

Abstract
Approximately 80% genes have an active expression in the brain and 40% of known genetic disorders affect the central nervous system. Revolutionary techniques like next-genome sequencing have solved the problem of the diagnostic odyssey in neurological genetic diseases. As several studies have shown the cost-effectiveness of next-genome sequencing compared to the older tests available in our diagnostic armamentarium, it becomes imperative to know about these genetic mutations and the tests available to diagnose them. We have tried to explain the basic concepts of genetics, the selection of available tests and their interpretation for neurophysicians.
Keywords: Exome, NGS, Sanger, WES
INTRODUCTION
The association of genetic factors with neurological disorders is well recognised. The molecular genetic technology has improved by leaps and bounds, which has led to discovery of novel diseases and their causative genes.[1] Eighty percent of the identified genes are actively expressed in the brain and 40% of all genetic disorders involve the nervous system in one way or another as per the OMIM database.[2] Diagnostic odyssey is common in neurological Mendelian disorders and in such scenarios genetic testing has proven to be cost effective.[3] Genetic heterogeneity associated with a phenotype [Figure 1] (mutations of different genes presenting with the same phenotype) and phenotypic pleiotropy associated with a genotype [Figure 2] (different phenotypic presentation because of mutation in a single gene) can confound the clinical presentation. Hence, it is important for the clinician to be well versed with the nitty-gritties of genetic testing.[1,3]
Figure 1.
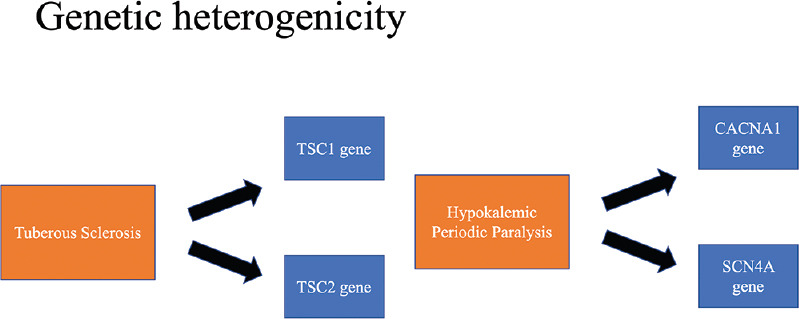
Genetic heterogeneity associated with a phenotype
Figure 2.
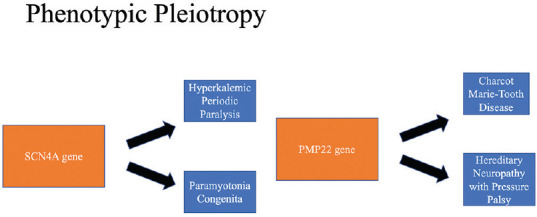
Phenotypic pleiotropy associated with a genotype
Genetic material ranges from chromosomes to a single unit of two complementary base nucleotide pairs in opposite deoxyribonucleic acid (DNA) strands. A genetic disorder can result from any defect in the range between these two extremes. As the size of the defect varies from >5 megabase to a single base pair, multiple genetic tests are available in the diagnostic armamentarium, as per the genomic variation.[1]
In this article, we will try to explain the basic concepts of genetic variations, the available genetic tests, and which test to order based on the phenotypic classification through case scenarios.
BASIC GENETICS
At the chromosomal level:
The complete chromosome can either be deleted or duplicated (monosomy or trisomy, respectively). This is known as chromosomal aneuploidy.
A part of chromosome can be deleted or duplicated, resulting in a chromosomal copy number variation (Eg: distal 18q deletion syndrome).
Variation in a single nucleotide can occur resulting in point mutations. These can be silent, missense or nonsense mutations.[1]
Selection of a genetic test depends on the site of defect. Chromosomal and sub-chromosomal pathologies can be identified by cytogenetics, whereas molecular methods are required for exon/gene level and single base pair mutations [Figure 3].
Figure 3.
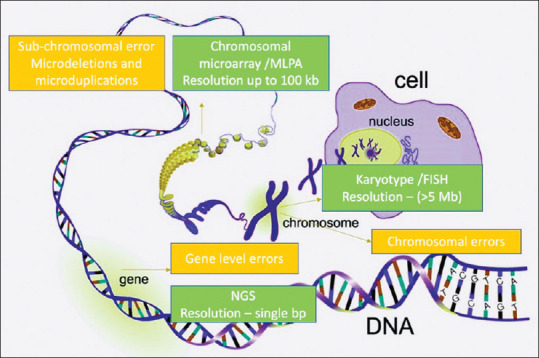
Genetic pathologies and the tests employed
To simplify concept, imagine the sentence “Give me the bat” as a chromosome, individual words as different genes and each letter as a single nucleotide.
GENETIC TESTS
-
Sanger sequencing
It is a single gene sequencing method in which DNA is sequenced in short portions to detect the mutation of interest (<1 kilobase). It can recognise point mutations, small deletions and insertions of few nucleotide base pairs. However, large deletions, copy number variations and nucleotide repeat expansions are not detected by Sanger sequencing. As it is a targeted mutation analysis, we look at a pre-specified region of a gene that has earlier been described as pathogenic. Therefore, a negative Sanger sequencing result rules out mutations only of the region of the gene investigated, and not of the entire gene per se.[4] This test is proven to be cost-effective when verifying a mutation amongst family members of a proband diagnosed with the same genetic mutation.[4]
-
Next genome sequencing
The revolutionary technique was developed in 2005 and has been continuously evolving since then. It is quicker and more cost-effective compared to other genetic analysis methods. Under the Human Genome Project, the human genome was sequenced for the first time in 2001, by Sanger sequencing (SS), but took 13 years and 2.7 billion United States Dollars (USD) to complete. Present-day next generation sequencing (NGS) can sequence the same in 2 weeks and costs around 4000 USD, illustrating the extraordinary progress in DNA sequencing.[5]
Simplyfing the concept
| Example | Type of error | Genetic tool useful |
|---|---|---|
| Givye me the bat Givye me the bat Givye me the bat |
Large deletion/ duplication | Karyotype/FISH |
| Givye me the bat Givye me the bat |
Smaller deletion/ duplication | FISH/chromosomal microarry,MLPA |
| Givye me the cat | Single nucleotide variation | DNA sequencing (Sanger /NGS) |
| Givye me the bat bat bat bat bat bat | Short tandem repeats | Fragment length analysis |
It involves isolation of genomic DNA followed by fragmentation into small nucleotide segments and attachment of artificial linkers to them. Fragments corresponding to exome sequences are thereafter isolated and sequenced in parallel on a slide utilising an in-situ amplification method using fluorescent dyes. Finally, with the help computerised bioinformatic analysis, target sequences are aligned to reference sequences in the human genome, and any difference is estimated, and the variant list is produced. It is capable of detecting point mutations, small insertions/deletions and splice site mutations.[6] Variants noted can be benign polymorphisms (not associated with any disease) or pathogenic mutations. ACMG recommends that these variants be classified as pathogenic, likely pathogenic, variants of unknown significance (VUS), likely non-pathogenic and non-pathogenic.[2]
VUS is not an insignificant finding and signifies that the variation documented for the gene of interest is new/rare in the population, and has not yet been documented to have any association with genetic disease,[6] but could be pathogenic. As the database of pathogenic mutations is constantly updated, VUS results can be re-interpreted if ascertained to be pathogenic. The presence of serum biomarkers, positive testing of family members, trio studies (sequencing of proband and patents) and functional studies can further verify the pathogenicity of VUS.[2]
Other technical terms that a physician should be aware of while interpreting NGS results are: depth of coverage and completeness of gene coverage. Depth of coverage is average times each exon is viewed (A depth of coverage of 20-fold entails that each nucleotide of that exome was observed 20 times on average). It's an estimate of certainty and reduces the risk of false-negative results.[7] Breadth of coverage is percentage of bases sequenced at particular depth.[7] Increased breadth reduces depth. Although while sequencing DNA samples depth is often prespecified, not all parts of the exome are sequenced equally. Sometimes mutation of concern can exist in part of the gene where coverage is limited. Increasing depth can improve coverage. The possibility of incompleteness of gene coverage should always be considered in case of a negative NGS result when the likelihood of positivity was high.[6]
NGS can also identify pathogenic sequences not related to the patient's phenotype or clinical diagnosis.[3] These are known as incidental or secondary findings and have ethical implications for the treating physician.[6]
NGS can be used as targeted panel sequencing, whole-exome sequencing (WES) and whole-genome sequencing (WGS).
A) Targeted gene panel sequencing
It targets all the exons on a pre-specified number of genes. Genetic heterogeneity with a well-defined disease phenotype has considerable detection rate by targeted gene panel sequencing.[2] The physician should be confident about the disease phenotype and should have the essential genetic knowledge of the disease before choosing the appropriate genes for this targeted sequencing panel. The usual diagnostic rates range from 10% to 40%.[3] Its major advantages include the lowest false negative results (because of high coverage depth), reduced cost and less data production for bioinformatics analysis. Also, since we mostly look at pathogenic mutations in a limited number of genes, the possibility of encountering VUS and incidental pathogenic mutations (not related to the patient's phenotype) are lesser than WES and WGS, reducing ethical issues and result complexity.[2]
However, the possibility of detecting novel mutation is low as we mostly look for sequence variations that have been previously authenticated as pathogenic constituting an important limitation.[3]
B) Whole exome sequencing (WES)
As suggested by its name, it sequences all protein-coding regions (exomes) of the genome (about 2%).[2] It covers nearly all disease-causing mutations and is useful in diseases where phenotypic presentation is diverse like cerebellar ataxias and neurodevelopmental disorders.[3] As WES includes entire genome sequencing, more data is produced for analysis, which takes more time for diagnosis (and money) and results in more VUS, incidental pathogenic findings and ethical dilemmas. Incomplete gene coverage is another problem as only around 90% exome is covered adequately.[3] Even on increasing depth (higher cost), coverage cannot be complete. This test should be considered in a patient strongly suspected to have a genetic aetiology, but whose targeted gene panel results are negative.[3]
C) Whole-genome sequencing (WGS)
It is the most comprehensive sequencing method that covers the entire genome (approximately 3 billion base pairs of DNA). WGS data analysis is cumbersome for clinical indication because a large number of variants (nearly 4,00,000) remain for interpretation even after ruling out benign variants. It is mainly used in research settings. However, few studies have shown some theoretical advantages of WGS over WES, such as uniform depth coverage, detection of copy number variation and possible ability to detect short tandem repeats.
Another factor that limits the use of WGS is less information available about variants in intergenic regions and introns. Hence, targeted gene panels and WES are the most commonly used techniques of NGS for a genetic diagnosis.
D) Chromosomal microarray
It is a form of comparative genomic hybridization which is used for detecting deletion, duplications and copy number variations. It is the first line test for developmental and intellectual disorders, autism spectrum disorders and congenital anomalies. Therefore,it is commonly prescribed by paediatric neurologists and is complimentary to NGS.
Table 1 and Table 2 illustrate the best and alternative tests to be conducted in various genetic alterations.
Table 1.
Preferred genetic test for Chromosomal abnormalities[1]
| Type of genetic mutation | Best test | Alternative test |
|---|---|---|
| Chromosomal aneuploidy E.g.: trisomy 21 | Karyotype (>5mb) | Chromosomal microarray |
| Chromosomal translocation E.g.: (t[1,7]) | Chromosomal microarray (5 mb - 100 kb) | Karyotype, Fluorescent in-situ hybridization |
| Chromosomal copy number variation E.g.: Distal 18q deletion syndrome | Chromosomal microarray | Karyotype (if >5mb) |
Table 2.
Preferred genetic test for Gene abnormalities[1]
| Type of genetic mutation | Best test | Alternative test |
|---|---|---|
| Gene and exon-level deletions and duplication E.g.: HNPP, DMD | Multiplex ligation dependent probe amplification | Fluorescent in-situ hybridization (FISH) |
| Short tandem repeat E.g.: Myotonic dystrophy | Fragment analysis | |
| Single nucleotide variant E.g.: CADASIL, Fabry’s | Next generation sequencing | Sanger’s |
| Indel E.g.: DYT1 | Next generation sequencing | Sanger’s |
CASE-BASED EXAMPLES
Case 1
A 47-year-old gentleman presented with progressive history of imbalance while walking and dysarthria for the past 10 years. His elder brother had similar complaints. His examination revealed a cerebellar ataxia and oculomotor apraxia. Electrophysiological studies revealed a sensory motor neuropathy (axonal) affecting all four limbs. Investigations revealed elevated alfa-fetoprotein (AFP) levels. These findings can be observed both in ataxia telangiectasia and ataxia with oculomotor apraxia type 2 (late onset cerebellar ataxia with oculomotor apraxia with raised AFP).
As we were aware of the genetic mutation of interest, we ordered a Sanger sequencing in the patient and his family members (proband, affected sibling, unaffected sibling and parents). It revealed a SETX: c.6029A>G, p.N2010S mutation in the senataxin gene both in the proband and affected sibling.
This case illustrates the importance of a phenotype, description which enabled us to narrow down our differentials and made us to choose Sanger sequencing was the first test. As described previously, this was rapid and cost-effective.
Case 2
A 20-year-old lady, born out of non-consanguineous marriage, presented with progressive gait ataxia at 9 years of age. She developed hand incoordination at 12 years of age and dysarthria at the age of 18 years. Examination revealed pes cavus, spasticity of both lower limbs and brisk deep tendon reflexes. Electrophysiological study was suggestive of a demyelinating sensory motor polyneuropathy. Multiple siblings had similar complaints in family, though heterogeneity was present in terms of age of onset and clinical presentation. As Friedreich's ataxia (FA) is the most common cause of autosomal recessive ataxia in this age group, she was initially screened for loss of function mutations in the frataxin (FXN) gene located on chromosome 9q13, but was not found. Thereafter, a targeted gene panel for ataxia yielded one homozygous insertion change resulting in a frameshift in SACS gene. This mutation is responsible for Autosomal Recessive Spastic Ataxia of Charlevoix Saguenay type (ARSACS).
All juvenile, autosomal recessive, ataxias should initially be screened for FA mutation. If negative, as in this case, one should proceed to a targeted gene panel based on the disease (ataxia in this case).
Case 3
A 15-year-old boy presented with progressive intellectual decline for the past 5 years. His elder sister had similar complaints when she was his age (10 years back) and was not investigated and is presently bed bound and unable to care for herself. Examination revealed impaired frontal and temporal lobe functions, grade 2 spasticity in lower limbs, Medical Research Council grade 4/5 power at hip, knee and ankle, brisk knee jerk, ankle clonus and an extensor plantar response. Magnetic resonance imaging (MRI) brain revealed bilateral symmetric T2-FLAIR hyperintensities in the centrum semiovale and MRI spine was normal. Based on positive family history and imaging findings, a neuro-metabolic disorder was suspected (leukodystrophy, hereditary spastic paraparesis, mitochondrial disorders and homocysteine re-methylation defects). As our list of differentials was broad, WES was ordered for both the patients, which revealed a homozygous c. 394C>T pathogenic mutation in the MMACHC gene (exon 3), confirming the diagnosis of Cobalamin C disease. Thereafter, Sanger sequencing revealed the same mutation in his sister as well. He was treated with parenteral methyl cobalamin with around 80% improvement in his symptoms. The sister, however, did not improve (probably due to burnt out disease).
This case highlights the benefit of early, prompt and aggressive investigation into a seemingly untreatable genetic disease. WES was the modality chosen as the list of differentials was broad and one of the aetiologies was likely to get missed on targeted testing.
CONCLUSIONS
A detailed and systematic clinical evaluation forms the foundation for the selection of genetic test. A multidisciplinary approach including geneticist, pathologist, physician and genetic counsellor should be used for diagnosis of clinically heterogeneous phenotype. Knowledge about the use of newer techniques (i.e. NGS) and interpretation of its results can help to diagnose genetic diseases, not diagnosed by older techniques. NGS still requires good clinical examination, as it's not the gene alone that brings a diagnosis but the appropriateness of clinical phenotype and genotype. Financial constraints, however, remain a significant barrier in most patients.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Silveira-Moriyama L, Paciorkowski AR. Genetic diagnostics for neurologists. Contin Lifelong Learn Neurol. 2018;24:18–36. doi: 10.1212/CON.0000000000000556. [DOI] [PubMed] [Google Scholar]
- 2.Rexach J, Lee H, Martinez-Agosto JA, Németh AH, Fogel BL. Clinical application of next-generation sequencing to the practice of neurology. Lancet Neurol. 2019;18:492–503. doi: 10.1016/S1474-4422(19)30033-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Klein CJ, Foroud TM. Neurology individualized medicine: When to use next-generation sequencing panels. Mayo Clin Proc. 2017;92:292–305. doi: 10.1016/j.mayocp.2016.09.008. [DOI] [PubMed] [Google Scholar]
- 4.Fogel BL. Genetic and genomic testing for neurologic disease in clinical practice. Handbook of Clinical Neurology. 1st ed. Vol. 147. Elsevier B.V; 2018. pp. 11–22. Available from: http://dx.doi.org/10.1016/B978-0-444-63233-3.00002-6 . [DOI] [PubMed] [Google Scholar]
- 5.Keogh MJ, Chinnery PF. Next generation sequencing for neurological diseases: New hope or new hype? Clin Neurol Neurosurg. 2013;115:948–53. doi: 10.1016/j.clineuro.2012.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fogel BL, Satya-Murti S, Cohen BH. Clinical exome sequencing in neurologic disease. Neurol Clin Pract. 2016;6:164–76. doi: 10.1212/CPJ.0000000000000239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sims D, Sudbery I, Ilott NE, Heger A, Ponting CP. Sequencing depth and coverage: Key considerations in genomic analyses. 2014. Available from: www.nature.com/reviews/genetics . [DOI] [PubMed]