

Abstract
Objective:
Patients with melanoma and early stable disease (SD) with pembrolizumab have unclear prognosis. We present post hoc analyses of long-term outcomes for patients with early SD, partial response (PR) or complete response (CR) with pembrolizumab.
Patients and methods:
Patients who received pembrolizumab in the KEYNOTE-001 and KEYNOTE-006 studies and had SD, PR or CR at weeks 12 or 24 were included.
Results:
Of 294 patients in the week 12 analysis, 107 (36.4%) had SD at week 12, of whom 7 (6.5%) had a best overall response of CR, 43 (40.2%) had PR and 57 (53.3%) had SD. Forty-eighte–month overall survival (OS) rates were 95.2%, 73.0% and 47.7%, respectively, for patients with CR, PR and SD at week 12. Similar results were observed in the 241 patients in the week 24 analysis. Forty-eight–month OS rates were 72.1% for patients with SD at week 12 followed by subsequent response and 75.0% for patients with PR at week 12 followed by no change in response or progression. Thirty-six–month and 48-month OS rates were 11.6% and not reached, respectively, for patients with SD at week 12 followed by progression before week 24.
Conclusions:
A substantial proportion of patients (46.7%) with early (week 12) SD with pembrolizumab achieved subsequent PR or CR. Patients with SD at week 12 and subsequent CR/PR had similar survival to those who maintained PR. In contrast, patients with SD at week 12 and subsequent progression had poor survival outcomes. These findings may guide treatment decisions for patients achieving early SD.
Trial registration:
Clinicaltrials.gov: NCT01295827 (KEYNOTE-001); NCT01866319 (KEYNOTE-006).
Keywords: Melanoma, Pembrolizumab, Programmed death 1, PD-1
1. Introduction
Despite increased education and awareness of risk factors for melanoma, the number of cases diagnosed each year continues to rise [1]. Historically, the prognosis for advanced melanoma was poor, but survival has improved significantly with the introduction of targeted therapies and immune checkpoint inhibitors [2]. For patients with advanced BRAF wild-type melanoma, the preferred first-line regimens are pembrolizumab, nivolumab or nivolumab with ipilimumab [3]. For the 50–60% of patients with BRAF-mutant melanoma, BRAF and MEK inhibitor combination therapy is also an option if early response is needed [1,3]. Although immunotherapy has improved survival in advanced melanoma, predictive factors associated with long-term response remain to be elucidated [4].
Pembrolizumab is a first-line standard of care option for unresectable stage IIIeIV or metastatic melanoma [3,5,6]. Pembrolizumab has shown durable antitumour activity in advanced melanoma in several trials, including KEYNOTE-001 and KEYNOTE-006 [7,8]. KEYNOTE-001, a phase I trial, evaluated pembrolizumab in patients with locally advanced or metastatic solid tumours, including melanoma [7]. The results from the melanoma cohorts showed that pembrolizumab was well tolerated and had durable antitumour activity, including long-term survival benefit, in patients with treatment-naive or previously treated disease. At five-year follow-up, the objective response rate (ORR) in the overall melanoma population was 34%, and the median overall survival (OS) was 23.8 months [7]. A greater proportion of patients with complete response (CR) had ongoing response compared with patients who had partial response (PR) in the total (89% vs. 63%) and treatment-naive (92% vs. 73%) populations [7].
KEYNOTE-006, a phase III trial, evaluated two regimens of pembrolizumab versus ipilimumab in patients with advanced melanoma [9]. The primary analysis showed significant benefit with pembrolizumab and durable benefit on long-term follow-up [9,10]. In the five-year follow-up analysis, the median OS was 32.7 months with pembrolizumab versus 15.9 months with ipilimumab, and the ORR was 42% versus 17% [8]. Of patients who completed two years of pembrolizumab, 76% with CR, 77% with PR and 54% with stable disease (SD) had an ongoing response.
These trials showed that most patients who achieve CR or PR with pembrolizumab experience durable benefit; however, a better understanding of the prognosis for pembrolizumab-treated patients who have an assessment of SD is needed. This analysis evaluates the outcome of patients with melanoma who received pembrolizumab and had an assessment of SD, PR or CR at week 12 or week 24 in the KEYNOTE-001 and KEYNOTE-006 trials.
2. Materials and methods
2.1. Study design and participants
KEYNOTE-001 evaluated pembrolizumab in patients with locally advanced or metastatic carcinoma, melanoma or non-small cell lung carcinoma [7,11]. For the melanoma cohort, eligible patients were required to have ipilimumab-pretreated or ipilimumab-naive melanoma and to have received ≤2 lines of systemic treatment for metastatic or locally advanced melanoma.
KEYNOTE-006 compared two dose schedules of pembrolizumab with ipilimumab in patients with unresectable stage III or IV melanoma who had received ≤1 prior systemic therapy for advanced disease [8,9]. Patients with BRAF-mutant melanoma were required to have received prior BRAF inhibitor therapy unless they met specific criteria. Detailed methods for these trials have been reported previously [11,12]. Eligibility criteria, programmed death ligand 1 (PD-L1) status and baseline patient characteristics are listed in the Supplementary Methods.
2.2. Ethics
The KEYNOTE-001 and KEYNOTE-006 studies were conducted in accordance with the protocol, Good Clinical Practice standards, the Declaration of Helsinki and all local regulations. The protocols and amendments were approved by the relevant institutional review boards or ethics committees at each participating institution. All patients provided written informed consent.
2.3. Procedures
In KEYNOTE-001, patients received pembrolizumab 2 mg/kg every 3 weeks (Q3W), 10 mg/kg Q2W or 10 mg/kg Q3W. In KEYNOTE-006, patients received pembrolizumab 10 mg/kg Q2W, 10 mg/kg Q3W or four doses of ipilimumab 3 mg/kg Q3W (for details see Supplementary Methods). In both studies, response at 12, 18 and 24 weeks was assessed as per Response Evaluation Criteria in Solid Tumours v1.1 by independent central review, and the best overall response (BOR) with confirmation was used.
2.4. Statistical analysis
Post hoc analysis of long-term outcomes is presented for patients with an assessment of SD, PR or CR 12 and 24 weeks after randomisation. Patients were included in the week 12 or 24 analysis populations if they had a single time point assessment of CR, PR or SD at week 12 or 24, respectively, and had not experienced disease progression or were censored before week 12 or 24. Patients were not required to have a week 12 assessment for inclusion in the week 24 analysis population.
The analysis included patients with melanoma who were treatment naive or who had received BRAF or MEK inhibitors as their only prior therapy. Patients who had previously received ipilimumab in KEYNOTE-001 were excluded. Data from pembrolizumab dose groups were pooled.
The association of baseline characteristics and clinical response was evaluated using the chi-square test of independence. The data cutoff was 1 September 2017, for KEYNOTE-001, and 4 December 2017, for KEYNOTE-006.
3. Results
3.1. Patients
In KEYNOTE-001 and KEYNOTE-006, there were 643 patients treated with pembrolizumab who had treatment-naive disease or had received BRAF inhibitors as their only prior therapy (Table 1). Of these, 294 (45.7%) were included in the week 12 analysis and 241 (37.5%) were included in the week 24 analysis. Of the 294 patients in the week 12 population, 23 (7.8%) had CR, 164 (55.8%) had PR and 107 (36.4%) had SD (Fig. 1, Supplementary Fig. 1). Of the 241 patients included in the week 24 population, 42 (17.4%) had CR, 160 (66.4%) had PR and 39 (16.2%) had SD.
Table 1.
Pembrolizumab-treated patients included in the analysis.
| Patients, n | Week 12 analysis |
Week 24 analysis |
||||
|---|---|---|---|---|---|---|
| KEYNOTE-001 | KEYNOTE-006 | Total | KEYNOTE-001 | KEYNOTE-006 | Total | |
| Treatment naive or prior BRAFi | 182 | 461 | 643 | 182 | 461 | 643 |
| Progressed or censored before time point | 82 | 202 | 284 | 97 | 240 | 337 |
| Missing response at landmark | 7 | 22 | 29 | 9 | 35 | 44 |
| Response other than SD, PR or CRa | 7 | 29 | 36 | 3 | 18 | 21 |
| Patients included in analysis, n | 86 | 208 | 294 | 73 | 168 | 241 |
| Treatment naive | 79 | 177 | 256 | 68 | 144 | 212 |
| Prior BRAFi only | 7 | 31 | 38 | 5 | 24 | 29 |
BRAFi, BRAF inhibitor; CR, complete response; PD, progressive disease; PR, partial response; SD, stable disease.
Included patients with non-CR/non-PD, not available, not done or unconfirmed progression.
Fig. 1.

Subsequent response for patients with SD, PR or CR at week 12 in the week 12 analysis population. Response was assessed by independent central review in KEYNOTE-001 and in KEYNOTE-006. CR, complete response; PD, progressive disease; PR, partial response; SD, stable disease.
At baseline, most patients in the week 12 and week 24 populations had an Eastern Cooperative Oncology Group performance status (ECOG PS) of 0 and M1c disease (Table 2). Most patients (87.1%, week 12; 88.0%, week 24) had treatment-naive disease.
Table 2.
Baseline characteristics of patients included in the analysis.
| Characteristic, n (%) | Week 12 analysis population; n = 294 |
Week 24 analysis population; n = 241 |
|---|---|---|
| Sex | ||
| Male | 203 (69.0) | 172 (71.4) |
| Female | 91 (31.0) | 69 (28.6) |
| Age | ||
| <65 years | 150 (51.0) | 121 (50.2) |
| ≥65 years | 144 (49.0) | 120 (49.8) |
| Tumour size a | ||
| <2.5 cm | 67 (22.8) | 61 (25.3) |
| 2.5 to <5 cm | 90 (30.6) | 63 (26.1) |
| 5 to <10 cm | 69 (23.5) | 60 (24.9) |
| ≥10 cm | 68 (23.1) | 57 (23.7) |
| BRAF status (all patients) | ||
| Wild type | 187 (63.6) | 160 (66.4) |
| Mutant | 103 (35.0) | 79 (32.8) |
| Unknown | 4 (1.4) | 2 (0.8) |
| BRAF status (previously untreated patients) | ||
| Wild type | 185 (72.3) | 158 (74.5) |
| Mutant | 68 (26.5) | 53 (25.0) |
| Unknown | 3 (1.2) | 1 (0.5) |
| PD-L1 tumour status b | ||
| Negative | 33 (11.2) | 26 (10.8) |
| Positive | 207 (70.4) | 168 (69.7) |
| Unknown | 54 (18.4) | 47 (19.5) |
| ECOG PS | ||
| 0 | 217 (73.8) | 180 (74.7) |
| 1 | 77 (26.2) | 61 (25.3) |
| Lactate dehydrogenase level | ||
| Normal | 212 (72.1) | 179 (74.3) |
| Elevated | 77 (26.2) | 57 (23.6) |
| Unknown | 5 (1.7) | 5 (2.1) |
| Metastasis stage | ||
| M0/M1A/M1B | 98 (33.3) | 84 (34.9) |
| M1C | 196 (66.7) | 157 (65.1) |
ECOG PS, Eastern Cooperative Oncology Group performance status; PD-L1, programmed death ligand 1.
Baseline tumour size was measured by adding the sum of the longest dimensions of all measurable baseline target lesions.
PD-L1 positivity was defined as membranous staining in at least 1% of tumour cells.
In the week 12 analysis, of the 164 patients with an assessment of PR at week 12, 49 (29.9%) had a BOR of CR, 108 (65.9%) had a BOR of PR and 7 (4.2%) had a BOR of SD. Of the 107 patients with an initial assessment of SD at week 12, 7 (6.5%) had a BOR of CR, 43 (40.2%) had a BOR of PR and 57 (53.3%) had a BOR of SD. The median time for patients with SD at week 12 to evolve into PR or CR was 12.1 weeks (range, 0.1–98.6) and 12.1 weeks (range, 3.9–131.0), respectively. Of patients with SD at week 12, 23 (21.5%) experienced PD by week 24 and 45 (42.1%) experienced PD after week 24.
In the week 24 analysis, of the 160 patients with an assessment of PR at week 24, 32 (20.0%) had a BOR of CR. Of the 39 patients with SD at week 24, 1 (2.6%) had a BOR of CR, 13 (33.3%) had a BOR of PR and 25 (64.1%) had a BOR of SD. The median time for patients with SD at week 24 to evolve into PR or CR was 12.1 weeks (range, 6.1–86.1) and 120.1 weeks, respectively. Of patients with SD at week 24, 20 (51.3%) developed PD after week 24.
3.2. Association between baseline characteristics and response
Baseline tumour size, PD-L1 status, ECOG PS and metastatic stage were associated with week 12 response (Table 3). Patients with small tumours at baseline (<2.5 cm: CR, 73.9%; PR, 19.5%; SD, 16.8%), a baseline ECOG PS of 0 (CR, 95.6%; PR, 71.9%; SD, 72.0%) and stage M0/M1a/M1b disease (CR, 65.2%; PR, 29.9%; SD, 31.8%) were more likely to have CR at week 12 than PR or SD. Patients with positive PD-L1 tumours were more likely to have CR or PR at week 12 than SD (CR, 89.5%; PR, 91.2%; SD, 77.6%). Sex, baseline tumour size, ECOG PS and metastatic stage were associated with week 24 response (Table 3). As observed with week 12 data, patients with small tumours at baseline (<2.5 cm: CR, 66.7%; PR, 16.9%; SD, 15.4%), a baseline ECOG PS of 0 (CR, 90.5%; PR, 70.0%; SD, 76.9%) and stage M0/M1a/M1b disease (CR, 54.82%; PR, 28.79%; SD, 38.5%) were more likely to have CR at week 24 than PR and SD. Patients who were female (CR, 59.5%; PR, 77.5%; SD, 59.0%) and had stage M1c disease (CR, 45.2%; PR, 71.3%; SD, 61.5%) were more likely to have PR at week 24 than CR or SD.
Table 3.
Association between baseline characteristics and response in the week 12 and week 24 analysis populations.a
| Characteristic, n (%) | Week 12 assessment population |
Week 24 assessment population |
||||
|---|---|---|---|---|---|---|
| SD; n = 107 | PR; n = 164 | CR; n = 23 | SD; n = 39 | PR; n = 160 | CR; n = 42 | |
| Sex | ||||||
| Male | 75 (70.1) | 114 (69.5) | 14 (60.9) | 16 (41.0) | 36 (22.5) | 17 (40.5) |
| Female | 32 (29.9) | 50 (30.5) p = 0.673 |
9 (39.1) | 23 (59.0) | 124 (77.5) p = 0.01 |
25 (59.5) |
| Tumour size, b | ||||||
| <2.5 | 18 (16.8) | 32 (19.5) | 17 (73.9) | 6 (15.4) | 27 (16.9) | 28 (66.7) |
| 2.5 to <5 | 36 (33.6) | 49 (29.9) | 5 (21.7) | 14 (35.9) | 39 (24.4) | 10 (23.8) |
| 5 to <10 | 31 (29.0) | 37 (22.5) | 1 (4.4) | 14 (35.9) | 43 (26.9) | 3 (7.1) |
| ≥10 | 22 (20.6) | 46 (28.1) p < 0.001 |
0 | 5 (12.8) | 51 (31.9) p < 0.001 |
1 (2.4) |
| PD-L1 tumour status c | ||||||
| Positive | 66 (77.6) | 124 (91.2) | 17 (89.5) | 22 (75.9) | 114 (87.7) | 32 (91.4) |
| Negative | 19 (22.4) | 12 (8.8) p < 0.05 |
2 (10.5) | 7 (24.1) | 16 (12.3) p = 0.17 |
3 (8.6) |
| ECOG PS | ||||||
| 0 | 77 (72.0) | 118 (71.9) | 22 (95.6) | 30 (76.9) | 112 (70.0) | 38 (90.5) |
| 1 | 30 (28.0) | 46 (28.1) p < 0.05 |
1 (4.4) | 9 (23.1) | 48 (30.0) p < 0.05 |
4 (9.5) |
| Metastatic stage | ||||||
| M0/M1a/M1b | 34 (31.8) | 49 (29.9) | 15 (65.2) | 15 (38.5) | 46 (28.7) | 23 (54.8) |
| M1c | 73 (68.2) | 115 (70.1) p < 0.01 |
8 (34.8) | 24 (61.5) | 114 (71.3) p < 0.01 |
19 (45.2) |
CR, complete response; ECOG PS, Eastern Cooperative Oncology Group performance status; PD-L1, programmed death ligand 1; PR, partial response; SD, stable disease.
Association of baseline characteristics and clinical assessment was evaluated using the chi-square test of independence. p values are not adjusted for multiplicity.
Baseline tumour size was measured by adding the sum of the longest dimensions of all measurable baseline target lesions.
Among those with PD-L1eevaluable tumours (week 12: SD, n = 85; PR, n = 136; CR, n = 19. Week 24: SD, n = 29; PR, n = 130; CR, n = 35). PD-L1 positivity was defined as membranous staining in at least 1% of tumour cells.
3.3. Survival
In the overall week 12 population, the 24-, 36- and 48-month OS rates were 79.6%, 73.6% and 65.6%, respectively (Table 4). Response at week 12 was correlated with longer subsequent OS than SD at week 12, with 48-month OS rates of 95.2%, 73.0% and 47.7% for patients with CR, PR and SD, respectively (Fig. 2A). In the week 24 population, the overall 24-, 36- and 48-month OS rates were 88.3%, 81.8% and 77.5%, respectively. Response at week 24 was also associated with longer subsequent OS than SD at week 24, with 48-month OS rates of 97.6%, 75.2% and 66.0% for patients with CR, PR and SD, respectively (Fig. 2B).
Table 4.
Estimated OS rates by response in the week 12 or week 24 analysis populations.
| OS rate, % (95% CI) | Week 12 analysis populationa |
Week 24 analysis populationb |
||||
|---|---|---|---|---|---|---|
| 24-month | 36-month | 48-month | 24-month | 36-month | 48-month | |
| Overall | 79.6 (74.5–83.7) | 73.6 (68.1–78.3) | 65.6 (59.3–71.2) | 88.3 (83.6–91.8) | 81.8 (76.3–86.2) | 77.5 (71.2–82.7) |
| SD | 66.4 (56.6–74.4) | 57.5 (47.5–66.3) | 47.7 (36.6–58.1) | 82.0 (65.9–91.0) | 73.5 (56.2–84.8) | 66.0 (47.0–79.5) |
| PR | 85.4 (79.0–89.9) | 80.4 (73.4–85.7) | 73.0 (64.7–79.6) | 87.5 (81.3–91.7) | 79.7 (72.6–85.2) | 75.2 (67.0–81.6) |
| CR | 100.0 (100.0–100.0) | 100.0 (100.0–100.0) | 95.2 (70.7–99.3) | 97.6 (83.9–99.7) | 97.6 (83.9–99.7) | 97.6 (83.9–99.7) |
CI, confidence interval; CR, complete response; OS, overall survival; PR, partial response; SD, stable disease.
OS rate from week 12.
OS rate from week 24.
Fig. 2.
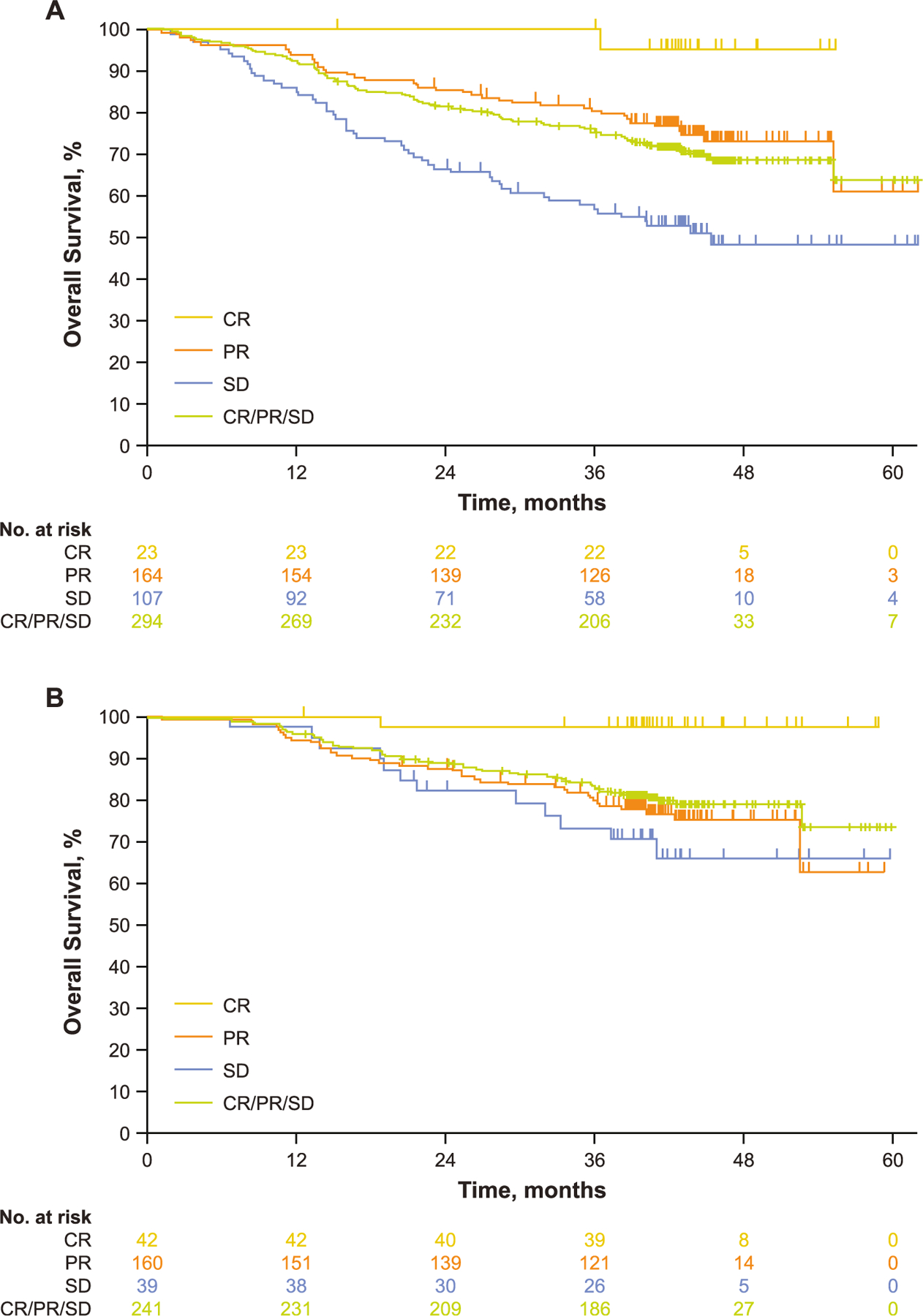
Kaplan-Meier estimates of OS (A) by week 12 response in the week 12 analysis population.a (B) by week 24 response in the week 24 analysis population.b CR, complete response; OS, overall survival; PR, partial response; SD, stable disease. aOS rate from week 12. bOS rate from week 24.
The OS of patients with SD and PR at week 12 by subsequent response was also assessed. Among patients with a week 12 assessment of SD, followed by subsequent response (PR/CR) or ongoing SD (no subsequent response, no subsequent progression), the estimated 48- month OS rates were 72.1% and 46.9%, respectively (Table 5). Among patients with a week 12 assessment of PR followed by subsequent CR, or ongoing PR, the estimated 48-month OS rates were 93.8% and 75.0%, respectively. The 48-month OS rates for patients with a week 12 assessment of SD or PR followed by progression were not estimable, and ongoing patients were censored before 48 months (Table 5).
Table 5.
Estimated OS rates by pattern of response in the week 12 analysis population.
| OS rate, %a (95% CI) | 24-month | 36-month | 48-month |
|---|---|---|---|
| SD at week 12 assessment | |||
| Followed by PR/CR at week 18 or 24 | 88.3 (66.6–92.1) | 77.8 (60.4–88.2) | 72.1 (54.4–83.9) |
| Followed by no response/no progressionb at week 18 or 24 | 70.8 (55.8–81.6) | 64.1 (48.7–76.0) | 46.9 (28.9–63.0) |
| Followed by progression before week 24 | 30.4 (13.5–49.3) | 11.6 (2.3–29.1) | NE |
| PR at week 12 assessment | |||
| Followed by CR at week 18 or 24 | 93.8 (63.2–99.1) | 93.8 (63.2–99.1) | 93.8 (63.2–99.1) |
| Followed by no change in response/no progressionb at week 18 or 24 | 88.1 (81.4–92.6) | 82.8 (75.3–88.2) | 75.0 (65.9–82.0) |
| Followed by progression before week 24 | 41.7 (15.2–66.5) | 33.3 (10.3–58.8) | NE |
CI, confidence interval; CR, complete response; NE, not estimable; OS, overall survival; PR, partial response; SD, stable disease.
OS rate from week 12.
Included patients with no subsequent change in response or disease progression, patients with missing subsequent response data and patients censored due to being lost to follow-up.
Survival outcomes were poorest for patients with a week 12 response of SD who subsequently progressed (Fig. 3). Observational comparisons of baseline characteristics among patients with SD at week 12 by subsequent response showed that patients with subsequent PD were more likely to have BRAF-mutant disease (SD followed by response, 41.7%; ongoing SD, 35.4%; SD followed by PD, 60.9%), have received prior BRAF inhibitor therapy only (SD followed by response, 8.3%; ongoing SD, 14.6%; SD followed by PD, 26.1%) and be <65 years (SD followed by response, 44.4%; ongoing SD, 50.0%; SD followed by PD, 69.6%) compared with patients with SD at week 12 and followed by response or ongoing SD (Table 6).
Fig. 3.
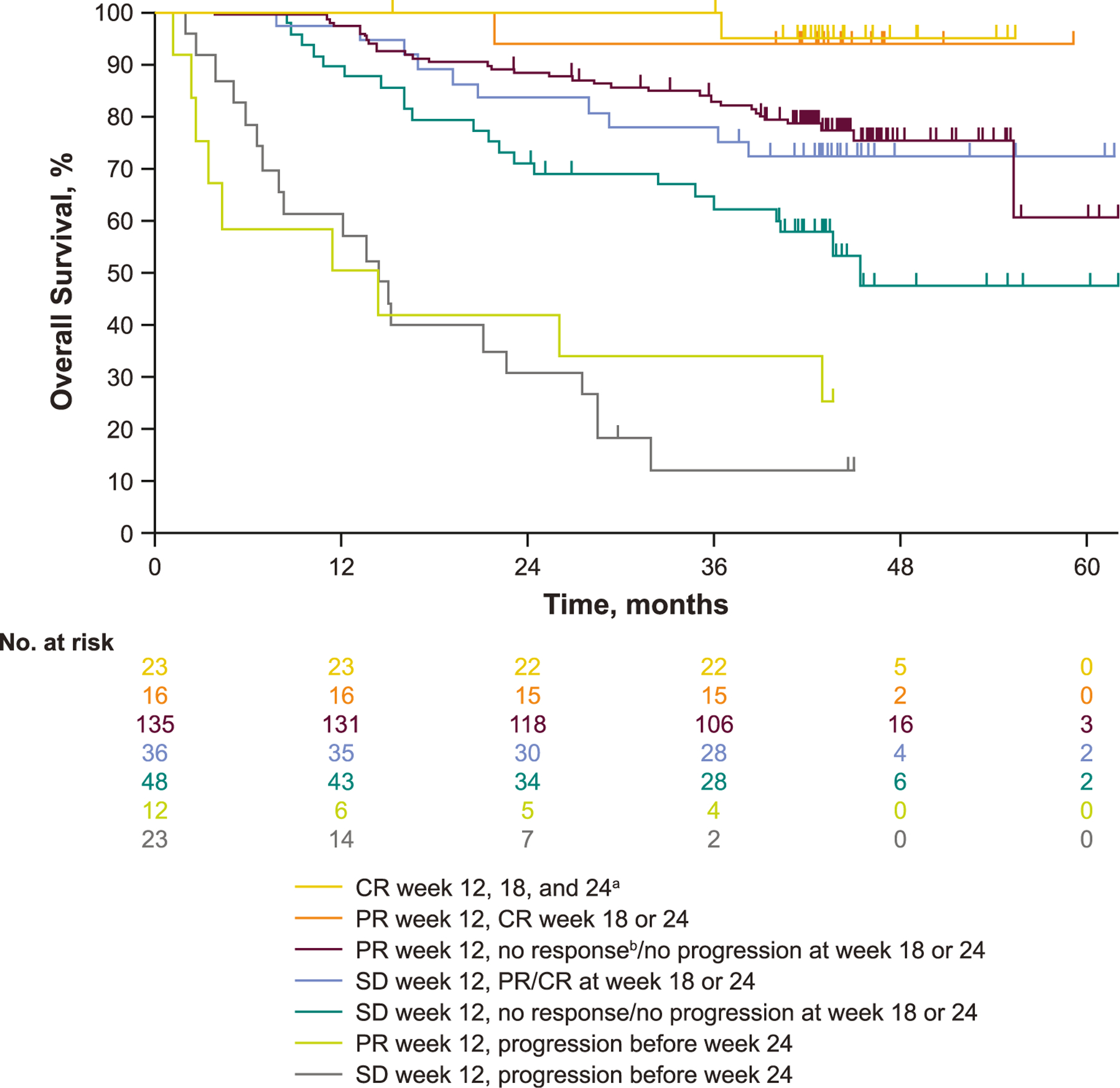
Kaplan-Meier estimate of OS from week 12 by subsequent response in patients with PR or SD at week 12 in the week 12 analysis population. CR, complete response; OS, overall survival; PR, partial response; SD, stable disease. aAll patients with CR at week 12 for whom data were available continued to have CR at weeks 18 and 24. bPatients had PR at week 12 and no subsequent change in response and no progression at week 18 or 24.
Table 6.
Baseline characteristics of patients with a week 12 response of SD by subsequent response.
| Characteristic, n (%) | SD followed by CR/PR; n = 36 |
SD followed by SD; n = 48 |
SD followed by PD; n = 23 |
|---|---|---|---|
| Prior lines | |||
| Treatment naive | 33 (91.7) | 41 (85.4) | 17 (73.9) |
| Prior BRAFi only | 3 (8.3) | 7 (14.6) | 6 (26.1) |
| Sex | |||
| Male | 30 (83.3) | 30 (62.5) | 15 (65.2) |
| Female | 6 (16.7) | 18 (37.5) | 8 (34.8) |
| Age | |||
| <65 years | 16 (44.4) | 24 (50.0) | 16 (69.6) |
| ≥65 years | 20 (55.6) | 24 (50.0) | 7 (30.4) |
| Tumour size,a cm | |||
| <2.5 cm | 8 (22.2) | 6 (12.5) | 4 (17.4) |
| 2.5 to <5 cm | 10 (27.8) | 19 (39.6) | 7 (30.4) |
| 5 to <10 cm | 10 (27.8) | 14 (29.2) | 7 (30.4) |
| ≥10 cm | 8 (22.2) | 9 (18.7) | 5 (21.7) |
| BRAF status (all patients) | |||
| Wild type | 21 (58.3) | 31 (64.6) | 8 (34.8) |
| Mutant | 15 (41.7) | 17 (35.4) | 14 (60.9) |
| Unknown | 0 | 0 | 1 (4.3) |
| BRAF status (previously untreated patients) | |||
| Wild type | 21 (63.6) | 31 (75.6) | 8 (47.1) |
| Mutant | 12 (36.4) | 10 (24.4) | 8 (47.1) |
| Unknown | 0 | 0 | 1 (5.8) |
| PD-L1 tumour status b | |||
| Negative | 7 (19.4) | 9 (18.8) | 3 (13.1) |
| Positive | 24 (66.7) | 27 (56.2) | 15 (65.2) |
| Unknown | 5 (13.9) | 12 (25.0) | 5 (21.7) |
| ECOG PS | |||
| 0 | 26 (72.2) | 32 (66.7) | 19 (82.6) |
| 1 | 10 (27.8) | 16 (33.3) | 4 (17.4) |
| Lactate dehydrogenase level | |||
| Normal | 28 (77.8) | 33 (68.7) | 18 (78.3) |
| Elevated | 8 (22.2) | 14 (29.2) | 5 (21.7) |
| Unknown | 0 | 1 (2.1) | 0 |
| Metastasis stage | |||
| M0/M1A/M1B | 11 (30.6) | 15 (31.3) | 8 (34.8) |
| M1C | 25 (69.4) | 33 (68.7) | 15 (65.2) |
BRAFi, BRAF inhibitor; CR, complete response; ECOG PS, Eastern Cooperative Oncology Group performance status; PD-L1, programmed death ligand 1; PR, partial response; SD, stable disease.
Baseline tumour size was measured by adding the sum of the longest dimensions of all measurable baseline target lesions.
PD-L1 positivity was defined as membranous staining in at least 1% of tumour cells.
4. Discussion
This retrospective analysis showed distinct outcomes in patients with advanced melanoma who are treated with pembrolizumab and have an early assessment of SD, PR or CR. As expected, the best survival outcomes were observed in patients with a week 12 or week 24 assessment of CR. Notably, however, a significant proportion of patients with early (week 12 or 24) SD went on to have a response with pembrolizumab. Among patients with SD at week 12, almost half (46.7%) achieved a BOR of CR (6.5%) or PR (40.2%) with continued treatment. Similarly, more than one third of patients (35.9%) with SD at week 24 had a BOR of CR (2.6%) or PR (33.3%) with continued pembrolizumab. Interestingly, patients with SD at week 12 had lower 48-month OS compared with patients who had SD at week 24 (47.7% vs. 66.0%); this may partly be due to a greater proportion of patients with primary resistance and/or slow-growing tumours being included in the former group.
Patients with an assessment of SD at week 12 followed by subsequent response had similar survival outcomes to patients with an assessment of PR at week 12 followed by no change in response or progression (48-month OS, 72.1% vs. 75.0%, respectively). Although patients with an assessment of SD at week 12 followed by no response or progression had worse survival outcomes than patients with SD at week 12 with subsequent response, approximately half of patients (46.9%) in the former group were still alive at 48 months. As expected, patients with SD at week 12 who subsequently progressed had the poorest outcomes. Analysis of baseline characteristics among patients with initial SD showed that patients with subsequent progression were more likely to have BRAF-mutant disease and to have received prior BRAF inhibitor therapy and were more likely to be younger than those with subsequent response or ongoing SD.
The current treatment paradigm for advanced melanoma involves discontinuation of immune checkpoint inhibitors because of unacceptable toxicity or disease progression [3]. In situations in which patients experience disease progression, switching to an alternative therapy is recommended. However, it is important to confirm disease progression before switching to a different therapy as there are limited effective therapies after checkpoint inhibitor failure [3]. In this analysis, a week 12 or 24 assessment of SD was still predictive of long-term OS benefit for a substantial proportion of patients, with 47.7% and 66.0% of patients, respectively, estimated to be alive at 4 years. These results argue against prematurely switching therapy in patients with early SD with pembrolizumab. An alternative approach may be the addition of other agents to pembrolizumab that may increase the proportion of patients with early SD or elicit response to such combination therapy. Several ongoing clinical trials are investigating the antitumour activity and safety of combinations of various agents with PD-1 inhibitors [13–18]. Alternatively, predictive markers such as circulating tumour DNA or imaging with 18F-fluorodeoxyglucose positron emission tomography can be used to identify early responses to PD-1 inhibitors in metastatic melanoma, which may help differentiate between patients with SD who are likely to gain durable clinical benefit from those whose disease is most likely to progress [19–22].
Our results also demonstrated that baseline tumour size, ECOG PS and metastatic stage were consistently associated with response. Patients with small baseline tumours, an ECOG PS of 0 and less disseminated disease (M0/M1a/M1b) were more likely to have CR at week 12 or week 24 than PR or SD.
This analysis was limited by its retrospective nature and the pooling of results from two trials with differing patient populations and differences in prior treatments between the studies. Approximately 30% of patients included were from KEYNOTE-001, which required patients with BRAF-mutant melanoma to have received prior BRAF and/or MEK inhibitors. In contrast, patients with BRAF-mutant melanoma in KEYNOTE-006 could be BRAF inhibitor naive if they had normal lactate dehydrogenase, non-symptomatic disease and absence of rapid progression. Furthermore, patients in KEYNOTE-001 could have received ≤2 prior lines of therapy, whereas patients in KEYNOTE-006 could have received 1 prior therapy. Patients in KEYNOTE-001 may therefore have had more advanced disease and differences in tumour biology [23] than those in KEYNOTE-006. KEYNOTE-001 was also an open-label study with multiple expansion cohorts enrolled without randomisation, which may have confounded this analysis.
Thus, prospective evaluation of outcomes in patients with melanoma who respond early to pembrolizumab is warranted. Earlier intervention with additional or subsequent therapy in patients with SD may provide optimal responses to PD-1 inhibitors, restoring the antitumour activity. In addition, biomarkers are needed to identify responders early during treatment.
5. Conclusions
These results indicate that a substantial proportion of patients who have SD during the first six months of treatment go on to achieve PR or CR with continued pembrolizumab therapy and have promising long-term survival. As expected, the longest survival was observed in patients with CR, followed by patients with early PR who went on to achieve subsequent CR. Patients who had early PR and sustained PR had the next best survival. Notably, patients with SD at week 12 who went on to have PR or CR exhibited similar long-term survival to patients with an early and sustained PR. Patients with SD at week 12 and no subsequent response or progression had poorer outcomes than these groups. The worst outcomes were observed in patients with PR or SD at week 12 who developed progression within 6 months of initiating treatment. The current findings may help guide future trial design and clinical decisions for patients with advanced melanoma who have an initial assessment of SD with pembrolizumab.
Supplementary Material
Acknowledgements
The authors thank the patients and their families and caregivers for participating in this trial and all investigators and site personnel.
Funding
Funding for this research was provided by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA, and Memorial Sloan Kettering Cancer Center support grant P30 CA008748. The sponsor collaborated with academic authors in the study design and in the collection, analysis and interpretation of the results. The authors had final responsibility for the decision to submit the manuscript.
Medical writing assistance
Medical writing and/or editorial assistance was provided by Jemimah Walker, PhD, and Doyel Mitra, PhD, of ApotheCom (Yardley, PA). This assistance was funded by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA.
Footnotes
Conflict of interest statement
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: O.H. is a consultant advisor for Aduro, Akeso, Amgen, Array, BeiGene, BMS, Genentech, GSK, Immunocore, Incyte, Janssen, Merck, NextCure, Novartis, Sanofi, Regeneron, Seattle Genetics, Tempus and Zelluna. He is a speaker for Array, BMS, Novartis, Pfizer, Sanofi and Regeneron. He has contracted research for his institution from Aduro, Akeso, Amgen, Arcus, Array, BMS, CytomX, Exelixis, Genentech, GSK, Immunocore, Incyte, Iovance, Merck, Merck Serono, Moderna, NextCure, Novartis, Regeneron, Sanofi, Seattle Genetics, Torque and Zelluna. S.J.D., B.H.M. and E.J. are employees of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA, and S.J.D. owns stock in Merck & Co., Inc., Kenilworth, NJ, USA. A.A. is a consultant advisor for Amgen, BMS, Merck, Novartis, Pierre Fabre, Roche, Sanofi and Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA. M.S.C. is a consultant advisor for Amgen, BMS, Eisai, IDEAYA, MSD, Merck Serono, Nektar, Novartis, Pierre Fabre, QBiotics, OncoSec, Regeneron, Roche and Sanofi. He has also received honoraria from BMS, MSD and Novartis. T.M. has served on advisory boards for Merck, BMS and OncoSec. A.D. has received grants from BMS, Merck, Pfizer/Array and Xencor. He also served on an advisory board meeting for Xencor. J.J.G. has served on advisory boards for BMS, Merck, MSD, Novartis, Pfizer, Pierre Fabre, Roche, Sanofi and Sun Pharma. He has also received personal fees from BMS, MSD, Novartis and Pierre Fabre for travel as well as personal fees for serving as a speaker for MSD, Novartis and Pierre Fabre. P.H. has no disclosures to report. F.S.H. is a consultant advisor for Aduro, Bristol Myers Squibb, Eisai, EMD Serono, Genentech/Roche, Idera, Merck, Novartis, Pieris Pharmaceutical, Sanofi and Takeda. He reports grants and royalties paid to his institution by Bristol Myers Squibb and Novartis, as well as personal fees from Gossamer. He also holds equity in Apricity, Bicara, Pionyr and Torque. He has served on advisory boards for Apricity, Bicara, Bioentre, Checkpoint Therapeutics, Compass Therapeutics, Iovance, Pionyr, Surface and Torque. His pending patents include Methods for Treating MICA-Related Disorders (#20100111973; includes royalties), Angiopoietin-2 Biomarkers Predictive of Anti-Immune Checkpoint Response (#20170248603), Compositions and Methods for Identification, Assessment, Prevention, and Treatment of Melanoma Using PD-L1 Isoforms (#20160340407), Therapeutic Peptides (#20160046716, #20140004112, #20170022275, #20170008962), Methods of Using Pembrolizumab and Trebananib and Anti-Galectin Antibody Biomarkers Predictive of Anti-Immune Checkpoint and Anti-Angiogenesis Responses (#20170343552). He has been issued patents for Tumor Antigens and Uses Thereof (#7250291), Therapeutic Peptides (#9402905), Vaccine Compositions and Methods for Restoring NKG2D Pathway Function Against Cancers (#10279021) and Antibodies that Bind to MHC Class I Polypeptide-Related Sequence A (#10106611). A.M.J. has received institutional support from MSD. L.M. has no disclosures to report. A.R. is a consultant for Amgen, Chugai, Merck, Novartis, Nurix, Sanofi and Vedanta. He is a current scientific advisory board member and stockholder in 4C Biomed, Apricity, Arcus, Compugen, Highlight, ImaginAb, Isoplexis, Lutris Pharma, MapKure, Merus, PACT Pharma, Rgenix and Tango Therapeutics and is a past scientific advisory board member and stockholder in Advaxis, Cytomx, Five Prime, Kite-Gilead and RAPT. He has also received grants paid to his institution from Agilent and Bristol Myers Squibb. C.R. is a consultant advisor for Amgen, AstraZeneca, BMS, Merck, MSD, Novartis, Pierre Fabre, Roche and Sanofi. J.S. has no disclosures to report. J.S.W. has received grants paid to his institution from Merck. He has been issued a PD-1 biomarker patent with Biodesix unrelated to the submitted work. J.D.W. is a non-compensated consultant for Merck. He is also a consultant for Amgen, Apricity, Arsenal IO, Ascentage Pharma, Astellas, AstraZeneca, Bayer, BeiGene, Boehringer Ingelheim, Bristol Myers Squibb, Celgene, Chugai, Eli Lilly, Elucida, F Star, Georgiamune, Imvaq, Kyowa Hakko Kirin, Linneaus, Merck, Neon Therapeutics, Polynoma, Psioxus, Recepta, Sellas, Serametrix, Surface Oncology, Syndax, Syntalogic, Takara Bio, Tizona Pharmaceuticals, Trieza, Truvax and Werewolf Therapeutics. He reports equity in Adaptive Biotech, Apricity, Arsenal IO, Belgene, Georgiamune, Imvaq, Linneaus and Tizona Pharmaceuticals. He has received research grants from Bristol Myers Squibb and Sephora. He has licensed patents for xenogeneic DNA vaccines (Merial; includes royalties), a myeloid-derived suppressor cell (MDSC) assay (Serametrix; includes royalties), an antiePD-1 antibody (Agenus), anti-CTLA4 antibodies (Agenus) and anti-GITR antibodies and method of use thereof (Agenus/Incyte). He has also been issued patents for alphavirus replicon particles expressing Newcastle disease viruses for cancer therapy, phosphatidylserine targeting agents and uses thereof for adoptive T-cell therapies, immuno-suppressive follicular help-like T cells modulated by immune checkpoint blockade, identifying and treating subjects at risk for checkpoint blockade therapy–associated colitis, CAR+ T cells targeting differentiation antigens as means to treat cancer, anti-CD40 agonist mAB fused to monophosphoryl lipid A (MPL) for cancer therapy and engineered vaccinia viruses for cancer immunotherapy. G.V.L. is consultant advisor for Aduro Biotech Inc, Amgen Inc, Array Biopharma inc, Boehringer Ingelheim International GmbH, Bristol Myers Squibb, Evaxion Biotech A/S, Hexel AG, Highlight Therapeutics S.L., Merck Sharp & Dohme, Novartis Pharma AG, OncoSec, Pierre Fabre, QBiotics Group Limited, Regeneron Pharmaceuticals Inc, SkylineDX B.V., and Specialised Therapeutics Australia Pty Ltd.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ejca.2021.08.013.
Data sharing statement
Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA (MSD) is committed to providing qualified scientific researchers access to anonymised data and clinical study reports from the company’s clinical trials for the purpose of conducting legitimate scientific research. MSD is also obligated to protect the rights and privacy of trial participants and, thus, has a procedure in place for evaluating and fulfilling requests for sharing company clinical trial data with qualified external scientific researchers. The MSD data sharing website (available at: http://engagezone.msd.com/ds_documentation.php) outlines the process and requirements for submitting a data request. Applications will be promptly assessed for completeness and policy compliance. Feasible requests will be reviewed by a committee of MSD subject matter experts to assess the scientific validity of the request and the qualifications of the requestors. In line with data privacy legislation, submitters of approved requests must enter into a standard data-sharing agreement with MSD before data access is granted. Data will be made available for request after product approval in the United States and European Union or after product development is discontinued. There are circumstances that may prevent MSD from sharing requested data, including country- or region-specific regulations. If the request is declined, it will be communicated to the investigator. Access to genetic or exploratory biomarker data requires a detailed, hypothesis-driven statistical analysis plan that is collaboratively developed by the requestor and MSD subject matter experts; after approval of the statistical analysis plan and execution of a data-sharing agreement, MSD will either perform the proposed analyses and share the results with the requestor or will construct biomarker covariates and add them to a file with clinical data that is uploaded to an analysis portal so that the requestor can perform the proposed analyses.
References
- [1].Schadendorf D, van Akkooi ACJ, Berking C, Griewank KG, Gutzmer R, Hauschild A, et al. Melanoma. Lancet 2018; 392(10151):971–84. [DOI] [PubMed] [Google Scholar]
- [2].Luke JJ, Flaherty KT, Ribas A, Long GV. Targeted agents and immunotherapies: optimizing outcomes in melanoma. Nat Rev Clin Oncol 2017;14(8):463–82. [DOI] [PubMed] [Google Scholar]
- [3].National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology. cutaneous melanoma Version 3.2020. Plymouth Meeting, PA: National Comprehensive Cancer Network; 2020. [Google Scholar]
- [4].Strudel M, Festino L, Vanella V, Beretta M, Marincola FM, Ascierto PA. Melanoma: prognostic factors and factors predictive of response to therapy. Curr Med Chem 2020;27(17):2792–813. [DOI] [PubMed] [Google Scholar]
- [5].Michielin O, van Akkooi A, Ascierto P, Dummer R, Keilholz U. Cutaneous melanoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2019;30(12): 1884–901. [DOI] [PubMed] [Google Scholar]
- [6].Garbe C, Amaral T, Peris K, Hauschild A, Arenberger P, Bastholt L, et al. European consensus-based interdisciplinary guideline for melanoma. Part 2: treatment e update 2019. Eur J Canc 2020;126:159–77. [DOI] [PubMed] [Google Scholar]
- [7].Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, et al. Five-year survival outcomes for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. Ann Oncol 2019;30(4):582–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Robert C, Ribas A, Schachter J, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus ipilimumab in advanced melanoma (KEYNOTE-006): post-hoc 5-year results from an open-label, multicentre, randomised, controlled, phase 3 study. Lancet Oncol 2019;20(9):1239–51. [DOI] [PubMed] [Google Scholar]
- [9].Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med 2015;372(26):2521–32. [DOI] [PubMed] [Google Scholar]
- [10].Schachter J, Ribas A, Long GV, Arance A, Grob J-J, Mortier L, et al. Pembrolizumab versus ipilimumab for advanced melanoma: final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006). Lancet 2017;390(10105): 1853–62. [DOI] [PubMed] [Google Scholar]
- [11].Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med 2013;369(2):134–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015;372(4):320–30. [DOI] [PubMed] [Google Scholar]
- [13].Tomivosertib (eFT-508) in combination with PD-1/PD-L1 inhibitor therapy. ClinicalTrials.gov indentifier: NCT03616834. Updated November 2. 2020. https://clinicaltrials.gov/ct2/show/NCT03616834. [Accessed 6 April 2021].
- [14].Phase I-Ib/II study of MBG453 as single agent and in combination with PDR001 in patients with advanced malignancies. ClinicalTrials.gov identifier: NCT02608268. 2021. Updated February 24, https://clinicaltrials.gov/ct2/show/NCT02608268. [Accessed 4 April 2021].
- [15].A phase 1 study of TSR-022, an anti-TIM-3 monoclonal antibody, in patients with advanced solid tumors (AMBER). ClinicalTrials.gov identifier: NCT02817633. 2020. Updated November 27, https://clinicaltrials.gov/ct2/show/NCT02817633. [Accessed 4 April 2021].
- [16].A study of LY3321367 alone or with LY3300054 in participants with advanced relapsed/refractory solid tumors. Clinical-Trials.gov identifier: NCT03099109. 2020. Updated August 19, https://clinicaltrials.gov/ct2/show/NCT03099109. [Accessed 4 April 2021].
- [17].Safety and efficacy of LAG525 single agent and in combination with PDR001 in patients with advanced malignancies. Clinical-Trials.gov identifier: NCT02460224. 2021. Updated February 24, https://clinicaltrials.gov/ct2/show/NCT02460224. [Accessed 4 April 2021].
- [18].Tolcher AW, Hafez N, Yamamoto N, Park J, Grempler R, Lucarelli AG, et al. A phase Ia/Ib, dose-escalation/expansion study of BI 907828 in combination with BI 754091 and BI 754111 in patients (pts) with advanced solid tumors. J Clin Oncol 2020;38(15 suppl):TPS3660. [Google Scholar]
- [19].Lee JHJ, Carlino MS, Menzies AM, Odegaard JI, Lefterova M, Scolyer RA, et al. Circulating tumor DNA (ctDNA) using Guardant360 to predict response in BRAF V600 WT metastatic melanoma (MM) patients (pts) receiving immune checkpoint inhibitors (ICI). J Clin Oncol 2020;38(15 suppl):10050. [Google Scholar]
- [20].Lee JH, Long GV, Menzies AM, Lo S, Guminski A, Whitbourne K, et al. Association between circulating tumor DNA and pseudoprogression in patients with metastatic melanoma treated with anti-programmed cell death 1 antibodies. JAMA Oncol 2018;4(5):717–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Tan AC, Emmett L, Lo S, Liu V, Kapoor R, Carlino MS, et al. FDG-PET response and outcome from anti-PD-1 therapy in metastatic melanoma. Ann Oncol 2018;29(10):2115–20. [DOI] [PubMed] [Google Scholar]
- [22].Lee JH, Long GV, Boyd S, Lo S, Menzies AM, Tembe V, et al. Circulating tumour DNA predicts response to anti-PD1 antibodies in metastatic melanoma. Ann Oncol 2017;28(5): 1130–6. [DOI] [PubMed] [Google Scholar]
- [23].Winder M, Virós A. Mechanisms of drug resistance in melanoma. Handb Exp Pharmacol 2018;249:91–108. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.