

Abstract
Microbial infections are most often controlled by host inflammatory responses that are initiated by innate immune receptors after recognition of conserved microbial products. As inflammation can also lead to pathology, tissues that are exposed to microbial products such as the intestinal epithelium, are subject to stringent regulatory mechanisms to prevent indiscriminate signaling through innate immune receptors. The enteric pathogen Salmonella Typhimurium, which requires intestinal inflammation to sustain its replication in the intestinal tract, uses effector proteins of its type III secretion systems to trigger an inflammatory response without the engagement of innate immune receptors. Furthermore, Salmonella Typhimurium utilizes a different set of effectors to restrict the inflammatory response in order to preserve the host’s homeostasis. The Salmonella-host interface is a remarkable example of the unique balance that emerges from the co-evolution of a pathogen and its host.
Introduction
Salmonella enterica constitutes a major public health concern and it is estimated to cause more than 300,000 annual deaths, mostly in developing countries1,2. Based on its surface antigenic composition, Salmonella enterica is classified into hundreds of serovars3,4. Some serovars (e. g. S. enterica serovar Typhi and serovar Paratyphi) are host adapted to humans, where they cause a systemic infection known as typhoid or paratyphoid fever and are therefore referred to as “typhoidal” serovars5–7. Other serovars such as S. Typhimurium, have a broad host range and in humans, most often cause self-limiting gastroenteritis and are referred to as “non-typhoidal” serovars8. Intestinal inflammation is central for the pathology that follows infection with non-typhoidal Salmonellae9.
In the context of infectious diseases, inflammation is often seen as a central host response aimed at the expulsion of an invading pathogen. Indeed, the inflammatory response is the most prominent outcome of the stimulation of innate immune receptors that have evolved to detect bacterial-associated molecular patterns abundantly displayed by bacterial pathogens10–12. However, in the case of Salmonella Typhimurium infections, it has become clear that the inflammatory response is essential for the ability of this pathogen to colonize the intestinal tract13,14. It is well established that the resident intestinal microbiota provides a powerful barrier that restricts infection by bacterial pathogens15–17. Although the mechanisms by which the resident microbiota exerts this powerful restrictive effect are incompletely understood and likely multi-factorial, it is clear that the dysbiosis that follows intestinal inflammation results in a breakdown of the colonization barrier13,18,19. It has also become clear that intestinal inflammation results in the availability of nutrients that are otherwise not accessible in the uninflamed gut10,13. Therefore, the stimulation of intestinal inflammation allows S. Typhimurium to compete with the resident microbiota and secure carbon sources and electron acceptors essential to sustain its metabolism and its replication in the gut13,14,20,21. Consequently, in the case of S. Typhimurium the inflammatory response can be best viewed as a pathogen-orchestrated host response to secure its replication rather than as a host-initiated response aimed at the expulsion of the pathogen. In this article the mechanisms by which S. Typhimurium triggers inflammation in the intestinal tract through the activities of effector proteins delivered by its type III secretion systems will be discussed. Mechanisms orchestrated by the pathogen’s type III secretion systems aimed at recovering the host’s homeostasis after the inflammatory response will be also covered. For other aspects of the biology of Salmonella in the intestinal tract, including its interaction with the resident microbiota, readers should consult other excellent reviews22,23.
Interaction of Salmonella Typhimurium with the intestinal epithelium
Non-typhoidal Salmonellae such as S. Typhimurium are most often acquired through the consumption of contaminated food or water24. Although the acidity of the stomach constitutes an effective barrier against this pathogen25, consumption of a large enough inoculum or contaminated food with buffering capacity may result in a productive infection leading to overt disease. After its oral acquisition, Salmonella travels down the intestinal tract reaching the large intestine where most of its replication is thought to take place. Much of what is known about S. Typhimurium pathogenesis has been learned using the mouse model of infection26. The disease presentation in mice is significantly different from human disease since in this animal model, S. Typhimurum causes systemic infection. Nevertheless, at least some of the basic concepts learned from this model system are likely applicable to the understanding of human disease. After reaching the large intestine, S. Typhimurim uses its flagella and chemotactic systems to reach a location in close proximity to the intestinal epithelium27. Contact with the intestinal epithelium leads to the activation of the Salmonella’s type III protein secretion system (T3SS) encoded within its pathogenicity island 1 (SPI-1) (Text Box 1 and Figure 1)28,29, which results in the delivery of several bacterial effector proteins with the capacity to modulate various host processes (for an extensive review of the Salmonella T3SS effectors and their activities see30,31). The main outcome of this initial interaction is the stimulation of host-cell responses that leads to the internalization of bacteria and the transcriptional reprogramming of the infected cell, ultimately leading to inflammation (Figure 2) (see below)32–36. More specifically, the activation of Rho-family GTPases, in particular Rac1, by the effector proteins SopE, SopE2, and SopB leads to actin-cytoskeleton rearrangements and macropinocytosis, resulting in bacterial internalization37–40. Other effectors, such as the actin nucleator SipA also contribute to the internalization process41. Once internalized in a membrane-bound compartment, Salmonella modulates vesicle trafficking through the activities of effectors largely encoded by a second T3SS encoded within its pathogenicity island 2 (SPI-2), whose expression is stimulated by the intracellular environment42. Modulation of vesicle trafficking allows Salmonella to avoid innate immune responses resulting in the sculpting of an intracellular niche permissive for its survival and replication. However, the bulk of the bacterial load in the intestine derives not from the intracellular pool but from the expansion of the luminal pool of bacteria13. Indeed, the stimulation of the inflammatory response initiated by the activities of the T3SS effectors (see below) and subsequently amplified by the engagement of the innate immune system allows Salmonella to overcome the rather stringent colonization resistance mechanisms that are derived from the presence of the resident microbiota. Intestinal inflammation results in dysbiosis and the depletion of resident bacterial species that antagonize the replication of luminal Salmonella at least in part by competing for essential nutrients. In addition, the inflammatory response allows Salmonella to have access to nutrients and electron acceptors that are otherwise unavailable in uninflamed tissues and that are necessary to support its replication (for excellent reviews on this aspect of Salmonella pathogenesis see22,43). Ultimately, the acquired immune response mounted by the infected host results in the elimination of the pathogen and the recovery of the host’s homeostasis. Although in the mouse S. Typhimurium quickly becomes systemic and most often leads to death26, in most other healthy hosts, infections with non-typhoidal Salmonellae are self-limiting and do not become systemic44. In recent years, however, the emergence of variants of non-typhoidal Salmonellae capable of causing systemic disease have been reported45.
Test Box 1. Type III protein secretion systems (T3SS).
Complex molecular machines evolved by many bacterial pathogens to modulate host cell processes through the delivery of bacterially-encoded effector proteins directly into the target host cells (Figure 3).
Figure 1.
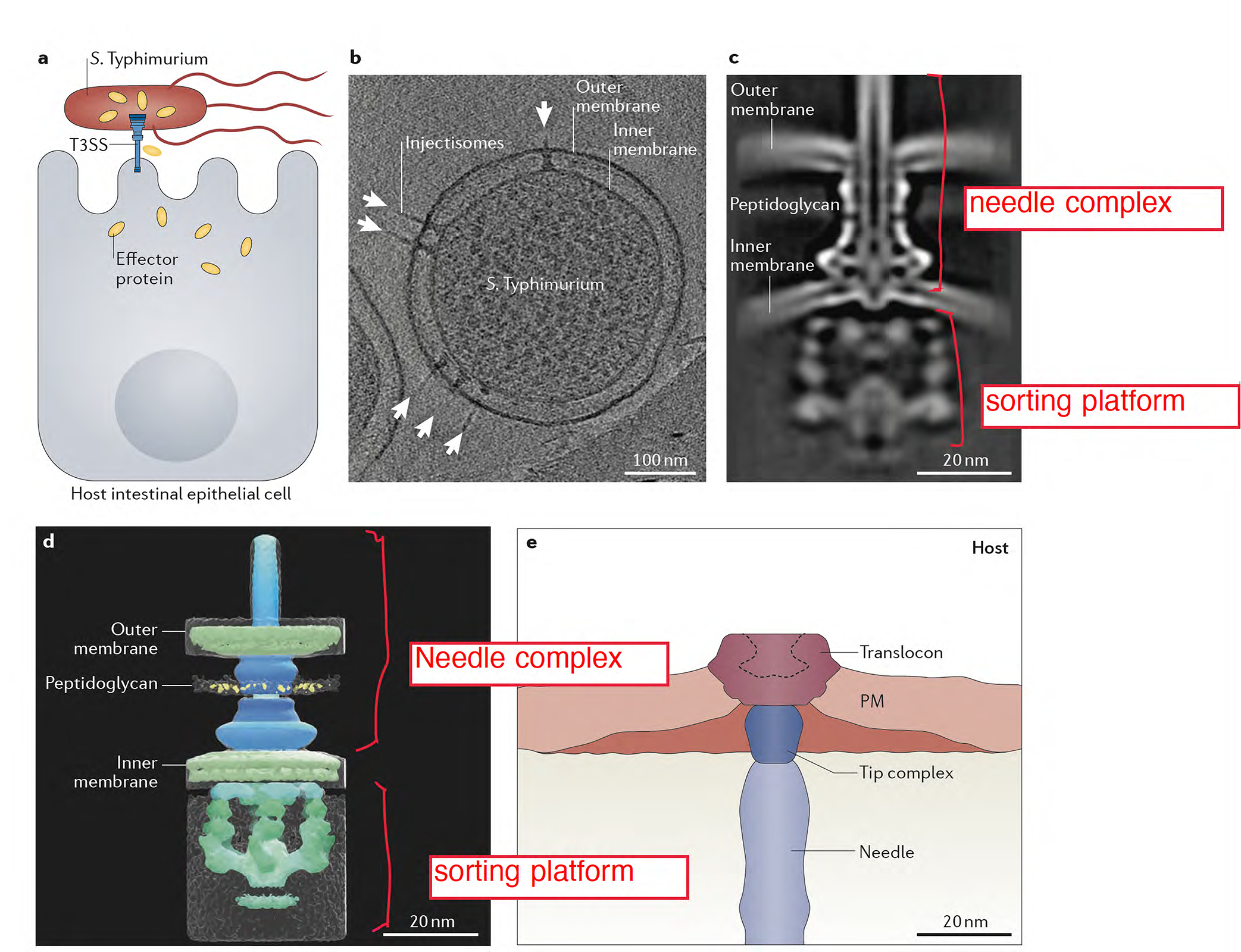
The type III protein secretion machine encoded by S. Typhimurium within its pathogenicity island 1. (A) Diagram depicting S. Typhimurium delivering effector proteins through its T3SS. (B) Electron micrograph of a S. Typhimurium cells showing multiple T3SS injectisomes (arrows). (C and D) Cryo electron microscopy images of the T3SS machine in situ. A central section (C) and 3-D surface rendering (D) of the T3SS injectisome are shown (adapted from108). (E) Cross section of the interface between S. Typhimurium and host cells as revealed by cryo electron tomography (adapted from109).
Figure 2. Model for the interaction of S. Typhimurium with the intestinal epithelium.
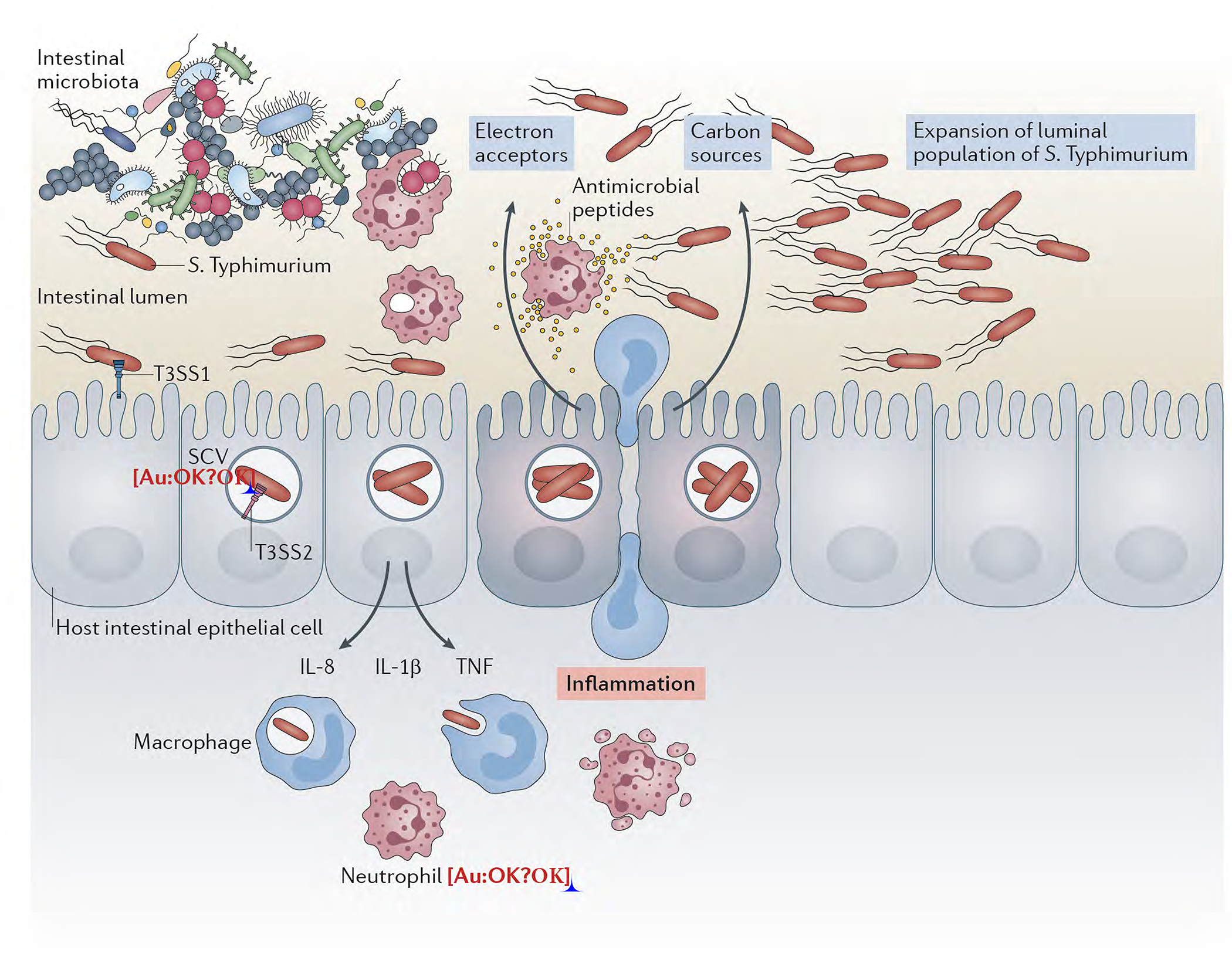
After gaining access to the host via the oral route, S. Typhimurium reaches the large intestine where with the help of motility, makes contact with the intestinal epithelium resulting in the activation of the type III secretion system encoded within its pathogenicity island 1 (T3SS-1). Effector proteins delivered by this system trigger cell responses that result in bacterial internalization and the production of pro-inflammatory cytokines. The intracellular environment provides the cues for S. Typhimurium to express another type III protein secretion system encoded within its pathogenicity island 2 (T3SS-2), which allows the pathogen to avoid innate immune defense mechanisms and replicate within cells. The production of pro-inflammatory cytokines by the infected cells starts a cascade of events that lead to the recruitment of inflammatory cells. The tissue inflammatory response alters the intestinal lumen environment resulting the depletion of the resident microbiota and the availability of nutrients and electron acceptors that fuel the replication of the luminal population of S. Typhimurium.
Stimulating intestinal inflammation: type III secretion at work
Unlike most other tissues, where the presence of bacterial products capable of stimulating innate immune receptors can trigger inflammation, the intestinal tract presents a challenge to those pathogens that rely on the inflammatory response to sustain their replication. Indeed, the presence in the intestinal tract of an abundance of microbial products derived from the resident microbiota with the potential to stimulate innate immune receptors, demands for the intestinal epithelium to be subject to stringent negative regulatory mechanisms that can prevent the pathology that could result from the indiscriminate firing of these receptor11,46–50. In fact, mis-regulation of those mechanisms can result in chronic inflammatory conditions such as Crohn’s or inflammatory bowel disease. Consequently, to initiate an inflammatory response in the gut, S. Typhimurium cannot rely on the stimulation of innate immune receptors by conserved bacterial products (e. g LPS, peptidoglycan, flagellin) that, like many other bacteria, it possesses in abundance. Rather, it uses specific adaptations that allow this pathogen to trigger inflammation bypassing those receptors (Figure 3). Given its central role in pathogenesis, the mechanisms by which Salmonella trigger intestinal inflammation have been a long-standing question in the field and, at times, have been the subject of some controversy. More than two decades ago, before innate immune receptors came into the fore-front, it was already shown that S. Typhimurium could stimulate MAP kinases and NF-κB signaling in cultured intestinal epithelial cells, and that stimulation of these responses resulted in the production of pro-inflammatory cytokines35,51. More importantly, it was shown then that stimulation of these responses was strictly dependent on the activity of the T3SS encoded within SPI-135,51. These findings were the first indication that S. Typhimurium has evolved specific adaptations to be able to trigger inflammation in the intestinal track. However, later on, when the sensing mechanisms of the innate immune system had already become center stage, studies showed that the transcriptional responses stimulated by S. Typhimurium in cultured epithelia cells resembled those stimulated by innate immune receptors36. The requirement of a functional SPI-1 T3SS to stimulate these responses presumably eliminated the possibility that the pro-inflammatory responses were triggered by conserved agonists of innate immune receptors (i. e. LPS, peptidoglycan, flagellin, etc) abundantly present in S. Typhimurium. However, several studies suggested that components of the SPI-1 T3SS itself (e. g. the needle and inner rod components) may be recognized by innate immune receptors52,53. These observations raised the possibility that the inflammatory responses that followed S. Typhimurium infection could be the result of the recognition of the type III secretion machine by the innate immune system. However, the ability of S. Typhimurium to stimulate inflammatory signaling was shown to be strictly dependent on the function of 3 specific effector proteins of the SPI-1 T3SS: SopE, SopE2 and SopB (see below)36,39. Consequently, a mutant lacking these three effectors was shown to be unable to trigger inflammatory signaling. Since this mutant encodes a wild type SPI-1 T3SS machine, these findings in principle ruled out the hypothesis that the inflammatory response that follows S. Typhimurium infection is the result of the recognition of components of the secretion machine by innate immune receptors. However, these observations resulted in a conundrum: how could S. Typhimurium through the delivery of its effector proteins SopE, SopE2, and SopB trigger “innate immune-like” signaling without engaging innate immune receptors? The answer to this conundrum would require a better understanding of the mechanisms by which the SPI-1 T3SS effector proteins stimulate these responses (Figure 2).
Figure 3. Model for the S. Typhimurium pro- and anti-inflammatory signaling in the intestinal tract through its type III secretion effectors.
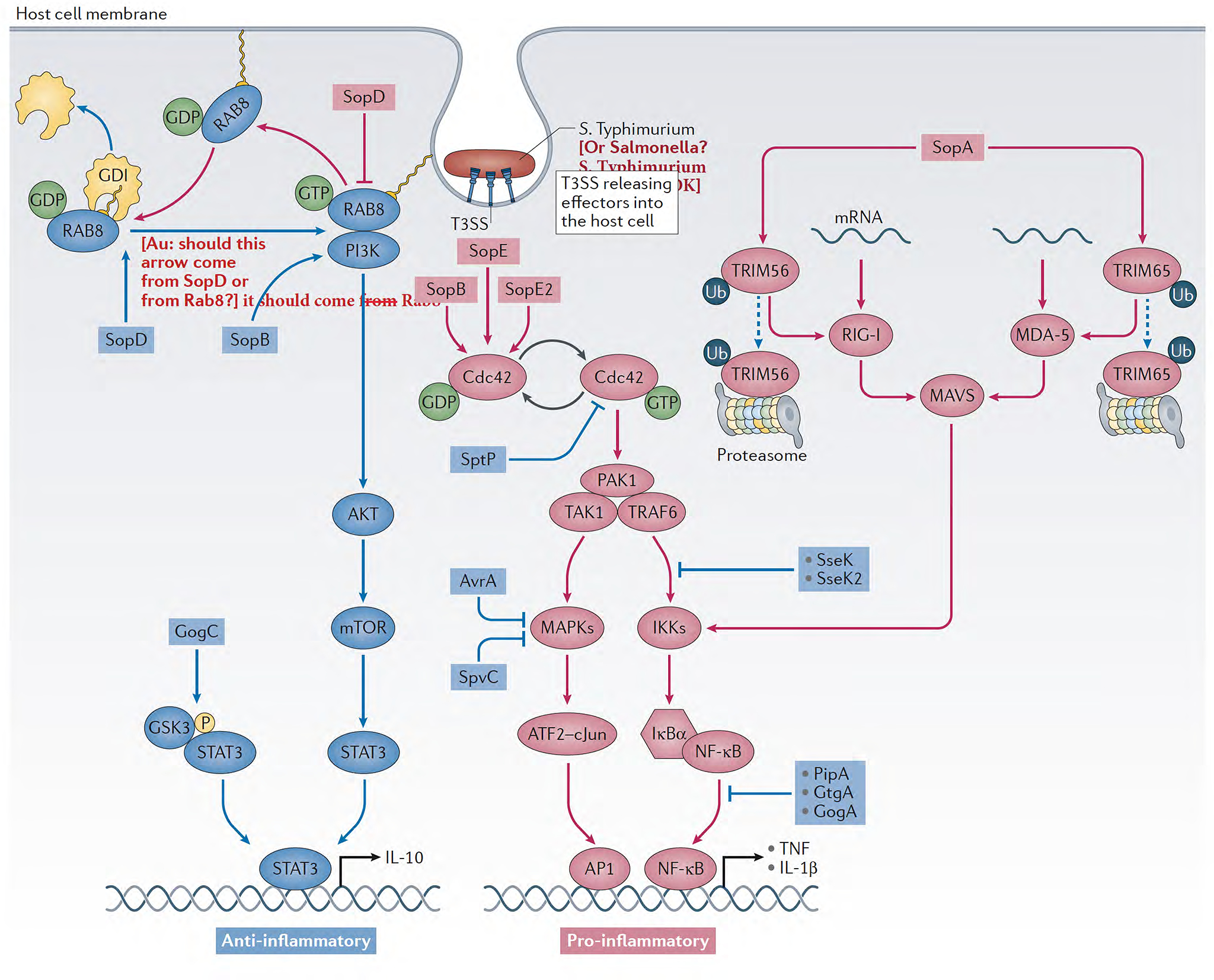
Pro- and anti-inflammatory signaling pathways are depicted in red and green, respectively. The effector proteins and their place of action are noted.
The SPI-T3SS effectors SopE and SopE2 are guanine nucleotide exchange factors (GEFs) for the Rho-family GTPases Rac1 and Cdc4237,54. SopB, which is a phosphoinositide phosphatase55, can also activate Rho-family GTPases although not by direct action on the GTPases but through the induction of phosphoinositide fluxes that result in the activation endogenous GEFs for these Rho-family GTPases39. By activating Rac1, these effectors mediate actin-cytoskeleton rearrangements that lead to bacterial internalization into host cells39. In addition, by activating Cdc42, these effectors also stimulate MAP kinase and NF-κB signaling that ultimately results in the production of pro-inflammatory cytokines35,39,51. Although these findings provided major insight into the mechanisms by which Salmonella triggers inflammation, these observations could not explain the similarities between the transcriptional responses induced by Salmonella with those induced by the stimulation of innate immune receptors as no connection between Cdc42 and canonical innate immune signaling mechanisms had been reported. Subsequent studies proposed that the activation of Rac1 by the S. Typhimurium effectors per se through unknown mechanisms is sensed as a “danger associated molecular pattern” by the innate immune receptor NOD1 leading to NF-κB activation and pro-inflammatory transcriptional response56. However, this proposal was not consistent with previous observations indicating that removal of Cdc42 abolished S. Typhimurium stimulation of inflammatory signaling in cultured cells, even though the absence of Cdc42 does not affect the ability of S. Typhimurium to activate Rac1 or to gain access to host cells39. These observations were also inconsistent with previous reports indicating that removal of Rip236 or Caspase 1 and 1157, which are critical components of the NOD1/inflammasome pathway36,57, do not affect the ability of S. Typhimurium to stimulate intestinal inflammation in mice. These issues were finally clarified when it was shown that stimulation of Cdc42 by the S. Typhimurium T3SS effector proteins SopE, SopE2, and SopB leads to the activation of the Cdc42-effector p21-activated kinase (PAK1) and the subsequent formation of a non-canonical signaling complex composed of PAK1,TRAF6, and TAK158. Removal of PAK1, TRAF6, or TAK1 from various cell lines abrogated the ability of S. Typhimurium to stimulate inflammatory signaling. Furthermore, oral administration of a highly specific inhibitor of all group I PAKs (PAK1, PAK2, and PAK3) drastically reduced the inflammatory response and the replication of S. Typhimurium in the intestinal tract without affecting its ability to invade cells58. It is well documented that TRAF6 and TAK1 are critical components of a signal transduction hub downstream from multiple Toll like receptors59,60. These observations provided a mechanistic explanation for the similarities between the Salmonella-induced pro-inflammatory transcriptional responses and those that generally follow the stimulation of innate immune receptors. Therefore, by engaging innate immune signaling pathways downstream from the actual receptors, Salmonella is able to stimulate a response that shares great similarity with the responses stimulated by the activation of canonical innate immune receptors, while avoiding the negative regulatory mechanisms that prevent the activation of these receptors in the intestinal tract (Figure 3).
Blocking the inflammatory response in the intestine by inhibiting p21-activated kinases resulted in a drastic reduction in the number of S. Typhimurium in the intestinal tract58, which is consistent with the requirement of intestinal inflammation for bacterial replication in the intestinal lumen. However, this inhibiting effect was not observed in animals that had been pre-treated with streptomycin to deplete the resident microbiota. These results are consistent with previous observation indicating that in the absence of the competing microbiota S. Typhimurium does not need intestinal inflammation to sustain its replication13. In contrast, blocking of p21-activated kinases in the intestinal epithelium resulted in an increase in bacterial load in systemic tissues58. These observations indicate that while intestinal inflammation is critically important for the replication of S. Typhimurium within the intestine, this response is also central for the host to anatomically restrict the pathogen and prevent its access to deeper tissues.
Although SopE, SopE2 and SopB are essential for the initiation of the inflammatory response that follows S. Typhimurium infection, two other effector proteins contribute to its amplification. One of these effectors is SopA, which was originally identified as an effector required for the efficient stimulation of intestinal inflammation in a cow model of infection61. Subsequent studies showed that SopA is a HECT-type E3 ubiquitin ligase that preferentially uses the host’s UbcH5a, UbcH5c and UbcH7 E2 components of the ubiquitination machinery62. The similarity with eukaryotic HECT ubiquitin ligases was later corroborated by its crystal structure, which showed that, despite very little sequence similarity, SopA shares structural architectural features with its eukaryotic counterparts62. Functional and biochemical studies showed that SopA exerts its pro-inflammatory activity by ubiquitinating the TRIM-family ubiquitin ligases TRIM56 and TRIM65, stimulating downstream signaling63. TRIM proteins are a large family of E3 ubiquitin ligases that have been implicated in a variety of function64–66. More specifically, TRIM56 has been shown to modulates innate immune responses by ubiquinating and activating STING, a major component of the RIG-I signaling pathway that leads to inflammation67. TRIM65, on the other hand, interacts with MDA563, a member of the RIG-I-like Receptor (RLR) protein family68,69, stimulating downstream signaling that also results in interferon-β expression and inflammation. RLRs such as RIG-I itself and MDA5, are essential components of microbial RNA-sensing pathways68. However, it is unclear whether the ability of SopA to modulate RLRs signaling is enhanced by the presence of microbial nucleic acids. Nevertheless, it has been reported that during infection of non-phagocytic cells the mRNA from S. Typhimurium can be sensed by the RIG-I pathway70. It should be noted that it has also been reported that the SopA-mediated ubiquitination of TRIM56 and TRIM65 leads to their degradation71. Although this activity would be incompatible with the well documented pro-inflammatory role of SopA, it is possible that it may contribute to the recovery of host homeostasis subsequent to the inflammatory response (see below).
Like SopA, the Salmonella type III effector protein SopD synergizes with other effectors to stimulate inflammation72,73. Recent studies have illuminated the mechanisms by which this effector protein stimulates the inflammatory response74. Because inflammation can lead to tissue damage, innate immune receptors are most often linked to anti-inflammatory pathways that help the recovery of host homeostasis11,46–50. One such anti-inflammatory pathway operating downstream of cell surface-localized Toll like receptors is strictly dependent on Rab8. This signaling mechanism results in the activation of Phosphoinositide 3-kinase (PI3-kinase) and protein kinase B (also known as Akt), which ultimately leads to the biasing of cytokine production toward an anti-inflammatory program75–77. In addition, the SPI-T3SS effector SopB, which is a phosphoinositide phosphatase, can also activate this Rab8-dependent anti-inflammatory pathway by fluxing phosphoinositides and thereby activating PI-3 kinase and Akt (see below)74. SopD antagonizes this anti-inflammatory response by directly targeting Rab8 as a specific GTPase activating protein (GAP)74. Therefore, by inhibiting an anti-inflammatory pathway, SopD effectively acts as a pro-inflammatory effector protein. Consequently, similar to SopE, SopE2 and SopB, SopA, and SopD can also stimulate inflammation by targeting hard-wired innate immune inflammatory signaling without the need to engage innate immune receptors.
The role of the inflammasome in Salmonella-induced intestinal inflammation
The inflammasomes are cytosolic signaling platforms that can sense and coordinate the response to the presence of pathogen-associated molecules in the cell cytoplasm78–80. Depending on their mechanisms of activation, they are classified as canonical and non-canonical. Canonical inflammasomes, which include the NLRP1, NLRP3, NLRC4, Pyrin and AIM2 inflammasomes, are generally activated by conserved microbial products resulting in the activation of Caspase-1. The non-canonical inflammasome is activated by the direct sensing of LPS by Caspase 11 in mice or the human orthologues Caspase 4 and 5. Activation of both types of inflammasomes lead to similar types of responses that include the stimulation of the production of pro-inflammatory cytokines and a form of programmed cell known as pyroptosis (extensively reviewed in81–83). The ability of S. Typhimurium to stimulate pyroptosis in macrophages through the activation of this signaling platform in a T3SS-dependent manner has been long recognized84,85. In addition, inflammasome signaling has also been shown to be operational in intestinal epithelial cells and play a role during Salmonella infection86,87. The mechanisms by which S. Typhimurium activates the inflammasome through the activity of its SPI-1 T3SS are not fully understood and most likely multi factorial. Since a functional SPI-1 T3SS machine (though not its effectors) is required for S. Typhimurium to activate the inflammasome, it is likely that the deployment of the type III secretion translocon on the eukaryotic cell membrane by itself leading to ion fluxes may be the trigger of its activation. When over-expressed in cells the needle filament and inner rod components of the T3SS machine have been shown to activate the inflammasome52,53. However, since these components are essential for type III secretion function it has been challenging to ascertain the physiological significance of these cell culture observations as mutations of these components would affect type III secretion function and therefore deployment of the translocon. Flagellin, the building subunit of the flagellar filament, has also been shown to activate the inflammasome88. Although it is clear that the inflammasome is important in controlling S. Typhimurium systemic infection86,87, its specific contribution to the stimulation of intestinal inflammation appears to be secondary, at least in the context of the mouse model of infection. Consistent with this notion, the ability of S. Typhimurium to stimulate intestinal inflammation is unaltered in mice simultaneously deficient in Caspase 1 and Caspase 1157, or in Rip2 36, which are essential component of inflammasome signaling. It has been reported that activation of the inflammasome leads to the extrusion of intestinal epithelial cells harboring Salmonella89. Therefore, rather that contributing to inflammation, activation of the inflammasome may help the host to recover homeostasis after the inflammatory response triggered by Salmonella by reducing bacterial numbers through epithelial shedding.
Actively promoting cell homeostasis: the Yin and Yang of Salmonella-induced inflammation
It is often overlooked that pathogens that have sustained long standing association with their hosts, have evolved specific mechanisms not just to ensure their replication but also to preserve the host’s homeostasis. This concept may appear counterintuitive at first glance, as research tends to emphasize mechanisms of pathogenesis. This is particularly the case when it comes to inflammation, since microbial factors aimed at dumping down the inflammatory response to preserve host homeostasis are often viewed as “virulence factors” that are aimed at thwarting the host’s defense response. This concept is eloquently illustrated by the battery of T3SS effectors that S. Typhimurium has specifically evolved to counter the activities of pro-inflammatory effector counterparts (see below) (Fig. 3 and Table 1). Removal of the antagonistic effectors results in increased pathology and virulence90–92, which demonstrates the fact that preservation of the host homeostasis through virulence limitation is central to the ecology and the evolution of this pathogen. Antagonistic effectors utilize at least two general mechanisms to antagonize the inflammatory response: 1) directly counter signaling pathways triggered by agonistic, pro-inflammatory effectors; and 2) actively stimulate anti-inflammatory pathways. Among the first group is the SPI-1 T3SS effector SptP, which is a GTPase activating protein for the Rho-family GTPases Cdc42, Rac1 and Rho93, thus opposing the pro-inflammatory activity the effectors SopE and SopE2, which are GTP exchange factors for the same Rho-family GTPases37,54. By limiting the activation of Cdc42 in particular, SptP limits the inflammatory response to Salmonella helping the host to recover homeostasis93. Another subset of effectors, PipA, GtgA, and GogA, proteolitically target the NF-κB transcription factors RelA and RelB, effectively limiting the inflammatory response to S. Typhimurium92. Consistent with their biochemical activity, removal of these three effectors from S. Typhimurium resulted in a significant increase in intestinal inflammation and increase lethality in a mouse model of infection92. Similarly, the effector proteins SseK1 and SseK3 inactivate NF-κB signaling by transferring N-acetylglucosamine to specific arginine residues in the death domains of several key proteins in this signaling pathway94,95. Additional examples of effectors that directly target inflammatory signaling components in a negative regulatory manner are SpvD, AvrA and SpvC. SpvD inhibits NF-κB activation by interfering with the nuclear translocation of RelA through interactions with the exportin Xpo2, which mediates nuclear-cytoplasmic recycling of importins96. AvrA suppresses c-JUN N-terminal kinase (JNK) signaling through acetylation of the upstream kinases mitogen-activated receptor kinase kinases 4 and 7 (MKK4/7)97,98. SpvC, on the other hand, is a phosphothreonine lyase that directly targets ERK1/2 and p38 by irreversibly removing phosphate groups from phosphothreonine residues99,100. As predicted by their biochemical activities, S. Typhimurium mutants lacking either AvrA or SpvC induced a more pronounced intestinal inflammation in a mouse model of infection90,91.
Table 1:
Pro- and anti-inflammatory type III secretion effectors in Salmonella Typhimurium
| Pro-inflammatory effectors | Anti-inflammatory effectors | ||
|---|---|---|---|
| Name | Function | Name | Function |
| SopE, SopE2 | GEFs for Rho-family GTPases37,54 (stimulate NF-κB through non-canonical PAK1/TRAF6/TAK1 signaling58) | SptP | GAP for Rho-family GTPases93 |
| SopB | Phosphoinositide phosphatase55 (activates endogenous GEFS for Rho Family GTPases39) | SopB | Phosphoinositide phosphatase55 (activates PI3K-dependent anti-inflammatory pathways74) |
| SopA | E3 ubiquitin ligase62 (activates RIG-I and MDA-5 signaling through ubiquitination of TRIM56 and TRIM6563) | SopA | E3 ubiquitin ligase62 (antagonizes RIG-I and MDA-5 signaling through ubiquitine-mediated degradation of TRIM56 and TRIM6571) |
| SopD | GAP for Rab8 (neutralizes a Rab8-dependent anti-inflammatory pathway)74 | SopD | GDI-dissociation factor for Rab8 (activates a Rab8-dependent anti-inflammatory pathway)74 |
| GogC (SteE, SarA, PagJ) | Stimulates STAT3-dependent anti-inflammatory signaling103,104 | ||
| SpvD | Inhibits RelA nuclear translocation96. | ||
| PipA, GtgA, GogA | Proteases for NF-κB transcription factors RelA and RelB (inhibit NF-κB-dependent transcription)92 | ||
| AvrA | Acetylates MKK4 and MKK7 (inhibits JNK signaling)97,98 | ||
| SpvC | Phosphothreonine lyase for ERK1/2 and p38 (inhibits MAPK signaling)99,100. | ||
| SseK, SseK2 | N-acetylglucosamine transferase for DEAD-domain containing proteins (inhibits NF-κB signaling)94,95 | ||
The second group of effectors help to preserve host homeostasis by actively stimulating anti-inflammatory pathways. For example, by fluxing phosphoinositides with its phosphoinositide phosphatase enzymatic activity, SopB stimulates the activation of a Rab8-dependent, PI3K/Akt/mTOR signaling pathway that operates downstream of Toll-like receptors74. This pathway leads to the production of the anti-inflammatory cytokine IL-10 thus promoting host recovery after and innate immune response75–77. Interestingly, the same pathway is targeted by the effector protein SopD but through a completely different mechanism74. This effector works by stimulating the dissociation of Rab8 from its cognate GDP-dissociation inhibitor (GDI), which leads to the GTP loading of this GTPase and the subsequent stimulation of the PI3K/Akt/mTOR anti-inflammatory pathway. In essence, the SopD activity is equivalent to that of eukaryotic GDI-displacement factors (GDF), which activate Rab GTPase by removing them from their cognate GDIs thus targeting them to the membrane for recycling and activation101,102. Therefore, in the case of SopD, both pro-(see above) and anti-inflammatory activities are encoded within the same effector.
Another example of this group of effectors is GogC (also known as SteE, SarA, or PagJ), which targets signal transducer and activator of transcription 3 (STAT3)103,104. This signaling protein is involved in many cell biological processes including signaling pathways that direct the recovery of homeostasis after an inflammatory response105,106. S. Typhimurium is a potent activator of this signaling pathway, which is required for its efficient intracellular growth107. The STAT3 activation mechanism is non-canonical as it does not require the Jak kinases. Instead, the mechanism requires the host kinase GSK-3, which phosphorylates GogC leading to the formation of a GogC/STAT3 complex and the activation of this signaling cascade103,104.
Concluding remarks
The mechanisms by which S. Typhimurium triggers intestinal inflammation through the action of its T3SS effectors are now well understood. The studies of these mechanisms have provided insight not only on the pathogenesis of Salmonella infection but also of the underlying mechanisms that lead to some chronic intestinal inflammatory illnesses such as Crohn’s or inflammatory bowel disease. Given the central role played by intestinal inflammation in the pathogenesis of Salmonella infections, it is possible that the knowledge of the detailed mechanisms by which this pathogen modulates inflammation could serve as the bases for the development of novel anti-infectants targeting relevant host pathways or effector proteins. In fact, it has been shown that oral administration of an inhibitor of host PAK kinases, which are essential for the initiation of the inflammatory response to S. Typhimurium, effectively blocked the ability of this pathogen to trigger intestinal inflammation and replicate with the intestinal tract58. However, addition of the inhibitor resulted in increased bacterial replication in systemic tissues. These findings illustrate the challenge of targeting a host response that is required for both, pathogen replication and host defense. The mechanisms by which S. Typhimurium modulates the inflammatory response are a remarkable example of the complex adaptations that emerge from long-standing host-pathogen associations.
Acknowledgments
I apologize to many colleagues whose important work I could not discuss or cite due to space limitations. I thank Maria Lara-Tejero for critical review of this manuscript. Work in my laboratory is supported by NIH Grants R01AI114618, R01AI055472 and R01AI030492. I declare no financial interest relevant to this article or the research conducted in my laboratory.
References
- 1.Shannon E et al. The Global Burden of Nontyphoidal Salmonella Gastroenteritis. Clinical Infectious Diseases 50, 882–889 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Buckle G, Walker C & Black R Typhoid fever and paratyphoid fever: Systematic review to estimate global morbidity and mortality for 2010. J Glob Health. 2, 010401 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Popoff M, Bockemühl J & Brenner F Supplement 1998 (no. 42) to the Kauffmann-White scheme. Res Microbiol. 151, 63–65 (2000). [DOI] [PubMed] [Google Scholar]
- 4.Brenner FW, Villar RG, Angulo FJ, Tauxe R & Swaminathan B Salmonella Nomenclature. Journal of Clinical Microbiology 38, 2465–2467, doi: 10.1128/jcm.38.7.2465-2467.2000 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.House D, Bishop A, Parry C, Dougan G & Wain J Typhoid fever: pathogenesis and disease. Curr. Opin. Infect. Dis. 14, 573–578 (2001). [DOI] [PubMed] [Google Scholar]
- 6.Dougan G & Baker S Salmonella enterica serovar Typhi and the pathogenesis of typhoid fever. Annu Rev Microbiol. 68, 317–336 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Parry C, Hien TT, Dougan G, White N & Farrar J Typhoid fever. N Engl J Med. 347, 1770–1782 (2002). [DOI] [PubMed] [Google Scholar]
- 8.Hohmann E Nontyphoidal salmonellosis. Clin Infect Dis. 32, 263–269 (2001). [DOI] [PubMed] [Google Scholar]
- 9.Rivera-Chávez F & Bäumler A The Pyromaniac Inside You: Salmonella Metabolism in the Host Gut. Annu Rev Microbiol. 69, 31–48 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Akira S, Uematsu S & Takeuchi O Pathogen recognition and innate immunity. Cell 124, 783–801 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Creagh E & O’Neill L TLRs, NLRs and RLRs: a trinity of pathogen sensors that co-operate in innate immunity. Trends Immunol. 27, 352–357 (2006). [DOI] [PubMed] [Google Scholar]
- 12.Medzhitov R Toll-like receptors and innate immunity. Nature Rev Immunol. 1, Nature Rev Immunol. (2001). [DOI] [PubMed] [Google Scholar]
- 13.Stecher B et al. Salmonella enterica serovar typhimurium exploits inflammation to compete with the intestinal microbiota. PLoS Biol. 5, 2177–2189 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Winter S et al. Gut inflammation provides a respiratory electron acceptor for Salmonella. Nature 467, 426–429 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Buffie C & Pamer E Microbiota-mediated colonization resistance against intestinal pathogens. Nat Rev Immunol. 13, 790–801 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rangan K & Han H Biochemical mechanisms of pathogen restriction by intestinal bacteria. Trends Biochem Sci. 42, 887–898 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Olsan E et al. Colonization resistance: The deconvolution of a complex trait. J Biol Chem. 292, 8577–8581 (2017. ). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lupp C et al. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe 2, 204–2012 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Zeng M, Inohara N & Nuñez G Mechanisms of inflammation-driven bacterial dysbiosis in the gut. Mucosal Immunol. 10, 18–26 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thiennimitr P et al. Intestinal inflammation allows Salmonella to use ethanolamine to compete with the microbiota. Proc Natl Acad Sci U S A. 108, 17480–17485 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Santos R et al. Life in the inflamed intestine, Salmonella style. Trends Microbiol. 17, 498–506 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rogers A, Tsolis R & Bäumler A Salmonella versus the Microbiome. Microbiol Mol Biol Rev. 85, e00027–00019 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fattinger S, Sellin M & Hardt W Epithelial inflammasomes in the defense against Salmonella gut infection. Curr Opin Microbiol. 59, 86–94 (2021). [DOI] [PubMed] [Google Scholar]
- 24.Ohl ME & Miller SI Salmonella: a model for bacterial pathogenesis. Annu. Rev. Med. 52, 259–274. (2001). [DOI] [PubMed] [Google Scholar]
- 25.Giannella RA, Broitman SA & N., Z. Gastric acid barrier to ingested microorganisms in man: studies in vivo and in vitro. Gut 13, 251–256 (1972). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsolis RM, Xavier MN, Santos RL & AJ. B How to become a top model: impact of animal experimentation on human Salmonella disease research. I. Infect Immun. 79, 1806–1814 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stecher B et al. Flagella and chemotaxis are required for efficient induction of Salmonella enterica serovar Typhimurium colitis in streptomycin-pretreated mice. Infect Immun. 72, 4138–4150 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Galán JE & Curtiss III R Cloning and molecular characterization of genes whose products allow Salmonella typhimurium to penetrate tissue culture cells. Proc. Natl. Acad. Sc. USA 86, 6383–6387 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Galán J, Lara-Tejero M, Marlovits T & Wagner S Bacterial type III secretion systems: specialized nanomachines for protein delivery into target cells. Annu Rev Microbiol. 68, 415–438 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Figueira R & Holden D Functions of the Salmonella pathogenicity island 2 (SPI-2) type III secretion system effectors. Microbiology 158, 1147–1161 (2012). [DOI] [PubMed] [Google Scholar]
- 31.LaRock D, Chaudhary A & Miller S Salmonellae interactions with host processes. Nat Rev Microbiol. 13, 191–205 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takeuchi A Electron microscopic studies of experimental Salmonella infection. 1. Penetration into the intestinal epithelium by Salmonella typhimurium. Am. J. Pathol. 50, 109–136 (1967). [PMC free article] [PubMed] [Google Scholar]
- 33.Galán JE Salmonella interaction with host cells: Type III Secretion at Work. Annu. Rev. Cell Dev. Biol. 17, 53–86 (2001). [DOI] [PubMed] [Google Scholar]
- 34.Hume P, Singh V, Davidson A & Koronakis V Swiss Army Pathogen: The Salmonella Entry Toolkit. Front Cell Infect Microbiol. 7, 348–358 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hobbie S, Chen LM, Davis R & Galán JE Involvement of the mitogen-activated protein kinase pathways in the nuclear responses and cytokine production induced by Salmonella typhimurium in cultured intestinal cells. J. Immunol. 159, 5550–5559 (1997). [PubMed] [Google Scholar]
- 36.Bruno VM et al. Salmonella Typhimurium type III secretion effectors stimulate innate immune responses in cultured epithelial cells. PLoS Pathog 5, e1000538, doi: 10.1371/journal.ppat.1000538 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hardt W-D, Chen L-M, Schuebel KE, Bustelo XR & Galán JE Salmonella typhimurium encodes an activator of Rho GTPases that induces membrane ruffling and nuclear responses in host cells. Cell 93, 815–826 (1998). [DOI] [PubMed] [Google Scholar]
- 38.Patel JC & Galan JE Manipulation of the host actin cytoskeleton by Salmonella--all in the name of entry. Curr Opin Microbiol 8, 10–15, doi: 10.1016/j.mib.2004.09.001 (2005). [DOI] [PubMed] [Google Scholar]
- 39.Patel JC & Galan JE Differential activation and function of Rho GTPases during Salmonella-host cell interactions. J Cell Biol 175, 453–463, doi: 10.1083/jcb.200605144 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou D, Chen LM, Hernandez L, Shears SB & Galan JE A Salmonella inositol polyphosphatase acts in conjunction with other bacterial effectors to promote host cell actin cytoskeleton rearrangements and bacterial internalization. Mol Microbiol 39, 248–259 (2001). [DOI] [PubMed] [Google Scholar]
- 41.Zhou D, Mooseker M & Galán JE Role of the S. typhimurium actin-binding protein SipA in bacterial internalization. Science 283, 2092–2095 (1999). [DOI] [PubMed] [Google Scholar]
- 42.Jennings E, Thurston T & Holden D Salmonella SPI-2 Type III Secretion System Effectors: Molecular Mechanisms And Physiological Consequences. Cell Host Microbe. 22, 217–231 (2017). [DOI] [PubMed] [Google Scholar]
- 43.Wotzka S, Nguyen B & Hardt W Salmonella Typhimurium Diarrhea Reveals Basic Principles of Enteropathogen Infection and Disease-Promoted DNA Exchange. Cell Host Microbe. 21, 443–454 (2017). [DOI] [PubMed] [Google Scholar]
- 44.Stevens M, Humphrey T & Maskell D Molecular insights into farm animal and zoonotic Salmonella infections. Philos Trans R Soc Lond B Biol Sci. 364, 2709–2723 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Feasey N, Dougan G, Kingsley R, Heyderman R & Gordon M Invasive non-typhoidal salmonella disease: an emerging and neglected tropical disease in Africa. Lancet 379, 2489–2499 (2012. ). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee J, Mo J, Shen C, Rucker A & Raz E Toll-like receptor signaling in intestinal epithelial cells contributes to colonic homoeostasis. Curr Opin Gastroenterol. 23, 27–31 (2007). [DOI] [PubMed] [Google Scholar]
- 47.Kelly D, Conway S & Aminov R Commensal gut bacteria: mechanisms of immune modulation. Trends Immunol. 26, 326–333 (2005). [DOI] [PubMed] [Google Scholar]
- 48.Eckmann L Sensor molecules in intestinal innate immunity against bacterial infections. Curr. Opin. Gastroenterol. 22, 95–101 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shibolet O & Podolsky D TLRs in the Gut. IV. Negative regulation of Toll-like receptors and intestinal homeostasis: addition by subtraction. Am J Physiol Gastrointest Liver Physiol. 292, G1469–1473 (2007). [DOI] [PubMed] [Google Scholar]
- 50.Lang T & Mansell A The negative regulation of Toll-like receptor and associated pathways Immunol Cell Biol. 85, 425–434 (2007). [DOI] [PubMed] [Google Scholar]
- 51.Chen LM, Hobbie S & Galan JE Requirement of CDC42 for Salmonella-induced cytoskeletal and nuclear responses. Science 274, 2115–2118 (1996). [DOI] [PubMed] [Google Scholar]
- 52.Miao E et al. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc Natl Acad Sci U S A. 107, 3076–3080 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jessen D et al. Type III secretion needle proteins induce cell signaling and cytokine secretion via Toll-like receptors. Infect Immun. 82, 2300–2309 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Friebel A et al. SopE and SopE2 from Salmonella typhimurium activate different sets of RhoGTPases of the host cell. J Biol Chem. 276, 34035–34040 (2001). [DOI] [PubMed] [Google Scholar]
- 55.Norris FA, Wilson MP, Wallis TS, Galyov EE & Majerus PW SopB, a protein required for virulence of Salmonella dublin, is an inositol phosphate phosphatase. Proc. Natl. Acad. Sc. U. S. A. 95, 14057–14059 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Keestra A et al. Manipulation of small Rho GTPases is a pathogen-induced process detected by NOD1. Nature 496, 233–237 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lara-Tejero M et al. Role of the caspase-1 inflammasome in Salmonella typhimurium pathogenesis. J Exp Med 203, 1407–1412, doi: 10.1084/jem.20060206 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sun H, Kamanova J, Lara-Tejero M & Galán JE Salmonella stimulates pro-inflammatory signalling through p21-activated kinases bypassing innate immune receptors. Nat Microbiol. 3, 1122–1130 ( 2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Walsh M, Lee J, Choi Y & Ibrahim M Tumor necrosis factor receptor-associated factor 6 (TRAF6) regulation of development, function, and homeostasis of the immune system. Immunol Rev. 266, 72–92 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ajibade A, Wang H & Wang R Cell type-specific function of TAK1 in innate immune signaling. Trends Immunol. 34, 307–316 (2013). [DOI] [PubMed] [Google Scholar]
- 61.Wood MW et al. The secreted effector protein of Salmonella dublin, SopA, is translocated into eukaryotic cells and influences the induction of enteritis. Cell Microbiol 2, 293–303 (2000). [DOI] [PubMed] [Google Scholar]
- 62.Zhang Y, Higashide W, McCormick B, Chen J & Zhou D The inflammation-associated Salmonella SopA is a HECT-like E3 ubiquitin ligase. Mol Microbiol. 62, 786–793. (2006). [DOI] [PubMed] [Google Scholar]
- 63.Kamanova J, Sun H, Lara-Tejero M & Galán J The Salmonella Effector Protein SopA Modulates Innate Immune Responses by Targeting TRIM E3 Ligase Family Members. PLoS Pathog 12, e1005552. (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rajsbaum R, García-Sastre A & Versteeg G TRIMmunity: the roles of the TRIM E3-ubiquitin ligase family in innate antiviral immunity. J Mol Biol. 426, 1265–1284 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yap M & Stoye J TRIM proteins and the innate immune response to viruses. Adv Exp Med Biol. 770, 93–104 (2012). [DOI] [PubMed] [Google Scholar]
- 66.Ikeda K & Inoue S TRIM proteins as RING finger E3 ubiquitin ligases. Adv Exp Med Biol. 770, 27–37 (2012). [DOI] [PubMed] [Google Scholar]
- 67.Tsuchida T et al. The ubiquitin ligase TRIM56 regulates innate immune responses to intracellular double-stranded DNA. Immunity 33, 765–776 (2010). [DOI] [PubMed] [Google Scholar]
- 68.Dixit E & Kagan J Intracellular pathogen detection by RIG-I-like receptors. Adv Immunol. 117, 99–125 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Reikine S, Nguyen J & Modis Y Pattern Recognition and Signaling Mechanisms of RIG-I and MDA5. Front Immunol. 23, 5:342 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schmolke M et al. RIG-I detects mRNA of intracellular Salmonella enterica serovar Typhimurium during bacterial infection. MBio 5, e01006–01014 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fiskin E et al. Structural basis for the recognition and degradation of host TRIM proteins by Salmonella effector SopA. Nat Commun. 8, 14004 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jones MA et al. Secreted effector proteins of Salmonella dublin act in concert to induce enteritis. Infection & Immunity 66, 5799–5804 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang S et al. The Salmonella enterica serotype typhimurium effector proteins SipA, SopA, SopB, SopD, and SopE2 act in concert to induce diarrhea in calves. Infect. Immun. 70, 3843–3855 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lian H et al. The Salmonella Effector Protein SopD targets Rab8 to positively and negatively modulate the inflammatory response. Nature Microbiol. 10.1038/s41564-021-00866-3 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wall A et al. Small GTPase Rab8a-recruited Phosphatidylinositol 3-Kinase γ Regulates Signaling and Cytokine Outputs from Endosomal Toll-like Receptors. J Biol Chem. 292, 4411–4422 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Luo L et al. TLR Crosstalk Activates LRP1 to Recruit Rab8a and PI3Kγ for Suppression of Inflammatory Responses. Cell Rep. 24, Cell Rep. 2018 Sep 2011;2024(2011):3033–3044. (2018). [DOI] [PubMed] [Google Scholar]
- 77.Tong S, Wall A, Hung Y, Luo L & Stow J Guanine nucleotide exchange factors activate Rab8a for Toll-like receptor signalling. Small GTPases 7, 1–17 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.de Vasconcelos N & Lamkanfi M Recent Insights on Inflammasomes, Gasdermin Pores, and Pyroptosis. Cold Spring Harb Perspect Biol. 12, a036392 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.P., B. Recognition of Intracellular Bacteria by Inflammasomes. Microbiol Spectr. 7, doi: 10.1128/microbiolspec.BAI-0003-2019 (2019). [DOI] [PubMed] [Google Scholar]
- 80.Hayward J, Mathur A, Ngo C & Man S Cytosolic Recognition of Microbes and Pathogens: Inflammasomes in Action. Microbiol Mol Biol Rev. 82, e00015–00018 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Deets K & Vance R Inflammasomes and adaptive immune responses. Nat Immunol. doi: 10.1038/s41590-021-00869-6., I (2021). [DOI] [PubMed] [Google Scholar]
- 82.Ta A & Vanaja S Inflammasome activation and evasion by bacterial pathogens. Curr Opin Immunol. 68, 125–133 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.von Moltke J, Ayres J, Kofoed E, Chavarría-Smith J & Vance R Recognition of bacteria by inflammasomes. Annu Rev Immunol. 31, 73–106 (2013). [DOI] [PubMed] [Google Scholar]
- 84.Monack DM, Raupach B, Hromockyj AE & Falkow S Salmonella typhimurium invasion induces apoptosis in infected macrophages. Proc. Natl. Acad. Sci. U. S. A. 93, 9833–9838 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chen LM, Kaniga K & Galan JE Salmonella spp. are cytotoxic for cultured macrophages. Mol Microbiol 21, 1101–1115 (1996). [DOI] [PubMed] [Google Scholar]
- 86.Fattinger S, Sellin M & Hardt W Epithelial inflammasomes in the defense against Salmonella gut infection. Curr Opin Microbiol. 59, 86–94 (2020). [DOI] [PubMed] [Google Scholar]
- 87.Crowley S, Knodler L & Vallance B Salmonella and the Inflammasome: Battle for Intracellular Dominance. Curr Top Microbiol Immunol. 397:43–67., 43–67 (2016). [DOI] [PubMed] [Google Scholar]
- 88.Miao E et al. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat Immunol. 7, 569–575 (2006). [DOI] [PubMed] [Google Scholar]
- 89.Knodler L et al. Dissemination of invasive Salmonella via bacterial-induced extrusion of mucosal epithelia. Proc Natl Acad Sci U S A. 107, 17733–17738 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Haneda T et al. Salmonella type III effector SpvC, a phosphothreonine lyase, contributes to reduction in inflammatory response during intestinal phase of infection. Cell Microbiol. 14, 485–499 (2012). [DOI] [PubMed] [Google Scholar]
- 91.Lu R et al. Chronic effects of a Salmonella type III secretion effector protein AvrA in vivo. PLoS One. 5(e10505 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sun H, Kamanova J, Lara-Tejero M & Galán J A Family of Salmonella Type III Secretion Effector Proteins Selectively Targets the NF-κB Signaling Pathway to Preserve Host Homeostasis. PLoS Pathog. 12, e1005484. (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fu Y & Galan JE A salmonella protein antagonizes Rac-1 and Cdc42 to mediate host-cell recovery after bacterial invasion. Nature 401, 293–297, doi: 10.1038/45829 (1999). [DOI] [PubMed] [Google Scholar]
- 94.Newson J et al. Salmonella Effectors SseK1 and SseK3 Target Death Domain Proteins in the TNF and TRAIL Signaling Pathways. Mol Cell Proteomics. 18, 1138–1156 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Günster R, Matthews S, Holden D & Thurston T SseK1 and SseK3 Type III Secretion System Effectors Inhibit NF-kappaB Signaling and Necroptotic Cell Death in Salmonella-Infected Macrophages. Infect Immun. 85, e00010–00017 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rolhion N et al. Inhibition of Nuclear Transport of NF-κB p65 by the Salmonella Type III Secretion System Effector SpvD. PLoS Pathog. 12, e1005653 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jones R et al. Salmonella AvrA Coordinates Suppression of Host Immune and Apoptotic Defenses via JNK Pathway Blockade. Cell Host Microbe 3, 233–244 (2008). [DOI] [PubMed] [Google Scholar]
- 98.Du F & Galan JE Selective inhibition of type III secretion activated signaling by the Salmonella effector AvrA. PLoS Pathog 5, e1000595, doi: 10.1371/journal.ppat.1000595 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Li H et al. The phosphothreonine lyase activity of a bacterial type III effector family. Science, 1000–1003 (2007). [DOI] [PubMed] [Google Scholar]
- 100.Mazurkiewicz P et al. SpvC is a Salmonella effector with phosphothreonine lyase activity on host mitogen-activated protein kinases. Mol Microbiol. 67, 1371–1383 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sivars U, Aivazian D & Pfeffer S Yip3 catalyses the dissociation of endosomal Rab-GDI complexes. Nature 425, 856–859 (2003). [DOI] [PubMed] [Google Scholar]
- 102.Yamashita T & Tohyama M The p75 receptor acts as a displacement factor that releases Rho from Rho-GDI. Nat Neurosci. 6, 461–467 (2003). [DOI] [PubMed] [Google Scholar]
- 103.Panagi I et al. Salmonella Effector SteE Converts the Mammalian Serine/Threonine Kinase GSK3 into a Tyrosine Kinase to Direct Macrophage Polarization. Cell Host Microbe. 27, 41–53 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gibbs K et al. The Salmonella Secreted Effector SarA/SteE Mimics Cytokine Receptor Signaling to Activate STAT3. Cell Host Microbe. 27, 129–139 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Leppkes M, Neurath M, Herrmann M & Becker C Immune deficiency vs. immune excess in inflammatory bowel diseases-STAT3 as a rheo-STAT of intestinal homeostasis. J Leukoc Biol. 99, 57–66 (2016). [DOI] [PubMed] [Google Scholar]
- 106.Hillmer E, Zhang H, Li H & Watowich S STAT3 signaling in immunity. Cytokine Growth Factor Rev. 31, 1–15 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hannemann S, Gao B & Galán J Salmonella modulates host cell gene expression to promote its intracellular growth. PLoS Pathog. 9, e1003668 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hu B, Lara-Tejero M, Kong Q, Galán J & Liu J In Situ Molecular Architecture of the Salmonella Type III Secretion Machine. Cell 168, 1065–1074 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Park D et al. Visualization of the type III secretion mediated Salmonella-host cell interface using cryo-electron tomography. Elife 7, e39514 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]