

Abstract
Proteome profiling is a powerful tool in biological and biomedical studies, starting with samples at bulk, single cell, or single cell type levels. Reliable methods for extracting specific cell type proteome are in need, especially for the cells (e.g., neurons) that cannot be readily isolated. Here, we present an innovative proximity labeling (PL) strategy for single cell type proteomics of mouse brain, in which TurboID (an engineered biotin ligase) is used to label almost all proteins in a specific cell type. This strategy bypasses the requirement of cell isolation and includes five major steps: (i) constructing recombinant adeno-associated viruses (AAV) to express TurboID driven by cell type-specific promoters, (ii) delivering the AAV to mouse brains by direct intravenous injection, (iii) enhancing PL labeling by biotin administration, (iv) purifying biotinylated proteins followed by on-bead protein digestion, and (v) quantitative tandem-mass-tag (TMT). We first confirmed that TurboID can label a wide range of cellular proteins in human HEK293 cells and optimized the single cell type proteomics pipeline. To analyze specific brain cell types, we generated recombinant AAVs to co-express TurboID and mCherry proteins, driven by neuron- or astrocyte-specific promoters, and validated the expected cell expression by co-immunostaining of mCherry and cellular markers. Subsequent biotin purification and TMT analysis identified ~10,000 unique proteins from a few micrograms of protein samples with excellent reproducibility. Comparative and statistical analysis indicated these PL proteomes contain cell type specific cellular pathways. Although proximity labeling was originally developed for studying protein-protein interactions and subcellular proteomes, we extend it to efficiently tag the entire proteomes of specific cell types in the mouse brain using TurboID biotin ligase. This simple, effective in vivo approach should be broadly applicable to single cell type proteomics.
Keywords: single cell type proteomics; proximity labeling; AAV, TMT; mass spectrometry; proteomics; proteome; brain tissue; neuron; astrocyte
Introduction
Quantifying proteomes at bulk and single cell levels provides crucial information to dissect the molecular mechanisms of development, aging and human disease, such as Alzheimer’s disease1–2. The latest development in mass spectrometry (MS) enables unprecedented deep analysis of proteome (~10,000 proteins) in cells/tissues, usually at the bulk level3–6. The bulk proteomic studies, however, analyze the proteome of a population of cells or a mixture of heterogeneous cell types from tissues7. Tracing proteins to specific cell types may be challenging, and the proteins from rare cell types are often masked in the bulk analysis. Single cell proteomics offers an excellent solution, but the minute sample (usually sub-nanogram) only allows the analysis of ~1,000-2,000 proteins per cell at current stage8. Instead, the analysis of the proteome in a single cell type (termed single cell type proteomics) provides a balanced compromise between cell type specificity and proteome coverage9, with enhanced MS methods to analyze sub-microgram protein mixtures10.
Proteomic analysis of single cell or single cell type in the brain is scarce, largely due to technical limitations in brain cell isolation9. Traditionally, intact single cells have to be isolated through ex vivo manipulations through cell dissociation with chemical/mechanical treatments, and fluorescence-activated cell sorting (FACS). The diverse types of cells in the brain form a dense network with chemical, electrical, and physical connectivity, so that small cells (e.g., microglia) can be sorted without severe damage, but large cells of complex morphologies (e.g., neurons) may not survive in this process. Laser capture microdissection (LCM) may be another method to procure single cells from brain slides under microscope11. To skip the step of cell isolation from the brain, newly synthesized proteins in neurons can be specifically labeled by a noncanonical amino acid through an engineered translation machinery, such as bioorthogonal noncanonical amino acid tagging (BONCAT)12, and stochastic orthogonal recoding of translation (SORT)13. The labeled noncanonical amino acid is converted to a biotin tag by click chemistry, allowing the isolation of neuronal proteome from whole brain lysate by affinity purification for MS analysis. However, the translation-based labeling appears to be a slow process, requiring noncanonical amino acids that might impact cellular proteomes.
More recently, enzyme-catalyzed proximity labeling (PL) has emerged as a sensitive approach to label interacting proteins and subcellular proteomes in vivo14. When expressing a target protein fused with a promiscuous enzyme (e.g., BioID/BioID2/TurboID or APEX/APEX2)15–19 in cells, the promiscuous enzyme can biotinylate biomolecules spatially proximal to the target (~10 nm)20. TurboID17 and APEX219 are next generation enzymes engineered to improve labeling efficiency from the original sequences of BioID (from a mutated biotin-ligase BirA from E. coli)15 and APEX (from a modified plant ascorbate peroxidase)18, respectively. In principle, BioID/TurboID directly uses ATP and biotin to label proteins, while APEX/APEX2 biotinylates proteins with the biotin-phenol substrate under oxidized condition (e.g., H2O2)14. For example, BioID was used to map the subcellular locations of more than 4,000 proteins based on 192 subcellular markers in HEK293 cells21. When the promiscuous enzyme is expressed alone usually as a negative control in the PL analysis, the enzyme diffuses in the cell and can label many cellular proteins. Based on this property, APEX alone was used to analyze tissue-specific proteome in Drosophila22, C. elegans23 and mouse24 in ex vivo experiments, but the catalytic condition for APEX limits its application to mammals in vivo.
In this study, we present a strategy to directly label cell type specific proteome by TurboID in the mouse brain in vivo. We first confirmed that free TurboID can effectively label proteins across most of the subcellular locations in HEK293 cells. Next, we combined the advantages of cell type specific expression of TurboID and the red fluorescent protein mCherry25, as well as an engineered AAV-PHP.eB virus that can infect brain cells through intravenous injection26. Using a highly sensitive MS pipeline, more than 10,000 proteins were analyzed for the neuronal and astrocytic proteomes. Thus, we provide a noninvasive, time-efficient method for deep proteome characterization of single-cell types, which can be applied to any mouse models for neuroscience research.
Methods
AAV vectors and Animals
The engineered AAV-PHP.eB capsid plasmid was obtained from California Institute of Technology to enable efficient transduction of the brain cells through systemic delivery26–27. The TurboID was developed from yeast display-based directed evolution17, fused with T2A “self-cleaving” peptide28 and mCherry25. The expression was driven by a modified neuronal promoter for mouse calcium/calmodulin-dependent protein kinase type II subunit alpha (simplified as CaMKII)29 or an astrocytic promoter for glial fibrillary acidic protein (GFAP)30. C57BL/6J mice used in this study were purchased from The Jackson Laboratory and maintained in the Animal Resources Center at St. Jude Children’s Research Hospital according to the Guidelines for the Care and Use of Laboratory Animals. All animal procedures were approved by Institutional Animal Care and Use Committee (IACUC).
Biotin labeling of proteins in HEK293 cells and mouse brain
Recombinant AAVs were generated and titered following the reported protocol26. The AAV viruses were produced by co-transfection of three plasmids: (i) the pHelper plasmid encoding adenoviral replication proteins; (ii) the pUCmini-iCAP-PHP.eB plasmid encoding engineered PHP.eB capsid protein; (iii) 3xFLAG-TurboID-mCherry pAAV plasmids with different promoters (synthesized DNA into the backbone of the pAAV plasmid, VectorBuilder). The titer of AAVs was measured by PCR reactions using an internal standard with known titer.
HEK293 cell labeling was performed with the previously reported protocol17. The cells were cultured under standard growth media of DMEM (Cellgro) supplemented with 10% (w/v) fetal bovine serum (Thermo Scientific), and transfected with Lipofectamine2000 (Life Technologies) and ~10 μg 3xFLAG-TurboID-mCherry pAAV plasmids with the CaMKII promoter (synthesized by VectorBuilder) for one 10 cm dish of cells in serum-free media for ~4 h. Then the media was replaced with fresh serum-containing media and incubated for ~2 days. The TurboID transfected cells were labeled by supplementing 100 μM biotin for 10 min before harvest.
To investigate the expression of endogenous biotinylated proteins with biotin supplement, a time course study was conducted with HEK293 cells. The cultured cells were treated with biotin (final concentration of 100 μM in the culture media), and harvested at different time points (10 min, 1 hrs, and 6 hrs). Then the cells were lysed by an SDS sample buffer (50 mM Tris, pH 6.8, 2% SDS, 10% glycerol, 0.1% Bromophenol blue, and 100 mM DTT), and analyzed by western blotting with streptavidin HRP (Cat#434323, Thermo Fisher Scientific).
C57BL/6J mice (~12-month-old) were briefly anesthetized using isoflurane, and 3E11 viral genomes per animal (~100 μl) were delivered into the retro-orbital sinus. The virus dosage was estimated by the previous report (1E11 to 1E12 viral genomes per mouse)26. The animals were then recovered and maintained for three weeks, followed by subcutaneous injection with biotin for one week and euthanization for analysis. The biotin dosage was based on a previous study31: 24 mg/kg daily, equivalent to 500 μl of 5 mM biotin per mouse (~25 g body weight). No obvious toxicity of AAV and biotin injection was observed in these treated mice.
Immunostaining of HEK293 cells and mouse brain
To stain the HEK293 cells, the 3xFLAG-TurboID-mCherry transfected cells were fixed with 4% paraformaldehyde for 15 min. Endogenous peroxidase activity was quenched by 3% H2O2 and 0.1%NaN3 for 30 min. Cells were blocked with 5% Donkey serum for 30 min, incubated with the primary FLAG antibody (1:200 dilution, Cat#MA1-142, Thermo Fisher Scientific) for 1 h, the 2nd antibody (donkey-anti-rat-HRP, 1:500 dilution, Cat# 712-035-153, Jackson ImmunoResearch) with DAPI (1 μg/ml, D21490, Thermo Fisher Scientific) for 1 h. Anti-FLAG signal was processed by tyramide signal amplification by FITC-tyramide (1:80, PerkinElmer) for 5 min. Final images were obtained by Zeiss LSM 780 confocal microscopy.
The staining method for the mouse brain was performed as previously reported6. Briefly, brain tissue samples were fixed with 10% formalin, dehydrated, and embedded in paraffin for section (10 μm). The sections were deparaffinized with CitriSolv Hybrid, rehydrated with isopropanol, followed by antigen retrieval with 10 mM citric buffer (pH 6.0). After quenching endogenous peroxidase activity by 1% H2O2 in PBS, sections were blocked with 10% donkey serum in PBS with 0.3% Triton X-100 (PBST) for 30 min at 21°C, incubated sequentially with mCherry primary antibody (chicken polyclonal, Abcam Cat# ab205402, 1:200 dilution) at 4°C overnight and the horseradish peroxidase-conjugated secondary antibody for 1 h at 21°C, and stained with Cyanine 3 tyramide (PerkinElmer) for 2-10 min. For double-immunofluorescence staining, NeuN antibody (rabbit polyclonal, Abcam Cat# ab104225, 1:100 dilution) GFAP antibody (rabbit polyclonal, Millipore Cat# 3429094, 1:100 dilution), and the secondary antibody conjugated with Alexa flour 488 (Jackson ImmunoResearch Laboratories, 1:500 dilution) were used. The slides were mounted with Prolong gold antifade mounting media and imaged by Zeiss Axioscan Z1 and by Zeiss LSM 780 confocal microscopy.
Purification of biotinylated proteins from HEK293 cells and mouse brain
HEK293 cells (~10 million) or mouse cortex (~50 mg) were lysed in 0.5 ml lysis buffer (50 mM HEPES, pH 8.5, fresh 8 M urea, 0.5% sodium deoxycholate (NaDoc), and 0.1% SDS) with glass beads in Bullet Blender. The protein concentration was measured by BCA (bicinchoninic acid assay) with BSA as a standard. The samples were diluted to adjust the protein concentration to 2 mg/ml and urea to 6 M, and followed by centrifugation at 21,000 × g for 10 min. The supernatant was used for affinity purification. First, streptavidin beads (~50 μl beads in ~100 μl slurry, GE Cat# 17-5113-01, Millipore Sigma) were preequilibrated in a binding buffer (50 mM HEPES, pH 8.5, fresh 6 M urea, 0.5% NaDoc, and 0.1% SDS) in a chromatography column (Bio-rad Cat# 731-1550). Then the cleared lysates were loaded and flowed by gravity. The column was washed with the binding buffer (> 20 bed volumes) three times32.
In-gel and on-bead digestion of biotinylated proteins
For in-gel digestion, biotinylated HEK293 proteins on streptavidin beads were further washed twice with binding buffer (> 20 bed volumes, 50 mM HEPES, pH 8.5, 0.5% NaDoc, and 0.1% SDS) to remove urea, and subsequently eluted with an elution buffer (~6 bed volumes, 50 mM HEPES, pH 8.5, 2% SDS, and 20 mM biotin) at 95 °C for 10 min. Eluted proteins were run on a very short SDS gel followed by in-gel digestion33 with protein LoBind microcentrifuge tubes (Eppendorf)10. The resulting peptides were analyzed by the optimized LC-MS/MS platform34. In addition, the eluted proteins were analyzed on a long SDS gel followed by silver staining.
For on-bead digestion, biotinylated HEK293 proteins on streptavidin beads were washed twice with the binding buffer (> 20 bed volumes, 50 mM HEPES, pH 8.5, fresh 6 M urea, and 0.5% NaDoc) to remove SDS. Similar to in-solution digestion35, the beads were then incubated with a digestion buffer (~2 bed volumes, 50 mM HEPES, pH 8.5, fresh 8 M urea, and 0.5% NaDoc) with LysC (Wako, 1:100 w/w) for 3 h at 21 °C with rotation, diluted to reduce urea to 2 M, further digested with trypsin (Promega, 1:50 w/w) overnight at 21 °C. The supernatant was transferred to a protein LoBind microcentrifuge tube (Eppendorf)10. Residual peptides in the beads were extracted with ~6 bed volumes of 60% acetonitrile and 0.1% TFA twice, combined with the on-bead digestion supernatant, acidified and desalted for the LC-MS/MS analysis.
TMT labeling and LC/LC-MS/MS analysis
To reduce sample loss, we performed on-bead digestion and direct TMT labeling. To optimize the protocol, the TMTpro zero reagents were titrated with various TMT/streptavidin bead ratios (μg/μl) during the labeling for 1 h at 37 °C. The sample aliquots before and after labeling were compared by LC–MS/MS. To ensure complete labeling, the TMT/streptavidin bead ratio (μg/μl) of 10:1 was used in the following experiments. Briefly, TMT reagents (500 μg per channel) were added to label digested peptides from streptavidin beads (50 μl of bed volume) for 1 h at 37 °C and then quenched by 0.5% hydroxylamine35–36. The supernatant was saved, and the beads were extracted with ~6 bed volumes of 60% acetonitrile and 0.1% TFA twice, and then combined with the supernatant. Finally, all TMT labeled samples were pooled by the equal volume, desalted, and fractionated by an offline basic pH RPLC (XBridge C18 column (3.5 μm particle size, 2.1 mm × 15 cm, Waters) buffer A: 10 mM ammonium formate, pH 8.0; buffer B: 90% AcN, 10 mM ammonium formate, pH 8.0). A total of 160 fractions were collected every min in a gradient of 15-42% buffer and concatenated into 80 fractions. Each fraction was injected into a Q Exactive HF Orbitrap MS (Thermo Fisher Scientific) with a self-packed column (75 μm × 15 cm with 1.9 μm C18 resin from Dr. Maisch GmbH, heated at 65°C to reduce backpressure). Peptides were analyzed in a 60 min gradient at 250 nl/min (buffer A: 0.2% formic acid, 3% DMSO; buffer B: buffer A plus 65% AcN). The MS1 parameters included 60,000 resolution, 450-1600 m/z range, 1 x 106 AGC, and 50 ms maximal ion time; and MS2 parameters were 60,000 resolution, starting from 120 m/z, 1 x 105 AGC, 110 ms maximal ion time, 20 data-dependent MS2 scans, 1.0 m/z isolation window with 0.2 m/z offset, 32 normalized collision energy in HCD, and 10 s dynamic exclusion.
Protein identification and quantification by MS raw data
All data were analyzed using the JUMP software suite, a tag-based hybrid search engine to improve sensitivity37. MS/MS spectra were searched against the protein database generated by combining Swiss-Prot, TrEMBL, and UCSC databases and removing redundancy (human: 83,955 entries; mouse: 59,423 entries). The target protein sequences were reversed to generate decoys to evaluate the false discovery rate (FDR)38. All spectra were corrected by known mass information of the TMT reporter ions, and then allowed 6 ppm mass tolerance for precursor ions and 15 ppm for product ions. Other settings included full tryptic, two maximal missed cleavages, static modifications of Cys carbamidomethylation (+57.02146) and TMT labeling of Lys and N-termini (+229.16293 for TMT11 or +304.20715 for TMTpro), dynamic modification of Met oxidation (+15.99491). The putative PSMs were grouped by precursor ion states and filtered by matching scores (Jscore and ΔJn) and mass accuracy to reduce protein FDR below 1%. For the peptides shared by multiple homologous proteins, the peptides were assigned to the protein of most abundance (i.e., with the maximal PSM number) based on the rule of parsimony. Then the protein quantification was performed using the TMT reporter ion intensities and y1-ion-based correction of TMT data to alleviate the effect of ratio compression39.
Statistical methods
To identify differentially expressed (DE) proteins, the analysis was largely performed as previously described40. In general, the experimental variances were first analyzed based on the replicated measurements by modeling with a Gaussian distribution to evaluate standard deviation (SD). The p values of intergroup comparisons were obtained through the moderated t-test; and the FDR values were derived from Benjamini-Hochberg (BH) correction for multiple testing. DE proteins were defined by the selected cutoffs of changed magnitude (e.g., 3 SD) and FDR (e.g., 0.05).
Enrichment of subcellular localizations and pathways
The enrichment analysis was performed by the over-representation analysis (ORA) method41 against the gene ontology (GO) cellular component annotations42. Before the analysis of DE proteins, all identified proteins by MS were mapped to their corresponding Entrez Gene IDs43 based on gene symbols. These proteins were further consolidated to a unique protein list (one protein per gene by removing protein isoforms with relatively smaller fold changes) and used as the background for the enrichment analysis. Human and mouse GO annotations were extracted by the OrgDb packages from Bioconductor44 and were used for the analyses of HEK293 cells and mouse brains, respectively. The up- and down-regulated protein sets were processed separately, and the P values was calculated based on the hypergeometric test. All P values were further adjusted by the BH procedure for multiple testing. Enriched cellular components (e.g., subcellular localizations) with FDR (adjusted P value) < 0.05 were considered statistically significant.
Data availability
All MS raw files in this study are available in the MassIVE database with the accession number of MSV000088866 (https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp).
Results
Proximity labeling of entire cellular proteome by TurboID in mammalian cells
Although proximity labeling has initially been developed to probe protein-protein interactions14, we reason that free promiscuous enzymes can label most if not all proteins encountered in cells. To confirm this, we expressed the newly engineered TurboID with rapid labeling kinetics17 in human HEK293 cells by TurboID plasmid transfection under several conditions: (i) control without transfection, (ii) TurboID proximity labeling (PL), and (iii) TurboID expression with biotin supplement (PL/biotin) (Fig. 1A). We first investigated the location of expressed TurboID protein by immunofluorescence staining, which showed that TurboID protein diffused throughout the cell, but with slightly less distribution in the nuclei, maybe due to the lack of strong nuclear localization signal sequences (Supporting Fig. S1). After cell lysis and biotin affinity purification by streptavidin beads, the isolated proteins were analyzed on a silver stained SDS gel (Fig. 1B). Compared to very few weak bands detected in the control samples, many protein bands were found in the TurboID PL samples and their intensities were significantly enhanced with biotin supplement, confirming that free TurboID is capable of modifying a large number of proteins in the mammalian cells.
Figure 1.
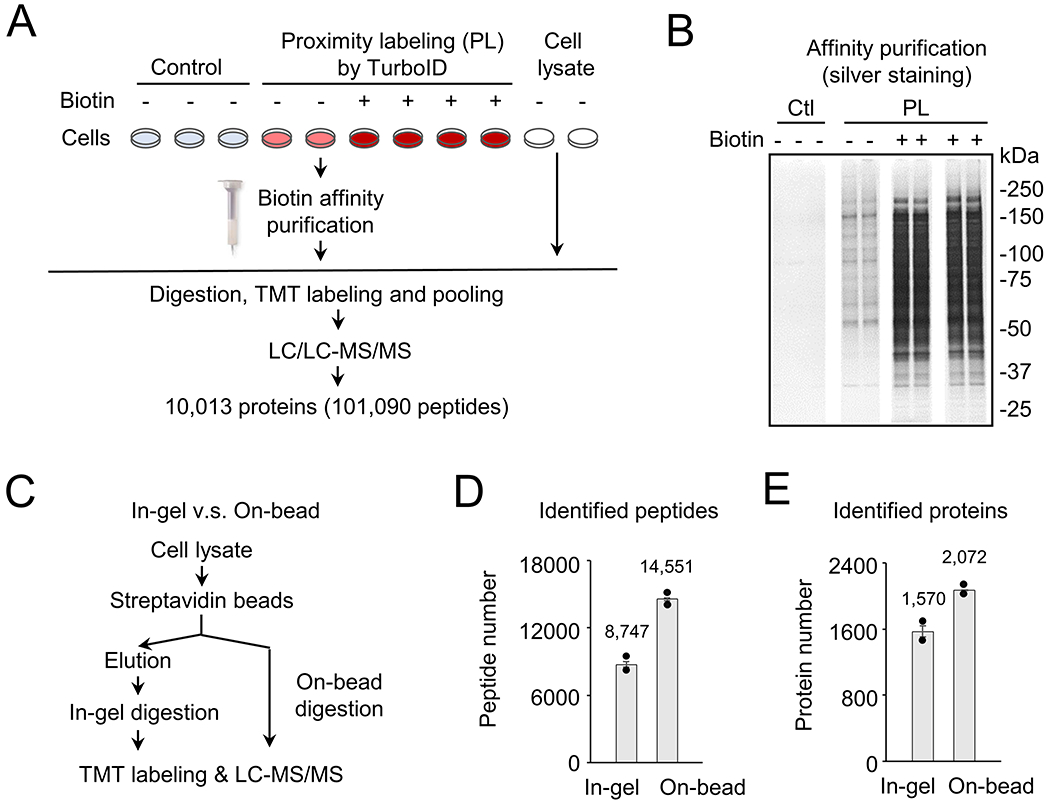
Characterization of proteins by TurboID-mediated proximity labeling (PL) in HEK293 cells. A, Scheme of analyzing PL proteins in HEK293 cells under different conditions. The cells were cultured and transfected with control and TurboID-expression plasmids. Additional biotin (100 μM) was added into the culture media to enhance biotin labeling. This experiment was performed with replicates. Biotinylated proteins were purified and analyzed by a large-scale TMT-LC/LC-MS/MS analysis. B, Silver-stained gel images to show affinity purified biotinylated proteins under different conditions: control (ctl), PL samples with or without additional biotin. The image was assembled from the same gel. C, Comparison of in-gel and on-bead digestion protocols in a separate pilot study. The biotinylated proteins from HEK293 cells (TurboID with biotin supplement) were subjected to affinity purification followed by the in-gel protocol or the on-bead digestion protocol, and protein identification by the LC-MS/MS analysis. D, Comparison of unique peptides by the in-gel and on-bead digestion methods. Replicated data points were shown by the black dots. E, Comparison of unique proteins by the in-gel and on-bead digestion methods. Replicated data points were shown by the black dots.
During the affinity purification, it is often difficult to elute all biotinylated proteins due to extremely tight binding of biotin to streptavidin45. To improve the protein recovery, we examined a direct on-bead LysC/trypsin digestion protocol (simplified as “on-bead”), and compared with the traditional protocol including protein elution, SDS gel electrophoresis, and in-gel protein digestion (simplified as “in-gel”) (Fig. 1C). Clearly, the “on-bead” protocol led to the identification of 66.4% more peptides, and 32.0% more proteins than the “in-gel” protocol (Fig. 1D and 1E). Therefore, the optimized “on-bead” protocol was selected in the subsequent experiments.
To fully evaluate the potential for labeling the cellular proteome by free TurboID, we profiled biotinylated proteins in HEK293 cells under different conditions (control, PL, and PL/Biotin) and compare them with whole cell lysate by quantitative mass spectrometry (e.g., TMT-LC/LC-MS/MS)35. While a standard TMT analysis requires the amount of 100 micrograms of protein per sample, we recently developed a new TMT protocol using a minimal 0.5 micrograms of protein per sample10. Here we refined this new protocol to couple it with biotin affinity purification, “on-bead” digestion and optimized TMT labeling (Supporting Fig. S2) to improve protein/peptide recovery and the depth of proteome analysis. Although only a few micrograms of protein were estimated to be labeled by TurboID from ~10 million HEK293 cells, we managed to identify and quantify a total of 10,013 proteins from these samples (101,090 peptides, protein FDR < 0.01, Fig. 1A, Supporting Tables S1 and S2). The dataset includes over three times as many proteins, compared to those which were quantified in previous single cell type studies (<3,000 proteins)12–13, 24.
We then performed a series of analyses to evaluate the quality of this PL-mediated proteomic study. Clustering analyses (Fig. 2A) indicated obvious differences among different sample groups. As expected, the PL/Biotin group showed changes to 9,931 proteins (FDR < 0.05) compared to the control group (99.2% of the profiled proteins, 9,931/10,013). Interesting, we observed endogenous biotinylation proteins of high abundance, such as ACACA, ACACB, MCCC1, PC and PCCA24, which were not significantly altered upon biotin supplement (Supporting Fig. S3) or the expression of TurboID (Fig. 2B). In contrast, when comparing the PL/Biotin group with total cell lysate (normalized by the median of identified proteins), only 3,199 proteins showed differences (FDR < 0.05, Fig. 2C, Supporting Tables S2), while the remaining 6,814 (68.0%) proteins had similar levels. Further comparison of peptide MS signals from the PL/Biotin and cell lysate samples suggests a strong correlation (r = 0.87, Fig. 2D). In the 3199 changed proteins, 1,528 proteins were decreased in the PL/Biotin group, mainly enriched in cell surface and organelles, such as mitochondria and the lumens of endoplasmic reticulum (ER), lysosome, vesicle and Golgi (Fig. 2E, Supporting Tables S3), while 1,671 proteins were increased in the PL/Biotin group, with enrichment in cytoplasmic region and cytoskeleton cellular compartments, such as actin, lamellipodium, intermediate filament and microtubule (Fig. 2F, Supporting Tables S4). Despite some biases in TurboID-mediated labeling that may be due to uneven cellular distribution of the TurboID enzyme (e.g., the lower level in the nucleus than that in the cytoplasm, Supporting Fig. S1), and inaccessibility of certain organelles because of the lack of organelle-targeting sequences, the majority of PL proteome was comparable with whole cell proteome in this MS analysis.
Figure 2.
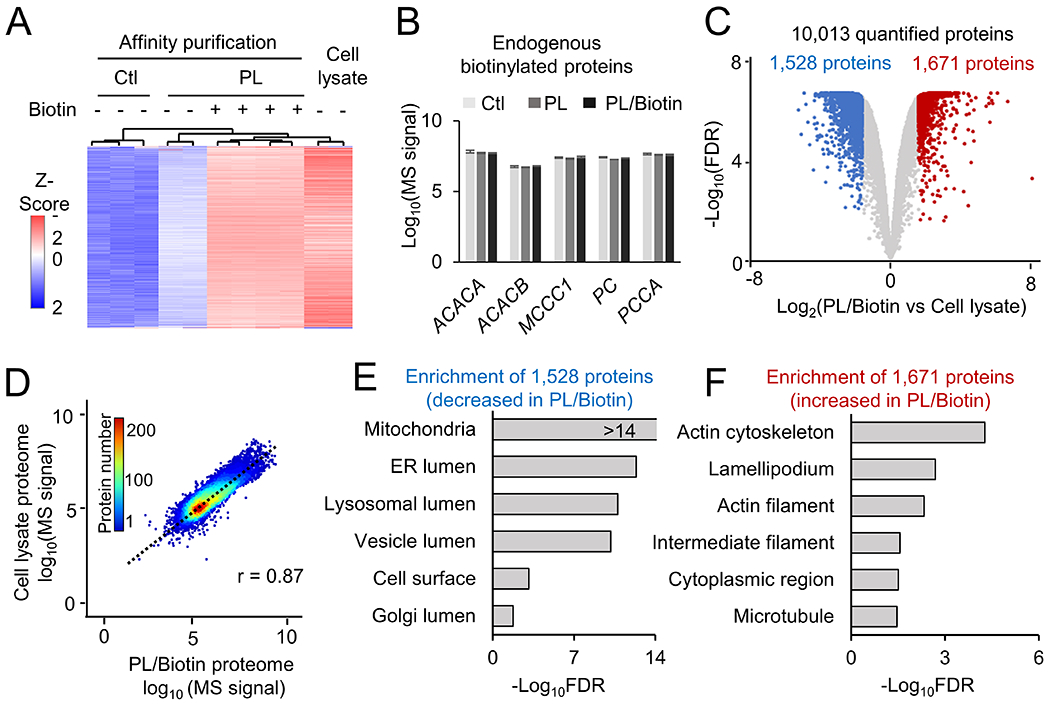
Analysis of single cell type proteome in HEK293 cells by biotin labeling and TMT-MS. A, Heatmap of identified proteins under different conditions. B Quantification of endogenous biotinylated proteins under each condition. C, Volcano plot to show the comparison of total cell lysate proteome to the TurboID PL proteome with biotin addition. The cutoffs were FDR of 0.05, and fold change of 3 standard deviation (SD). Shown in colored dots are altered proteins. D, Correlation of TurboID PL/Biotin proteome with cell lysate proteome. The scatter plot shows the Log10 scale of each protein intensity. E-F, Pathway enrichment analysis of DE proteins in TurboID+biotin (PL/Biotin) compared to lysates based on GO database, with selected pathways shown.
Single cell type proximity labeling mediated by engineered AAVs
Recognizing the potential of TurboID-based proximity labeling of cellular proteome, we attempted to develop a strategy for single cell type proteomics in the mouse brain (Fig. 3A). Our integrated strategy included (i) the novel AAV-PHP.eB for efficient, noninvasive gene delivery to the adult mouse brain26, (ii) TurboID expression driven by a cell type specific promoter (e.g., CaMKII promoter in neurons29, or GFAP promoter in astrocytes46), (iii) TurboID tagged with 3xFLAG at its N-terminus, and mCherry at its C-terminus25, and (iv) a fused T2A sequence between 3xFLAG-TurboID and mCherry, which induces ribosome skipping to translate two proteins independently28. The recombinant gene and protein sequences were shown in Supporting Table S5. The recombinant AAV was constructed, validated by sequencing, and administrated to the mice by retro-orbital injection. After three weeks, the mice were supplemented with additional biotin, and harvested after one additional week for imaging and MS analyses.
Figure 3.
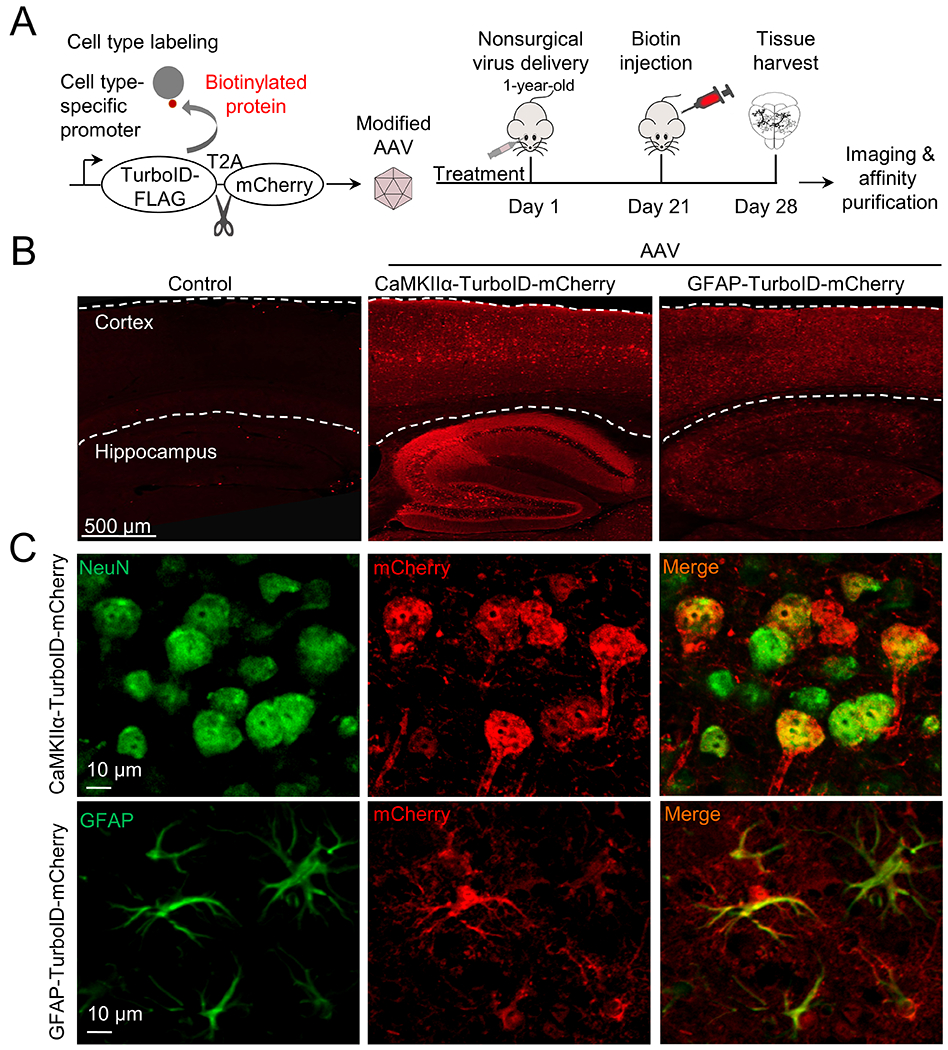
AAV-mediated expression of TurboID protein in specific cell types in the mouse brain. A, The workflow of AAV-TurboID labeling. We used AAV-PHP.eB to package single-stranded recombinant AAV genomes that express TurboID tagged with FLAG at N-terminus and mCherry at C-terminus. The T2A sequence was inserted between TurboID and mCherry for self-cleavage. The cell type-specific expression was controlled by promoters. The recombinant viruses were delivered to mouse brains, followed by biotin administration and brain harvest for immunostaining and proteomic analyses. B, Detection of cell type-specific expression by co-translated mCherry throughout the brain. C, Double immunofluorescence staining shows the specificity of cell type-specific expression. NeuN and GFAP are cellular markers for neurons and astrocytes, respectively.
We validated the expression of the AAV construction by fluorescence imaging of mCherry (Fig. 3B), and the expression patterns driven by CaMKII promoter and GFAP promoter were completely distinct. To further confirm the cell type specific expression, we performed double immunostaining of mCherry and cellular markers (NeuN for neurons and GFAP for astrocytes, Fig. 3C). As expected, CaMKII promoter led to the recombinant protein expression in NeuN-labeled neurons, and GFAP promoter drove the recombinant protein expression in the astrocytes.
Single cell type proteomics by AAV-mediated biotinylation and TMT-LC/LC-MS/MS
We then analyzed the single cell type proteomes in three sets of brain samples: negative controls without virus infection, infection with CaMKII-PL or GFAP-PL virus (Fig. 4A). After three weeks of infection, the mice were subcutaneously injected with biotin for one week and harvested. The brain cortices co-expressing 3xFLAG-TurboID and mCherry were harvested and lysed, followed by biotin affinity purification, on-bead digestion and TMT-LC/LC-MS/MS analysis. This highly sensitive protocol led to the identification and quantification of 10,294 proteins (101,756 peptides, protein FDR < 0.05, Supporting Table S1 and S6). Sample reproducibility was demonstrated by the results of the replicates in clustering analysis (Fig. 4B), correlation analysis (e.g., comparing MS signals between two CaMKII replicates, r = 0.99, Fig. 4C), and null analysis (e.g., comparing two pairs of replicates, Fig. 4D). Compared to the negative control (no virus infection), the CaMKII-PL showed the enrichment of 9,919 proteins (FDR < 0.05, 96.4% of the profiled proteins, 9,919/10,294, Fig. 5A, Supporting Table S6). Similar results were obtained for the GFAP-PL. These results indicate a strong effect of TurboID-mediated biotinylation of brain proteome with high reproducibility.
Figure 4.
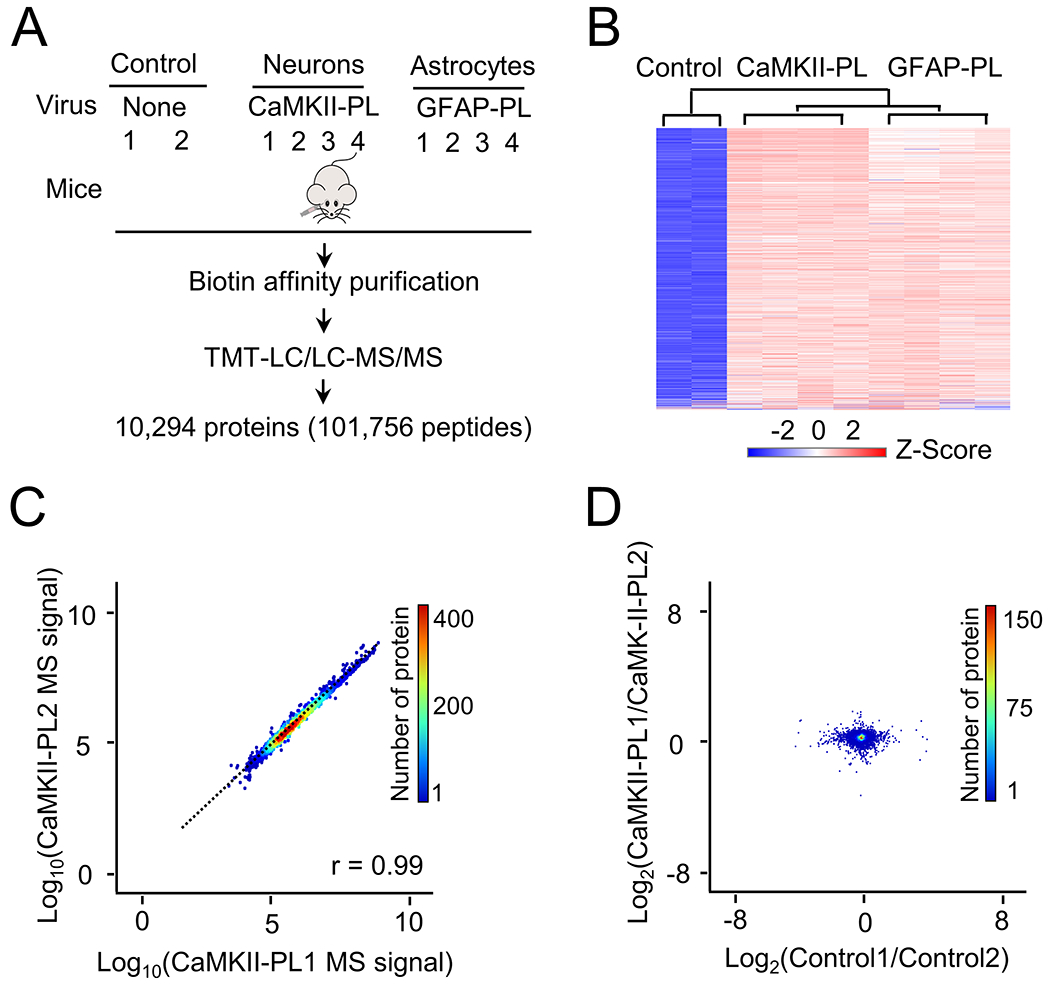
Analysis of single cell type proteome in the mouse brain by biotin labeling and TMT-MS. A, The analysis workflow with different cell types and negative controls. B, Heatmap of identified proteins by TMT-LC/LC-MS/MS, consistent with different conditions. C, A pair of biological replicate example to show the consistency of MS analysis (data from Supporting Table S6). D, Null (intragroup) comparison to show the small variation of individual proteins. Two control samples and two CaMKII PL samples are selected as examples.
Figure 5.
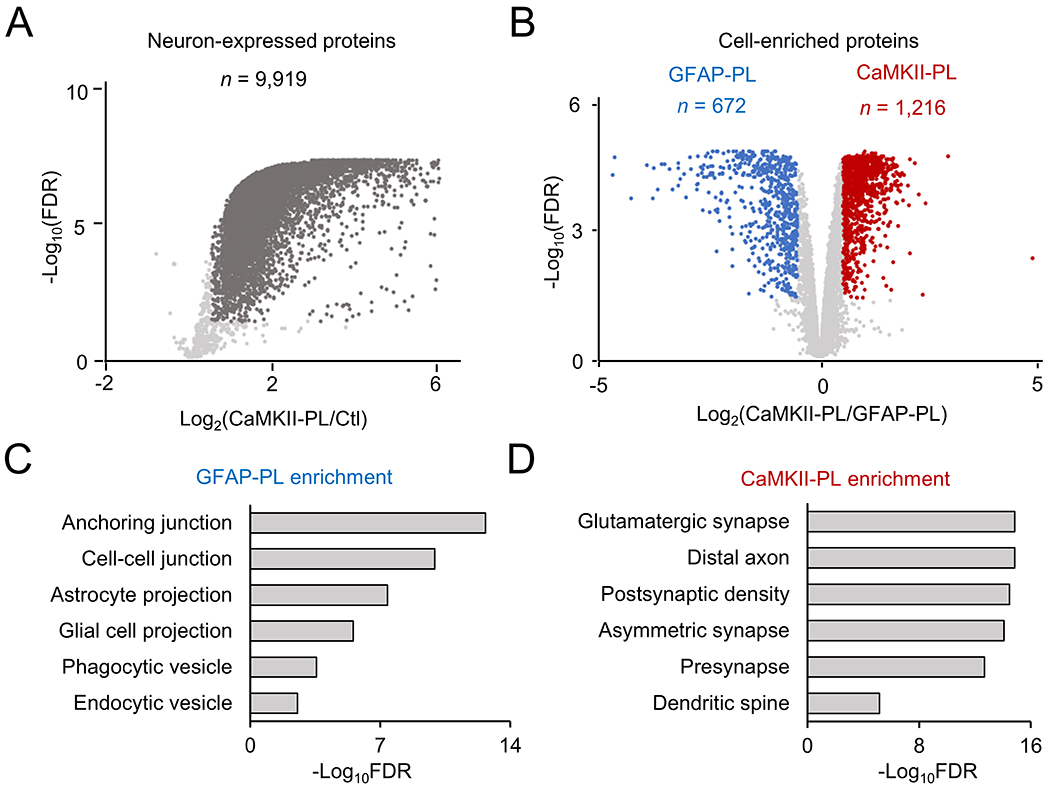
Characterization of single cell type proteome in the mouse brain by statistical analysis. A, Volcano plot to show the comparison of negative control and CaMKII-PL proteome. The cutoffs were FDR of 0.05, and fold change of 3 standard deviation (SD). Highlighted dots: proteins enriched in the CaMKII-PL proteome. B, Similar volcano plot to show the comparison of GFAP-PL and CaMKII-PL proteomes. C-D, Pathway enrichment analysis of GFAP-PL- and CaMKII-PL-enriched proteins, with selected pathways shown.
We then compared the CaMKII-PL and GFAP-PL samples to define cell type specific proteins (FDR < 0.05, Fig. 5B, Supporting Table S7). A total of 672 proteins were decreased in the CaMKII-PL, but increased in the GFAP-PL brain, which are enriched in astrocytic cellular compartments, including anchoring and cell-cell junctions, astrocyte and glial cell projections, and phagocytic and endocytic vesicles (Fig. 5C, Supporting Table S8). For example, the highly enriched proteins included known astrocytic markers, such as GFAP46, vimentin (VIM)47, excitatory amino acid transporter 2 (EAA2 protein from Slc1a2 gene)48, N-myc downregulated gene 2 (NDRG2)49, aldehyde dehydrogenase family 1 member L1 (ALDG1L1)50, and Aquaporin 4 (Aqp4)51.
Conversely, 1,216 proteins were increased in the CaMKII-PL brain, which are highly enriched in neuron-related cellular compartments, such as glutamatergic synapse, asymmetric synapse, presynapse, postsynaptic density, distal axon, and dendritic spine (Fig. 5D, Supporting Table S9). For example, the highly enriched neuronal proteins included microtubule-associated protein 2 (MAP2) and Tau (MAPT)52, neurofilament heavy, medium and light chains (NEFH, NEFM and NEFL)53, presynaptic proteins54 of synapsin 1 (SYN1), synapsin 2 (SYN2) and synaptotagmin 7 (SYT7), and postsynaptic proteins55, such as PSD-95 (DLG4), PSD-93 (DLG2), GKAP family (DLGAP1, DLGAP2, DLGAP3, and DLGAP4), SHANK family (SHANK1, SHANK2, and SHANK3). Overall, our proteomic data are highly consistent with cell-specific expression of TurboID and the labeling of cell type specific proteome.
Discussion
We report a novel cell type specific in vivo proteomic labeling approach, in which the engineered biotin ligase, TurboID, is expressed in the desired cell type in the mouse brain using a noninvasive AAV-based strategy. Cell type specificity is largely achieved by the expression of TurboID under specific promoters, which enables the biotin labeling of the corresponding cell type proteome to bypass the physical step of cell isolation. Although only neuron- and astrocyte-specific promoters are examined in our proof-of-principle studies, it is anticipated that the strategy can be applied to other cell types and other animals. It should be mentioned that the engineered AAV-PHP.eB capsid also contributes to cell type- or tissue-specificity, as the virus is delivered to the brain through intravenous injection26. Indeed, AAV capsid engineering is a highly active field for AAV-mediated gene therapy to improve expression and tissue specificity56. As AAV capsids have in vivo cell type or tissue tropism, capsid evolution can enhance the cell type- and tissue-specific expression57, and more cell type specific AAV capsid variants have recently been developed to cross the blood–brain barrier in mice58. Thus, our single cell type proteomics strategy allows the flexibility of selecting appropriate recombinant AAV for different cell types of interest.
This TurboID-based approach enables the profiling of more than 10,000 proteins from each brain cell type, which markedly improves the proteome coverage, since <5,000 proteins were reported in brain cell types by co-translational labeling methods12–13 or recent Cre-dependent APEX AAV strategy24. As regulatory proteins of key cellular functions are usually present at low abundance and are often missed in shallow proteomics studies, the proteome coverage is an important parameter to evaluate proteomics approaches. The high proteome coverage of our approach is largely attributed to the combination of several factors: (i) robust biotinylation by TurboID with supplemental biotin, (ii) on-bead digestion of biotinylated proteins and TMT labeling, (iii) highly sensitive microgram-scale TMT-LC/LC-MS/MS protocol10, and (iv) a newly developed JUMP software suite for protein identification and quantification37, 39, 59.
Conclusion
Here, we show for the first time the deep profiling of >10,000 proteins from neurons or astrocytes by an integrated strategy, which combines nonsurgical AAV-mediated proximity labeling by TurboID, biotin-based affinity purification, and microgram-scale TMT-LC/LC-MS/MS in a streamlined protocol. The strategy has further been utilized to investigate cell-specific protein changes in a commonly used mouse model. Overall, our investigation provides a solid and effective approach to dissect different cell type proteomes of the mouse brain in vivo, which is a highly potential method of general application in neuroscience research.
Supplementary Material
Acknowledgments
We thank all other lab and center members for discussion and technical support. We also thank V. Gradinaru (Caltech) for providing the AAV PHP.eB capsid plasmid, D. Vanderwall for editing, M. Bauler and M. Wielgosz at St. Jude Vector Development and Production Core for the large scale AAV preparation, St. Jude Cell and Tissue Imaging Center, and St. Jude Animal Resources Center. This work was partially supported by National Institutes of Health grants R01AG047928, R01AG053987, RF1AG064909, RF1AG068581, U54NS110435, U19AG069701, and American Lebanese Syrian Associated Charities (ALSAC). The MS analysis was performed in the Center of Proteomics and Metabolomics at St. Jude Children’s Research Hospital, partially supported by NIH Cancer Center Support Grant (P30CA021765).
Abbreviations:
- AAV
adeno-associated virus
- PL
proximity labeling
- MS
mass spectrometry
- TMT
tandem mass tags
- LC
liquid chromatography
Footnotes
Reference
- 1.Bai B; Vanderwall D; Li Y; Wang X; Poudel S; Wang H; Dey KK; Chen PC; Yang K; Peng J, Proteomic landscape of Alzheimer’s Disease: novel insights into pathogenesis and biomarker discovery. Mol Neurodegener 2021, 16 (1), 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rayaprolu S; Higginbotham L; Bagchi P; Watson CM; Zhang T; Levey AI; Rangaraju S; Seyfried NT, Systems-based proteomics to resolve the biology of Alzheimer’s disease beyond amyloid and tau. Neuropsychopharmacology 2021, 46 (1), 98–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aebersold R; Mann M, Mass-spectrometric exploration of proteome structure and function. Nature 2016, 537 (7620), 347–355. [DOI] [PubMed] [Google Scholar]
- 4.Stewart E; McEvoy J; Wang H; Chen X; Honnell V; Ocarz M; Gordon B; Dapper J; Blankenship K; Yang Y; Li Y; Shaw TI; Cho JH; Wang X; Xu B; Gupta P; Fan Y; Liu Y; Rusch M; Griffiths L; Jeon J; Freeman BB 3rd; Clay MR; Pappo A; Easton J; Shurtleff S; Shelat A; Zhou X; Boggs K; Mulder H; Yergeau D; Bahrami A; Mardis ER; Wilson RK; Zhang J; Peng J; Downing JR; Dyer MA; St. Jude Children’s Research Hospital-Washington University Pediatric Cancer Genome, P., Identification of Therapeutic Targets in Rhabdomyosarcoma through Integrated Genomic, Epigenomic, and Proteomic Analyses. Cancer cell 2018, 34 (3), 411–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang H; Diaz AK; Shaw TI; Li Y; Niu M; Cho JH; Paugh BS; Zhang Y; Sifford J; Bai B; Wu Z; Tan H; Zhou S; Hover LD; Tillman HS; Shirinifard A; Thiagarajan S; Sablauer A; Pagala V; High AA; Wang X; Li C; Baker SJ; Peng J, Deep multiomics profiling of brain tumors identifies signaling networks downstream of cancer driver genes. Nat Commun 2019, 10 (1), 3718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bai B; Wang X; Li Y; Chen PC; Yu K; Dey KK; Yarbro JM; Han X; Lutz BM; Rao S; Jiao Y; Sifford JM; Han J; Wang M; Tan H; Shaw TI; Cho JH; Zhou S; Wang H; Niu M; Mancieri A; Messler KA; Sun X; Wu Z; Pagala V; High AA; Bi W; Zhang H; Chi H; Haroutunian V; Zhang B; Beach TG; Yu G; Peng J, Deep Multilayer Brain Proteomics Identifies Molecular Networks in Alzheimer’s Disease Progression. Neuron 2020, 105 (6), 975–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yu J; Peng J; Chi H, Systems immunology: Integrating multi-omics data to infer regulatory networks and hidden drivers of immunity. Curr Opin Sys Biol 2019, 15, 19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perkel JM, Single-cell proteomics takes centre stage. Nature 2021, 597, 580–582. [DOI] [PubMed] [Google Scholar]
- 9.Wilson RS; Nairn AC, Cell-Type-Specific Proteomics: A Neuroscience Perspective. Proteomes 2018, 6 (4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu D; Yang S; Kavdia K; Sifford JM; Wu Z; Xie B; Wang Z; Pagala VR; Wang H; Yu K; Dey KK; High AA; Serrano GE; Beach TG; Peng J, Deep Profiling of Microgram-Scale Proteome by Tandem Mass Tag Mass Spectrometry. J Proteome Res 2021, 20 (1), 337–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu Y; Dou M; Piehowski PD; Liang Y; Wang F; Chu RK; Chrisler WB; Smith JN; Schwarz KC; Shen Y; Shukla AK; Moore RJ; Smith RD; Qian WJ; Kelly RT, Spatially Resolved Proteome Mapping of Laser Capture Microdissected Tissue with Automated Sample Transfer to Nanodroplets. Mol Cell Proteomics 2018, 17 (9), 1864–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alvarez-Castelao B; Schanzenbacher CT; Hanus C; Glock C; Tom Dieck S; Dorrbaum AR; Bartnik I; Nassim-Assir B; Ciirdaeva E; Mueller A; Dieterich DC; Tirrell DA; Langer JD; Schuman EM, Cell-type-specific metabolic labeling of nascent proteomes in vivo. Nat Biotechnol 2017, 35 (12), 1196–1201. [DOI] [PubMed] [Google Scholar]
- 13.Krogager TP; Ernst RJ; Elliott TS; Calo L; Beranek V; Ciabatti E; Spillantini MG; Tripodi M; Hastings MH; Chin JW, Labeling and identifying cell-specific proteomes in the mouse brain. Nat Biotechnol 2018, 36 (2), 156–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qin W; Cho KF; Cavanagh PE; Ting AY, Deciphering molecular interactions by proximity labeling. Nat Methods 2021, 18 (2), 133–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roux KJ; Kim DI; Burke B; May DG, BioID: A Screen for Protein-Protein Interactions. Curr Protoc Protein Sci 2018, 91, 19 23 1–19 23 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim DI; Jensen SC; Noble KA; Kc B; Roux KH; Motamedchaboki K; Roux KJ, An improved smaller biotin ligase for BioID proximity labeling. Mol Biol Cell 2016, 27 (8), 1188–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Branon TC; Bosch JA; Sanchez AD; Udeshi ND; Svinkina T; Carr SA; Feldman JL; Perrimon N; Ting AY, Efficient proximity labeling in living cells and organisms with TurboID. Nat Biotechnol 2018, 36 (9), 880–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rhee HW; Zou P; Udeshi ND; Martell JD; Mootha VK; Carr SA; Ting AY, Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science 2013, 339 (6125), 1328–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lam SS; Martell JD; Kamer KJ; Deerinck TJ; Ellisman MH; Mootha VK; Ting AY, Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat Methods 2015, 12 (1), 51–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim DI; Birendra KC; Zhu W; Motamedchaboki K; Doye V; Roux KJ, Probing nuclear pore complex architecture with proximity-dependent biotinylation. Proc Natl Acad Sci U S A 2014, 111 (24), E2453–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Go CD; Knight JDR; Rajasekharan A; Rathod B; Hesketh GG; Abe KT; Youn JY; Samavarchi-Tehrani P; Zhang H; Zhu LY; Popiel E; Lambert JP; Coyaud E; Cheung SWT; Rajendran D; Wong CJ; Antonicka H; Pelletier L; Palazzo AF; Shoubridge EA; Raught B; Gingras AC, A proximity-dependent biotinylation map of a human cell. Nature 2021, 595 (7865), 120–124. [DOI] [PubMed] [Google Scholar]
- 22.Chen CL; Hu Y; Udeshi ND; Lau TY; Wirtz-Peitz F; He L; Ting AY; Carr SA; Perrimon N, Proteomic mapping in live Drosophila tissues using an engineered ascorbate peroxidase. Proc Natl Acad Sci U S A 2015, 112 (39), 12093–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reinke AW; Mak R; Troemel ER; Bennett EJ, In vivo mapping of tissue- and subcellular-specific proteomes in Caenorhabditis elegans. Sci Adv 2017, 3 (5), e1602426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dumrongprechachan V; Salisbury RB; Soto G; Kumar M; MacDonald ML; Kozorovitskiy Y, Cell-type and subcellular compartment-specific APEX2 proximity labeling reveals activity-dependent nuclear proteome dynamics in the striatum. Nat Commun 2021, 12 (1), 4855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shaner NC; Steinbach PA; Tsien RY, A guide to choosing fluorescent proteins. Nat Methods 2005, 2 (12), 905–9. [DOI] [PubMed] [Google Scholar]
- 26.Chan KY; Jang MJ; Yoo BB; Greenbaum A; Ravi N; Wu WL; Sanchez-Guardado L; Lois C; Mazmanian SK; Deverman BE; Gradinaru V, Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat Neurosci 2017, 20 (8), 1172–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Naso MF; Tomkowicz B; Perry WL; Strohl WR, Adeno-associated virus (AAV) as a vector for gene therapy. BioDrugs 2017, 31 (4), 317–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu Z; Chen O; Wall JBJ; Zheng M; Zhou Y; Wang L; Vaseghi HR; Qian L; Liu J, Systematic comparison of 2A peptides for cloning multi-genes in a polycistronic vector. Sci Rep 2017, 7 (1), 2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dittgen T; Nimmerjahn A; Komai S; Licznerski P; Waters J; Margrie TW; Helmchen F; Denk W; Brecht M; Osten P, Lentivirus-based genetic manipulations of cortical neurons and their optical and electrophysiological monitoring in vivo. Proc Natl Acad Sci U S A 2004, 101 (52), 18206–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee Y; Messing A; Su M; Brenner M, GFAP promoter elements required for region-specific and astrocyte-specific expression. Glia 2008, 56 (5), 481–93. [DOI] [PubMed] [Google Scholar]
- 31.Spence EF; Dube S; Uezu A; Locke M; Soderblom EJ; Soderling SH, In vivo proximity proteomics of nascent synapses reveals a novel regulator of cytoskeleton-mediated synaptic maturation. Nat Commun 2019, 10 (1), 386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Udeshi ND; Pedram K; Svinkina T; Fereshetian S; Myers SA; Aygun O; Krug K; Clauser K; Ryan D; Ast T, Antibodies to biotin enable large-scale detection of biotinylation sites on proteins. Nature methods 2017, 14 (12), 1167–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bai B; Hales CM; Chen PC; Gozal Y; Dammer EB; Fritz JJ; Wang X; Xia Q; Duong DM; Street C; Cantero G; Cheng D; Jones DR; Wu Z; Li Y; Diner I; Heilman CJ; Rees HD; Wu H; Lin L; Szulwach KE; Gearing M; Mufson EJ; Bennett DA; Montine TJ; Seyfried NT; Wingo TS; Sun YE; Jin P; Hanfelt J; Willcock DM; Levey A; Lah JJ; Peng J, U1 small nuclear ribonucleoprotein complex and RNA splicing alterations in Alzheimer’s disease. Proc Natl Acad Sci U S A 2013, 110 (41), 16562–16567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu P; Duong DM; Peng J, Systematical Optimization of Reverse-Phase Chromatography for Shotgun Proteomics. J Proteome Res 2009, 8 (8), 3944–3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bai B; Tan H; Pagala VR; High AA; Ichhaporia VP; Hendershot L; Peng J, Deep profiling of proteome and phosphoproteome by isobaric labeling, extensive liquid chromatography, and mass spectrometry. Methods in enzymology 2017, 585, 377–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang H; Dey KK; Chen PC; Li Y; Niu M; Cho JH; Wang X; Bai B; Jiao Y; Chepyala SR; Haroutunian V; Zhang B; Beach TG; Peng J, Integrated analysis of ultra-deep proteomes in cortex, cerebrospinal fluid and serum reveals a mitochondrial signature in Alzheimer’s disease. Mol Neurodegener 2020, 15 (1), 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang X; Li Y; Wu Z; Wang H; Tan H; Peng J, JUMP: a tag-based database search tool for peptide identification with high sensitivity and accuracy. Mol Cell Proteomics 2014, 13 (12), 3663–3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peng J; Elias JE; Thoreen CC; Licklider LJ; Gygi SP, Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC-MS/MS) for large-scale protein analysis: the yeast proteome. J Proteome Res 2003, 2, 43–50. [DOI] [PubMed] [Google Scholar]
- 39.Niu M; Cho JH; Kodali K; Pagala V; High AA; Wang H; Wu Z; Li Y; Bi W; Zhang H; Wang X; Zou W; Peng J, Extensive Peptide Fractionation and y1 Ion-Based Interference Detection Method for Enabling Accurate Quantification by Isobaric Labeling and Mass Spectrometry. Anal Chem 2017, 89 (5), 2956–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tan H; Yang K; Li Y; Shaw TI; Wang Y; Blanco DB; Wang X; Cho JH; Wang H; Rankin S; Guy C; Peng J; Chi H, Integrative Proteomics and Phosphoproteomics Profiling Reveals Dynamic Signaling Networks and Bioenergetics Pathways Underlying T Cell Activation. Immunity 2017, 46 (3), 488–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boyle EI; Weng S; Gollub J; Jin H; Botstein D; Cherry JM; Sherlock G, GO::TermFinder--open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics 2004, 20 (18), 3710–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ashburner M; Ball CA; Blake JA; Botstein D; Butler H; Cherry JM; Davis AP; Dolinski K; Dwight SS; Eppig JT; Harris MA; Hill DP; Issel-Tarver L; Kasarskis A; Lewis S; Matese JC; Richardson JE; Ringwald M; Rubin GM; Sherlock G, Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet 2000, 25 (1), 25–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maglott D; Ostell J; Pruitt KD; Tatusova T, Entrez Gene: gene-centered information at NCBI. Nucleic Acids Res 2011, 39 (Database issue), D52–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gentleman RC; Carey VJ; Bates DM; Bolstad B; Dettling M; Dudoit S; Ellis B; Gautier L; Ge Y; Gentry J; Hornik K; Hothorn T; Huber W; Iacus S; Irizarry R; Leisch F; Li C; Maechler M; Rossini AJ; Sawitzki G; Smith C; Smyth G; Tierney L; Yang JY; Zhang J, Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 2004, 5 (10), R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weber PC; Ohlendorf DH; Wendoloski JJ; Salemme FR, Structural origins of high-affinity biotin binding to streptavidin. Science 1989, 243 (4887), 85–8. [DOI] [PubMed] [Google Scholar]
- 46.Nolte C; Matyash M; Pivneva T; Schipke CG; Ohlemeyer C; Hanisch UK; Kirchhoff F; Kettenmann H, GFAP promoter‐controlled EGFP‐expressing transgenic mice: a tool to visualize astrocytes and astrogliosis in living brain tissue. Glia 2001, 33 (1), 72–86. [PubMed] [Google Scholar]
- 47.Potokar M; Morita M; Wiche G; Jorgacevski J, The Diversity of Intermediate Filaments in Astrocytes. Cells 2020, 9 (7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Su ZZ; Leszczyniecka M; Kang DC; Sarkar D; Chao W; Volsky DJ; Fisher PB, Insights into glutamate transport regulation in human astrocytes: cloning of the promoter for excitatory amino acid transporter 2 (EAAT2). Proc Natl Acad Sci U S A 2003, 100 (4), 1955–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Flugge G; Araya-Callis C; Garea-Rodriguez E; Stadelmann-Nessler C; Fuchs E, NDRG2 as a marker protein for brain astrocytes. Cell Tissue Res 2014, 357 (1), 31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cahoy JD; Emery B; Kaushal A; Foo LC; Zamanian JL; Christopherson KS; Xing Y; Lubischer JL; Krieg PA; Krupenko SA; Thompson WJ; Barres BA, A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci 2008, 28 (1), 264–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Papadopoulos MC; Verkman AS, Aquaporin water channels in the nervous system. Nat Rev Neurosci 2013, 14 (4), 265–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dehmelt L; Halpain S, The MAP2/Tau family of microtubule-associated proteins. Genome Biol 2005, 6 (1), 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Khalil M; Teunissen CE; Otto M; Piehl F; Sormani MP; Gattringer T; Barro C; Kappos L; Comabella M; Fazekas F; Petzold A; Blennow K; Zetterberg H; Kuhle J, Neurofilaments as biomarkers in neurological disorders. Nat Rev Neurol 2018, 14 (10), 577–589. [DOI] [PubMed] [Google Scholar]
- 54.Sudhof TC, The synaptic vesicle cycle: a cascade of protein-protein interactions. Nature 1995, 375 (6533), 645–53. [DOI] [PubMed] [Google Scholar]
- 55.Cheng D; Hoogenraad CC; Rush J; Ramm E; Schlager MA; Duong DM; Xu P; Wijayawardana SR; Hanfelt J; Nakagawa T; Sheng M; Peng J, Relative and absolute quantification of postsynaptic density proteome isolated from rat forebrain and cerebellum. Mol. Cell. Proteomics 2006, 5 (6), 1158–70. [DOI] [PubMed] [Google Scholar]
- 56.Li C; Samulski RJ, Engineering adeno-associated virus vectors for gene therapy. Nat Rev Genet 2020, 21 (4), 255–272. [DOI] [PubMed] [Google Scholar]
- 57.Bedbrook CN; Deverman BE; Gradinaru V, Viral Strategies for Targeting the Central and Peripheral Nervous Systems. Annu Rev Neurosci 2018, 41, 323–348. [DOI] [PubMed] [Google Scholar]
- 58.Ravindra Kumar S; Miles TF; Chen X; Brown D; Dobreva T; Huang Q; Ding X; Luo Y; Einarsson PH; Greenbaum A; Jang MJ; Deverman BE; Gradinaru V, Multiplexed Cre-dependent selection yields systemic AAVs for targeting distinct brain cell types. Nat Methods 2020, 17 (5), 541–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li Y; Wang X; Cho JH; Shaw TI; Wu Z; Bai B; Wang H; Zhou S; Beach TG; Wu G; Zhang J; Peng J, JUMPg: An Integrative Proteogenomics Pipeline Identifying Unannotated Proteins in Human Brain and Cancer Cells. J Proteome Res 2016, 15 (7), 2309–2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All MS raw files in this study are available in the MassIVE database with the accession number of MSV000088866 (https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp).