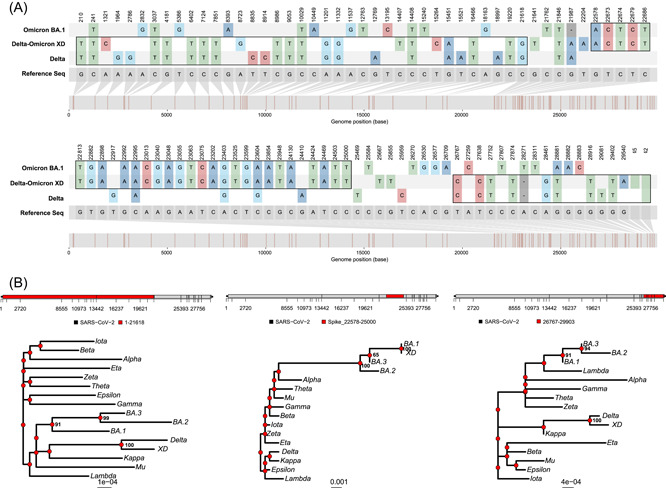
Pairwise nucleotide comparison and phylogenetic analysis of Delta–Omicron XD. (A) Pairwise nucleotide comparison of Delta, Delta–Omicron XD and Omicron BA.1 against the reference sequence (Wuhan‐Hu‐1). (B) Phylogenetic analysis of different portions of the first reported sequence of Delta–Omicron XD has been compared with VOCs, VOIs, and Omicron subvariants. VOCs, variants of concerns; VOIs, variants of interest.
Multiple putative recombination lineages have been raised by researchers in issues online (https://github.com/cov-lineages/pango-designation/issues), and then have been designated as lineages starting with the letter “X” by Pango nomenclature, including the lineage XD (Issue 444), XE (Issue 454), XF (Issue 445), XG (Issue 447), XH (Issue 448), XJ (Issue 449), XK (Issue 460), XL (Issue 464), XM (Issue 472), XS (Issue 471), and more. Here, we selected one qualified sequence for each putative recombinant lineage from the GISAID database and compared the mutations between the sequences (XD:EPI_ISL_10819657, XE:EPI_ISL_11835954, XF:EPI_ISL_9027096, XG:EPI_ISL_9270822, XH:EPI_ISL_9120017, XJ:EPI_ISL_12624883, XK:EPI_ISL_10864857, XL:EPI_ISL_10480052, XM:EPI_ISL_10307024, XS:EPI_ISL_10998033) and their parental lineages (BA.1:EPI_ISL_7605614, BA.2:EPI_ISL_9423053, Delta:EPI_ISL_9615237, BA.1.1:EPI_ISL_9715759). The mosaic genome structures shown in Figure 3A indicate that the lineage BA.1 (or BA.1.1) prefers to preserve the 5′‐terminal of the SARS‐CoV‐2 genome while the lineage BA.2 almost keeps the 3′‐terminal when they recombination with each other. Additionally, the recombination breakpoints were mostly distributed among the ORF1ab gene.
Figure 3.
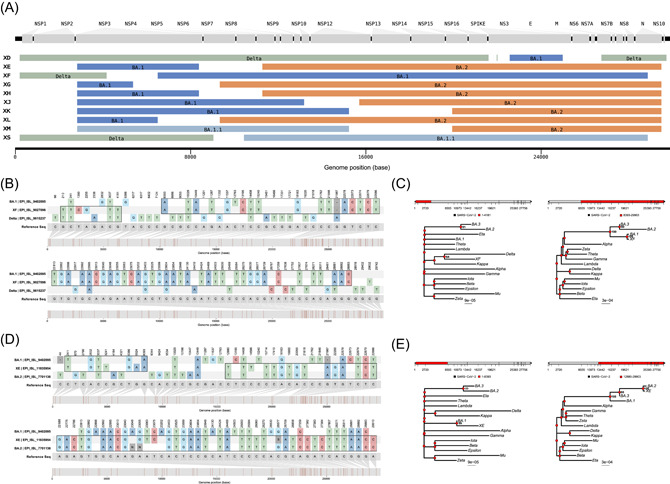
Pairwise nucleotide comparison and phylogenetic analysis of Delta–Omicron XF and XE. (A) Sequences of putative recombinants raised by Pango nomenclature exhibit the mosaic genomes structures composed of parental lineages. Tracts colored green, dark blue, pale blue, and orange represent the lineage Delta, BA.1, BA.1.1, and BA.2, respectively. Gaps represent the vague positions for recombinant breakpoints, there are no lineage‐specific mutations within these regions. The parental lineages for each putative recombinant are noted on the corresponding bar. (B) Pairwise nucleotide comparison of Delta, XF, and Omicron BA.1 against the reference sequence (Wuhan‐Hu‐1). (C) Phylogenetic analysis of different portions of the reported sequence of XF has been compared with VOCs, VOIs, and Omicron subvariants. (D) Pairwise nucleotide comparison of XE, Omicron BA.1, and BA.2 against the reference sequence (Wuhan‐Hu‐1). (E) Phylogenetic analysis of different portions of the first reported sequence of XE has been compared with VOCs, VOIs, and Omicron subvariants. VOCs, variants of concerns; VOIs, variants of interest.
We have also analyzed the XF and XE variants. Phylogenetic analysis of the entire XF sequence shared a high likelihood with the BA.1 (Figure 3B). Phylogenetic analysis of XF (EPI_ISL_9027096) against BA.1 (EPI_ISL_9402095) and Delta (EPI_ISL_9615237) suggested a single recombination event at the site: 4181nt–8393nt (Figure 3C). Sequence comparison of the entire XE sequence shared high sequence similarity with the BA.2 (Figure 3D). Phylogenetic analysis of XE (EPI_ISL_11835954) against BA.1 (EPI_ISL_9402095) and BA.2 (EPI_ISL_7701136) showed a single recombination event between BA.1 and BA.2 at the site: 8393nt–12880nt (Figure 3E). Thus, the Delta–Omicron XD is likely a recombination of the Delta (AY.4) and Omicron BA.1. In addition to XD, XF is also a potential combination of Delta and Omicron, while XE combines Omicron subvariant BA.1 and BA.2.
3.2. Omicron BA.3 shares mutations with BA.1 and BA.2
The discovery of these inter‐variant and intra‐variant Omicron‐related recombinants drives us to hypothesize other potential recombination events within the Omicron sublineages. There are currently five main sub‐variants in the Omicron lineage (Figure 4A); BA.1 and BA.2 sequences were the most prevalent in the past few months (Figure 5A). Due to their newly identified and relatively short prevalence time, insufficient sequencing data is available for BA.4 and BA.5. Therefore, they were excluded from this analysis. To examine the relationship between the BA.1‐3, we first performed a phylogenetic analysis for each subvariant from the GISAID to determine the most ancestral genomes known while ensuring high quality and coverage (Figure 4B). BA.1 (EPI_ISL_9402095), BA.2 (EPI_ISL_7701136), and BA.3 (EPI_ISL_8616600) were selected for multiple sequences alignment analysis and phylogenetic analysis (Figure 4B). Due to a large number of shared mutations between BA.1‐3 and the mixed existence of two BA.1/BA2 feature mutations in the Spike protein of BA.3, it cannot be inferred directly that the BA.3 is a recombinant of current BA.1 and BA.2 (Figure 4D). However, the number of unique single nucleotide polymorphisms (SNPs) and shared (between two genomes) SNPs (Figure 5B) provides extra clues. BA.1 and BA.2 showed similar numbers of unique and shared SNPs, during BA.3 showed only three unique SNPs but significantly higher (21) shared SNPs (Figure 4C). The unique SNPs suggest that BA.3 is newer than BA.1‐2, but more shared mutations with BA.1‐2 imply close genetic distance. Therefore, we hypothesize that BA.3 may have been recombined from a more ancestorial BA.1 and BA.2 (Figure 5C). To test this hypothesis, we further examined the mutations in the BA.3 Spike protein (PDB: 7TB4) and found that potential recombination events may occur in the NTD and S2 regions (Figure 5D,E). The BA.3 Spike protein mutations are like other Omicron subvariants and can increase susceptibility against antibodies, vaccines, and serological treatments (Figure 5F). Taken together, our data showed that BA.3 may have gained many of its SNPs from BA.1 and BA.2, but not from the currently circulating lineages.
Figure 4.
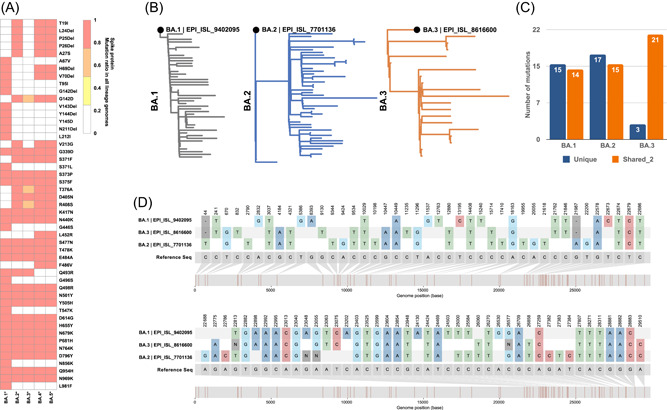
Epidemiology assessment of the Omicron variants. (A) Global mutation prevalence of subvariants of Omicron and other existing subclades in South Africa. (B) Phylogenetic analysis of Omicron subvariants (BA.1, BA.2, and BA.3) to determine the most ancestral sequence. (C) The total number of unique mutations from each subvariant and the number of shared mutations between two subvariants. (D) Nucleotide comparison of BA.1, BA.2, and BA.3 against the reference sequence (Wuhan‐hu‐01).
Figure 5.
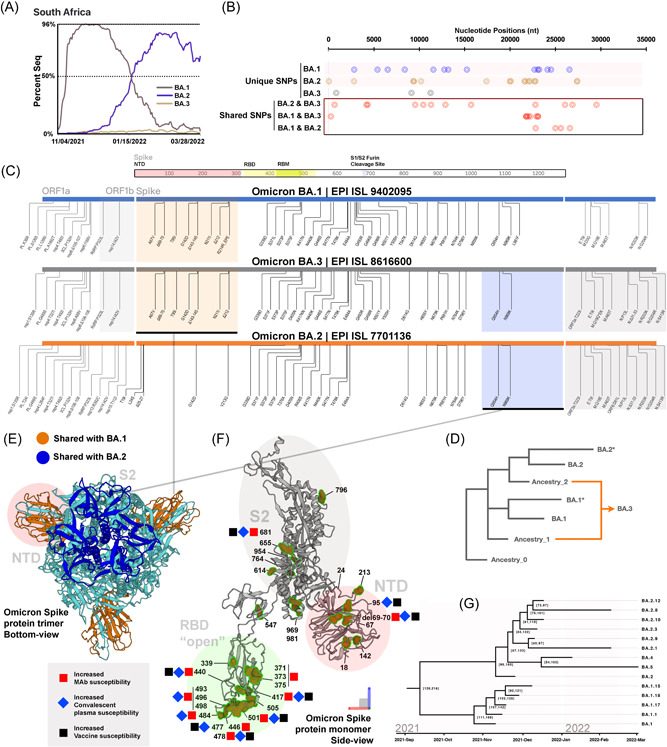
Omicron BA.3 shares mutations with BA.1 and BA.2. (A) Data obtained from GISAID showed the percent of sequence isolated from Omicron subvariant in South Africa between November 4, 2021 and March 28, 2022. (B) Unique and shared mutations of BA.1, BA.2, and BA.2. (C) Graphical illustration of amino acid comparison between Omicron subvariants, emphasizing Spike gene. Similar shared amino acid mutations were grouped and highlighted in orange (BA.1 and BA.3) and blue (BA.2 and BA.3). (D) Graphical illustration of potential ancestral lineages of Omicron BA.3. Hypothetical ancestry lineages were determined by comparing the ancestral BA.3 sequence against BA.1 and BA.2. (E) Shared mutations from BA.1 and BA.2 were illustrated using Omicron BA.1 Spike structure (PDB: 7TB4). Regions in the Spike protein were highlighted for NTD (red), RBD (green), and S2 (grey). (F) Mutations of all BA.3 sequences were labeled using the same structure. Sites are previously shown to have altered the susceptibility treatments, and vaccines were highlighted next to the amino acid. (G) The molecular clock of the Omicron main subvariants.
3.3. Molecular clock analysis of Omicron BA.1, 2, 4, and 5
The origin of the Omicron variant remains a mystery. To infer the possible divergent time of the current identified BA.1‐5, a Bayesian phylogenetic analysis was performed on the dominant sub‐variant of Omicron via BEAST v1.10.4. BA.3 was excluded from this analysis because it was inferred to be a potential recombinant of BA.1 and BA.2 (Figure 5G). Our data suggest that the first Omicron variant is likely to have originated before September 2021, followed by the formation of BA.1 in October and that of BA.2 in November. These findings could explain that BA.2 showed a selective advantage over BA.1 but became the dominant lineage later than BA.1. The newly identified BA.4 and BA.5 have a close relationship with BA.2. However, they were very different from other minor subvariants of BA.2, suggesting these subvariants are more likely to evolve from the ancestorial BA.2.
4. DISCUSSION
The SARS‐CoV‐2 is likely to continue spreading worldwide with the periodic emergence of new lineages. While mutations in the viral genomes are mainly responsible for the evolution of new lineages, recombination between two different viral lineages can undoubtedly accelerate the viral evolution process. Recently, a study by Dr. Bedford's lab suggested that SARS‐CoV‐2 recombination is a widespread phenomenon. 16 The present study has examined possible recombination events between multiple inter and intra‐variants. We showed that Delta‐Omicron XD isolated from France in early 2022 has the backbone of Delta variant with a segment of Omicron BA.1 from the Spike gene corresponding to the RBD to the 3a gene, spanning around 3.5 K in length. As a result of such recombinations, the Delta‐Omicron XD has more Spike protein mutations than the Delta variant with the potential to increase the infectivity and evade antibody and vaccine‐mediated protection. However, further studies are needed to determine whether XD is as infectious and efficient at immune escaping as Omicron variant while causing similar disease severity as Delta variant. We further showed that recombination might have also occurred in the XF and XE variants.
The cocirculation of multiple lineages in one place in the same period was not rare, especially when the Alpha variant outcompeted previous lineages in early 2021 and the Delta variant outcompeted Alpha and other lineages in the middle of 2021. However, recombination of different variants has been detected mainly after the Omicron variant. Thus, why were recombinants seen more frequently between the Omicron and other variants? The high genetic divergence of the Omicron variant compared with different lineages is an apparent reason. This could lower the required sensitivity of the detecting software and engage more robust recombination analysis. On the other hand, the wide‐spreading and regional divergence of the Omicron variant could also promote the recombination of Omicron because this process provides more opportunities for co‐infections. Although not confirmed, it is interesting to explore if Omicron subvariants with similar genetic distances could prolong these subvariants' co‐exist in hosts and, therefore, provide a suitable environment for recombination.
Despite the inter‐recombination of BA.1 and BA.2 (XE), Our study in Omicron sub‐variants also provided novel insight into the genetic evolution of BA.3. We found that the BA.3 seemed to have a later origination time. However, most mutations of BA.3 are either shared with BA.2 or with BA.3, suggesting a potential recombination event with an Omicron subvariant that is even older than the selected sequences. Since the recombination will interfere with the inference of the phylogenetic analysis, recombination analysis will need to be deployed before phylogeny.
There were several limitations to the current study. Firstly, the biased subsampling of SARS‐CoV‐2 samples could hamper a comprehensive analysis of the virus evolution. The lack of detailed and continuous sampling in South Africa makes tracing Omicron ancestry more difficult. Secondly, the inference of the original Omicron relies on the hypothesis that the Omicron variant comes from direct evolution rather than multiple recombinations of Alpha, Delta, or other variants. Thus, the erroneous results may be drawn if the Omicron variant was derived from multiple recombinations. Thirdly, there are a limited number of BA.3 sequences at the time of the study, and no direct evidence could be provided to confirm the recombination of BA.3 without the sequences of ancestral BA.1 and ancestral BA. Furthermore, we also didn't account for potential APOBEC deamination which led to C‐to‐T transitions. The current result was a relatively reasonable hypothesis of the observed phenomenon.
The current landscape of the COVID‐19 pandemic raises new questions on the molecular mechanisms responsible for the rapidly evolving SARS‐CoV‐2 variants. Our studies have provided evidence on the contribution of both inter‐variant and intra‐variant recombination of SARS‐CoV‐2 viral genomes in developing recently emerged novel variants such as XD, XE, XF, and BA3. Such recombination events can accelerate the evolution of highly infectious and disease‐causing lineages and may weaken the protection by current vaccines.
AUTHOR CONTRIBUTIONS
All authors contributed to the study designing, data analysis, and manuscript writing. Lulan Wang, Hang‐Yu Zhou, and Jia‐Ying Li performed the analysis and wrote the manuscript. Genhong Cheng and Aiping Wu co‐supervised this study. All authors have revised and approved the final draft of the paper.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
ACKNOWLEDGMENTS
This project is supported by the Research Funds from US National Institute of Health funds (AI069120, AI158154, AI149718, and AI155232), the UCLA AIDS Institute, and UCLA David Geffen School of Medicine – Eli and Edythe Broad Center of Regenerative Medicine and Stem Cell Research Award Program, Microbial Pathogenesis Training Grant (AI7323‐31). National key research and development program (2021YFC2301305); the CAMS Innovation Fund for Medical Sciences (2021‐I2M‐1‐061); the National Natural Science Foundation of China (92169106); the Non‐profit Central Research Institute Fund of Chinese Academy of Medical Sciences (2021‐PT180‐001); China postdoctoral science foundation grants (2020T130007ZX); Suzhou science and technology development plan (szs2020311); the Youthful Teacher Project of Peking Union Medical College (3332019114). We greatfully acknowledge all data contributors, i.e. the Authors and their Originating laboratories responsible for obtaining the specimens, and their Submitting laboratories for generating the genetic sequence and metadata and sharing via the GISAID Initiative, on which this research is based.
Wang L, Zhou H‐Y, Li J‐Y, et al. Potential intervariant and intravariant recombination of Delta and Omicron variants. J Med Virol. 2022;94:4830‐4838. 10.1002/jmv.27939
Lulan Wang, Hang‐Yu Zhou, and Jia‐Ying Li contributed equally to the study.
Contributor Information
Aiping Wu, Email: wap@ism.cams.cn.
Genhong Cheng, Email: gcheng@mednet.ucla.edu.
DATA AVAILABILITY STATEMENT
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
REFERENCES
- 1. Lauring AS, Tenforde MW, Chappell JD, et al. Clinical severity of, and effectiveness of mRNA vaccines against, covid‐19 from omicron, delta, and alpha SARS‐CoV‐2 variants in the United States: prospective observational study. BMJ. 2022;376:e069761. 10.1136/bmj-2021-069761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Viana R, Moyo S, Amoako DG, et al. Rapid epidemic expansion of the SARS‐CoV‐2 Omicron variant in Southern Africa. Nature. 2022;603(7902):679‐686. 10.1038/s41586-022-04411-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang L, Cheng G. Sequence analysis of the emerging SARS‐CoV‐2 variant O in South Africa. J Med Virol . 2021;1728‐1733. 10.1002/jmv.27516 [DOI] [PubMed]
- 4. Cao Y, Wang J, Jian F, et al. Omicron escapes the majority of existing SARS‐CoV‐2 neutralizing antibodies. Nature. 2022;602(7898):657‐663. 10.1038/s41586-021-04385-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ou J, Lan W, Wu X, et al. Tracking SARS‐CoV‐2 Omicron diverse spike gene mutations identifies multiple inter‐variant recombination events. Signal Transduct Target Ther. 2022;7(1):138. 10.1038/s41392-022-00992-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Simon‐Loriere E, et al. Rapid characterization of a Delta‐Omicron SARS‐CoV‐2 recombinant detected in Europe. Res. Sq . 2022. 10.21203/rs.3.rs-1502293/v1 [DOI]
- 7. Lacek KA, Rambo‐Martin BL, Batra D, et al. Identification of a novel SARS‐CoV‐2 Delta‐Omicron recombinant virus in the United States. bioRxiv . Published online January 1, 2022. 2022.03.19.484981 10.1101/2022.03.19.484981 [DOI]
- 8. Shu Y, McCauley J. GISAID: global initiative on sharing all influenza data ‐ from vision to reality. Euro Surveill: Bull. 2017;22(13):30494. 10.2807/1560-7917.ES.2017.22.13.30494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular biology and evolution. 2013;30(4):772‐780. 10.1093/molbev/mst010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen C, Nadeau S, Yared M, Voinov P, Xie N, Roemer C, Stadler T. CoV‐Spectrum: Analysis of Globally Shared SARS‐CoV‐2 Data to Identify and Characterize New Variants. Bioinformatics. 2021;38:1735‐1737. 10.1093/bioinformatics/btab856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nguyen LT, Schmidt HA, Von Haeseler A, Minh BQ. IQ‐TREE: a fast and effective stochastic algorithm for estimating maximum‐likelihood phylogenies. Molecular biology and evolution. 2015;32(1):268‐274. 10.1093/molbev/msu300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yu G, Smith DK, Zhu H, Guan Y, Lam TTY. ggtree: an R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods in Ecology and Evolution. 2017;8(1):28‐36. 10.1111/2041-210X.12628 [DOI] [Google Scholar]
- 13. Etherington GJ, Dicks J, Roberts IN. Recombination Analysis Tool (RAT): a program for the high‐throughput detection of recombination. Bioinformatics. 2005;21:278‐281. 10.1093/bioinformatics/bth500 [DOI] [PubMed] [Google Scholar]
- 14. Tzou PL, Tao K, Pond SLK, Shafer RW. Coronavirus resistance database (CoV‐RDB): SARS‐CoV‐2 susceptibility to monoclonal antibodies, convalescent plasma, and plasma from vaccinated persons. PLoS One. 2022;17(3):e0261045. 10.1371/journal.pone.0261045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Suchard MA, Lemey P, Baele G, Ayres DL, Drummond AJ, Rambaut A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018;4(1):vey016. 10.1093/ve/vey016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Müller NF, Kistler KE, Bedford T Recombination patterns in coronaviruses. bioRxiv. Published online January 1, 2021. 10.1101/2021.04.28.441806 [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.