

Abstract
Pseudomonas putida GJ31 contains an unusual catechol 2,3-dioxygenase that converts 3-chlorocatechol and 3-methylcatechol, which enables the organism to use both chloroaromatics and methylaromatics for growth. A 3.1-kb region of genomic DNA of strain GJ31 containing the gene for this chlorocatechol 2,3-dioxygenase (cbzE) was cloned and sequenced. The cbzE gene appeared to be plasmid localized and was found in a region that also harbors genes encoding a transposase, a ferredoxin that was homologous to XylT, an open reading frame with similarity to a protein of a meta-cleavage pathway with unknown function, and a 2-hydroxymuconic semialdehyde dehydrogenase. CbzE was most similar to catechol 2,3-dioxygenases of the 2.C subfamily of type 1 extradiol dioxygenases (L. D. Eltis and J. T. Bolin, J. Bacteriol. 178:5930–5937, 1996). The substrate range and turnover capacity with 3-chlorocatechol were determined for CbzE and four related catechol 2,3-dioxygenases. The results showed that CbzE was the only enzyme that could productively convert 3-chlorocatechol. Besides, CbzE was less susceptible to inactivation by methylated catechols. Hybrid enzymes that were made of CzbE and the catechol 2,3-dioxygenase of P. putida UCC2 (TdnC) showed that the resistance of CbzE to suicide inactivation and its substrate specificity were mainly determined by the C-terminal region of the protein.
Microbial degradation of most chlorinated aromatics occurs via chlorocatechols as intermediates. These chlorocatechols are usually further degraded via a modified ortho-cleavage pathway. This pathway involves an intradiol dioxygenase which cleaves the aromatic ring at the ortho position. Dechlorination occurs further along the pathway (43).
An alternative route for the degradation of catechol derivatives is the so-called meta-cleavage pathway. This pathway is mostly involved in the degradation of methylated aromatics. It involves an extradiol dioxygenase that cleaves the catechol ring at the 2,3 position, yielding a 2-hydroxymuconic semialdehyde derivative (47). Many extradiol dioxygenases have been cloned and sequenced, and the crystal structures of two of them were determined (11, 44). These and most other extradiol dioxygenases have a two-domain structure, in which the C-terminal domain contains the active site. The enzymes usually require Fe2+ as a cofactor (9).
When extradiol dioxygenases are confronted with 3-chlorocatechol (3CC), they usually become inactivated (e.g., see references 2, 5, 10, 13, 15, and 22). This inactivation might be caused by the strong chelating activity of 3CC, which removes the Fe2+ cofactor of the enzyme (22), or by suicide inactivation of the enzyme due to the formation of a reactive intermediate or product, such as an acylchloride (5). Inactivation of the catechol 2,3-dioxygenases results in a low turnover capacity of the enzyme with 3CC that is insufficient for growth (50).
It was assumed for a long time that it was impossible to metabolize 3-chlorinated catechols via the meta-cleavage pathway, because they would inactivate the extradiol dioxygenase. However, we recently described a Pseudomonas putida strain that does degrade chlorobenzene via 3CC by this pathway (19, 25). The strain contains a novel chlorocatechol 2,3-dioxygenase (CbzE) that can efficiently cleave 3CC at the 2,3 position, leading to simultaneous ring cleavage and dechlorination. In this paper we describe the cloning and sequence of the cbzE gene. Furthermore, we determined the substrate ranges and characteristics of 3CC conversion of several catechol 2,3-dioxygenases and of constructed hybrid enzymes.
MATERIALS AND METHODS
Bacterial strains and plasmids.
P. putida GJ31 was isolated on chlorobenzene; its characteristics were described previously (25). Burkholderia cepacia G4 (32) contains a catechol 2,3-dioxygenase (TomB) that is involved in the conversion of catechol and 3-methylcatechol (3MC) (36, 45). Escherichia coli NM522(pWW15.3201) contains a 2.1-kb EcoRI fragment carrying the gene for C23OII of P. putida MT15 (20) in pUC18 and was a gift from P. A. Williams (University of Wales, Bangor, Wales, United Kingdom). Plasmid pTDN1-1018 contains a 2.1-kb SstI-HindIII fragment from P. putida UCC2 in pHG327 on which tdnC is localized (39) and was a gift from N. C. McClure (Flinders University of South Australia, Adelaide, Australia). pAW31 was derived from pEMBL9 and contains xylTE of P. putida mt-2 (3, 7).
E. coli JM101 (53) was used for cloning and construction of hybrid dioxygenases, and E. coli BL21(DE3) (48) was used to express catechol 2,3-dioxygenases that were cloned behind a T7 promoter. pBluescript SK+ (Stratagene, La Jolla, Calif.) was used as a cloning vector, and pGEF+ (made from pGELAF+ by deleting dhlA [42]) was used to make translational fusions of (hybrid) catechol 2,3-dioxygenase genes behind a T7 promoter.
DNA isolation and hybridization and cloning of the chlorocatechol 2,3-dioxygenase gene.
Total DNA from P. putida GJ31 was isolated by the method of Ausubel et al. (4). Plasmid DNA was isolated from P. putida GJ31 by a modified method of Kado and Liu (18) as described by Duetz et al. (8). Southern hybridization and chemiluminescent detection of plasmid DNA or genomic DNA that was digested with PstI were done with, as the probe, a digoxigenin-labeled DNA fragment that was generated by PCR according to the instructions of the manufacturer of the kit (Boehringer, Mannheim, Germany). This fragment was obtained by amplification of a DNA segment of P. putida GJ31 with degenerated primers that were designed against the N-terminal amino acid sequence (SIMRVGHVSI NVMDMAAAVK HYENVLGLKT TMQDNAGNVY LKK) of CbzE (19) (5′-AAXGTZATGGAXATGGC-3′; X = C/T, Z = G/A/T/C) and a conserved C-terminal region (YFFDP) of catechol 2,3-dioxygenases (5′-GGYTCYAAYAAYTA-3′; Y = A/G). A PstI fragment of 3.1 kb of P. putida DNA that hybridized with this probe was isolated from a 0.8% agarose gel and ligated in pBluescript SK+, after which the ligation mixture was transformed into electroporation-competent E. coli JM101 cells. Transformants were plated on Luria broth (LB) agar plates containing 100 μg of ampicillin ml−1, 40 μg of 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside ml−1, and 0.4 mM isopropyl-β-d-thiogalactopyranoside (IPTG). White colonies were screened for catechol 2,3-dioxygenase activity by spraying them with a 10 mM 4-chlorocatechol (4CC) solution. Positive colonies turned yellow owing to conversion of 4CC to a chlorinated 2-hydroxymuconic semialdehyde derivative.
Sequence analysis.
Cycle sequencing (29) was performed on double-stranded DNA with the Amersham Thermo Sequenase cycle sequencing kit with 7-deaza-dGTP and 5′-Cy5 fluorescent primers. Sequence reactions were run on the Pharmacia ALF-Express automatic sequencing machine (Amersham Pharmacia Biotech, Uppsala, Sweden).
DNA and protein databases were screened for homologous proteins with the BLAST program (1). Multiple sequence alignments were made in ClustalW of Lasergene for Windows (DNAstar, Inc., Madison, Wisconsin), and the percent identity between two proteins was calculated by the program as 100 × consensus length/(consensus length + mismatches + gaps). Putative ribosome binding sites were identified by using the sequences of the 3′ ends of 16S rRNA of E. coli and Pseudomonas aeruginosa (46).
Enzyme essays.
Crude cell extracts of B. cepacia G4, E. coli NM522(pWW15.3201), E. coli JM101(pTDN1-1018), E. coli JM101(pAW31), and E. coli JM101(pBScbzE) were used to measure catechol 2,3-dioxygenase activities of TomB, C23OII, TdnC, XylE, and CbzE, respectively. Crude cell extracts were made from overnight cultures that were grown on LB containing 100 μg of ampicillin ml−1 (LB-Amp) and 0.4 mM IPTG in the case of the recombinant E. coli strains. B. cepacia G4 was grown overnight on mineral medium (26) containing 20 mM acetate and 2 mM phenol at 30°C. Two hours before the cells were harvested, 2 mM phenol was added to ensure good induction of TomB (45). Cells were harvested by centrifugation and washed with ice-cold 0.1 M Tris-HCl (pH 7.5) containing 0.1 mM 1,4-dithiothreitol. After cells were resuspended in a small volume of dithiothreitol, they were disrupted by sonication and centrifuged for 30 min in an Eppendorf centrifuge (10,000 × g, 4°C). A fresh Fe(II) (NH4)2(SO4)2 · 6H2O solution was added to a concentration of 0.1 mM to the clear supernatant, after which the supernatant was used as a source of crude cell extract. The protein content of the extract was determined with Coomassie brilliant blue, with bovine serum albumin as a standard.
Catechol 2,3-dioxygenase activities in crude cell extract were measured spectrophotometrically as described previously (25) with different catechols at 0.1 mM. The conversion of catechol, 3MC, 4-methylcatechol (4MC), and 4CC to their corresponding meta-cleavage products was monitored at 375, 388, 382, and 379 nm, respectively, with extinction coefficients of 36,000, 16,800, 31,500, and 39,600 liter mol−1 cm−1, respectively (40, 41). The meta conversion of 3CC to 2-hydroxymuconic acid was monitored at 290 nm. The extinction coefficient of the product is 12,500 liter mol−1 cm−1 (19).
The turnover capacity with 3CC was determined for several catechol 2,3-dioxygenases. It represents the amount of 3CC that can be converted per amount of enzyme (nanomoles per unit of activity with catechol). This was measured by adding a defined amount of crude cell extract to an assay mixture, which contained 115 to 140 μM 3CC in phosphate buffer (25), unless indicated otherwise. The concentration of 3CC was measured over time by reversed-phase high-performance liquid chromatography. For this, a Merck-Hitachi L-6200 pump (Merck-Hitachi, Darmstadt, Germany) was used that was connected to a LiChrosorb RP18 column (Chrompack, Bergen op Zoom, The Netherlands). The samples were injected directly from the incubation mixture with a Marathon-Basic autosampler (Spark-Holland, Emmen, The Netherlands) and eluted with water containing 25% (vol/vol) acetonitrile and 0.2% (wt/vol) H3PO4. 3CC was detected at 210 nm with a Merck-Hitachi L-4200 UV-visible light detector, while acquisition of the data was done with PC integration package software (Kontron Instruments, Milan, Italy).
For most enzymes, the catechol 2,3-dioxygenase was rapidly inactivated within the first one to two minutes of the incubations, and substrate-independent loss of enzyme activity was negligible during this short period. For these catechol 2,3-dioxygenases, the number of data points that could be obtained was insufficient to be fitted to a mathematical description of the depletion of 3CC, and the turnover capacity with 3CC was calculated by dividing the amount of 3CC that was converted by the amount of enzyme that was added via the crude cell extract. The latter value was determined as the amount of activity with catechol that was present in the extract.
When the conversion of 3CC continued for a longer period, such as with CbzE, significant substrate-independent inactivation occurred during the incubation period. The spontaneous loss of enzyme activity was described with the equation dE/dt = kE, and substrate conversion-dependent inactivation was described with dE/dt = pdS/dt. Provided that the concentration range of 3CC was high enough to allow full saturation of the enzyme throughout the conversion, the turnover of 3CC can be described by dS/dt = VmaxE. Solving these differential equations gives equation 1:
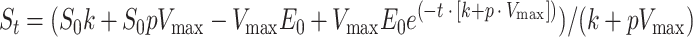 |
1 |
In this equation, St and S0 are the concentrations (micromolar) of 3CC at time t and zero (minutes), respectively; Vmax is the dimensionless maximal specific conversion rate of 3CC relative to the rate with catechol; E0 is the initial activity of the catechol 2,3-dioxygenase in the assay mixture (units of activity with catechol per liter); p is the coefficient that describes inactivation of the enzyme as a result of the conversion of 3CC, which is expressed as the amount of enzyme activity that is inactivated per amount of 3CC that is converted (units per micromole); and k is a first-order inactivation constant (minute−1) that describes aspecific enzyme inactivation. Including the latter constant in the calculations was especially important for CbzE and hybrids thereof since CbzE is rather unstable, although substrate-dependent inactivation is relatively minor. To determine the rate constant for substrate-independent inactivation, an amount of extract that was equal to the amount used in the turnover measurements was incubated in phosphate buffer without 3CC. The remaining catechol 2,3-dioxygenase activity was measured over time with catechol, and k was obtained from the slope of the logarithmic plot of the catechol 2,3-dioxygenase activity versus time.
The values for Vmax and p were estimated from the 3CC depletion curves by fitting to the equation with the program Scientist for Windows 2.0 (Micromath Scientific Software, Salt Lake City, Utah). The turnover capacity was calculated by Vmax/(k + pVmax).
Protein expression and construction of hybrid catechol 2,3-dioxygenases.
The genes encoding CbzE and TdnC were amplified with PCR by using primers against the start and end of the genes. The N-terminal primers contained an NcoI site (P1, 5′-GCTGCTCCATGGGTATTATGAGAGTTGGC-3′ for cbzE; P2, 5′-GCTGCTCCATGGGTGTACTGAGAAT-3′ for tdnC; NcoI sites underlined). The C-terminal primers contained a BamHI site (P3, 5′-GACGTCGGATCCTCATGTGTACACATC-3′ for cbzE; P4, 5′-GACGTCGGATCCTCAGGTATAGACGTC-3′ for tdnC; BamHI sites underlined, stop codons shown in bold). To ensure high fidelity of the PCR product, Pwo DNA polymerase or the Expand High Fidelity PCR system was used according to instructions of the manufacturer (Boehringer, Mannheim, Germany). Due to the introduction of an NcoI site in cbzE, the second codon of cbzE was changed to encode a glycine instead of a serine. The NcoI-BamHI fragments were cloned between the NcoI and BamHI sites of pGEF+.
Protein expression was done in E. coli BL21(DE3). For this, constructs of pGEF+ containing the desired genes were transformed to E. coli BL21(DE3) and the cells were grown overnight on LB-Amp agar plates at 30°C. The next day, the transformants were collected from the plates and used to inoculate 100 ml of LB-Amp. The cultures were grown at 15°C for 2 days, after which the cells were harvested and used to prepare crude cell extracts as described above. The amounts of soluble and insoluble catechol 2,3-dioxygenase that were produced by E. coli BL21(DE3) were estimated by comparing the protein profile of crude cell extract with the protein profile of whole cells on a 12.5% polyacrylamide gel containing sodium dodecyl sulfate (SDS) as described by Schanstra et al. (42). Insoluble protein aggregates (inclusion bodies) are absent from crude cell extracts because they are removed by centrifugation (30 min, 10,000 × g, 4°C), while they remain visible in the protein profiles obtained with whole cells.
Hybrid catechol 2,3-dioxygenases were made of CbzE and TdnC by using PCR fusion reactions (54). For this, fragments of cbzE and tdnC were amplified with PCR by using primers that generated overlapping regions. The fragments were fused together by PCR using P1 or P2 as the forward primer and P3 or P4 as the reverse primer. The sequences of the other primers that were used were as follows: P5, 5′-GGTCGAGCCAGTGCGCCCCGGC-3′; P6, 5′-GATCCAACCAGTGCGCGCCCGC-3′; P7, 5′-GCGCACTGGTTGGATC-3′; P8, 5′-GCGCACTGGCTCGACC-3′; P9, 5′-GTCGCCCTGGGGGCCGACGGTCAA-3′; P10, 5′-TGAGCCATCGGGGCCCACCAGCAC-3′; P11, 5′-GGCCCCGATGGCTCA-3′; P12, 5′-GGCCCCCAGGGCGAC-3′; P13, 5′-TTCTTGGCCATGAC-3′; P14, 5′-GTCATGGCCAAGAA-3′. The nucleotides of the primers that are complementary to tdnC are shown in italics, and the nucleotides that are complementary to cbzE are underlined. This way, fusions were created between amino acid positions 147 and 148, 189 and 190, and 240 and 241 of CbzE and TdnC. The PCR fusion products were cloned behind the T7 promoter of pGEF+, and the hybrid enzymes were expressed in E. coli BL21(DE3). The hybrid genes that were made with PCR fusions were checked with restriction analyses and cycle sequencing.
Nucleotide sequence accession number.
The sequence obtained in this study has been submitted to GenBank and is available under accession no. AF109307.
RESULTS AND DISCUSSION
Cloning and sequencing of a genomic DNA fragment encoding a chlorocatechol 2,3-dioxygenase.
The N-terminal amino acid sequence of the chlorocatechol 2,3-dioxygenase (CbzE) of P. putida GJ31 was determined previously and showed homology with other catechol 2,3-dioxygenases (19). This sequence was used together with a conserved C-terminal sequence (YFFDP) from homologous catechol 2,3-dioxygenases to design degenerate oligonucleotide primers. A 761-bp PCR product was obtained, the nucleotide sequence of which corresponded with the N-terminal amino acid sequence of CbzE. This PCR product was used as a probe in Southern hybridization experiments, and a 3.1-kb PstI fragment of genomic DNA of P. putida GJ31 was found to hybridize with the probe. This fragment was cloned in pBluescript SK+ to give pBScbzE.
The nucleotide sequence of the 3.1-kb fragment was determined (GenBank accession no. AF109307). A schematic representation of the open reading frames (ORFs) that were identified is given in Fig. 1. The cbzE gene starts at position 1319 and encodes a polypeptide of 314 amino acids with a calculated molecular mass of 34,951 Da, which corresponds to the experimentally determined mass of 33.4 kDa of CbzE (19). The N-terminal amino acid sequence that was deduced from cbzE was identical to the N-terminal sequence of 43 amino acids that was determined with the purified CbzE protein (19), except for the last amino acid. In front of cbzE is a putative ribosome binding site (AGGAG).
FIG. 1.

A schematic representation of the organization of the ORFs that were identified on the 3.1-kb PstI fragment. The PstI cloning sites and a XhoI site that was confirmed by digestion are shown. The positions of the start codons that were identified are indicated, as well as the sizes of the three complete ORFs. The position where a deletion of monooxygenase genes that corresponded to tbmBCDE (17) may have occurred is indicated with an arrow.
Downstream of cbzE, a fragment that encodes an ORF of 154 amino acids was found, which was preceded by a Shine-Dalgarno-like sequence, GGA. This ORF (CbzX) was 41% identical to CmpX of Sphingomonas sp. strain HV3 (GenBank accession no. Z84817). The cmpX gene is part of an operon encoding a meta-cleavage pathway for catechol. Its function is unknown (55).
Further downstream, the beginning of an ORF (CbzG) was found that is similar to many 2-hydroxymuconic semialdehyde dehydrogenases, of which DmpC of P. putida CF600 (GenBank accession no. P19059) (35) was one of the most similar enzymes, having 56% identity with CbzG. These enzymes are responsible for oxidation of the carbonyl group that is present in the meta-cleavage products that are formed when catechols are converted by catechol 2,3-dioxygenases. CbzG most likely has the same function. The corresponding genes are found in operons that encode meta-cleavage pathways (e.g., see references 23, 52, and 55). The cbzG gene continues until the PstI cloning site and is preceded by a putative ribosome binding site (GAGAG) (Fig. 1).
Upstream of cbzE, an ORF of 119 amino acids is localized (CbzT), which is also behind a putative ribosome binding site (AAGG) (Fig. 1). The derived amino acid sequence shows up to 40% identity with chloroplast-type ferredoxins that are known to play a role in maintaining the iron ion of catechol 2,3-dioxygenases in the reduced (Fe2+) state (16, 37). The genes that encode these ferredoxins are found in front of the genes that encode catechol 2,3-dioxygenases (e.g., see references 12, 23, and 52), and cbzT probably encodes such a protein as well.
At the beginning of the cloned fragment is a segment of an ORF (ORF1) of 126 amino acids which has 61% identity with the C-terminal part of a transposase of Pseudomonas pseudoalcaligenes JS45 (GenBank accession no. AF028594) (unpublished data). This strain grows on nitrobenzene and uses a meta-cleavage dioxygenase for the conversion of the intermediate 2-aminophenol to 2-aminomuconic acid semialdehyde (24). ORF1 continues until the PstI cloning site. Presumably, the complete transposase gene is present in P. putida GJ31, but only the last part was cloned.
In between ORF1 and cbzT are two (incomplete) ORFs that are homologous to components of toluene and phenol monooxygenases. In front of cbzT is a fragment that starts at position 720 and encodes a sequence (ORF3) of 91 amino acids which is up to 50% identical to the C-terminal part of reductase components of these monooxygenases like TbmF of Pseudomonas sp. strain JS150 (GenBank accession no. L40033) (17). TbmF itself consists of 355 amino acids. This suggests that the N-terminal part of this gene was lost in P. putida GJ31, also, because no suitable start codon or putative ribosome binding site was found. Immediately upstream of ORF3 is an ORF (ORF2) the C-terminal part of which resembles another component of toluene and phenol monooxygenases. TbmA of the toluene/benzene-2-monooxygenase of Pseudomonas sp. strain JS150 (GenBank accession no. L40033) (17) is most similar to this part of ORF2, and it has a region of 23 amino acids (Pro-43 to Val-65) that contains 16 amino acids that are identical to the corresponding region in ORF2. The region between Pro-39 and Val-61 of DmpK of the phenol monooxygenase complex of Pseudomonas sp. strain CF600 (GenBank accession no. M37764) (34) is 57% identical to this region in ORF2. The ORF continues into the region that encodes ORF1. The N-terminal part of ORF2 lacks any homology with TbmA or with any other protein in the database, and it is unclear whether there is a real start codon for ORF2. A GTG is present at position 385, which is immediately behind the stop codon of ORF1 at position 381, but no putative ribosome binding site was found. Alternatively, an ATG is present at position 328, which is located inside the fragment that encodes ORF1. The components of the phenol and toluene monooxygenases are normally encoded by six genes that are located in an operon the last gene of which encodes the reductase component. In front of this reductase gene are the four genes that encode the three subunits of the hydroxylase component, and a small subunit that increases the oxidation rate of the aromatic substrate in vitro. The first gene of these operons encodes the TbmA-like component, to which the C-terminal end of ORF2 has similarity. The function of this component is unknown (17, 33, 34). The presence of two ORFs (ORF2 and ORF3) that are similar to parts of TbmA and TbmF, which are encoded by the first and the last gene of the toluene monooxygenase operon in Pseudomonas sp. strain JS150 (17), suggests that recombinations have occurred in this region of the genome of P. putida GJ31. Strain GJ31 presumably does not contain an intact toluene monooxygenase since it most likely uses a dioxygenase for growth on aromatic compounds (25).
The results show that the cbzTEXG genes are clustered. The genes that encode the enzymes of meta-cleavage pathways are usually located in operons (e.g., see references 38 and 52), and the other enzymes that belong to the meta-cleavage pathway of P. putida GJ31 might also be located near cbzTEXG, but they were not cloned with the 3.1-kb fragment. The presence of a part of a transposase-like ORF (ORF1) on the border of the cloned fragment could indicate that the meta-cleavage pathway genes of P. putida GJ31 are located on some kind of transposable element, which has been observed with other meta-cleavage pathway operons as well (52).
Plasmid localization of cbzE.
Plasmid isolations revealed that P. putida GJ31 contains a large plasmid which hybridized with the probe against cbzE. When P. putida GJ31 was grown on benzoate, mutants arose that could no longer grow on chlorobenzene and toluene but that were still able to grow on benzoate and benzene by an ortho-cleavage pathway (25). The plasmid of the mutant (P. putida GJ31M1) was slightly smaller than the wild-type plasmid and no longer hybridized with the probe against cbzE (results not shown). These observations indicate that the meta-cleavage pathway genes are plasmid encoded.
Similarity of CbzE sequence to those of other catechol 2,3-dioxygenases.
ClustalW amino acid sequence alignments showed that CbzE was most similar to the two-domain, iron-containing extradiol dioxygenases that cleave monocyclic diols (9). On the basis of phylogenetic analysis, Eltis and Bolin (9) proposed that this family consisted of five subfamilies, and CbzE was most homologous to the members of subfamily C. The identity of CbzE with the other members of the subfamily ranged from 72% for TdnC of P. putida UCC2 (GenBank accession no. X59790) (unpublished data) and a catechol 2,3-dioxygenase of B. cepacia AA1 (GenBank accession no. U47111) (unpublished data) to 51% for TbuE of Ralstonia pickettii PKO1 (GenBank accession no. U20258) (23). The characterization of most extradiol dioxygenases was not sufficient to attribute any biochemical or functional properties to the various subfamilies (9), although it was suggested that subfamily C might represent a group that has an increased affinity for catechol and molecular oxygen (9, 23). However, this now seems unlikely since the recent determination of the sequence of the catechol 2,3-dioxygenase gene of B. cepacia G4 (tomB) (36) revealed that the TomB enzyme also belongs to the 1.2.C subfamily, while previous measurements showed that this enzyme does not have increased affinities for oxygen and catechols (23).
An alignment of CbzE with the other members of subfamily C, as well as XylE (GenBank accession no. V01161) (31) and BphC (GenBank accession no. X66122) (14), is given in Fig. 2. XylE and BphC are 38 and 15% identical to CbzE, respectively. The crystal structure of the latter enzyme is known (11), and the alignment of Fig. 2 is based on the structure-validated alignment that was made by Eltis and Bolin (9). The histidine and glutamate residues that are known to be involved in the binding of Fe2+ in extradiol dioxygenases are strictly conserved in all of the enzymes.
FIG. 2.

Structure-based amino acid sequence alignment of CbzE with those of other extradiol dioxygenases. The secondary structure elements are indicated below the sequence of BphC as open bars (for α helices) and arrows (for β strands). The alignment was based on the structure-validated alignment that was made by Eltis and Bolin (9). Residues that are strictly conserved within the catechol 2,3-dioxygenases are shown in boldface type, and the three amino acids that are known to be involved in the binding of Fe2+ are underlined. Sequences (GenBank accession numbers in parentheses): CbzE, chlorocatechol 2,3-dioxygenase of P. putida GJ31; TdnC, 3-methylcatechol 2,3-dioxygenase of P. putida UCC2 (X59790) (unpublished data); C23O, Catechol 2,3-dioxygenase of B. cepacia AA1 (U47111) (unpublished data); TomB, catechol 2,3-dioxygenase of B. cepacia G4 (36); Cdo2, catechol 2,3-dioxygenase II of P. putida MT15 (U01286) (unpublished data); TbuE, catechol 2,3-dioxygenase of R. pickettii PKO1 (U20258) (23); XylE, catechol 2,3-dioxygenase of P. putida mt-2 (V01161) (31); BphC, 2,3-dihydroxybiphenyl 1,2-dioxygenase of B. cepacia LB400 (X66122) (14).
Substrate range of several catechol 2,3-dioxygenases.
CbzE is the only catechol 2,3-dioxygenase described so far that is able to productively cleave 3CC at the 2,3 position. However, none of the enzymes that belong to the same subfamily as CbzE has been tested for the ability to convert 3CC. To find out whether this capacity was restricted to CbzE or whether it was a feature which was shared with the other members of the 1.2.C subfamily, we determined the substrate ranges of some other members of this group. For this, the initial rates of product formation were measured with various catechols for each catechol 2,3-dioxygenase by using cell extracts. CbzE and XylE were included as enzymes with good and very poor conversion of 3CC, respectively.
Large differences existed in the relative initial activities of the various enzymes (Table 1). TdnC and C23OII had the highest activities with 3MC, which corresponds with the data of McClure and Venables (27) and Keil et al. (20). TomB and XylE preferred unsubstituted catechol, although both enzymes occur in pathways that have methylcatechols as intermediates. Only CbzE was able to convert 3CC at a high relative rate and could sustain the conversion of 3CC much longer than the other enzymes. The relative activities of the recombinant enzyme correspond rather well with the relative activities that were measured with CbzE that was purified from P. putida GJ31 (19). Significant conversion of 3CC with C23OII and XylE was observed only when much higher amounts of enzyme were added. TdnC, TomB, and C23OII became completely inactivated within 20 s, while XylE was inactivated more slowly than the other catechol 2,3-dioxygenases of the 1.2.C subfamily (Fig. 3). The plots of the amounts of product formed in time yielded straight lines when catechol, 3MC, 4MC, or 4CC was converted by CbzE, while a gradual decrease in the product formation rate was observed with the other enzymes (Fig. 4).
TABLE 1.
Activities and turnover capacities with 3CC of several catechol 2,3-dioxygenases in crude cell extracts with various catechols
| Enzyme | Organism | Extract from strain: | Turnover capacity with 3CC (nmol/U)a | Activity with catechol (U/mg of protein) | Relative activity (%) with:
|
||||
|---|---|---|---|---|---|---|---|---|---|
| Catechol | 3MC | 4MC | 3CC | 4CC | |||||
| CbzE | P. putida GJ31 | E. coli JM101(pBScbzE) | 21,000 | 1.1 | 100 | 43 | 89 | 95 | 75 |
| TdnC | P. putida UCC2 | E. coli JM101(pTDN1-1018) | 220 | 6.4 | 100 | 110 | 49 | 6 | 15 |
| TomB | B. cepacia G4 | B. cepacia G4 | 60 | 0.7 | 100 | 58 | 57 | —b | 15 |
| C23OII | P. putida MT15 | E. coli NM522(pWW15.3201) | 100 | 13.4 | 100 | 170 | 107 | 4 | 60 |
| XylE | P. putida mt-2 | E. coli JM101(pAW31) | 60 | 65.7 | 100 | 41 | 55 | 1 | 57 |
Turnover capacities are expressed in nanomoles of 3CC/unit of catechol 2,3-dioxygenase activity with catechol.
—, No activity detectable.
FIG. 3.

Specific production of 2-hydroxymuconate (2HM) by several catechol 2,3-dioxygenases (C23Os) due to the conversion of 3CC (100 μM). The production of 2HM was measured spectrophotometrically at 290 nm. The amount of C23O that was added was determined as the amount of C23O activity with catechol. Lines: ———, CbzE; · ⋯, TdnC, —⋯—, C23OII; — —, XylE.
FIG. 4.

Formation of meta-cleavage products due to conversion of catechol (———), 3MC (⋯), and 4MC (— —) by CbzE (A), TdnC (B), and XylE (C). The production of the meta-cleavage compounds was measured spectrophotometrically at 375, 388, and 382 nm for catechol, 3MC, and 4MC, respectively. For CbzE, the product formation was linear in time with each catechol. For TdnC and XylE, the rates of product formation with 3MC and 4MC decreased over time due to inactivation of the enzyme.
The measurements were done at catechol concentrations (100 μM) well above the Michaelis-Menten constant (Km) of the catechol 2,3-dioxygenases since the Km values are generally below 10 μM (6, 19, 23, 49). This means that the decrease in the product formation rate cannot be caused by a reduction of the substrate concentration during the assays but is due to some inactivation. Harayama and coworkers already observed that XylE was susceptible to inactivation during conversion of 3MC and 4MC (6, 37), which most likely results from the oxidation of the Fe2+ cofactor (16, 37). Since this kind of inactivation was not observed for CbzE, this indicates that this enzyme not only is most resistant to inactivation by 3CC but also is able to resist inactivation by other catechols.
Turnover capacity with 3CC of several catechol 2,3-dioxygenases.
The amounts of 3CC that can be converted by the enzymes were measured (Table 1). The turnover capacity with 3CC of CbzE was as much as two orders of magnitude higher than the turnover capacities of TdnC, TomB, C23OII, and XylE, which shows that CbzE is indeed very resistant to inactivation during conversion of 3CC. During these measurements the conversion of 3CC by TomB, TdnC, and C23OII stopped within 1 min after the enzyme and substrate were mixed. For XylE, it took approximately 20 min before no further conversion took place (Fig. 5). The turnover capacity of XylE with 3CC was comparable to those with the other three enzymes. This means that XylE, which belongs to the 2.A subfamily of class 1 extradiol dioxygenases (9), converted 3CC at a much lower rate than the enzymes of the 2.C subfamily. Active XylE consists of four identical subunits, and the tetramer has a kcat of about 45,000 min−1 with catechol (30). With the turnover capacity of XylE of 64 nmol of 3CC per unit of activity with catechol (Table 1), this means that about 2,900 molecules of 3CC were converted per molecule of XylE (tetramer) in the crude cell extract. This value is in the same order of magnitude as the turnover of 1,000 that was previously determined for XylE by using oxygen depletion measurements with 3CC (50). For CbzE it took about 30 min before the enzyme stopped converting 3CC (Fig. 5).
FIG. 5.

Conversion of 3CC by CbzE and XylE. The open circles and squares show the concentrations of 3CC that were measured in the assay mixtures with CbzE and XylE, respectively. The lines show the 3CC depletion curves that were fitted through the data with equation 1.
Both CbzE and XylE were inactivated when they were incubated in phosphate buffer without 3CC. This was probably due to oxidation of the ferrous iron cofactor during the incubation. Because of this, the activities of extracts with CbzE and XylE were monitored over time when they were incubated in phosphate buffer in the absence of 3CC. A first-order decrease in the activities of the enzymes was observed in both cases with inactivation constants (k) of 0.028 min−1 (r2 = 0.93) and 0.0098 min−1 (r2 = 0.77) for CbzE and XylE, respectively. Because of this, part of the inactivation that was observed during 3CC conversion by both enzymes cannot be attributed to 3CC. The reciprocal of the coefficient p, which describes the inactivation of the catechol 2,3-dioxygenase due to conversion of 3CC, would give the turnover capacity of the enzymes when aspecific inactivation is absent. The value of p is 0.033 U/μmol for CbzE and 15.1 U/μmol for XylE, which means that the turnover capacities would be 44 and 3% higher without aspecific enzyme inactivation for CbzE and XylE, respectively.
Expression of CbzE and TdnC in pGEF+.
Both cbzE and tdnC were cloned behind the T7 promoter of pGEF+, to give pGFcbzE and pGFtdnC, respectively. The plasmid was transformed into E. coli BL21(DE3), which carries the T7 RNA polymerase gene behind the lac promoter which can be induced with IPTG. E. coli BL21(DE3) also expresses some T7 RNA polymerase without IPTG (48). When the lac promoter was induced with IPTG, a severe formation of inclusion bodies of CbzE and TdnC was observed when whole cells were analyzed on SDS-polyacrylamide gels. This did not occur in uninduced cells. Therefore, cells were grown without IPTG, and this resulted in expression levels that were comparable to that observed with pBScbzE in E. coli JM101 (Tables 1 and 2).
TABLE 2.
Activities and turnover capacities with 3CC of (hybrid) catechol 2,3-dioxygenasesa
| Enzyme | Turnover capacity with 3CC (nmol/U)b | Activity with catechol (U/mg of protein)c | Relative activity (%)
with:
|
||||
|---|---|---|---|---|---|---|---|
| Catechol | 3MC | 4MC | 3CC | 4CC | |||
| CbzE | 7,700 | 1.7 | 100 | 46 | 82 | 167 | 62 |
| TdnC | 80 | 9.8 | 100 | 105 | 47 | 9 | 16 |
| H1 | 170 | 0.5 | 100 | 158 | 78 | —d | 24 |
| H2 | 18,000 | 0.2 | 100 | 35 | 75 | 69 | 70 |
| H3 | 2,400e | 0.05 | 100 | 37 | 15 | 43 | 32 |
| H4 | 120e | 0.8 | 100 | 23 | 19 | — | 7 |
| H7 | NDf | 0.007 | 100 | 49 | 48 | — | 360 |
| H8 | 6,000e | 0.003 | 100 | 160 | 33 | 230 | 210 |
All assays were done with crude cell extracts prepared from catechol 2,3-dioxygenase genes expressed in E. coli with pGEF+.
Turnover capacities are expressed in nanomoles of 3CC/unit of catechol 2,3-dioxygenase activity with catechol.
The activities of H5, H6, H9, and H10 with catechol were <0.001 U/mg of protein.
—, No activity detectable.
The turnover capacity measurements were done with initial concentrations of 3CC of 25 μM for H3 and H8 and 90 μM for H4.
ND, not determined.
Upon cloning, the second amino acid of cbzE was changed from a serine to a glycine residue. The substrate range and turnover capacity with 3CC of CbzE that was expressed from pGFcbzE were somewhat different from the substrate range and turnover capacity of CbzE that was expressed from pBScbzE (Tables 1 and 2). When the turnover measurements were repeated with a lower initial concentration of 3CC (70 μM), the turnover capacity increased more than twice to 18,000 nmol/U, which indicates that the enzyme is very sensitive to high concentrations of 3CC. The increase in turnover capacity was approximately one-third for CbzE that was expressed from pBScbzE, which is much less. Also, the enzyme that was expressed from pGFcbzE was less stable in phosphate buffer. The first-order inactivation constants were 0.058 min−1 (r2 = 0.99) and 0.028 min−1 (r2 = 0.93) for CbzE expressed from pGFcbzE and pBScbzE, respectively (results not shown). Besides the Ser-2-to-Gly-2 mutation, the absence of CbzT in extracts from E. coli BL21(DE3)(pGFcbzE) could be responsible for this difference, since this protein is similar to ferredoxins that are involved in reactivation of catechol 2,3-dioxygenases (16, 37). Nevertheless, CbzE expressed from pGFcbzE had a turnover capacity with 3CC that was at least 35-fold larger than the turnover capacities of other catechol 2,3-dioxygenases, showing that the overexpressed enzyme is highly resistant to 3CC and that the resistance is due not only to CbzT or Gly-2. The substrate range of TdnC that was expressed from E. coli BL21(DE3)(pGFtdnC) was similar to that of pTDN1-1018. The turnover capacity with 3CC was smaller.
Substrate range and turnover capacity with 3CC of hybrid catechol 2,3-dioxygenases.
To find out which regions of CbzE and TdnC determined the substrate ranges, a number of hybrid catechol 2,3-dioxygenases genes were made with PCR fusion and cloned into pGEF+. The hybrids were designated H1 to H10 (Fig. 6).
FIG. 6.

Schematic representation of the construction of hybrid catechol 2,3-dioxygenases (H1 to H10). The primers (P1 to P14) that were used in PCRs to generate DNA fragments that contained overlapping regions for the PCR fusions are indicated with arrows. □, polypeptide fragments derived from CbzE; , polypeptide fragments derived from TdnC. The amino acids of CbzE and TdnC between which the fusions were made are indicated.
One monomer of catechol 2,3-dioxygenase consists of two separate domains that are thought to arise from a genetic duplication (9). The exchanges in hybrids H1 and H2 were made just in front of the start of domain 2, which is formed by the C-terminal half of the protein and starts at β strand I (Fig. 2). The differences in the specific activities of the various (hybrid) enzymes that were measured in crude cell extracts (Table 2) were mainly due to differences in the concentration of soluble (hybrid) catechol 2,3-dioxygenase in these extracts, which was confirmed by analysis on SDS-polyacrylamide gels. Therefore, activities are given relative to the value found with catechol, which is well oxidized by both TdnC and CbzE.
Hybrids H1 and H2 yielded active enzymes. The substrate ranges and turnover capacities with 3CC of H1 and H2 were comparable to the corresponding values of TdnC and CbzE, respectively (Tables 1 and 2). Besides, the product formation plots with catechol, 3MC, 4MC, and 4CC yielded straight lines for H2, which was also observed for CbzE (Fig. 4), while the curved lines that were obtained with H1 suggested that this hybrid was inactivated during conversion of these catechols in manner similar to that of TdnC (results not shown). These data indicate that both the substrate specificity and the susceptibility to inactivation were determined by the C-terminal domains of both enzymes. The active site of catechol 2,3-dioxygenases is located in that domain (9).
Attempts were made to elucidate which amino acids mainly determined the observed differences between both enzymes. This was done by exchanging smaller parts of the C-terminal domain, as is depicted in Fig. 6.
The hybrids H3 and H4, in which the exchanges were made in the beginning of the C-terminal domains, were normally active, although a large part of H3 that was produced was insoluble (Table 2). Compared with TdnC, the replacement of amino acids 148 to 189 of TdnC with amino acids of CbzE (H4) resulted in the loss of the substrate preference for 3MC. This hybrid was even worsened in its capacity to convert 3CC, although it obtained a part of CbzE. The complementary hybrid protein (H3), in which the corresponding fragment of CbzE was replaced by that of TdnC, did not have an improved relative rate for 3MC, while the rate by which 3CC was converted was reduced. Thus, the exchange of these peptide segments influenced the substrate specificity of the enzymes in such way that both hybrid enzymes lost their preference for substituted catechols. The amount of 3CC that could be converted by CbzE dropped significantly when amino acids 148 to 189 of CbzE were replaced by those of TdnC in H3. H4 also had a lowered turnover capacity (Table 2).
When the middle parts of the C-terminal domains (amino acids 190 to 240) were exchanged in H5 and H6, no activity could be detected in crude cell extracts of E. coli BL21(DE3) containing the constructed hybrid genes in pGEF+. This was also the case when the middle and last part of the domain (amino acids 190 to 314) were exchanged together in H9 and H10. The presence of protein inclusion bodies in whole cells was apparent from SDS-polyacrylamide gel electrophoresis, which means that the hybrid catechol 2,3-dioxygenases that were expressed were insoluble. Formation of inclusion bodies is thought to be caused by improper folding of enzymes (28), which indicates that the regions that were exchanged were not similar enough to yield properly folded hybrid enzymes.
When the last regions (amino acids 241 to 314) were exchanged, both hybrids H7 and H8 were soluble but had very low activities (Table 2). Therefore, the accuracy of the measurements of the substrate range of H7 and H8 was not very high. Besides, H7 was very rapidly inactivated when it converted methylated catechols. H8, which was equal to TdnC except for the last region, maintained a high relative activity with 3MC, while it appeared to have an improved relative rate for 3CC, although the conversion stopped within 30 s. The turnover capacity measurement also suggested that the ability to convert 3CC was improved for H8 as compared to the ability to convert TdnC. Besides, both hybrids seemed to have an improved relative rate with 4CC.
A mutant of XylE, in which a single amino acid in the last region of the C-terminal domain was substituted (Val-291 to Ile-291), had a fourfold increase in the turnover capacity with 3CC and other substituted catechols, whereas the affinity for 3CC was lowered (50). Like this mutant of XylE, CbzE has a valine at the corresponding position (Val-297) (Fig. 2), while all the other catechol 2,3-dioxygenases have the isoleucine at that place. The increased turnover capacity with 3CC of H8 compared with TdnC might be caused by a similar effect of this substitution, and this amino acid might play a role in the resistance of CbzE to inactivation with substituted catechols.
None of the hybrids H3, H4, H7, and H8 had an improved resistance to inactivation with the various catechols compared with TdnC, since the plots of the formation of product in time were always curved (results not shown).
The properties of other hybrid catechol 2,3-dioxygenases have been studied by Cerdan et al. (6), who constructed several hybrids of XylE and NahH. They concluded that the amino acid at position 250 (His-250 in XylE, Gln-250 in NahH) mainly determined the sensitivity of the enzyme to inhibition with 3MC and the relative kcat with this substrate. However, the amino acids of the catechol 2,3-dioxygenases of the 1.2.C subfamily that correspond to His-250 in XylE are always arginines (Arg-256), which means that this arginine cannot be responsible for the improved resistance of CbzE to inactivation with the various catechols.
Hybrid catechol 2,3-dioxygenases were also made of XylE of the TOL plasmid and XylE of P. aeruginosa JI104 by using a SalI site that both corresponding genes had in common. Although the enzymes are 94% identical, the substrate range of the former is much more relaxed than that of the latter, which is highly specific for catechol (21). The results indicated that the last 43 C-terminal amino acids probably determined the substrate specificities, since eight of the nine mutations that determined the substrate range of the hybrids are located in that region. The same strategy was used by Williams et al. (51), who made hybrids of two catechol 2,3-dioxygenases that were encoded by the TOL plasmid pWW53. The amino acid sequences of these enzymes are unknown, but these researchers also concluded that the C-terminal region determined the binding and catalytic specificity of the enzymes.
Mechanism of toxicity and resistance.
Although our results clearly showed that the substrate specificities and degrees of resistance to inactivation are determined by the C-terminal domains of catechol 2,3-dioxygenases, it remains to be established what mechanism underlies the inactivation with the various catechols and which biochemical properties are responsible for the resistance of the CbzE enzyme. For 3CC, irreversible suicide inactivation was observed with XylE (5), while Klečka and Gibson (22) observed reversible inactivation of TodE of P. putida F1, which was probably due to the chelation of the Fe2+ cofactor of TodE by 3CC. TodE is only distantly related to the catechol 2,3-dioxygenases used in this study (9), and its mechanism of inactivation seems to be different from that of XylE (5, 22).
When XylE was inactivated with 3CC, activity could not be restored by addition of Fe2+ and dithiothreitol (5), which suggests that the mechanism of inactivation does not involve the oxidation state or the loss of the iron cofactor. Another possible cause of the observed inactivation is the nucleophilic attack of one or more groups in the enzyme on an electrophilic intermediate in the reaction cycle. Preliminary experiments with XylE showed that the homodimeric protein band of native enzyme migrated slightly faster on SDS-polyacrylamide gels than that of enzyme that was inactivated with 3CC, provided that the samples were not boiled prior to application on the gel. Modification of XylE by 3CC also made the enzyme less susceptible to digestion with proteinase LysC. Complete digestion of dialyzed, inactivated XylE was possible only in the presence of 1 M urea, while native XylE could be digested in the absence of urea (unpublished results). These results might indicate that inactivation by 3CC involves a labile covalent modification which influences the multimeric form of XylE. The inactivation that was observed for XylE and NahH with methylated catechols (6) was also related to conversion of the catechols and not to the catechols themselves since the inactivation constants were usually similar at 50 and 300 μM.
The increased resistance of CbzE to inactivation by both methyl- and chlorocatechols might suggest that the mechanisms of inactivation by both compounds are similar. However, Hugo et al. (16) showed that the conversion of 4MC by XylE resulted in the oxidation of the catalytic iron. The oxidized iron (Fe3+) can be reduced by XylT, which resulted in a reactivation of the enzyme (16, 37). The inactivation of XylE by methylated catechols thus seems to be a reversible process, in contrast to the inactivation observed with 3CC (5). However, the EPR signal of the oxidized iron in XylE after inactivation by 4MC was clearly distinct from the signal of the catalytic iron that was aspecifically oxidized by H2O2 (16). The authors suggested that a putative ligand that resulted from the conversion of 4MC remained in the active site, which might even hinder complete reactivation by XylT (16). Perhaps such a ligand is also formed during the conversion of 3CC, which might bind in an irreversible manner and prevent the regeneration of the iron cofactor. It should be noted that the resistance of CbzE to 3CC is not exclusively caused by a modified interaction with CbzT, since increased resistance was also found with enzyme that was expressed from E. coli in the absence of CbzT, while purified CbzE also efficiently converts 3CC (19).
EPR studies of XylE that is inactivated with 3CC would provide more insight in the mechanism that causes the inactivation. This would also help us to understand the biochemical basis for the extraordinary resistance of CbzE to methyl- and chlorocatechols. If a specific group in the enzyme is attacked by the electrophiles that are formed during the reaction with 3CC, the resistance of CbzE to 3CC might result from a modification of this target. Alternatively, substitutions that lead to a more rapid conversion of the toxic intermediate could reduce the lifetime of the reactive electrophiles, which reduces the chance of damage of the enzyme and increases the resistance to inactivation.
ACKNOWLEDGMENTS
This work was financed by a grant from the Dutch IOP Environmental Biotechnology program and from the Deutsche Forschungsgemeinschaft (Re 659/7-1).
We acknowledge P. Terpstra for the sequence analyses and U. Dehmel, N. C. McClure, M. S. Shields, and P. A. Williams for kindly providing the strains and plasmids with the catechol 2,3-dioxygenases.
REFERENCES
- 1.Altschul S F, Madden T L, Schäffer A A, Zhang J, Zhang Z, Miller W, Lipman D J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arensdorf J J, Focht D D. Formation of chlorocatechol metacleavage products by a pseudomonad during metabolism of monochlorobiphenyls. Appl Environ Microbiol. 1994;60:2884–2889. doi: 10.1128/aem.60.8.2884-2889.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Assinder S J, Williams P A. The TOL plasmids: determinants of the catabolism of toluene and the xylenes. Adv Microb Physiol. 1990;31:1–69. doi: 10.1016/s0065-2911(08)60119-8. [DOI] [PubMed] [Google Scholar]
- 4.Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K. Current protocols in molecular biology. New York, N.Y: Green Publishing Associates, Inc.; 1994. [Google Scholar]
- 5.Bartels I, Knackmuss H-J, Reineke W. Suicide inactivation of catechol 2,3-dioxygenase from Pseudomonas putidamt-2 by 3-halocatechols. Appl Environ Microbiol. 1984;47:500–505. doi: 10.1128/aem.47.3.500-505.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cerdan P, Rekik M, Harayama S. Substrate specificity differences between two catechol 2,3-dioxygenases encoded by the TOL and NAH plasmids from Pseudomonas putida. Eur J Biochem. 1995;229:113–118. doi: 10.1111/j.1432-1033.1995.tb20445.x. [DOI] [PubMed] [Google Scholar]
- 7.Dehmel, U. Personal communication.
- 8.Duetz W A, Winson M K, van Andel J G, Williams P A. Mathematical analysis of catabolic function loss in a population of Pseudomonas putidamt-2 during non-limited growth on benzoate. J Gen Microbiol. 1991;137:1363–1368. doi: 10.1099/00221287-137-6-1363. [DOI] [PubMed] [Google Scholar]
- 9.Eltis L D, Bolin J T. Evolutionary relationships among extradiol dioxygenases. J Bacteriol. 1996;178:5930–5937. doi: 10.1128/jb.178.20.5930-5937.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haigler B E, Spain J C. Degradation of p-chlorotoluene by a mutant of Pseudomonassp. strain JS6. Appl Environ Microbiol. 1989;55:372–379. doi: 10.1128/aem.55.2.372-379.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Han S, Eltis L D, Timmis K N, Muchmore S W, Bolin J T. Crystal structure of the biphenyl-cleaving extradiol dioxygenase from a PCB-degrading pseudomonad. Science. 1995;270:976–980. doi: 10.1126/science.270.5238.976. [DOI] [PubMed] [Google Scholar]
- 12.Harayama S, Polissi A, Rekik M. Divergent evolution of chloroplast-type ferredoxins. FEBS Lett. 1991;285:85–88. doi: 10.1016/0014-5793(91)80730-q. [DOI] [PubMed] [Google Scholar]
- 13.Higson F K, Focht D D. Utilization of 3-chloro-2-methylbenzoic acid by Pseudomonas cepacia MB2 through the metafission pathway. Appl Environ Microbiol. 1992;58:2501–2504. doi: 10.1128/aem.58.8.2501-2504.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hofer B, Eltis L D, Dowling D N, Timmis K N. Genetic analysis of a Pseudomonaslocus encoding a pathway for biphenyl/polychlorinated biphenyl degradation. Gene. 1993;130:47–55. doi: 10.1016/0378-1119(93)90345-4. [DOI] [PubMed] [Google Scholar]
- 15.Hollender J, Dott W, Hopp J. Regulation of chloro- and methylphenol degradation in Comamonas testosteroniJH5. Appl Environ Microbiol. 1994;60:2330–2338. doi: 10.1128/aem.60.7.2330-2338.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hugo N, Armengaud J, Gaillard J, Timmis K N, Jouanneau Y. A novel [2Fe-2S] ferredoxin from Pseudomonas putidamt2 promotes the reductive reactivation of catechol 2,3-dioxygenase. J Biol Chem. 1998;273:9622–9629. doi: 10.1074/jbc.273.16.9622. [DOI] [PubMed] [Google Scholar]
- 17.Johnson G R, Olsen R H. Nucleotide sequence analysis of genes encoding a toluene/benzene-2-monooxygenase from Pseudomonassp. strain JS150. Appl Environ Microbiol. 1995;61:3336–3346. doi: 10.1128/aem.61.9.3336-3346.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kado C I, Liu S-T. Rapid procedure for detection and isolation of large and small plasmids. J Bacteriol. 1981;145:1365–1373. doi: 10.1128/jb.145.3.1365-1373.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaschabek S R, Kasberg T, Müller D, Mars A E, Janssen D B, Reineke W. Degradation of chloroaromatics: purification and characterization of a novel type of chlorocatechol 2,3-dioxygenase of Pseudomonas putidaGJ31. J Bacteriol. 1998;180:296–302. doi: 10.1128/jb.180.2.296-302.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Keil H, Lebens M R, Williams P A. TOL plasmid pWW15 contains two nonhomologous, independently regulated catechol 2,3-oxygenase genes. J Bacteriol. 1985;163:248–255. doi: 10.1128/jb.163.1.248-255.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kitayama A, Achioku T, Yanagawa T, Kanou K, Kikuchi M, Ueda H, Suzuki E, Nishimura H, Nagamune T, Kawakami Y. Cloning and characterization of extradiol aromatic ring-cleavage dioxygenases of Pseudomonas aeruginosaJI104. J Ferment Bioeng. 1996;82:217–223. [Google Scholar]
- 22.Klečka G M, Gibson D T. Inhibition of catechol 2,3-dioxygenase from Pseudomonas putidaby 3-chlorocatechol. Appl Environ Microbiol. 1981;41:1159–1165. doi: 10.1128/aem.41.5.1159-1165.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kukor J J, Olsen R H. Catechol 2,3-dioxygenases functional in oxygen-limited (hypoxic) environments. Appl Environ Microbiol. 1996;62:1728–1740. doi: 10.1128/aem.62.5.1728-1740.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lendenmann U, Spain J C. 2-Aminophenol 1,6-dioxygenase: a novel aromatic ring cleavage enzyme purified from Pseudomonas pseudoalcaligenesJS45. J Bacteriol. 1996;178:6227–6232. doi: 10.1128/jb.178.21.6227-6232.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mars A E, Kasberg T, Kaschabek S R, van Agteren M H, Janssen D B, Reineke W. Microbial degradation of chloroaromatics: use of the meta-cleavage pathway for mineralization of chlorobenzene. J Bacteriol. 1997;179:4530–4537. doi: 10.1128/jb.179.14.4530-4537.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mars A E, Prins G T, Wietzes P, de Koning W, Janssen D B. Effect of trichloroethylene on the competitive behavior of toluene-degrading bacteria. Appl Environ Microbiol. 1998;64:208–215. doi: 10.1128/aem.64.1.208-215.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McClure N C, Venables W A. Adaptation of Pseudomonas putidamt-2 to growth on aromatic amines. J Gen Microbiol. 1986;132:2209–2218. doi: 10.1099/00221287-132-8-2209. [DOI] [PubMed] [Google Scholar]
- 28.Mitraki A, King J. Protein folding intermediates and inclusion body formation. Biotechnology. 1989;7:690–697. [Google Scholar]
- 29.Murray V. Improved double-stranded DNA sequencing using the linear polymerase chain reaction. Nucleic Acids Res. 1989;17:8889. doi: 10.1093/nar/17.21.8889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakai C, Hori K, Kagamiyama H, Nakazawa T, Nozaki M. Purification, subunit structure, and partial amino acid sequence of metapyrocatechase. J Biol Chem. 1983;258:2916–2922. [PubMed] [Google Scholar]
- 31.Nakai C, Kagamiyama H, Nozaki M, Nakazawa T, Inouye S, Ebina Y, Nakazawa A. Complete nucleotide sequence of the metapyrocatechase gene on the TOL plasmid of Pseudomonas putidamt-2. J Biol Chem. 1983;258:2923–2928. [PubMed] [Google Scholar]
- 32.Nelson M J K, Montgomery S O, O’Neill O J, Pritchard P H. Aerobic metabolism of trichloroethylene by a bacterial isolate. Appl Environ Microbiol. 1986;52:383–384. doi: 10.1128/aem.52.2.383-384.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Newman L M, Wackett L P. Purification and characterization of toluene 2-monooxygenase from Burkholderia cepaciaG4. Biochemistry. 1995;34:14066–14076. doi: 10.1021/bi00043a012. [DOI] [PubMed] [Google Scholar]
- 34.Nordlund I, Powlowski J, Shingler V. Complete nucleotide sequence and polypeptide analysis of multicomponent phenol hydroxylase from Pseudomonassp. strain CF600. J Bacteriol. 1990;172:6826–6833. doi: 10.1128/jb.172.12.6826-6833.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nordlund I, Shingler V. Nucleotide sequences of the meta-cleavage pathway enzymes 2-hydroxymuconic semialdehyde dehydrogenase and semialdehyde hydrolase from PseudomonasCF600. Biochim Biophys Acta. 1990;1049:227–230. doi: 10.1016/0167-4781(90)90046-5. [DOI] [PubMed] [Google Scholar]
- 36.Oh J M, Kang E, Min K R, Kim C-K, Kim Y-C, Lim J-Y, Lee K-S, Min K-H, Kim Y. Structure of catechol 2,3-dioxygenase gene encoded in TOM plasmid of Pseudomonas putidaG4. Biochem Biophys Res Commun. 1997;234:578–581. doi: 10.1006/bbrc.1997.6680. [DOI] [PubMed] [Google Scholar]
- 37.Polissi A, Harayama S. In vivoreactivation of catechol 2,3-dioxygenase mediated by a chloroplast-type ferredoxin: a bacterial strategy to expand the substrate specificity of aromatic degradative pathways. EMBO J. 1993;12:3339–3347. doi: 10.1002/j.1460-2075.1993.tb06004.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Powlowski J, Shingler V. Genetics and biochemistry of phenol degradation by Pseudomonassp. CF600. Biodegradation. 1994;5:219–236. doi: 10.1007/BF00696461. [DOI] [PubMed] [Google Scholar]
- 39.Saint C P, McClure N C, Venables W A. Physical map of the aromatic amine and m-toluate catabolic plasmid pTDN1 in Pseudomonas putida: localization of a unique meta-cleavage pathway. J Gen Microbiol. 1990;136:615–625. doi: 10.1099/00221287-136-4-615. [DOI] [PubMed] [Google Scholar]
- 40.Sala-Trepat J M, Evans W C. The meta cleavage of catechol by Azotobacterspecies. 4-Oxalocrotonate pathway. Eur J Biochem. 1971;20:400–413. doi: 10.1111/j.1432-1033.1971.tb01406.x. [DOI] [PubMed] [Google Scholar]
- 41.Sala-Trepat J M, Murray K, Williams P A. The metabolic divergence in the meta cleavage of catechols by Pseudomonas putidaNCIB 10015. Physiological significance and evolutionary implications. Eur J Biochem. 1972;28:347–356. doi: 10.1111/j.1432-1033.1972.tb01920.x. [DOI] [PubMed] [Google Scholar]
- 42.Schanstra J P, Rink R, Pries F, Janssen D B. Construction of an expression and site-directed mutagenesis system of haloalkane dehalogenase in Escherichia coli. Protein Expression Purif. 1993;4:479–489. doi: 10.1006/prep.1993.1063. [DOI] [PubMed] [Google Scholar]
- 43.Schlömann M. Evolution of chlorocatechol catabolic pathways. Biodegradation. 1994;5:301–321. doi: 10.1007/BF00696467. [DOI] [PubMed] [Google Scholar]
- 44.Senda T, Sugiyama K, Narita H, Yamamoto T, Kimbara K, Fukuda M, Sato M, Yano K, Mitsui Y. Three-dimensional structures of free form and two substrate complexes of an extradiol ring-cleavage type dioxygenase, the BphC enzyme from Pseudomonassp. strain KKS102. J Mol Biol. 1996;255:735–752. doi: 10.1006/jmbi.1996.0060. [DOI] [PubMed] [Google Scholar]
- 45.Shields M S, Montgomery S O, Cuskey S M, Chapman P J, Pritchard P H. Mutants of Pseudomonas cepaciaG4 defective in catabolism of aromatic compounds and trichloroethylene. Appl Environ Microbiol. 1991;57:1935–1941. doi: 10.1128/aem.57.7.1935-1941.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shine J, Dalgarno L. Determinant of cistron specificity in bacterial ribosomes. Nature. 1975;254:34–38. doi: 10.1038/254034a0. [DOI] [PubMed] [Google Scholar]
- 47.Smith M R. The biodegradation of aromatic hydrocarbons by bacteria. Biodegradation. 1990;1:191–206. doi: 10.1007/BF00058836. [DOI] [PubMed] [Google Scholar]
- 48.Studier F W, Rosenberg A H, Dunn J J, Dubendorff J W. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 1990;185:60–89. doi: 10.1016/0076-6879(90)85008-c. [DOI] [PubMed] [Google Scholar]
- 49.Wallis M G, Chapman S K. Isolation and partial characterization of an extradiol non-haem iron dioxygenase which preferentially cleaves 3-methylcatechol. Biochem J. 1990;266:605–609. [PMC free article] [PubMed] [Google Scholar]
- 50.Wasserfallen A, Rekik M, Harayama S. A Pseudomonas putida strain able to degrade m-toluate in the presence of 3-chlorocatechol. Biotechnology. 1991;9:296–298. [Google Scholar]
- 51.Williams P A, Assinder S J, Shaw L E. Construction of hybrid xylEgenes between the two duplicate homologous genes from TOL plasmid pWW53: comparison of the kinetic properties of the gene products. J Gen Microbiol. 1990;136:1583–1589. doi: 10.1099/00221287-136-8-1583. [DOI] [PubMed] [Google Scholar]
- 52.Williams P A, Sayers J R. The evolution of pathways for aromatic hydrocarbon oxidation in Pseudomonas. Biodegradation. 1994;5:195–217. doi: 10.1007/BF00696460. [DOI] [PubMed] [Google Scholar]
- 53.Yanisch-Perron C, Vieira J, Messing J. Improved M13 phage cloning vectors and host strains: nucleotide sequences of M13mp18 and pUC19. Gene. 1985;33:103–109. doi: 10.1016/0378-1119(85)90120-9. [DOI] [PubMed] [Google Scholar]
- 54.Yolov A A, Shabarova Z A. Constructing DNA by polymerase recombination. Nucleic Acids Res. 1990;18:3983–3986. doi: 10.1093/nar/18.13.3983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yrjälä K, Paulin L, Romantschuk M. Novel organisation of catechol meta-pathway genes in Sphingomonassp. HV3 pSKY4 plasmid. FEMS Microbiol Lett. 1997;154:403–408. doi: 10.1111/j.1574-6968.1997.tb12674.x. [DOI] [PubMed] [Google Scholar]