

Sir,
Tay–Sachs disease (TSD) is one of the common glycolipid storage disorders with an incidence of 1 in 100,000 live births.[1] TSD (OMIM # 272800) is a result of biallelic pathogenic variants in the HEXA gene that causes deficiency of β-hexosaminidase A (HexA) enzyme (EC 3.2.1.52). This is further categorized into a classic infantile form, sub-acute juvenile form, and late-onset form, depending on the age of onset of symptoms. Notably, India has a TSD case mostly with the infantile phenotype[2] whereas juvenile or late-onset forms have been rarely reported.
Here we describe the second case of juvenile TSD from India along with a review of previously reported juvenile TSD cases having confirmed genetic study.
The proband is the first child born to a non-consanguineous couple and was referred to us at 12 years of age. He had a normal development till the age of 5 years and progressive deterioration of the learned skill with bilateral tremors thereafter. On presentation at our centre, he had gait ataxia, difficulty in climbing stairs, slurred speech, difficulty in getting up and down. Magnetic Resonance Imaging (MRI) scans of the brain showed mild cortical atrophic changes [Figure 1]. On eye examination, cherry red spot was absent. An IQ assessment study showed IQ level to be 33.3. The clinical presentation suggested a neurodegenerative disorder with a strong suspicion of TSD. Our differentials included Sandhoff disease, neuronal ceroid lipofuscinosis, Friedreich ataxia and late-onset spinal muscular atrophy.
Figure 1.
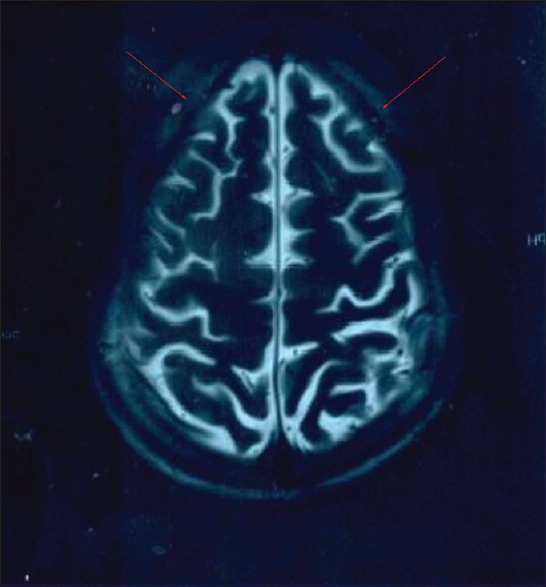
T2 weighted MRI images of the brain showing cortical atrophy
To confirm the clinical diagnosis, a lysosomal enzyme β-hexosaminidase A study was carried out from the leucocytes. The test showed β-hexosaminidase A activity to be 3.1 nmol/hr/mg protein, which was less than 10% of the normal range (62.7–659.4 nmol/hr/mg protein). Further, genomic DNA extracted from blood sample was used to carry out neurology gene panel study. This identified a compound heterozygous variant c.1496G>A (chr15-72637817) (p.Arg499His) in exon 13 and c.902T>G (chr15-72641504) (p.Met301Arg) in exon 8 of the HEXA gene. As per ACMG guidelines, these variants were classified as pathogenic and likely pathogenic, respectively. The results were validated in the proband and both parents by bidirectional sequencing of the coding region of the HEXA gene (ENST00000268097). This study confirmed the presence of both variants in compound heterozygous state in the proband and heterozygous state in both parents.
The juvenile form of TSD is a rare and progressive neurodegenerative disorder with a heterogeneous clinical course.[3] To date, 155 cases of juvenile TSD have been reported in the literature including a single case from India.[4] The mean age of onset was 5.24 ± 3.9 years. We found that dysarthria and gait ataxia are the most common clinical signs, seen in 96.5% and 93.1% of the cases, respectively.[3,5,6,7,8,9] In the present case also, there was a similar observation with an additional sign of bilateral tremors in hand at 5 years of age which has been seen in only 26.72% of the previously reported cases.[3] The MRI findings showing cortical atrophy in our case are consistent with those observed in other juvenile TSD cases.
Sandhoff et al.[10] have suggested an inverse correlation between the heterogeneity of onset and the residual activity of the β-hexosaminidase enzyme.[10] Patients with the juvenile forms of TSD may have 5-10% of wild-type enzyme activity that lies in the range of 2 to 9 nmol/hr/mg protein.[3,5,6,7,8,9] Though, previous large study of infantile cases and present case of juvenile TSD could not find a correlation between enzyme activity and the onset of disease.[2] In the present case, this could be due to presence of one heterozygous variant (c.902T>G) which is commonly associated with infantile TSD.
On literature review, we found, forty-one variants in HEXA to be observed with juvenile TSD [Table 1, Figure 2]. The two most common variants found in juvenile TSD are c.1496G>A (p.Arg499His) and c.533G>A (p.Arg178His) found in exon 13 and 5, respectively, in 25.4% of the 67 patients including the present case.[3,5,6,7,8,9]
Table 1.
Review of molecularly proven cases of juvenile TSD
| Exon/Intron | Variant | Type of variant | Ethnicity | Percentage of juvenile TSD patients with the variant |
|---|---|---|---|---|
| Exon 1 | c.1A>T (p.M1L) | Start loss | Multiple Ethnic Groups | 1.5% |
| Exon 1 | c.1A>G (p. M1V) | Start loss | African-American | 1.5% |
| Exon 1 | c.10T>C (p.S4P) | Missense | Multiple Ethnic Groups | 1.5% |
| Exon 1 | c.32T>C (p.L11P) | Missense | Japanese | 1.5% |
| Exon 1 | c.155C>A (p.S22X) | Nonsense | Spanish/Portuguese | 1.5% |
| Exon 1 | c.77G>A (p.W26X) | Nonsense | Multiple Ethnic Groups | 3% |
| Exon 1 | c.78G>A (p.W26X) | Nonsense | Cyprus | 1.5% |
| Exon 1 | c.109T>A (p.Y37N) | Missense | Multiple Ethnic Groups | 3% |
| Exon 1 | c.173G>A (p.C58Y) | Missense | NA | 1.5% |
| Exon 3 | c.409C>T (p.R137X) | Nonsense | Multiple Ethnic Groups | 4.5% |
| Intron 4 | c.459+5G>A | Non-Coding | Spanish | 3% |
| Exon 5 | c.509G>A (p.R170Q) | Missense | NA | 1.5% |
| Exon 5 | c.533G>A (p.R178H) | Missense | Multiple Ethnic Groups | 25.4% |
| Exon 5 | c.536A>G (p.H179R) | Missense | Spanish | 1.5% |
| Exon 5 | c.566G>A (p.R189H) | Missense | NA | 3% |
| Intron 5 | c.571_1G>T | Splicing | Japanese | 1.5% |
| Intron 6 | c.672+1G>A | Splicing | Multiple Ethnic Groups | 3% |
| Exon 7 | c.681C>A (p.Y227X) | Nonsense | NA | 1.5% |
| Exon 7 | c.736_737delG (p.A246R) | Missense | Spanish | 1.5% |
| Exon 7 | c.736G>A (p.A246T) | Missense | Korean | 1.5% |
| Exon 7 | c.749G>A (p.G250D) | Missense | Lebanese Maronite | 1.5% |
| Exon 7 | c.772G>C (p.D258H) | Missense | NA | 1.5% |
| Exon 7 | c.805G>A (p.G269S) | Missense | Multiple Ethnic Groups | 9% |
| Exon 8 | c.814G>A (p.G272R) | Missense | West Indian Origin | 1.5% |
| Exon 8 | c.902T>G (p.M301R) | Missense | Multiple Ethnic Groups | 1.5% (*Present case) |
| Exon 8 | c.972T>A (p.V324V) | Synonymous | Multiple Ethnic Groups | 1.5% |
| Exon 9 | c.1003A>T (p.I335F) | Missense | Spanish/Portuguese | 1.5% |
| Intron 9 | c.1073+1G>A | Splicing | Spanish | 6% |
| Intron 10 | c.1146+1G>A | Splicing | Spanish | 1.5% |
| Exon 11 | c.1195A>G (p.N399D) | Missense | West Indian Origin | 1.5% |
| Exon 11 | c.1274_1277dupTATC (p.Y427X) | Nonsense | Multiple Ethnic Groups | 17.9% |
| Exon 11 | c.1281T>A (p.Y427X) | Nonsense | India | 1.5% |
| Exon 11 | c.1305C>T (p.Y435Y) | Synonymous | Multiple Ethnic Groups | 6% |
| Exon 12 | c.1382G>T (p.G461V) | Missense | Multiple Ethnic Groups | 3% |
| Intron 12 | c.1421+5G>C | Non coding | Spanish | 1.5% |
| Exon 13 | c.1422G>C (p.W474C) | Missense | German-Dutch | 3% |
| Exon 13 | c.1496G>A (p.R499H) | Missense | Multiple Ethnic Groups | 25.4% (*Present case) |
| Exon 13 | c.1495C>T (p.R499C) | Missense | NA | 3% |
| Exon 13 | c.1511G>A (p.R504H) | Missense | Multiple Ethnic Groups | 4.5% |
| Exon 13 | c.1511G>T (p.R504L) | Missense | Argentina | 1.5% |
| Exon 14 | c.1529_1530del (p.R510H) | Missense | India | 1.5% |
Figure 2.

Schematic diagram of HEXA gene structure with functional domains showing the 41 reported variants in juvenile TSD
Both variants in the present case: c.1496G>A (p.Arg499His) and c.902T>G (p.Met301Arg) have been previously reported in the literature in multiple ethnicity. Interestingly, c.1496G>A has been observed in affected TSD patients from various ethnic backgrounds like Caucasian, Argentinean, Portuguese and Italian populations.[7,9] This variant is located outside the catalytic domain, and hence causes minor structural changes, which explains the late-onset clinical phenotype. While the other variant, c.902T>G (p.Met301Arg), has been reported only twice in the literature for infantile TSD. In one case, it was in homozygous state, whereas in another case, it was present in combination with another pathogenic HEXA variant p.Arg504His. This variant is located in the catalytic domain of the α- subunit of β-hexosaminidase A. Although the effect of this variant on the enzyme is unclear, it has been hypothesized that the association process of the two subunits (α and β) of the enzyme might be affected.[11]
Thus, based on the reported cases in the literature and present case, it is likely possible to predict the onset of symptoms and the disease severity depending on the mutation in HEXA gene and its subsequent effect on the residual β-hexosaminidase A activity. Hence, establishing genotype-phenotype correlation is critical to understand the patient prognosis and plan effective management of the condition.
Present case highlights the rarity of juvenile TSD in India and shows bilateral tremors as an early sign in this condition. The variant c.902T>G in the HEXA has been reported in infantile forms of TSD. Nonetheless, due to presence of c.1496G>A, a common variant in juvenile TSD, the index case has shown a milder phenotype with juvenile onset.
Financial support and sponsorship
This work was partly supported by Gujarat State Biotechnology Mission (GSBTM) (grant no: GSBTM/JDR &D/608/2020/459-461).
Conflicts of interest
There are no conflicts of interest.
Acknowledgements
We express our thanks to the patient and his parents for their cooperation. We thank Dr. Heli Shah for referring this case. We also thank Dr. Harsh Sheth for his partial help in the reading and making critical suggestions for the manuscript.
REFERENCES
- 1.Lew RM, Burnett L, Proos AL, Delatycki MB. Tay-Sachs disease: Current perspectives from Australia. Appl Clin Genet. 2015;8:19–25. doi: 10.2147/TACG.S49628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mistri M, Tamhankar PM, Sheth F, Sanghavi D, Kondurkar P, Patil S, et al. Identification of novel mutations in HEXA gene in children affected with tay sachs disease from India. PLoS One. 2012;7:e39122. doi: 10.1371/journal.pone.0039122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maegawa GHB, Stockley T, Tropak M, Banwell B, Blaser S, Kok F, et al. The natural history of juvenile or subacute GM2 gangliosidosis: 21 new cases and literature review of 134 previously reported. Pediatrics. 2006;118:e1550–62. doi: 10.1542/peds.2006-0588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Udwadia-Hegde A, Hajirnis O. Temporary efficacy of pyrimethamine in Juvenile-Onset Tay-Sachs disease caused by 2 unreported HEXA mutations in the Indian population. Child Neurol Open. 2017;4:2329048X16687887. doi: 10.1177/2329048X16687887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith NJ, Winstone AM, Stellitano L, Cox TM, Verity CM. GM2 gangliosidosis in a UK study of children with progressive neurodegeneration: 73 cases reviewed. Dev Med Child Neurol. 2012;54:176–82. doi: 10.1111/j.1469-8749.2011.04160.x. [DOI] [PubMed] [Google Scholar]
- 6.Sakurai M, Azuma J, Hamada Y, Yamamoto T, Sakai N. Early juvenile Tay-Sachs disease with atypical symptoms. Pediatr Int. 2019;61:611–3. doi: 10.1111/ped.13848. [DOI] [PubMed] [Google Scholar]
- 7.Rozenberg R, Kok F, Burin MG, Sá Miranda MC, Vasques C, Henriques-Souza AMM, et al. Diagnosis and molecular characterization of non-classic forms of Tay-Sachs disease in Brazil. J Child Neurol. 2006;21:540–4. doi: 10.1177/08830738060210061101. [DOI] [PubMed] [Google Scholar]
- 8.Ou L, Kim S, Whitley CB, Jarnes-Utz JR. Genotype-phenotype correlation of gangliosidosis mutations using in silico tools and homology modeling. Mol Genet Metab Rep. 2019;20:100495. doi: 10.1016/j.ymgmr.2019.100495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.King KE, Kim S, Whitley CB, Jarnes-Utz JR. The juvenile gangliosidoses: A timeline of clinical change. Mol Genet Metab Rep. 2020;25:100676. doi: 10.1016/j.ymgmr.2020.100676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sandhoff K. My journey in to the world of Sphingolipids and Spingolipidosis. Proc Jpn Acad Ser B Phys Biol Sci. 2012;88(10):554–582. doi: 10.2183/pjab.88.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheema H, Bertoli-Avella AM, Skrahina V, Anjum MN, Waheed N, Saeed A, et al. Genomic testing in 1019 individuals from 349 Pakistani families results in high diagnostic yield and clinical utility. NPJ Genom Med. 2020;5:44. doi: 10.1038/s41525-020-00150-z. [DOI] [PMC free article] [PubMed] [Google Scholar]