

Abstract
Betaine aldehyde dehydrogenase (BADH) (EC 1.2.1.8) catalyzes the last, irreversible step in the synthesis of the osmoprotectant glycine betaine from choline. In Pseudomonas aeruginosa this reaction is also an obligatory step in the assimilation of carbon and nitrogen when bacteria are growing in choline or choline precursors. We present here a method for the rapid purification to homogeneity of this enzyme by the use of ion-exchange and affinity chromatographies on 2′,5′-ADP–Sepharose, which results in a high yield of pure enzyme with a specific activity at 30°C and pH 7.4 of 74.5 U/mg of protein. Analytical ultracentrifugation, gel filtration, chemical cross-linking, and sodium dodecyl sulfate-polyacrylamide gel electrophoresis suggest that BADH from P. aeruginosa is a homodimer with 61-kDa subunits. The amino acid composition and the N-terminal sequence of 21 amino acid residues showed significant similarity with those of the enzymes from Xanthomonas translucens and Escherichia coli. Neither BADH activity nor BADH protein was found in cell extracts from bacteria grown in the absence of choline. In contrast to other BADHs studied to date, the Pseudomonas enzyme cannot use positively charged aldehydes other than betaine aldehyde as substrates. The oxidation reaction has an activation energy of 39.8 kJ mol−1. The pH dependence of the velocity indicated an optimum at pH 8.0 to 8.5 and the existence of two ionizable groups with macroscopic pK values of 7.0 ± 0.1 and 9.7 ± 0.1 involved in catalysis and/or binding of substrates. The enzyme is inactivated at 40°C, but activity is regained when the heated enzyme is cooled to 30°C or lower. At the optimum pH of 8.0, the enzyme is inactivated by dilution, but it is stable at pH 6.5 even at very low concentrations. Also, P. aeruginosa BADH activity is rapidly lost on removal of K+. In all cases studied, inactivation involves a biphasic process, which was dependent on the enzyme concentration only in the case of inactivation by dilution. NADP+ considerably protected the enzyme against these inactivating conditions.
It has been suggested that the virulence of the opportunistic pathogen Pseudomonas aeruginosa might be related to its ability to adapt to osmotic stress (45), a stressful condition which is frequently present at infection sites, such as in lungs of patients suffering from cystic fibrosis (30). It is known that P. aeruginosa is able to grow under osmotic stress, particularly in the presence of the osmoprotectant glycine betaine or compounds such as phosphatidylcholine, acetylcholine, phosphorylcholine, and choline (13, 27), which produce glycine betaine through the sequence of reactions shown in Fig. 1.
FIG. 1.

Production of glycine betaine. PLC, phospholipase C; PcPase, phosphorylcholine phosphatase; ChE, acetyl cholinesterase; CDH, choline dehydrogenase.
It is interesting that the glycine betaine precursors are very abundant at infection sites (39, 41); for instance, phosphatidyl choline constitutes 70% of the lung surfactant (58). Accordingly, the virulence of P. aeruginosa has been linked to the expression of phospholipase C (37), which is induced by phosphorylcholine, choline, and glycine betaine (28, 37). In addition, P. aeruginosa can grow on choline or glycine betaine as the sole carbon, nitrogen, and energy source (35); therefore, glycine betaine may play the dual role of an osmoprotectant and a metabolic intermediate in the catabolism of choline or choline precursors. In this respect, P. aeruginosa is similar to Xanthomonas translucens (34) and Sinorhizobium meliloti (2, 46) but is different from Escherichia coli, in which glycine betaine is involved only in the response of the bacteria to osmotic stress (24). Accordingly, appreciable levels of activity of betaine aldehyde dehydrogenase (BADH) (EC 1.2.1.8), the enzyme which catalyzes the final, irreversible step in the synthesis of glycine betaine, are found in P. aeruginosa cells grown in choline or choline precursors (35). Recently, Sage et al. (43) reported on a P. aeruginosa mutant deficient in BADH activity which is not able to grow either in choline or in glycine betaine plus choline, due to a toxic effect of the BADH substrate, betaine aldehyde, which accumulates in this mutant when choline is present.
BADH activity thus appears to be crucial for bacterial growth under the conditions of infection, i.e., osmotic stress plus an abundance of choline or choline precursors, and therefore appears to be a suitable target for antimicrobial agents. The development of selective antimicrobial agents would require much more information about the kinetic and physicochemical properties of the enzyme as well as easily available pure enzyme, which could be used to screen new compounds. Limited information on this enzyme was published more than two decades ago. The data were obtained with an enzyme purified by using a cumbersome and inefficient procedure which yielded enzyme with a very low specific activity (35). Recently, a different and much more efficient purification protocol was published (42), but it still involved four chromatographic steps, and the enzyme was not further characterized. Therefore, because of the importance of understanding the biochemical basis of the response of microorganisms to osmotic stress and the importance of possible future efforts in the development of drugs against P. aeruginosa, we found it of interest to develop a quick procedure for purifying BADH from this pathogen to homogeneity with high yields and to further investigate some of its physicochemical and kinetic properties. In addition, since N-terminal sequence data from the purified enzyme and antibodies against the pure enzyme would be valuable tools for cloning and expression of the corresponding gene, we determined the N-terminal sequence of and obtained monospecific polyclonal antibodies against this enzyme.
MATERIALS AND METHODS
Chemicals and biochemicals.
Betaine aldehyde chloride, glycine betaine (free base), choline chloride, NAD(P)+, NADPH, dithiothreitol (DTT), Tris, β-mercaptoethanol, urea, phenazine methosulfate, nitroblue tetrazolium, and Coomassie brilliant blue G and R were obtained from Sigma (St. Louis, Mo.). EDTA and glycerol were from Merck KGaA (Darmstadt, Germany). Ampholines and nitrocellulose membranes were from Bio-Rad (Hercules, Calif.), and Immobilon-PSQ membrane was from Millipore (Bedford, Mass.). 3-Dimethylsulfoniopropionaldehyde was a kind gift from A. D. Hanson (University of Florida, Gainesville). γ-Aminobutyraldehyde (diethylacetal form) was from Aldrich (Milwaukee, Wis.). γ-Aminobutyraldehyde chloride was freshly prepared from the diethylacetal form by the procedure described by Flores and Filner (15). Materials for column chromatography were purchased from Pharmacia Fine Chemicals (Uppsala, Sweden). Freund’s complete and incomplete adjuvants and horseradish peroxidase-linked goat anti-rabbit immunoglobulin G conjugate were from GIBCO BRL (Gaithersburg, Md.), and the enhanced chemiluminescence kit was from Pierce (Rockford, Ill.). All other chemicals, of analytical grade, were from standard suppliers.
Bacterial strains and culture conditions.
P. aeruginosa PAO1, kindly provided by M. L. Vasil (University of Colorado Health Sciences Center, Denver) was used in all experiments. Cells were grown aerobically at 37°C in liquid media. The basal medium used to grow the cells was essentially the M63 minimal medium described by Miller (32), except that, for maximum induction of BADH, we used 20 mM choline as the sole carbon and nitrogen source. The medium (1,800 ml) was inoculated with a seed culture (36 ml) in the log phase and was grown on a gyratory shaker (150 rpm) at 37°C until the stationary phase was reached. The cells from 1.8 liters of culture medium were harvested by centrifugation at 3,000 × g for 10 min and then resuspended in 90 ml of 50 mM potassium phosphate buffer (pH 6.5) containing 0.1 mM EDTA and 20 mM β-mercaptoethanol (buffer A).
Enzyme purification.
All operations were carried out at 4°C. The resuspended cells were disintegrated by sonic oscillation (90 s at 60 W) in a Branson (Danbury, Conn.) Sonifier Cell Disruptor. The slurry was centrifuged at 14,500 × g for 30 min. The supernatant (cell extract) was brought to 20% (wt/vol) sucrose (buffer B) and then applied to a Q-Sepharose Fast Flow column (1.8 by 6.5 cm) equilibrated with buffer B. The column was washed with the same buffer, and the enzyme was eluted with 110 ml of a linear salt gradient of 0 to 250 mM KCl in buffer B at a flow rate of 1 ml/min. Fractions with enzyme activity were pooled, and the pH was adjusted to 6.0 with diluted HCl. Following pH adjustment, the enzyme was applied to a 2′,5′-ADP–Sepharose column (1.1 by 3.5 cm) equilibrated with buffer C (10 mM potassium phosphate [pH 6.0] containing 5 mM DTT, 20% [wt/vol] sucrose, 0.1 mM EDTA, and 25 mM KCl). After washing with buffer C (40 ml), the enzyme was eluted at a flow rate of 0.5 ml/min with 75 ml of a linear pH gradient, from 6.0 to 8.5, of buffer C. The enzyme eluted at pH 6.9. Fractions with enzyme activity were pooled, aliquoted, and stored at −20°C.
Enzyme assay.
During the purification procedure, the BADH activity was assayed spectrophotometrically by monitoring the absorbance at 340 nm (NADPH formation) in a mixture (0.5 ml) consisting of 1.0 mM betaine aldehyde and 0.3 mM NADP+ in a 100 mM potassium phosphate buffer, pH 8.0 (standard assay). A PU 8710 spectrophotometer (Philips, Cambridge, United Kingdom) equipped with a kinetics software package was used for the assays, which were conducted at 30°C in 1.0-cm-path-length cuvettes. All assays were initiated by addition of the enzyme. Initial steady-state rates were determined from the initial, linear portions of reaction progress curves. The initial rate of betaine aldehyde oxidation was proportional to the enzyme concentration over a range of 0.06 to 2.3 μg of protein per ml of reaction mixture. Each determination was performed at least in duplicate. One unit of activity is defined as the amount of enzyme that catalyzes the formation of 1 μmol of NADPH per min in our standard assay. Kinetics studies were performed by using the same spectrophotometric assay but varying the concentration of substrates.
The effect of pH on the activity of purified BADH was measured over the pH range of 6.0 to 9.5 in either 100 mM potassium phosphate buffer (pH 6.0 to 8.0) or 100 nM potassium pyrophosphate buffer (pH 8.0 to 9.5) under otherwise standard assay conditions. pH stability was determined by assaying the residual activity at pH 8.0 after preincubation of the enzyme (0.5 μg/ml) in 100 mM potassium phosphate or potassium pyrophosphate buffer in a pH range of 6.0 to 9.5 for 2 min at 30°C.
Protein determination.
Protein concentrations were determined by the Coomassie G dye-binding technique of Bradford (6) with bovine serum albumin as a protein standard. For the pure enzyme, the protein concentration was also calculated as described by Scopes (44) by measuring its absorbance at 205 nm in 10 mM potassium phosphate buffer (pH 6.9) containing 0.1 mM EDTA, 20% (wt/vol) sucrose, and 25 mM KCl (buffer D). The two methods yielded protein concentrations that agreed very well. In column effluents, protein elution was monitored by measurements of A280.
Gel electrophoresis and Western blot analysis.
Acrylamide gel electrophoresis under dissociating conditions (sodium dodecyl sulfate-polyacrylamide gel electrophoresis [SDS-PAGE]) was performed with an 8% acrylamide resolving gel and a 4% acrylamide stacking gel as described by Laemmli (22). A MiniProtean II Electrophoresis Cell (Bio-Rad) was used, and the gels were stained with Coomassie brilliant blue (R-250) or with silver (36). Nondenaturing analytical electrofocusing in thin layers of polyacrylamide (8.4%, wt/vol) was done with slab gels prepared with 2.24% (wt/vol) ampholytes in the pH range of 3.0 to 10, 20% (vol/vol) glycerol, and 5 mM DTT. The gels were run at 250 V for 4 h with 20 mM NaOH in the cathode and 10 mM phosphoric acid in the anode. After focusing, the protein band exhibiting BADH activity was visualized by incubating the gel at 37°C with 100 mM potassium phosphate buffer (pH 8.0) containing 2.0 mM betaine aldehyde, 0.3 mM NADP+, 666 μg of nitroblue tetrazolium per ml, and 66 μg of phenazine methosulfate per ml. Denaturing analytical electrofocusing was carried out as previously described (48) except that Triton X-100 was used instead of Nonidet P-40. Protein bands were stained with Coomassie blue by standard methods.
Immunoblotting was carried out essentially by the method of Towbin et al. (50). The enzyme (0.25 or 38 μg of protein for the pure enzyme or cell extract, respectively) was subjected to SDS-PAGE and transferred to a nitrocellulose membrane by semidry blotting in a Mini Trans-Blot electrophoretic transfer cell (Bio-Rad) with a transfer buffer containing 25 mM Tris-HCl (pH 8.3), 192 mM glycine, and 10% (vol/vol) methanol. A polyclonal antibody raised against P. aeruginosa BADH at a dilution of 1:500 was used as the primary antibody. Anti-rabbit goat IgG (conjugated to horseradish peroxidase) at a dilution of 1:1,000 was used as the secondary antibody. Bound antibody was visualized by enhanced chemiluminescence accordingly to the manufacturer’s instructions.
Molecular mass analysis.
Gel filtration of the native purified enzyme was performed with a Biosep Sec 3,000 column (Phenomenex, Torrance, Calif.) connected to a high-pressure liquid chromatography system (Waters, Milford, Mass.) equipped with an automated sample injector. The column was equilibrated and eluted at room temperature with 100 mM sodium phosphate buffer (pH 6.9) containing 150 mM NaCl, 0.1 mM EDTA, and 5 mM DTT at a flux of 1 ml/min. Elution profiles were determined by recording the absorbance of the eluate at 280 nm. The molecular mass of the enzyme subunit was estimated by SDS-PAGE, which was carried out as described above.
Sedimentation equilibrium experiments were performed with an Optima XL-A analytical ultracentrifuge equipped with scanner optics (Beckman, Fullerton, Calif.). The protein sample solution (500 μg/ml) was exhaustively dialyzed against 0.1 M potassium phosphate buffer (pH 6.9) containing 10 mM β-mercaptoethanol, 0.1 mM EDTA, 25 mM KCl, and 20% (wt/vol) sucrose. A portion of the dialysate was retained and used as the reference solution. The samples were placed in cells fitted with conventional aluminum-filled Epon double-sector centerpieces and quartz windows. Sedimentation equilibrium experiments were carried out at 44,000 × g at 20°C. Scans were taken at 280 nm, with a spacing of 0.001 cm, in a step scan mode at intervals of 5 h. Sedimentation equilibrium was judged to have been reached when the difference in concentration distributions between consecutive scans was zero.
Cross-linking experiments.
BADH (0.25 mg/ml) was incubated in buffer C at 30°C. The reaction was initiated by the addition of glutaraldehyde to a final concentration of 32 mM and was stopped by the addition of glycine to a final concentration of 450 mM. In order to assess the degree of cross-linking of the protein, the reaction was stopped several times, and samples were withdrawn, mixed with the appropriate amount of Laemmli sample buffer, and subjected to SDS-PAGE. To estimate the relative cross-linked and non-cross-linked protein contents of the samples, the relative color density of each band was determined with a laser-beam densitometer (GSXL; Pharmacia).
Amino acid composition and N-terminal sequence.
After SDS-PAGE, proteins were electroblotted to Immobilon-PSQ essentially as described by Towbin et al. (50). Coomassie blue-stained protein bands were excised and submitted to the Biotechnology Resource Laboratory of the W.W. Keck Foundation, New Haven, Conn., for amino acid composition determination and N-terminal sequencing.
Protein absorption spectra.
Absorbance spectra were measured on a Philips PU 8710 spectrophotometer at 30°C with 10-mm-path-length cuvettes. Immediately before use, the purified protein was desalted twice in buffer D by the method of Penefsky (38) to remove any oxidized DTT which could be present in the sample. The spectra were read with the buffer used for desalting as control.
Fluorescence measurements.
Fluorescence measurements were carried out at 30°C on an LS50B luminescence spectrophotometer with a thermostated compartment (Perkin-Elmer, Norwalk, Conn.). Freshly desalted protein in buffer D was used. Fluorescence spectra were recorded with an excitation wavelength of 296 nm (2.5-nm bandwidth) and an emission wavelength of 300 to 400 nm (5-nm bandwidth). All solutions for fluorescence measurements were prepared with glass-distilled deionized water and filtered through 0.22-μm-pore-size Millipore filters before use. To minimize the inner filter effects, we used a microcell with a 2-mm path length placed eccentrically in the cell compartment (8).
Preparation of antiserum.
The 61-kDa protein band obtained after SDS-PAGE of fractions from the affinity chromatography step was excised from the gel and used as antigen. Rabbit antibodies were generated by a standard protocol consisting of subcutaneous injection with about 150 μg of protein emulsified in an equal volume of Freund’s adjuvant. After three booster injections with 100 μg of the same antigen, the rabbit was bled and the serum was checked for the ability to cross-react with homologous antigen.
Analysis of the data.
Data sets from the ultracentrifugation experiments were analyzed by using the nonlinear regression program NONLIN (21). The program fits to a reduced apparent molecular weight
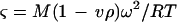 |
1 |
where M is the molecular weight, v is the partial specific volume, ρ is the solvent density, ω is the radial velocity, R is the gas constant, and T is the absolute temperature (kelvin). The partial specific volume of BADH (0.7392 ml/g), was estimated from the amino acid composition by the method of Cohn and Edsall (10). The density of the buffer was computed to be 1.07725 g/ml.
Initial-velocity data were analyzed by nonlinear regression calculations with a commercial computing program formulated with the algorithm of Marquardt (29). Apparent Km and Vmax values were obtained by fitting the initial-velocity data to the Michaelis-Menten equation. When substrate inhibition was observed, the data were fitted to the equation
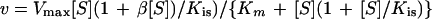 |
2 |
where v is the experimentally determined initial velocity, Vmax is the maximum velocity, [S] is the concentration of the variable substrate, β is the interaction factor that describes the effect of substrate inhibition on Vmax, Kis is the substrate inhibition constant, and Km is the Michaelis-Menten constant for the substrate.
Data for the pH profile were fitted by using
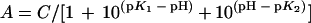 |
3 |
where A is the enzyme activity observed as a function of pH, C is the pH-independent value of A, and K1 and K2 are macroscopic acid dissociation constants reflected on the acid side or on the basic side of the pH profile, respectively.
Thermal inactivation data were fit to a double exponential reaction
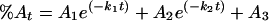 |
4 |
where %At is the percentage of activity remaining at time t; A1 and A2 are the percentages of activity lost in the fast and slow phases, respectively; k1 and k2 are the apparent first-order rate constants of the fast and slow phases, respectively; and A3 is the percentage of activity remaining at equilibrium. Reactivation data were fitted to the same equation.
The experimental data for enzyme inactivation by dilution and in the absence of K+ were fitted to the following equation, which is similar to equation 4 but represents the situation where, in the slow phase, the activity decayed not to a finite value but to zero:
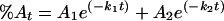 |
5 |
RESULTS AND DISCUSSION
Purification of BADH.
Growth of P. aeruginosa on choline as the sole carbon and nitrogen source resulted in the induction of significant intracellular levels of BADH activity (around 1 U per mg of protein in the cell extract), which was completely absent in cells grown in the standard glucose medium. These results are in full agreement with the previous finding of induction of the enzyme by choline in Pseudomonas (35) and other bacteria (24, 34).
Our purification scheme contains major modifications compared with those reported previously by others (35, 42), which result in a marked increase in the final yield and specific activity of the purified enzyme and in a significant decrease in the overall time involved in the procedure. Enzyme may be purified from the starting material in only 2 days. A crucial step in the purification is the affinity chromatography on 2′,5′-ADP–Sepharose, which resulted in a 19-fold increase in the specific activity over that in the previous step, with a typical recovery of 66%. The choice of this particular affinity matrix was based on the higher affinity of the enzyme for NADP+ than for NAD+. Preliminary attempts to purify BADH from P. aeruginosa by using the 5′-AMP–Sepharose affinity matrix, which has been successfully used in the purification of the NAD+-dependent BADHs from E. coli (14) and from the plant Amaranthus hypochondriacus (52), resulted in very poor yields and in an incomplete purification. Elution of the 2′,5′-ADP–Sepharose with ADP or NADP+ did not yield homogeneous enzyme. Complete purification of the enzyme was achieved by eluting the affinity column with a linear pH gradient as described in Materials and Methods. The purified enzyme was stable for up to 1 year when stored at −20°C in buffer D plus 5 mM DTT.
Results from a typical purification are summarized in Table 1. The modified procedure, described in this paper, for the purification of BADH from P. aeruginosa was reproducible and gave good yields of enzyme, with specific activities at 30°C of 156 and 74.5 U/mg of protein at pH 8.0 and 7.4, respectively. These values are considerably higher than those reported for BADH from human liver (9), leaves of plants (1, 52, 55), fungi (33), and Bacillus subtilis (3) but are similar to those for X. translucens (34) and E. coli (14) and lower than that for crab heart (12). In particular, the specific activity of BADH reported in this work is almost 100 times higher than that first reported for BADH from P. aeruginosa (35) but similar to that recently reported (42). Based on the initial BADH activity of the cell-free preparation and on the final specific activity of the pure enzyme, we conclude that BADH constitutes approximately 0.7% of the soluble protein in choline-grown cells at the stationary phase of growth. A similar result was obtained by Mori et al. (34) with the BADH from another member of the Pseudomonadaceae, X. translucens, which, like the enzyme from Pseudomonas, plays a catabolic role. The published purification procedure for the X. translucens enzyme (34) involved six steps and had a yield considerably lower than the one we report here for the purification of the enzyme from P. aeruginosa.
TABLE 1.
Purification of BADH from P. aeruginosaa
| Purification step | Total protein (mg) | Total activity (U) | Sp act (U/mg of protein) | Purifi-cation (fold) | Yield (%) |
|---|---|---|---|---|---|
| Cell extract | 172.0 | 178 | 1.0 | 1.0 | 100 |
| Q-Sepharose Fast Flow | 23.0 | 177 | 7.7 | 7.7 | 99 |
| 2′,5′-ADP–Sepharose | 0.8 | 117 | 146.3 | 146.3 | 66 |
Results are from a typical purification starting with 1.8 liters of bacterial culture in stationary phase.
The preparations of BADH were homogeneous and contained a nontruncated, nonmodified enzyme as established by several criteria: (i) the elution profile from the 2′,5′-ADP–Sepharose column revealed a single major and symmetrical protein peak with constant BADH specific activity (results not shown), (ii) the purified BADH gave a single sharp protein band with silver or Coomassie blue staining when analyzed under reducing conditions by SDS-PAGE and denaturing isoelectric focusing (Fig. 2A and B, lanes 2), and (iii) the estimated pI and molecular mass of the pure enzyme were identical to those found for the enzyme in cell extracts when subjected to isoelectric focusing (Fig. 2B) or to Western blot analysis (Fig. 2C). Taken together, these results show that the enzyme was not altered during the purification procedure either by chemical modification or by limited proteolysis.
FIG. 2.

Homogeneity and integrity of purified BADH from P. aeruginosa. (A) SDS-PAGE (8% gel) and silver staining of the purified BADH (0.44 μg of protein). Lane 1, enzyme purified with β-mercaptoethanol in buffer and loaded after mixing with a sample buffer without a reducing agent. Lane 2, same as lane 1 but with 5 mM DTT in the sample buffer. The following proteins were used as Mr standards: rabbit muscle myosin (205,000), E. coli β-galactosidase (116,000), rabbit muscle phosphorylase b (97,400), bovine plasma albumin (66,000), ovalbumin (45,000), and bovine erythrocyte carbonic anhydrase (29,000). (B) Nondenaturing isoelectric focusing gel of BADH in cell extracts (22 μg of protein) (lane 1) and denaturing isoelectric focusing gel of purified BADH (5.0 μg of protein) in the presence of 8 M urea and 5 mM DTT (lane 2). Lane 1 was activity stained, and lane 2 was stained with Coomassie blue. The following proteins were used as pI standards: human hemoglobin C (7.5), human hemoglobin A (7.1), human carbonic anhydrase (6.5), bovine carbonic anhydrase (6.0), β-lactoglobulin B (5.1), and phycocyanin (4.5). The pIs of the standards are shown on the left. (C) Western blots of purified BADH (0.25 μg of protein) (lane 1) and BADH in cell extracts from P. aeruginosa grown in choline (38 μg of protein) (lane 2). Samples were subjected to SDS-PAGE (8% gel), transferred to a nitrocellulose membrane, and probed with anti-Pseudomonas BADH antibody.
Physicochemical properties of the purified BADH.
The subunit molecular mass estimated by SDS-PAGE was 61 ± 0.7 kDa (mean ± standard error from five determinations) (Fig. 2B), a higher value than those reported for the subunits of BADH from E. coli (14) and X. translucens (34). When the enzyme was electrophoresed under denaturing conditions in the absence of β-mercaptoethanol in the sample buffer, protein bands with molecular masses higher than 61 kDa were not obtained, indicating the absence of any intersubunit disulfide linkage. However, intrasubunit disulfide bonds readily form, as demonstrated by the two additional protein bands with apparent molecular masses of 57.2 and 55.7 kDa observed when the SDS-PAGE was performed under nonreducing conditions (Fig. 2A, lane 1). The higher electrophoretic mobility of the intrachain-bonded product is most likely due to an incomplete unfolding of the protein by SDS, thereby altering its mobility.
Estimation of the molecular mass of the native BADH in potassium phosphate buffer, using both the pure enzyme and the enzyme in fresh cell extracts, by gel filtration chromatography resulted in a apparent molecular mass of 139 kDa. SDS-PAGE of the BADH eluted from the gel filtration column gave the same molecular mass of the subunit, 61 kDa, confirming that the enzyme was not degraded during the gel filtration process. In accordance with our data, Nagasawa et al. (35) estimated an apparent molecular mass for P. aeruginosa BADH of 145 kDa by using gel filtration chromatography on Sephadex G200. Unfortunately, those authors did not report any molecular mass for the subunits of this enzyme. Taking into account the subunit molecular mass of 61 kDa, which we found by SDS-PAGE, the estimated native molecular mass, 139 kDa, suggests a dimeric enzyme with identical subunits. Since the protein might migrate atypically upon SDS-PAGE or gel filtration, to investigate further the quaternary structure of P. aeruginosa BADH, we subjected the purified enzyme to chemical cross-linking with the bifunctional reagent glutaraldehyde (17), followed by SDS-PAGE, and to analytical ultracentrifugation.
As shown in Fig. 3A, two main bands were obtained in SDS-PAGE after treatment of the enzyme with glutaraldehyde. The first has the same mobility as found for the native enzyme (corresponding to 61 kDa), and the second has an apparent molecular mass of 159 kDa, which is higher than that expected for the dimer. After prolonged incubation, a third band with a mobility corresponding to a molecular mass of 52 kDa appeared. The relative amount of the cross-linked species at different reaction times was estimated by densitometry (Fig. 3B). As expected, the amount of the monomeric, non-cross-linked subunit decreased steadily with increasing reaction time, while the amounts of the cross-linked aggregate and the cross-linked subunit increased with time. Since the composition of the cross-linked aggregates is an indication of the stability of specific enzyme aggregates present in solution and since we did not find any protein band corresponding to aggregates with a molecular mass lower or higher than 159 kDa, these data suggest that the dimer is the fundamental unit of aggregation of the enzyme. Inter- or intrasubunit cross-linking most likely produces alterations in the conformational structure either of the aggregates or of the subunit and therefore produces altered Stokes radii or molecular volumes, which result in bands moving slower or faster than expected.
FIG. 3.

Cross-linking of BADH from P. aeruginosa. (A) BADH (0.25 mg/ml) and 32 mM glutaraldehyde were incubated at 30°C in 10 mM potassium phosphate buffer (pH 6.9) containing 0.1 mM EDTA, 20% (wt/vol) sucrose, and 25 mM KCl. The samples were analyzed by SDS-PAGE (8% gel) and silver staining. (B) Relative quantitation by densitometry of the non-cross-linked 61-kDa subunit (●), the cross-linked 159-kDa aggregate (■), and the cross-linked 52-kDa subunit (▴) bands.
The most accurate method of absolute molecular mass determination in solution is sedimentation equilibrium analysis in an analytical ultracentrifuge. Sedimentation equilibrium studies showed that native BADH is a dimeric protein. Nonideality was not observed, and the residuals revealed no systematic deviation (Fig. 4). The NONLIN program returned a ς value of 1.001 cm2. The molecular mass calculated by this method was 109,224 ± 825.
FIG. 4.

Concentration distribution of BADH during sedimentation equilibrium. The circles are measured values in a standard two-sector ultracentrifuge cell. The solid line is the result of fitting the data with a single-ideal-species model by using the nonlinear regression program NONLIN and equation 1. The residuals to the fit are shown in the top panel.
Taken together, the results of gel filtration, chemical cross-linking, analytical ultracentrifugation, and SDS-PAGE experiments suggest that BADH from P. aeruginosa is a homodimer. In this respect, it resembles the enzymes from B. subtilis (3) and plants (1, 52, 55) and differs from those from X. translucens (34), E. coli (14), fungi (33), and mammals (9), which are tetrameric.
The amino acid composition of the enzyme, estimated on the basis of a 61-kDa subunit, is shown in Table 2. Although the composition indicates variability for several residues compared with those known so far for BADH from bacterial sources, such as those of E. coli (23), X. translucens (34), S. meliloti (40), and B. subtilis (4), the enzymes are similar in overall percentages of acidic (including the corresponding amides), basic, hydrophobic, and aromatic residues (not shown).
TABLE 2.
Amino acid content of BADH from P. aeruginosa
| Amino acid | mol% | No. of residues per subunita |
|---|---|---|
| Asxb | 7.03 | 41 |
| Thr | 6.00 | 35 |
| Ser | 5.49 | 32 |
| Glxb | 13.21 | 77 |
| Pro | 4.63 | 27 |
| Gly | 10.46 | 61 |
| Ala | 12.00 | 70 |
| Val | 9.26 | 54 |
| Met | 1.89 | 11 |
| Ileu | 5.66 | 33 |
| Leu | 8.40 | 49 |
| Tyr | 2.92 | 17 |
| Phe | 3.95 | 23 |
| Lys | 2.74 | 16 |
| His | 0.86 | 5 |
| Arg | 4.46 | 26 |
| Cys | 1.03 | 6 |
| Trp | NDc | ND |
| Total | 583 |
Estimated by assuming a subunit molecular mass of 61 kDa.
The values include the corresponding amides.
ND, not determined.
Twenty of the first 21 amino acid residues of the enzyme were uniquely identified, indicating that the final product was a homogeneous protein preparation and that the enzyme subunits were of identical sequence. The following N-terminal sequence was found: AXFEEQKLYIGGRYVEASSGA. The second residue (designated X) could not be determined accurately. The comparison of this N-terminal sequence with those of different BADH molecules revealed 65, 50, 30, and 25% identities with the BADHs from X. translucens (34), E. coli (5), S. meliloti (40), and B. subtilis (4), respectively. Lower identity with plant BADHs (18, 25, 31, 56, 57) and no identity with animal BADHs (20, 26) were found. Although we compared only the N-terminal regions, the results of this comparison suggest evolutionary relationships between the enzymes from bacteria and plants.
The UV and visible absorption spectra, from 200 to 400 nm, of native P. aeruginosa BADH at pH 6.9 and 30°C show only a single peak at 279.2 nm, with no absorption in the visible region (not shown). The A280/A260 ratio was 1.95. Therefore, the enzyme neither is a flavoprotein nor contains bound NADP+ or NADPH. The extinction coefficient, ɛ2801% (1-cm path length), was 10.82 ± 0.32. This value represents the average from determinations with three different enzyme preparations.
Excitation of native BADH in phosphate buffer (pH 6.9) at 286 nm results in a single emission peak at about 332 nm (Fig. 5), which is characteristic of a class II tryptophan residue, i.e., of a partially exposed tryptophan residue at the surface of the protein (7). This fluorescence was 30% quenched in the presence of saturating concentrations of NADP+ (500 μM), while the emission maximum, λmax, showed only a small red shift (2 nm). This quenching is most likely the result either of ring stacking of the indole ring of a tryptophan with the purine base of the nucleotide, in a manner similar to that which has been observed with nucleic acids (49), or of Förster energy transfer (16) from the tryptophan residues to the bound nucleotide. At pH 8.0 we found spectra similar to those at pH 6.9 (results not shown).
FIG. 5.

Fluorescence emission spectra of BADH from P. aeruginosa. Fluorescence emission spectra in the absence (solid line) and presence (dashed line) of 0.5 mM NADP+ are shown. The enzyme (164 μg/ml) was in potassium phosphate buffer (pH 6.9) containing 0.1 mM EDTA, 20% (wt/vol) sucrose, and 25 mM KCl.
Antibody interaction.
As shown in Fig. 2C, Western blotting with crude homogenates from bacteria grown in 20 mM choline as the only carbon and nitrogen source revealed a single protein band, indicating that the antibody is monospecific. No signals were visible with crude homogenates from bacteria grown in 11 mM glucose and 15 mM (NH4)2SO4, which is consistent with the absence of BADH activity in these homogenates. Neither BADH from E. coli cell extracts nor pure enzyme from amaranth leaves interacted with the polyclonal anti-BADH from P. aeruginosa. In this experiment, care was taken to load a larger amount of whole protein in cell extracts from E. coli than in those from P. aeruginosa, considering the reported specific activities of the pure enzymes (reference 14 and this work).
Kinetic properties of the purified BADH.
The following studies were carried out in order to compare the main kinetic features of the P. aeruginosa BADH purified by us with those of the previously purified enzyme (35) and BADHs from other sources. Similar to other BADHs (3, 14, 52), the Pseudomonas enzyme obeyed Michaelis-Menten kinetics for the two substrates and exhibited substrate inhibition by the aldehyde. At 30°C, pH 8.0, and 0.5 mM NADP+, the observed substrate inhibition was partial, as in the case of amaranth BADH (53). Fitting the experimental data to equation 2 gives an apparent Ki for betaine aldehyde of 4.6 ± 1.4 mM and a β value of 0.55 ± 0.02. The enzyme showed a high degree of specificity at the binding site of the aldehyde, as previously suggested (35). We found that it could not catalyze the NADP+- or NAD+-dependent oxidation of other aldehydes, including those with a positive charge, such as γ-aminobutyraldehyde or 3-dimethylsulfoniopropionaldehyde, which have been recently shown to be good substrates of mammalian (9) and plant (51, 54) BADHs. The former aldehyde was found, however, to be an efficient inhibitor of the oxidation of betaine aldehyde, indicating that it can bind to the active site with high affinity. Since in betaine aldehyde the length of the chain between the positive charge and the carbonyl group is two carbons and one carbon shorter than those in γ-aminobutyraldehyde and 3-dimethylsulfoniopropionaldehyde, respectively, it is possible that the distance between the recognition subsites for the positive charge and the aldehyde function in the active site of Pseudomonas BADH imposes restrictions for the reaction to occur with aldehydes other than betaine aldehyde. The absence of reaction with γ-aminobutyraldehyde is consistent with the fact that Pseudomonas species possess an inducible NAD+-dependent aldehyde dehydrogenase specific for this aldehyde (19).
Regarding the nucleotide, we confirmed the previous report (35) indicating that the enzyme preferentially used NADP+ over NAD+, although both nucleotides can be used. In this respect BADH from Pseudomonas resembles the enzyme from X. translucens (34), but it is different from all other BADHs studied to date, which prefer NAD+ as the coenzyme (3, 9, 12, 14, 33, 52, 55). At 0.5 mM NADP+, 100 mM potassium phosphate buffer (pH 7.5), and 37°C, the apparent Km for betaine aldehyde, estimated by using several concentrations of the aldehyde from 0.1 to 2.0 mM, was 453 ± 52 μM. The apparent Km values for NADP+ and NAD+, determined with the same buffer and also at 37°C by holding the concentration of betaine aldehyde at 1.0 mM and varying the concentration of NADP+ in a range of 0.025 to 0.5 mM and that of NAD+ in a range of 0.05 to 1 mM, were 62 ± 7 and 229 ± 5 μM, respectively. These apparent Km values are in full agreement with those previously reported for the Pseudomonas BADH (35). The apparent Vmax values, estimated at a fixed 1.0 mM betaine aldehyde concentration with NADP+ or NAD+ as the varied substrate, were 121 ± 4 and 133 ± 4 U/mg of protein, respectively. These values are similar to those reported for the enzyme from X. translucens (34) and are as much as 3- and 50-fold greater than those of purified BADHs from E. coli (14) and plants (1, 52, 55), respectively.
The effect of pH on the activity of purified BADH was measured under the standard assay conditions over the pH range of 6.0 to 9.5, in which we found that the enzyme is stable (not shown). Maximum enzyme activity was obtained at pH 8.0 in Tris-HCl buffer (results not shown). This behavior contrasts with that of amaranth BADH, which is inhibited by Tris (52), but is similar to that of the B. subtilis enzyme (3). In the phosphate-pyrophosphate buffering system, the enzyme showed an optimum pH of 8.0 to 8.5. Similar to the case for other BADHs (34, 35, 52), the velocity-versus-pH profile is a bell-shaped curve (Fig. 6A). Two macroscopic pK values, 7.0 ± 0.1 on the acidic side and 9.7 ± 0.1 on the basic side, were estimated from a fit of the experimental data to equation 3. Given that the ionizable groups of NADP+ and betaine aldehyde have pKs different from those estimated in the experiments described above (11), it appears that the enzyme possesses two ionizable groups involved in catalysis and/or binding of substrates.
FIG. 6.

Effect of pH and temperature on the activity of BADH from P. aeruginosa. (A) Profile of velocity versus pH. Enzyme (0.5 μg/ml) activities were determined as described in Materials and Methods with 100 mM potassium phosphate (●) or 100 mM potassium pyrophosphate (○) buffer. The points are the experimentally determined values, and the line drawn through these points was calculated from the best fit of the data to equation 3. (B) Arrhenius plot of log of specific activity versus reciprocal of absolute temperature. The same amount of enzyme (2 μg/ml) was assayed under the standard conditions in 100 mM potassium phosphate buffer (pH 8.0) at the different fixed temperatures without preincubation.
The energy of activation of the reaction was measured by performing enzyme assays at various temperatures in a range of 12 to 40°C under otherwise-standard conditions. The Arrhenius plot of the data (Fig. 6B) was linear, and the energy of enzyme activation calculated from the slope of this plot was 39.8 kJ mol−1. This value is considerably lower (by 22 kJ mol−1) than the activation energy of the amaranth enzyme (52), which is consistent with the much higher specific activity of the P. aeruginosa enzyme.
Stability of the purified BADH.
The purified BADH (31 μg/ml) lost catalytic activity at 40°C in a biphasic inactivation process with an initial fast phase followed by a slower reaction (Fig. 7A). Eventually, an equilibrium between active and inactive forms is reached. A simple model consistent with these features of the thermal inactivation is A⇌I⇌I′, where A is the active form of the enzyme, which equilibrated fairly rapid with I, an inactive form. This then decays in an apparent reversible reaction to another inactive form of the enzyme, I′. For this model, the equation relating the change in concentration of A to time is equation 4, described in Materials and Methods. As can be seen, the fits of the experimental data to this equation are very good, giving apparent first-order rate constants of 0.142 ± 0.019 and 0.014 ± 0.007 min−1 for the fast and slow phases, respectively, and 28% of residual activity at equilibrium. Similar results were obtained with an enzyme 10-fold more concentrated (not shown). When the partially heat-inactivated enzyme is cooled to 30°C or lower, activity is regained in a time-dependent process, consisting of two clearly distinct phases, with apparent first-order rate constants of 0.106 ± 0.040 and 0.003 ± 0.001 min−1. The presence of saturating concentrations of NADP+ (500 μM) during incubation afforded considerable protection against thermal inactivation (Fig. 7A). When added to the incubation medium of a partially inactivated enzyme, NADP+ prevented further inactivation from occurring, but it did not reactivate the enzyme (results not shown).
FIG. 7.

Effect of temperature, dilution, and buffer composition on the stability of BADH from P. aeruginosa. (A) Kinetics of the reversible heat inactivation. The enzyme (31 μg/ml) was incubated at 40°C in 10 mM potassium phosphate buffer (pH 8.0) containing 0.1 mM EDTA, 5 mM β-mercaptoethanol, 20% (wt/vol) sucrose, and 25 mM KCl in the absence (●) or presence (■) of 0.5 mM NADP+. After the times indicated, 4-μl samples were withdrawn and immediately added to the standard assay mixture. Reactivation of the heated enzyme (▴) was achieved by cooling the partially inactivated enzyme at 30°C. (B) Kinetics of the inactivation by dilution. Enzyme was diluted to 0.3 (●, ○, and ▴) or 3 (■ and □) μg/ml in 100 mM potassium phosphate buffer at pH 8.0 (●, ○, ■, and □) or pH 6.5 (▴) in the absence (●, ■, and ▴) or presence (○ and □) of 0.38 mM NADP+. The enzyme was incubated at 30°C and assayed at various time points to determine the activity remaining, using the standard assay. (C) Kinetics of inactivation in the absence of K+ ions. Enzyme (31 μg/ml) was incubated at 30°C in 31 mM Tris-HCl buffer (pH 8.0) in the absence of KCl and NADP+ (●) or in the presence of 25 mM KCl (○), 200 mM KCl (■), 0.5 mM NADP+ (□), or 0.5 mM NADP+ plus 25 mM KCl (▴) and was assayed at various time points to determine the activity remaining. In all panels the results are plotted as percentages of that for the untreated control. The lines are the results of the best fits of the experimental data to equation 4 (A) or to equation 5 (B and C).
As shown in Fig. 7B, the diluted enzyme (0.3 μg/ml) slowly lost activity at pH 8.0 in 100 mM phosphate buffer. The experimental data were best fitted to equation 5, described in Materials and Methods. Therefore, the kinetics of the inactivation were consistent with a sequential three-state model like the one described above, although in this case the activity decayed not to a finite value but to zero, suggesting that the second, slow step is irreversible. The apparent first-order rate constants for the fast and slow phases were 0.95 ± 0.24 and 0.12 ± 0.06 min−1, respectively, and the amplitude of the fast phase was 70.5%. At 3 μg/ml after 9 h of incubation, the enzyme retained 76% of its original activity, which suggests that inactivation by dilution at a basic pH involves enzyme dissociation. At pH 6.5 the enzyme at 0.3 μg/ml was stable for up to 9 h, which would seem to indicate that the protonization of some ionizable group(s) with a pK in the neutral range plays an important role in the maintenance of the native oligomeric structure of the enzyme. Both the rate constant and the amplitude of the fast phase were decreased in the presence of NADP+, which seems to indicate that the nucleotide stabilizes the dimeric native form of the enzyme.
To further study the conditions for the stabilization of the enzyme, we tested the effect of buffer composition. The enzyme (0.45 μg/ml) is highly unstable in Tris-HCl buffers, in a concentration range of 10 to 125 mM and a pH range of 7.0 to 9.0 (results not shown). The low stability of the BADH in Tris-HCl buffers has been previously observed for the enzymes from E. coli (14) and X. translucens (34), but no further characterization of this inactivation was performed; therefore, we found it of interest to study this property of the enzyme. We determined the kinetics of inactivation in 31 mM Tris-HCl buffer (pH 8.0) and found that, again, the inactivation involved two kinetically distinct processes, i.e., a rapid initial phase followed by a slower second phase (Fig. 7C). The experimental data were best fitted to equation 5, giving apparent first-order rate constants of 3.2 ± 0.12 and 0.15 ± 0.03 min−1 for the fast and slow phases, respectively, and an amplitude of the fast phase of 89%. An important difference in the inactivation by dilution is that the kinetics of inactivation were not affected by a 10-fold change in enzyme concentration (not shown). The inclusion of 5 mM DTT in the incubation medium did not affect the kinetics of inactivation (results not shown), ruling out a possible oxidation of an essential sulfhydryl group(s) which might be more exposed in Tris than in phosphate buffer. As in the cases of inactivation by heat and by dilution, inactivation in Tris buffers was greatly prevented by NADP+, but concentrations of the nucleotide of as high as 1 mM failed to give total protection (not shown). Partial protection against inactivation in Tris buffer was also afforded by KCl, but, as in the case of the nucleotide, we could not get total protection with any concentration of this ion. On the contrary, at KCl concentrations of above 500 mM, an increase in the inactivation rate was observed (not shown). NaCl at concentrations equimolar to those of KCl did not protect the enzyme, which indicates that the effect of KCl was not due either to Cl− ions or to the increase in ionic strength of the incubation medium. Thus, K+ ions might play an important role in the stability of the enzyme, and inactivation in Tris-HCl buffer might be due to the absence of these ions. To test this possibility, the enzyme was preincubated in 125 mM sodium phosphate buffer under conditions otherwise identical to those described as above. Inactivation was also observed in the sodium phosphate buffer, although with a lower rate than in the Tris buffer (not shown), which indicates different effects of the phosphate and Tris ions on the stability of the enzyme. NADP+ and K+ have synergistic protective effects, and total protection was achieved by the presence of 0.5 mM NADP+ and 25 mM K+. Therefore, under our experimental conditions, both NADP+ and K+ are needed to stabilize the enzyme in Tris buffers. It is interesting than in phosphate buffers, K+ by itself afforded total protection (not shown). These findings suggest that P. aeruginosa BADH requires K+ ions for stability. No other known BADH has a requirement for K+ ions for stability, although those from E. coli (14), amaranth leaves (52), B. subtilis (3), and crab (12) are activated to some extent by these ions. Of all aldehyde dehydrogenases studied to date, only the mitochondrial isoenzyme of Saccharomyces cerevisiae is unstable in the absence of high concentrations of K+ (47). The role of K+ ions in the stability of the enzyme described in this paper would make BADH from P. aeruginosa a potential target for inhibition by nonessential monovalent cations and deserves further investigation.
In summary, BADH has been purified to homogeneity from the pathogen P. aeruginosa by a rapid and high-yield method, and some of its physicochemical and kinetic properties have been elucidated. It is hoped that the information presented here will facilitate the cloning and overexpression of the gene encoding this enzyme, which would facilitate future works on rational drug design and screening. BADH from Pseudomonas has been shown to possess certain distinctive and unique properties, such as the requirement for K+ ions for stability. The enzyme is also highly specific for betaine aldehyde, unlike BADHs from other systems. Further studies will be oriented toward identifying additional differences between this enzyme and mammalian aldehyde dehydrogenases, which might be useful for the design of selective inhibitors.
ACKNOWLEDGMENTS
This work was supported by a grant to R.A.M. from Consejo Nacional de Ciencia y Tecnología (CONACYT-2552P-N).
We are grateful to A. D. Hanson (University of Florida, Gainesville) for the kind gift of 3-dimethylsulfoniopropionaldehyde and to M. L. Vasil (University of Colorado, Denver) for kindly providing P. aeruginosa PAO1.
REFERENCES
- 1.Arakawa K, Takabe T, Sugiyama T, Akazawa T. Purification of betaine-aldehyde dehydrogenase from spinach leaves and preparation of its antibody. J Biochem. 1987;101:1485–1488. doi: 10.1093/oxfordjournals.jbchem.a122019. [DOI] [PubMed] [Google Scholar]
- 2.Bernard T, Perroud B, Pocard J A, Le Rudulier D. Variations in the response of salt-stressed Rhizobium strains to betaines. Arch Microbiol. 1986;143:359–364. [Google Scholar]
- 3.Boch J, Nau-Wagner G, Kneip S, Bremer E. Glycine betaine aldehyde dehydrogenase from Bacillus subtilis: characterization of an enzyme required for the synthesis of the osmoprotectant glycine betaine. Arch Microbiol. 1997;168:282–289. doi: 10.1007/s002030050500. [DOI] [PubMed] [Google Scholar]
- 4.Boch J, Kempf B, Bremer E. Synthesis of the osmoprotectant glycine betaine in Bacillus subtilis: characterization of the gbsAB genes. J Bacteriol. 1996;178:5121–5129. doi: 10.1128/jb.178.17.5121-5129.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boyd L A, Adam L, Pelcher L E, Mchughen A, Hirji R, Selvaraj G. Characterization of an Escherichia coli gene encoding betaine aldehyde dehydrogenase (BADH): structural similarity to mammalian ALDH’s and a plant BADH. Gene. 1991;103:45–52. doi: 10.1016/0378-1119(91)90389-s. [DOI] [PubMed] [Google Scholar]
- 6.Bradford M M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–256. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 7.Burstein E A, Vedenkina N S, Irkova M N. Fluorescence and the location of tryptophan residues in protein molecules. Photochem Photobiol. 1973;18:263–279. doi: 10.1111/j.1751-1097.1973.tb06422.x. [DOI] [PubMed] [Google Scholar]
- 8.Chen R F, Hayes J E., Jr Fluorescence assay of high concentrations of DPNH and TPNH in a spectrophotofluorometer. Anal Biochem. 1965;1:523–529. doi: 10.1016/0003-2697(65)90347-7. [DOI] [PubMed] [Google Scholar]
- 9.Chern M K, Pietruszko R. Human aldehyde dehydrogenase E3 isozyme is a betaine aldehyde dehydrogenase. Biochem Biophys Res Commun. 1995;213:561–568. doi: 10.1006/bbrc.1995.2168. [DOI] [PubMed] [Google Scholar]
- 10.Cohn E J, Edsall E T. Proteins, amino acids, and peptides as ions and dipolar ions. New York, N.Y: Reinhold Publishing Co.; 1943. pp. 374–377. [Google Scholar]
- 11.Dawson R M C, Elliot D C, Elliot W H, Jones K M. Data for biochemical research. 3rd ed. Oxford, United Kingdom: Clarendon Press; 1986. p. 11130. [Google Scholar]
- 12.Dragolovich J, Pierce S K. Characterization of partially purified betaine aldehyde dehydrogenase from horseshoe crab (Limulus polyphemus) cardiac mitochondria. J Exp Zool. 1994;270:417–425. [Google Scholar]
- 13.D’Souza-Ault M R, Smith L T, Smith G M. Roles of N-acetylglutaminyl-glutamine amide and glycine betaine in adaptation of Pseudomonas aeruginosa to osmotic stress. Appl Environ Microbiol. 1993;59:473–478. doi: 10.1128/aem.59.2.473-478.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Falkenberg P, Stroem A R. Purification and characterization of osmoregulatory betaine aldehyde dehydrogenase from Escherichia coli. Biochim Biophys Acta. 1990;1034:253–259. doi: 10.1016/0304-4165(90)90046-y. [DOI] [PubMed] [Google Scholar]
- 15.Flores H E, Filner P. Polyamine catabolism in higher plants:characterization of pyrroline dehydrogenase. Plant Growth Regul. 1985;3:277–291. [Google Scholar]
- 16.Förster T. Intermolecular energy transference and fluorescence. Ann Phys (Leipzig) 1948;2:55–75. [Google Scholar]
- 17.Habeeb A F S A, Hiramoto R. Reaction of protein with glutaraldehyde. Arch Biochem Biophys. 1968;126:16–26. doi: 10.1016/0003-9861(68)90554-7. [DOI] [PubMed] [Google Scholar]
- 18.Ishitani M, Nakamura T, Han S Y, Takabe T. Expression of the betaine aldehyde dehydrogenase gene in barley in response to osmotic stress and abscisic acid. Plant Mol Biol. 1995;27:307–315. doi: 10.1007/BF00020185. [DOI] [PubMed] [Google Scholar]
- 19.Jakoby W B, Fredericks J. Pyrrolidine and putrescine metabolism: γ-aminobutyraldehyde dehydrogenase. J Biol Chem. 1959;234:2145–2150. [PubMed] [Google Scholar]
- 20.Johansson K, El-Ahmad M, Ramaswamy S, Hjelmqvist L, Jornvall H, Eklund H. Structure of betaine aldehyde dehydrogenase at 2.1 Å resolution. Protein Sci. 1998;7:2106–2117. doi: 10.1002/pro.5560071007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson M L, Correia J J, Yphanthis D A, Halvorson H R. Analysis of data from the analytical ultracentrifuge by nonlinear least-squares techniques. Biophys J. 1981;36:575–578. doi: 10.1016/S0006-3495(81)84753-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Laemmli U K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 23.Lamarck T, Kaasen E, Eshoo M W, Falkenberg P, McDougall J, Stroem A R. DNA sequence and analysis of the bet genes encoding the osmoregulatory choline-glycine betaine pathway of Escherichia coli. Mol Microbiol. 1991;5:1049–1064. doi: 10.1111/j.1365-2958.1991.tb01877.x. [DOI] [PubMed] [Google Scholar]
- 24.Landalf B, Stroem A R. Choline-glycine betaine pathway confers a high level of osmotic tolerance in Escherichia coli. J Bacteriol. 1986;165:849–855. doi: 10.1128/jb.165.3.849-855.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Legaría J, Rajsbaum R, Muñoz-Clares R A, Villegas-Sepúlveda N, Simpson J, Iturriaga G. Molecular characterization of two genes encoding betaine aldehyde dehydrogenase from amaranth. Expression in leaves under short-term exposure to osmotic stress or abscisic acid. Gene. 1998;218:69–76. doi: 10.1016/s0378-1119(98)00381-3. [DOI] [PubMed] [Google Scholar]
- 26.Lin S W, Chen J C, Hsu L C, Hsieh C, Yoshida A. Human gamma-aminobutyraldehyde dehydrogenase (ALDH9): cDNA sequence, genomic organization, polymorphism, chromosomal localization, and tissue expression. Genomics. 1996;34:376–380. doi: 10.1006/geno.1996.0300. [DOI] [PubMed] [Google Scholar]
- 27.Lisa T A, Casale C H, Domenech C E. Cholinesterase, acid phosphatase, and phospholipase C of Pseudomonas aeruginosa under hyperosmotic conditions in a high-phosphate medium. Curr Microbiol. 1994;28:71–76. [Google Scholar]
- 28.Lucchesi G I, Lisa T A, Domenech C E. Choline and betaine as inducer agents of Pseudomonas aeruginosa phospholipase C activity in high phosphate medium. FEMS Lett. 1989;57:335–338. doi: 10.1016/0378-1097(89)90324-8. [DOI] [PubMed] [Google Scholar]
- 29.Marquardt D W. An algorithm for least-squares estimation of nonlinear parameters. J Soc Ind Appl Math. 1963;11:431–441. [Google Scholar]
- 30.Matthews L W S, Spector J L, Lemm J, Potter J L. Studies on pulmonary secretions. I. The over-all chemical composition of pulmonary secretions from patients with cystic fibrosis, bronchiectasis, and laryngectomy. Am Rev Respir Dis. 1963;88:199–204. doi: 10.1164/arrd.1963.88.2.199. [DOI] [PubMed] [Google Scholar]
- 31.McCue K F, Hanson A D. Salt-inducible betaine aldehyde dehydrogenase from sugar beet: cDNA cloning and expression. Plant Mol Biol. 1992;18:1–11. doi: 10.1007/BF00018451. [DOI] [PubMed] [Google Scholar]
- 32.Miller J H. Experiments in molecular genetics. 3rd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1977. pp. 431–435. [Google Scholar]
- 33.Mori N, Kawakami B, Hyakutome K, Tani Y, Yamada H. Characterization of betaine aldehyde dehydrogenase from Cylindrocarpon didymun M-1. Agric Biol Chem. 1980;40:3015–3016. [Google Scholar]
- 34.Mori N, Yoshida N, Kitamoto Y. Purification and properties of betaine aldehyde dehydrogenase from Xanthomonas translucens. J Ferment Bioeng. 1992;73:352–356. [Google Scholar]
- 35.Nagasawa T, Kawabata Y, Tani Y, Ogata K. Purification and characterization of betaine aldehyde dehydrogenase from Pseudomonas aeruginosa A-16. Agric Biol Chem. 1976;40:1743–1749. [Google Scholar]
- 36.Oakley B R. A simplified ultrasensitive silver stain for detecting proteins in polyacrylamide gels. Anal Biochem. 1980;105:361–363. doi: 10.1016/0003-2697(80)90470-4. [DOI] [PubMed] [Google Scholar]
- 37.Ostroff R M, Wretlind B, Vasil M L. Molecular comparison of a nonhemolytic and a hemolytic phospholipase C from Pseudomonas aeruginosa. J Bacteriol. 1990;172:5915–5923. doi: 10.1128/jb.172.10.5915-5923.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Penefsky H S. Reversible binding of Pi by beef heart mitochondrial adenosine triphosphatase. J Biol Chem. 1977;252:2891–2899. [PubMed] [Google Scholar]
- 39.Pesin S R, Candia O A. Acetylcholine concentration and its role in ionic transport by the corneal epithelium. Invest Ophthalmol Vis Sci. 1982;22:651–659. [PubMed] [Google Scholar]
- 40.Pocard J A, Vincent N, Boncompagni E, Smith L T, Poggi M C, Le Rudulier D. Molecular characterization of the bet genes encoding glycine betaine synthesis in Synorizobium meliloti 102F34. Microbiology. 1997;143:1369–1379. doi: 10.1099/00221287-143-4-1369. [DOI] [PubMed] [Google Scholar]
- 41.Rennick B R. Renal tubule transport of organic ions. Am J Physiol. 1981;240:F83–F89. doi: 10.1152/ajprenal.1981.240.2.F83. [DOI] [PubMed] [Google Scholar]
- 42.Russell R, Scopes R K. Use of hydrophobic chromatography for purification of the membrane-located choline dehydrogenase from a Pseudomonas strain. Bioseparation. 1994;4:279–284. [PubMed] [Google Scholar]
- 43.Sage A E, Vasil A I, Vasil M L. Molecular characterization of mutants affected in the osmoprotectant-dependent induction of phospholipase C in Pseudomonas aeruginosa. Mol Microbiol. 1997;23:43–56. doi: 10.1046/j.1365-2958.1997.1681542.x. [DOI] [PubMed] [Google Scholar]
- 44.Scopes R K. Measurement of protein by spectrophotometry at 205 nm. Anal Biochem. 1974;59:277–282. doi: 10.1016/0003-2697(74)90034-7. [DOI] [PubMed] [Google Scholar]
- 45.Shortridge V D, Lazdunski A, Vasil M L. Osmoprotectants and phosphate regulate expression of phospholipase C in Pseudomonas aeruginosa. Mol Microbiol. 1992;6:863–871. doi: 10.1111/j.1365-2958.1992.tb01537.x. [DOI] [PubMed] [Google Scholar]
- 46.Smith L T, Pocard J A, Bernard T, Le Rudulier D. Osmotic control of glycine betaine biosynthesis and degradation in Rhizobium meliloti. J Bacteriol. 1988;170:3142–3149. doi: 10.1128/jb.170.7.3142-3149.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sorger G J, Evans H J. Effects of univalent cations on the properties of yeast NAD+ acetaldehyde dehydrogenase. Biochim Biophys Acta. 1966;118:1–8. doi: 10.1016/s0926-6593(66)80139-x. [DOI] [PubMed] [Google Scholar]
- 48.Thurston C F, Henley L F. Isoelectric focusing under denaturing conditions. In: Walker J M, editor. Methods in molecular biology: new protein techniques. Clifton, N.J: The Humana Press Inc.; 1988. pp. 257–267. [DOI] [PubMed] [Google Scholar]
- 49.Toulmé J J, Heléne C. Specific recognition of single-stranded nucleic acids. J Biol Chem. 1977;252:244–249. [PubMed] [Google Scholar]
- 50.Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Trossat C, Rathinasabapathi B, Hanson A D. Transgenically expressed betaine aldehyde dehydrogenase efficiently catalyzes oxidation of dimethylsulfoniopropionaldehyde and omega-aminoaldehydes. Plant Physiol (Rockville) 1997;113:1457–1461. doi: 10.1104/pp.113.4.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Valenzuela-Soto E M, Muñoz-Clares R A. Purification and properties of betaine aldehyde dehydrogenase extracted from detached leaves of Amaranthus hypochondriacus L. subjected to water deficit. J Plant Physiol. 1994;143:145–152. [Google Scholar]
- 53.Vojtechová M, Rodríguez-Sotres R, Valenzuela-Soto E M, Muñoz-Clares R A. Substrate inhibition by betaine aldehyde of betaine aldehyde dehydrogenase from leaves of Amaranthus hypochondriacus L. Biochim Biophys Acta. 1997;1341:49–57. doi: 10.1016/s0167-4838(97)00059-9. [DOI] [PubMed] [Google Scholar]
- 54.Vojtechová M, Hanson A D, Muñoz-Clares R A. Betaine aldehyde dehydrogenase from amaranth leaves efficiently catalyzes the NAD+-dependent oxidation of dimethylsulfoniopropionaldehyde to dimethylsulfoniopropionate. Arch Biochem Biophys. 1997;337:81–88. doi: 10.1006/abbi.1996.9731. [DOI] [PubMed] [Google Scholar]
- 55.Weretilnyk E A, Hanson A D. Betaine aldehyde dehydrogenase from spinach leaves. Purification, in vitro translation of the mRNA, and regulation by salinity. Arch Biochem Biophys. 1989;271:56–63. doi: 10.1016/0003-9861(89)90255-5. [DOI] [PubMed] [Google Scholar]
- 56.Weretilnyk E A, Hanson A D. Molecular cloning of a plant betaine-aldehyde dehydrogenase, an enzyme implicated in adaptation to salinity and drought. Proc Natl Acad Sci USA. 1990;87:2745–2749. doi: 10.1073/pnas.87.7.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wood A J, Saneoka H, Rhodes D, Joly R J, Goldsbrough P B. Betaine aldehyde dehydrogenase in sorghum: molecular cloning and expression of two related genes. Plant Physiol. 1996;110:1301–1308. doi: 10.1104/pp.110.4.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wright J R, Clements J A. Metabolism and turnover of lung surfactant. Am Rev Respir Dis. 1987;136:426–444. doi: 10.1164/ajrccm/136.2.426. [DOI] [PubMed] [Google Scholar]