

Abstract
Conformational antibodies specific for amyloid-forming peptides and proteins are important for a range of biomedical applications, including detecting, inhibiting and potentially treating protein aggregation disorders ranging from Alzheimer’s to Parkinson’s diseases. Generation of anti-amyloid antibodies is greatly complicated by the complex, heterogeneous and insoluble nature of amyloid antigens. Here we describe systematic methods for isolating and affinity maturing anti-amyloid antibodies using yeast surface display. Magnetic-activated cell sorting is used to sort single-chain antibody libraries positively for binding to amyloid antigens and negatively against the corresponding disaggregated antigens to remove antibodies that bind in a conformation-independent manner. Isolated lead antibody clones with conformational specificity are affinity matured via targeted CDR mutagenesis and magnetic-activated cell sorting.
Keywords: conformational, conformation, yeast surface display, aggregate, aggregation, oligomer, fibril, fiber, directed evolution, Alzheimer’s, Parkinson’s
1. INTRODUCTION
While different classes of peptides and proteins ranging from hormones to enzymes conduct diverse biological functions, they all possess limited physical stability. In some cases, this limited physical stability results in polypeptide aggregation in diseases such as Alzheimer’s and Parkinson’s disease and in therapeutic drug formulations (1–3). This is particularly concerning because peptide and protein aggregates formed in vivo mediate cellular toxicity, while aggregates formed in vitro in therapeutic formulations mediate immunogenicity.
Therefore, there is significant need for molecular agents such as conformational antibodies for detecting, disrupting and mediating removal of polypeptide aggregates. There are two broad classes of approaches for generating such antibodies, namely in vivo (e.g., immunization and hybridoma technology or single B cell screening) and in vitro (e.g., naïve libraries sorted via phage or yeast surface display) methods. While both methods have strengths and weaknesses, the notable strengths of in vitro methods are the ability to control antigen presentation during antibody selection, including antigen concentration and higher order structure (4). Moreover, in vitro sorting methods enable both positive selections for antibody recognition of aggregated antigens and negative selections for lack of recognition of the same antigens in their disaggregated forms (5–9).
Nevertheless, in vitro antibody generation methods, including those against soluble and aggregated antigens, have a higher likelihood of yielding antibodies with suboptimal properties relative to antibodies generated using immunization (10). There are several reasons for this, but the most important one is likely the sophisticated quality control mechanisms used by the immune system to select antibody variants with high specificity and stability in addition to high affinity. However, there has been significant progress in recent years in developing in vitro methods for selecting high quality antibodies with properties that rival those of antibodies generated by the immune system (4). Here we report an optimized procedure (Fig. 1) for selecting and affinity maturing antibodies with high conformational and sequence specificity for amyloid antigens using yeast-displayed antibody libraries.
Figure 1. Overview of conformational anti-amyloid antibody isolation from yeast-displayed libraries.
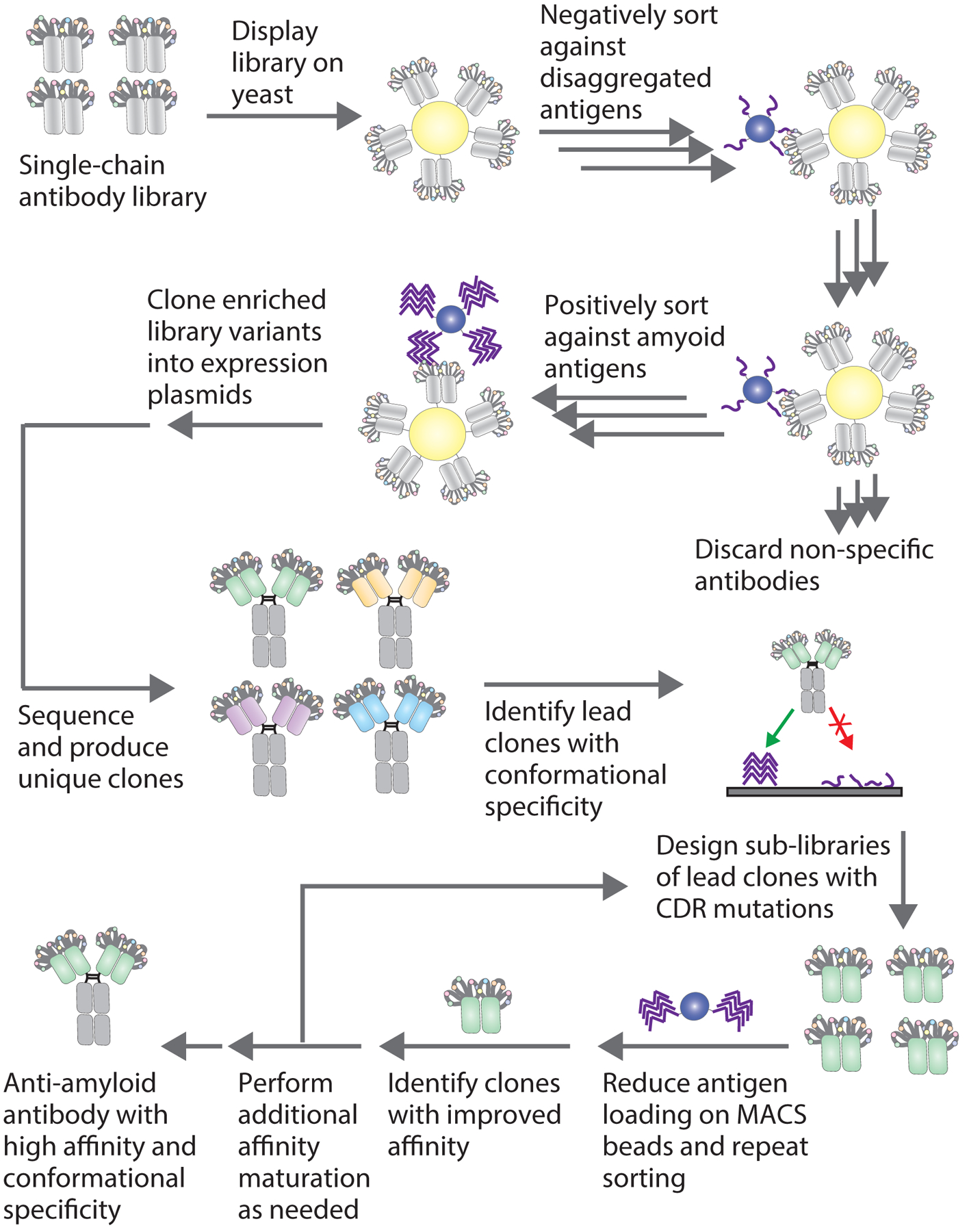
Single-chain antibody libraries displayed on yeast are sorted negatively to remove clones which bind to disaggregated antigen and positively to enrich clones which bind to amyloid aggregates. Negative selections can be performed using either magnetic-activated cell sorting (MACS) or fluorescence-activated cell sorting (FACS), and positive selections are performed with amyloid aggregates immobilized on beads using MACS. After several rounds of enrichment, the selected antibodies are cloned directly into Fc-fusion plasmids and expressed in mammalian cells. Individual antibody clones are analyzed using flow cytometry for affinity to amyloid fibrils and conformational binding to fibrils in the presence of disaggregated antigen. Clones identified with affinity and conformational specificity for the target amyloid fibrils are then affinity-matured through the preparation of yeast surface displayed sub-libraries. Sub-libraries are sorted stringently by reducing antigen loading or the number of antigen-coated beads to select for clones with improved affinity and conformational specificity.
2. MATERIALS
2.1. Initial library preparation and sorting
Yeast dropout plates: Add 15 gm agar to 800 mL DI water and autoclave using L20 cycle. Make supplemental solution by dissolving 20 gm glucose, 6.7 gm yeast nitrogen base (without amino acids) and 3.8 gm of tryptophan drop-out media in 200 mL DI water. Sterile filter supplemental solution into agar solution after the bottle containing agar solution has cooled sufficiently to pour and make plates. Store plates at 4 °C.
SDCAA media: Add 16.75 gm of sodium citrate, 4 gm anhydrous citric acid, 6.7 gm yeast nitrogen base (without amino acids), 5 gm acid casein peptone (casamino acids) and 20 gm glucose to 1000 mL DI water. Sterile filter with 0.22 μm filter and store at room temperature.
100 mg/mL Ampicillin: Add 1000 mg of ampicillin in 10 mL of DI water and dissolve by end-over-ending mixing for 10 min at room temperature. Filter through 0.22 μm filter, aliquot and store at −20 °C.
100 mg/mL Kanamycin: Add 1000 mg of kanamycin in 10 mL of DI water and dissolve by end-over-end mixing for 10 min at room temperature. Filter through 0.22 μm filter, aliquot and store at −20 °C.
Gene Pulser Xcell Electroporation Systems (Bio-Rad)
Penicillin-Streptomycin (pen-strep).
Yeast mini-prep kit (Zymo Research).
Bacterial strain: DH5α/XL-1 blue.
Q5 polymerase (New England Biolabs)
LB media: Dissolve 10 gm tryptone, 10 gm sodium chloride and 5 gm yeast extract in 1000 mL of DI water and autoclave using L20 cycle. Store at room temperature.
LB agar plates with 100 μg/mL of ampicillin: Add 10 gm tryptone, 10 gm sodium chloride, 5 gm yeast extract and 20 gm agar to 1000 mL of DI water and autoclave using L20 cycle. Add 1 mL ampicillin at 100 mg/mL after the bottle containing solution has cooled sufficiently to pour and make plates. Store plates at 4 °C.
Bacterial mini-prep kits.
Amyloid antigens.
Dynabeads: Streptavidin and tosyl activated beads (Invitrogen).
Magnet.
SDGCAA media: Add 8.56 gm sodium phosphate dibasic dihydrate, 6.76 gm sodium phosphate monobasic monohydrate, 6.7 gm yeast nitrogen base (without amino acids), 5 gm acid casein peptone (casamino acids), 2 gm glucose and 20 gm galactose in 1000 mL DI water. Sterile filter with 0.22 μm filter and store at room temperature. (See Note 1.)
Washing buffer: PBSB; Dissolve 1 gm BSA in 1000 mL PBS. Sterile filter with 0.22 μm filter and store at 4 °C
Blocking buffer: PBSB with 10% milk; Dissolve 500 mg dry milk powder in 5 mL washing buffer (PBSB) and mix with end-over-end mixing at room temperature for 10 min. Spin down milk solution at 2500 ×g for 5 min and move the supernatant to a fresh tube. (See Note 2.)
Streptavidin Alexa Fluor 647 (Molecular Probes).
Anti-Myc antibody (Cell Signaling Technology, 9B11). (See Note 3.)
Goat anti-mouse Alexa Fluor 488 (Invitrogen)
2.2. Antibody identification and characterization
2.2.1. Cloning antibody genes into mammalian expression plasmid
Yeast mini prep kits (Zymo Research).
Primers to clone antibody genes in mammalian Fc expression plasmids.
DNA polymerase.
dNTPs.
Restriction enzymes.
DNA ligase.
LB agar plates with 100 μg/mL ampicillin.
LB media with 100 μg/mL ampicillin.
Competent DH5α cells.
Bacterial mini-prep kits.
Plasmid sequencing primer.
2.2.2. Antibody expression, purification and characterization
HEK293–6E cells.
Passage media: F17 media supplemented with 0.1% w/v Kolliphor, 30 mL/L-Glutamine, and 0.5 mL/L G418.
Transfection media: F17 media without supplements
Kolliphor: Dissolve 100 gm Kolliphor in 1000 mL deionized (DI) water to make a 10% solution (w/v). Sterile filter with 0.22 μm filter.
1 mg/mL Polyethylenimine (PEI): Dissolve 500 mg of PEI in 500 mL of DI water. Sterile filter with 0.22 μm filter and store at 4 °C.
Yeastolate ultrafiltered (Gibco Difco)
Cell culture tubes or flasks.
Protein A agarose beads.
Filter columns.
Elution buffer: Dissolve 7.5 gm glycine in 800 mL of DI water. Adjust pH to 3.0 with 1 M HCl. Add DI water to a final volume of 1000 mL and sterile filter with 0.22 μm filter. Store at room temperature.
Neutralization buffer: Dissolve 121 gm Tris-HCl in 800 mL of DI water. Adjust pH to 8.0 with 1M NaOH. Add DI water to a final volume of 1000 mL and sterile filter with 0.22 μm filter. Store at room temperature. (See Note 4.)
Buffer exchange columns.
Protein storage buffer: Make 20 mM acetic acid solution in DI water. Make 20 mM potassium acetate by dissolving 1.96 gm potassium acetate in 1000 mL of DI. Add 20 mM acetic acid to 20 mM potassium acetate to a final pH 5.0. Sterile filter the buffer with 0.22 μm filter and store at room temperature.
Syringes.
Syringe filters.
Regeneration buffer: Dissolve 7.5 gm glycine in 800 mL of DI water. Adjust pH to 2.5 with 1 M HCl. Add DI water to a final volume of 1000 mL and sterile filter with 0.22 μm filter. Store at room temperature.
Resin storage buffer: PBS with 0.2% azide.
10% Bis-tris SDS-PAGE gels.
Size-exclusion chromatography (SEC) column.
SEC running buffer: PBS with 200 mM arginine. Dissolve 34.84 gm arginine in 1000 mL PBS. Adjust pH to 7.4 with 1 M HCl. De-gas the buffer and sterile filter with 0.22 μm filter.
2.2.3. Antibody binding
Streptavidin or tosyl activated beads (Invitrogen)
Goat anti-human Alexa Fluor 647.
Blocking buffer.
Washing buffer.
96-well plate.
2.2.4. Affinity maturation
Materials described in Section 2.1 and additional materials listed below.
PCR supplies.
Ethanol precipitation kit.
Pellet paint co-precipitant (Millipore Sigma)
Yeast strain EBY100.
Electroporation cuvettes (2 mm).
YPD media: Add 10 gm yeast extract, 20 gm peptone, 20 glucose to 1000 mL of DI water. Sterile filter with 0.22 μm filter and store at room temperature.
Electroporation buffer (1 M sorbitol and 1 mM CaCl2): Add 182 gm sorbitol and 111 mg CaCl2 to 1000 mL of DI water. Sterile filter with 0.22 μm filter and keep it on ice.
Sterile filtered DI water and keep it on ice. (See Note 5.)
Lithium acetate/1,4-Dithiothreitol (DTT) solution (0.1 M lithium acetate and 10 mM DTT): Add 1.98 gm lithium acetate and 462 mg of DTT to 300 mL of DI water. Sterile filter with 0.22 μm filter and use immediately. (See Note 6.)
Sorbitol (1 M): Add 9.1 gm sorbitol to 50 mL DI water. Sterile filter with 0.22 μm filter and store at room temperature.
3. METHODS
3.1. Library preparation and sorting
3.1.1. Antigen bead preparation
For biotinylated antigens, use streptavidin dynabeads. For non-biotinylated antigens, use tosyl dynabeads.
For non-biotinylated antigens: Add 100 μg of antigen to a final volume of 500 μL in PBS and sonicate for 5 min on ice (30 s on, 30 s off) at 100% power with probe sonicator. Sonication is necessary to break up large fibril aggregates into smaller, more homogeneous species to improve the efficiency of immobilization of aggregates on the beads. Pipette 41 μL (8×107 tosyl beads; 107 beads for one selection) beads from stock to an autoclaved 1.5 mL tube and put the tube on magnet. After a few minutes, pipette out the storage solution. Take the tube off the magnet and wash the beads with 1 mL PBS [or other buffer without primary amine (e.g., Tris)] and put the tube back on the magnet. Wash the beads twice with PBS. Post-washing, re-suspend the beads in 500 μL of PBS and add 500 μL of sonicated antigen (final volume of 1000 μL, 107 beads/125 μL). Mix by end-over-end mixing for ~48 h at room temperature and store them at 4 °C until future use.
For biotinylated antigens: Assemble amyloid fibrils by doping biotinylated monomer (1–10%) with unlabeled monomer (90–99%). Purify fibrils by ultracentrifuging them at 221,000 ×g for 1 h at 4 °C. Resuspend the fibrils in the same volume of buffer prior to sedimentation. Pipette 200 μL (8×107 streptavidin beads, 107 beads for one selection) and wash them twice with washing buffer (PBSB) as described above and re-suspend them in 1 mL PBSB. Sonicate fibrils for 2 min (10 s on, 30 s off) at 100% power on ice. Add sonicated fibrils to beads and PBSB to a final volume of 3200 μL (400 μL/107 beads). Mix by end-over-end mixing for ~48 h at room temperature and store them at 4 °C for future use. (See Note 7.)
3.1.2. Library preparation and screening
Grow/recover the library in 200 mL SDCAA media supplemented with 100 μg/mL ampicillin, 100 μg/mL kanamycin and 100x pen-strep at 30 °C for 16–18 h with agitation (225 RPM)
Measure OD by reading absorbance at 600 nm and calculate the density of the culture. It is recommended to induce cells in early log phase (OD of 0.5–1). Take cell volume corresponding to desired OD and centrifuge at 2500 ×g for 5 min. Discard supernatant and re-suspend cells in SDGCAA and move to a shake flask with 200 mL SDGCAA media supplemented with all three antibiotics. Induce library at 20 °C for 36–40 h with agitation.
On the day of screening, calculate OD of induced culture. We estimate ~3×107/mL cells for an OD600 of 1. For screening 109 cells, pipette volume corresponding to 109 cells in 50 mL conical tubes and wash cell pellet twice by centrifuging cells at 2500 ×g for 5 min with 25 mL washing buffer (PBSB). For screening 107 cells, pipette volume corresponding to 107 cells in 1.5 mL tube and wash cell pellet twice by centrifuging cells at 16000 ×g for 1 min with 1 mL washing buffer (PBSB).
On the day of screening, prepare 107 beads for sorting immediately before use. For non-biotinylated antigens, pipette 125 μL antigen immobilized beads into a 1.5 mL tube and place it on magnet. Take off the residual solution and wash beads twice with 1 mL 10 mM glycine for 5 min each followed by a wash with 1 mL PBSB. For biotinylated antigens, pipette 400 μL antigen immobilized beads into a 1.5 mL tube and place it on magnet. Take off the residual solution and wash beads twice with 1 mL PBSB.
3.1.3. Sorting
Cell and bead preparation is described above.
Positive selection against fibrils (See Note 8.): For screening 109 cells, mix cells and beads in a 15 mL tube. Add PBSB to a final volume of 4.5 mL. Add 0.5 mL blocking buffer to obtain a final concentration of 1% milk in 5 mL of solution and incubate tube with end-over-end mixing at room temperature for 3 h. For screening 107 cells, perform steps as above except the incubation is performed in a 1.5 mL tube with a final volume of 1 mL. Post-incubation, put the tubes on the magnet and pipette out unbound cells and discard them. Wash the beads with ice-cold 10 mL washing buffer if screening 109 cells or 1 mL washing buffer if screening 107 cells once. Again, put the tubes on the magnet and pipette out unbound cells and discard them. Move the beads to shake flasks containing 50 mL SDCAA media supplemented with ampicillin (100 μg/mL), kanamycin (100 μg/mL) and pen-strep (100x). Plate dilutions (1x and 10x) on yeast drop-out plates to calculate number of cells retained in selection. Grow cells for 36–48 h at 30 °C at 225 RPM. For the next round, induce cells in 5–50 mL SDGCAA culture as described above.
Negative selection against monomeric (disaggregated) peptide or protein by MACS: Immobilize the disaggregated monomer on beads and prepare the beads for sorting as described above. Incubate beads and cells for 1–3 h at room temperature with end-over-end mixing. Post-incubation, place the tube on the magnet. Take off the unbound cells and move them to a new tube containing fresh beads. It is recommended to perform negative selection 2–3 times in each round to remove sequence-specific antibodies efficiently. After multiple negative selections, take the unbound cells and add them to a tube containing beads immobilized with fibril to perform a positive selection.
Negative selection against monomeric (disaggregated) peptide or protein by FACS: Prepare the cells for sort as described above. Add biotinylated disaggregated monomer to the cells at a desired concentration (10–1000 nM) and anti-Myc antibody (final concentration ~0.3 μg/mL) in a final volume of 200 μL (of PBSB) and incubate for 1–3 h. Post-incubation, wash the cells once with ice-cold PBSB. Incubate with goat anti-mouse Alexa Fluor 488 (final concentration of ~10 μg/mL) and streptavidin Alexa Fluor 647 (final concentration of ~2 μg/mL) on ice for 4 min. Post-secondary incubation, wash cells once with ice-cold PBSB and sort on flow cytometer. It is recommended to gate and collect 20–50% of antibody displaying population of cells that do not bind or weakly bind monomer. Collect and grow the cells in SDCAA media. Induce cells for next round as described above.
3.2. Antibody identification and characterization
Several clones are isolated and tested for binding to amyloid fibrils during the characterization stage. Antibodies are produced in a soluble format using a mammalian expression system and tested as Fc-fusion proteins in order to evaluate affinity and conformational specificity for immobilized fibrils. Antibodies isolated from a scFv library are initial tested as scFv-Fc fusions, and antibodies isolated from scFab or Fab libraries are initially tested as mAbs. Antibodies are characterized using flow cytometry to evaluate affinity and conformational specificity.
3.2.1. Antibody library sub-cloning
Extract DNA plasmids from sorted yeast libraries with yeast mini-prep kits.
PCR amplify antibody genes from a yeast mini-prep, digest and ligate them into mammalian Fc expression vectors.
Transform the ligation mixture into competent DH5α cells and plate them on LB agar plates supplemented with 100 μg/mL of ampicillin. Incubate the plate at 37 °C for 16–20 h.
Pick 10–30 colonies, grow each in ~5 mL LB media supplemented with 100 μg/mL ampicillin for 16–20 h at 37 °C at 225 RPM.
Isolate plasmid DNA from bacterial cells with mini-prep kits. Sequence the plasmids with Sanger sequencing to identify the antibody sequences.
3.2.2. Antibody expression and purification
Maintain and passage HEK293–6E cells at density of 1.8–2 million/mL in passage media. Incubator conditions should be 37 °C, 225 rpm, with 5% CO2.
Add 15 μg of plasmid to 3 mL pre-warmed un-supplemented media. Add 45 μg PEI and vortex the mixture briefly for 5–10 s. Incubate the solution at room temperature for 10–20 min and add it to 25 mL of cell culture.
Add 750 μL of yeastolate (20% w/v), 4–48 h after transfection and allow the cells to grow for another 3–5 d.
Harvest the media by centrifuging the cells at 3500 ×g for 40 min and move the supernatant to a new 50 mL tube.
Add 0.5–1 mL Protein A resin (~30 mL of supernatant) and gently rock the tube overnight at 4 °C.
Collect the resin from media by passing it through filter column under vacuum and wash the resin with 50–100 mL PBS.
Incubate the antibody with elution buffer (0.1 M glycine, pH 3.0) at room temperature for 15 min and collect the buffer by centrifugation.
Neutralize with neutralization buffer (1 M Tris, pH 8.0) or buffer exchange the antibody into the desired buffer.
Check antibody purity by SDS-PAGE and size exclusion chromatography (SEC). If % monomeric antibody is <90%, it is recommended to perform SEC to collect the monomeric protein fraction.
Filter the protein through 0.22 μm filter and measure the concentration by determining absorbance at 280 nm.
Aliquot the proteins and store them at −80 ᵒC.
3.2.3. Antibody binding analysis
Take antigen-coated beads (105 beads/sample; preparation described in Section 3.1.1) and block with blocking buffer (PBSB with 10% milk) for streptavidin beads or 10 mM glycine (pH 7.4) for tosyl activated beads at room temperature for 1 h.
Post-blocking, wash the beads once with washing buffer (PBSB).
Thaw antibody solution and centrifuge at maximum speed for 5 min on table-top centrifuge. Move the supernatant to fresh tube and measure the concentration. (See Note 9.)
Incubate antigen-coated beads with antibody (at desired dilutions) in binding buffer (PBSB with 1% milk) in a final volume of 200 μL in a 96-well plate for 2–3 h at room temperature at 350 RPM.
For antibody conformational specificity analysis, incubate antigen-coated beads, antibody (at desired concentration) and disaggregated antigen monomer (range of concentration) in binding buffer (PBSB with 1% milk) in a final volume of 200 μL in a 96-well plate for 2–3 h at room temperature at 350 RPM.
Post-incubation, centrifuge the plate at 2500×g for 5 min, wash the beads once by resuspending in 200 μL of ice-cold PBSB, and centrifuging it again.
Incubate beads with 200 μL of goat anti-human Fc Alexa Fluor 647 (final concentration of ~5 μg/mL) on ice for 4 min (protect from light).
After secondary incubation, wash the beads once more with ice-cold PBSB and analyze by flow cytometry.
For flow cytometry, first evaluate forward versus side scatter (from 488 nm laser) and gate on the singlet population (Fig. 2A). Next, for the gated population, evaluate the fluorescence histogram (Fig. 2B) and evaluate the mean fluorescence intensity of the signal for each concentration to obtain a binding curve.
Figure 2. Flow cytometry-based analysis of antibody affinity and conformational specificity.
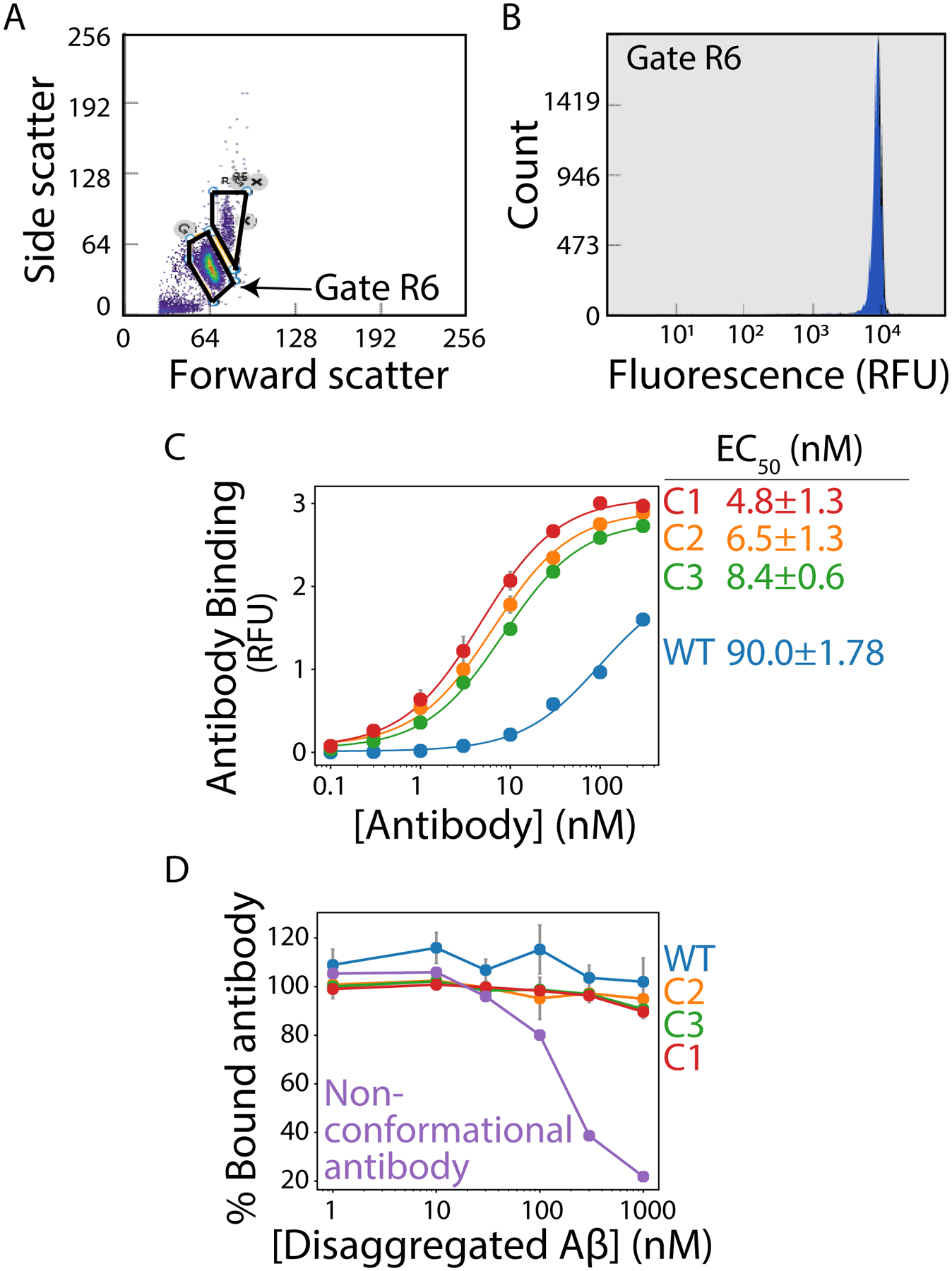
Amyloid fibrils are immobilized on micron-sized magnetic beads and binding of soluble antibodies is evaluated using flow cytometry. (A) Flow cytogram displaying forward scatter (488 nm) versus side scatter (488 nm) results, with the singlet population of fibril-coated beads highlighted in the R6 gate. (B) Histogram of the counts of binding events in the R6 gate as a function of the antibody binding signal detected via fluorescence measurements (647 nm). (C) Relative binding of antibodies to Aβ fibrils as a function of the concentration of wild-type (WT) and affinity-matured (C1, C2 and C3) antibodies. The results are mean signals from the histogram shown in (B). (D) Antibody binding to fibrils in the presence of disaggregated Aβ. Each antibody (30 nM) was pre-incubated with different concentrations of disaggregated Aβ, and then the antibodies were evaluated for their ability to recognize Aβ fibrils. A control (non-conformational antibody) displays decreased binding to immobilized fibrils as the concentration of disaggregated Aβ is increased.
3.3. Affinity maturation
3.3.1. Design of sub-libraries
The overall approach is summarized in Figure 3, which is adapted from a previous publication (8, 9, 11).
Annotate the sequence of the antibody to affinity mature with Kabat numbering. The ANARCI (antigen receptor numbering and receptor classification) program offers a command line interface for this as well as an online interface that outputs the annotation in a useful format (12). (http://opig.stats.ox.ac.uk/webapps/newsabdab/sabpred/anarci/)
First choose which CDRs to diversify. Typically, two to three CDRs are mutated depending on the length. (See Note 10.)
The amino acid frequencies of each position are taken from the abYsis database (13). The distributions are species dependent and the appropriate species (e.g., human) should be chosen for each application. (http://abysis.org/abysis/searches/distributions/distributions.cgi)
Ranking CDR sites for mutagenesis: For each residue of the CDRs, evaluate the level of variability of each position. Variability is defined as the average frequency of the most common amino acid at that position in antibody repertoires. For example, the most common amino acid at position H29 for human antibodies is Phe with an average frequency of 72%. Thus, this site is usually conserved (>50% Phe). If the frequency of the WT amino acid is > 50% or the WT amino acid is Arg, His, Lys or Cys, exclude this site from diversification. If the frequency of Tyr and Asp are at least 2% at this position, force inclusion of these amino acids in the diversification. If the frequency of Tyr or Asp are at least 2% at this position, force inclusion of the more common amino acid in the diversification. (See Note 11.) Rank the resulting positions amenable to diversification in order of increasing variability.
Selection of degenerate codons: To generate a library of 106 to 107 variants, seven to nine positions need to be mutated to five different amino acids (wild type and five mutations). For each of the nine most variable sites, choose the degenerate codons that encode the required amino acids (e.g., WT, Tyr, Asp). Remove any codons that encode for more than six amino acids. (See Note 12.) Remove any codons that encode for Arg, His, Lys or a stop codon. For the remaining codons, calculate the natural diversity coverage, which is defined as the sum of the average frequencies of the encoded amino acids for a given position in natural antibody repertories. For example, if position H52 is being mutated and Tyr is the wild type, the codon DMT would encode for Ala, Tyr, Ser, Thr, Asn and Asp and would give a natural diversity coverage of 78.5%. Choose the codon with the maximal natural diversity coverage. If there are multiple codons, use codon usage for the host organism to choose the most favorable codon.
Designing primers for generating libraries: For each mutated CDR, design a primer that encodes for the entire CDR with the degenerate codons. Include 18–30 base pairs of DNA on each end of the CDR. Use the 18–30 base pair regions before and after each CDR as primers to amplify and connect each fragment of DNA.
Figure 3. Overview of the design of antibody sub-libraries for affinity maturation.
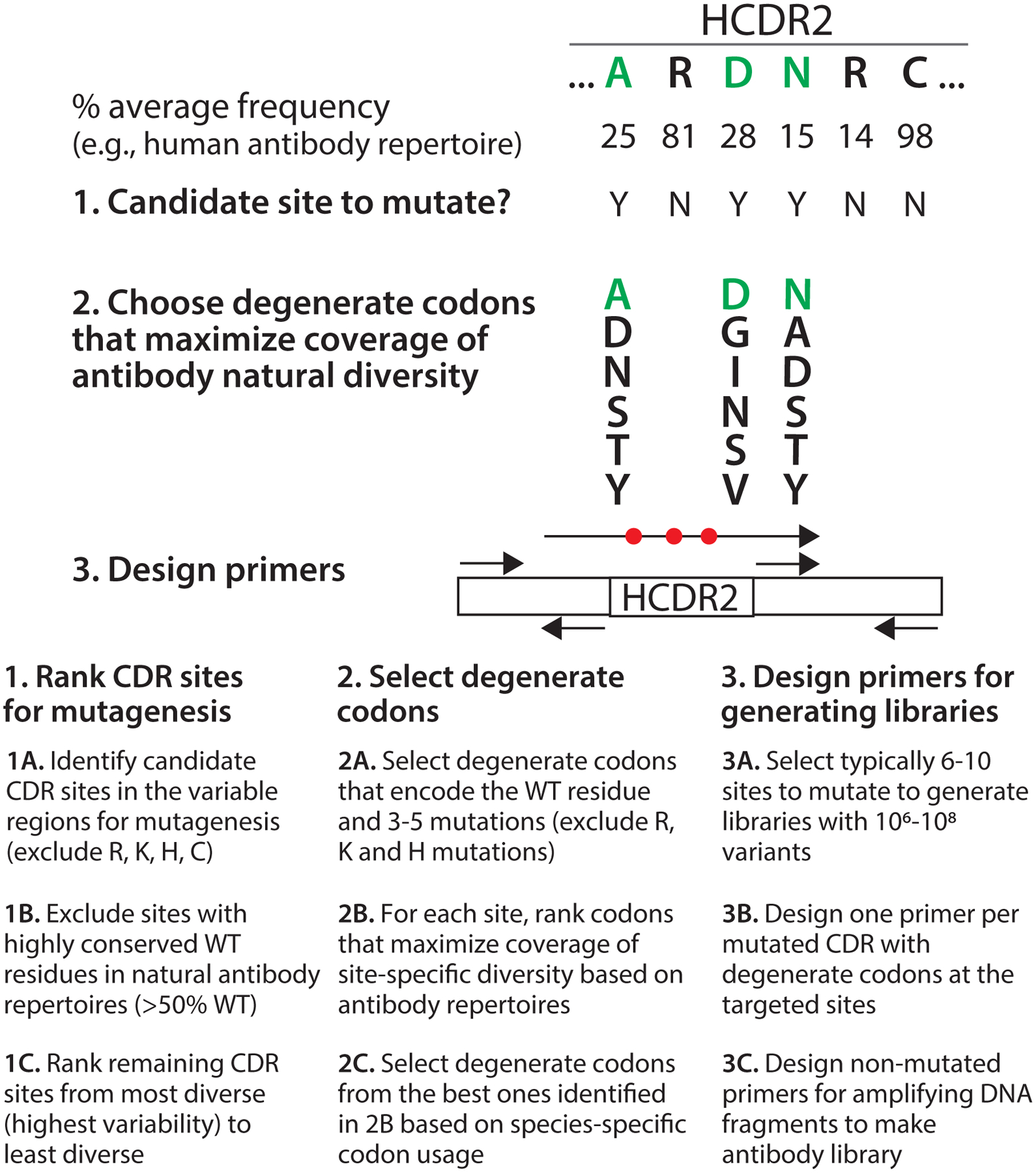
The design of sub-libraries for affinity maturation involves three major steps. First, the CDR positions to mutate are chosen based on the variability of each CDR position in natural antibody repertoires. The sites are ranked from most variable (most attractive for mutagenesis) to least variable (least attractive for mutagenesis), and highly conserved positions (>50% WT on average in natural antibody repertoires) are eliminated from consideration. Moreover, a subset of CDR sites are also excluded from consideration if their WT residues are Arg, Lys, His or Cys. Second, for the selected ~6–10 CDR sites with the highest variability, degenerate codons are chosen that encode the WT residue as well as 3–5 other amino acids based on maximizing the sum of the average site-specific frequencies of each encoded residue in natural antibody repertoires (referred to as natural diversity coverage). Degenerate codons with Arg, Lys and His are excluded. The libraries are designed to typically encode 106-108 variants. If there are multiple possible degenerate codons that encode the same set of amino acid mutations, codon selection is based on species-specific codon usage (e.g., S. cerevisiae codon usage). Third, mutagenic primers with degenerate codons and amplification primers without mutations are designed. One mutagenic primer is designed for each mutated CDR that contains the site-specific degenerate codons and 18–22 base pairs of framework DNA on both ends of the mutated CDR. Three additional primers are required for generating sub-libraries with a single mutated CDR. Typically two or three CDRs are mutated when generating sub-libraries for affinity maturation.
3.3.2. Preparing libraries
The following protocol for library transformation (beginning with Step 11) is adapted from a previous publication (14) with minor changes.
Prepare plasmid for the wild-type antibody in the yeast surface display plasmid.
Amplify regions of antibody that will not be diversified using standard PCR. Use of proof-reading enzymes like Q5 DNA polymerase or an equivalent proof-reading DNA polymerase is recommended.
Run the PCR product on a 1% agarose gel and purify the desired band by gel extraction.
Using amplified constant regions as templates, perform PCR with primers containing designed degenerate codons for each diversified CDR loop separately. The region of the primer containing degenerate codons should overhang the template DNA, such that the region complementary to the degenerate codons is not present during the PCR. A proof-reading polymerase and PCR cycles identical to that used in Step 2 may be used. (See Note 13.)
Repeat Step 4 as necessary to obtain diversity in all desired CDRs. The PCR product from a previous PCR incorporation of degenerate codons or from amplification of the constant regions may be used as the template as necessary.
Run the PCR product on a 1% agarose gel and purify the desired band by gel extraction.
Perform an overlap PCR to assemble DNA encoding all regions of the antibody as well as 30–50 base pairs of overlap with the vector at both the 5’ and 3’ ends. This overlap enables assembly of the plasmid via homologous recombination after transformation into yeast.
Run the PCR product on a 1% agarose gel and purify the desired band by gel extraction.
Double digest the backbone, incubate with calf intestine alkaline phosphatase (CIP), and purify by gel electrophoresis. (See Note 14.)
Mix 12 μg of PCR insert with 4 μg of linear backbone in a 1.5 mL tube. Ethanol precipitate the DNA mixture with pellet paint co-precipitant. Uncap the tube and wrap it in aluminum foil to dry by placing it in under vacuum or in a chemical hood.
Two days prior to transformation, start a 5 mL culture of EBY100 in YPD supplemented with 100 μg/mL ampicillin, 100 μg/mL kanamycin and 100x pen-strep. Grow cells overnight at 30 °C at 225 RPM.
The day before transformation, inoculate a 50 mL culture of EBY100 in YPD supplemented with 100 100 μg/mL ampicillin, 100 μg/mL kanamycin and 100x pen-strep from the 5 mL culture grown the previous day. Grow cells overnight at 30 °C at 225 RPM.
The day of transformation, inoculate an EBY100 culture in YPD supplemented with 100 μg/mL ampicillin, 100 μg/mL kanamycin and 100x pen-strep from the 50 mL culture grown overnight. This culture should be seeded at an OD of 0.3. The volume of this culture may be scaled up depending upon the number of libraries that will be transformed. For every library that will be transformed, prepare 50 mL of EBY100.
Grow the cells in a shaker incubator at 30 °C at 225 RPM until the OD reaches ~1.6. This step will take approximately 5–6 h.
Add 50 mL of EBY100 culture to 50 mL centrifuge tube. Centrifuge at 2500 ×g for 5 min and discard supernatant.
Wash the cells by resuspending in 25 mL of filtered cold DI water. Centrifuge at 2500 ×g for 5 min and discard supernatant.
Wash the cells by resuspending in 25 mL of cold electroporation buffer. Centrifuge at 2500 ×g for 5 min and discard supernatant.
Resuspend cells in 25 mL of lithium acetate/DTT buffer. Incubate cells in a shaker incubator at 30 °C at 225 RPM for 15 min.
Centrifuge cells at 2500 ×g for 5 min and discard supernatant.
Wash the cells by resuspending in 25 mL of cold electroporation buffer. Centrifuge at 2500 ×g for 5 min and discard supernatant.
Resuspend cell pellet in 100–200 μL cold electroporation buffer such that the total volume of the cells and buffer does not exceed 400 μL.
Transfer cells to tube containing DNA prepared by ethanol precipitation and resuspend the DNA completely in the cell solution by through mixing.
Transfer cells to a 2 mm electroporation cuvette and keep them on ice for 5 min.
Electroporate cells at the following settings: 2500 V, 200 Ω, 25 μF.
Immediately after electroporation, resuspend cells in 1 mL of 1:1 YPD:sorbitol (using 1 M sorbitol). Return cuvette to ice, and repeat electroporation process for remaining libraries.
Transfer electroporated cells to a 14 mL culture tube, and add 1:1 YPD:sorbitol to reach a total volume of 8 mL.
Allow cells to recover in a shaker incubator at 30 °C and 225 RPM for 1 h.
Centrifuge cells at 2500 ×g for 5 min.
Resuspend cells in 1 mL of SDCAA. Take a small aliquot of cells at this stage to prepare serial dilutions for plating. Plate 104 and 105 serial dilutions on dropout plates and allow to grow at 30 °C for 2 d. Library size can be determined from the number of colonies. Typical transformational efficiencies vary between ~106 and ~107 transformants per μg of linearized vector.
Transfer remaining cells in SDCAA to a 1 L culture flask containing 200 mL of SDCAA supplemented with 100 μg/mL ampicillin, 100 μg/mL kanamycin and 100x pen-strep.
Grow library for 36–48 h at 30 °C at 225 RPM.
Regrow library in 200 mL of SDCAA for 16–18 h at 30 °C and 225 RPM. Library can be stored at this stage by centrifuging at 2500 xg, resuspending cells in 30% glycerol and 0.67% yeast nitrogen base, aliquoting into cryovials, and freezing at −80 °C.
Induce library in 200 mL of SDGCAA.
3.3.3. Sorting
Prepare magnetic beads as described in Section 3.1.1. For affinity maturation, it is recommended to reduce the antigen concentration during sorting. This can be achieved in two ways. One approach is to reduce the antigen loading on beads. 5- to 20-fold reductions in the mass of antigen immobilized on beads is recommended. Reduced antigen loading is recommended in the case where streptavidin beads are used. A stock of beads at lower antigen concentration may be prepared prior to beginning affinity maturation sorting as described in Section 3.1.1 and for sorts as described in Section 3.1.2. A second approach is to reduce the number of beads. It is recommended that 107 beads be used in the first few positive selections, and in subsequent rounds, the number of beads may be decreased incrementally to as low as 1 × 106 beads. Reducing the number of beads used during sorting is recommended in cases in which enrichment for background binding (i.e., antibodies which bind to streptavidin beads or glycine-blocked tosyl beads) has been noted to be an issue during initial sorting or preliminary affinity maturation sorting. This has been observed more frequently in cases in which tosyl beads have been used. A stock of beads may be prepared for this strategy following the same protocol as described in Section 3.1.1.
Depending on the antigen type and library designs, one technique might work better than the other. It is recommended to try both approaches and see which one works the best.
Perform positive and negative selections as described in Section 3.1.3. (See Note 15.)
3.3.4. Clone evaluation of affinity and conformational specificity
The evaluation of the affinity and conformational specificity of affinity-matured clones is evaluated generally as described in Section 3.2.3. However, the multivalency of fibril antigens can mask intrinsic differences in affinity for affinity matured variants. Therefore, it is recommended to reduce the antigen concentration on the beads for affinity evaluation if required. 5- to 20-fold reductions in antigen loading may be tested in order to determine the appropriate loading condition. At an appropriate loading concentration, a sigmoidal binding curve should be obtained when a range of antibody concentrations are examined.
Prepare beads at low antigen loading, as described in Section 3.3.3.
Take desired number of beads (105 beads/sample) and block with blocking buffer (PBSB with 10% milk) for streptavidin beads or 10 mM glycine (pH 7.4) for tosyl-activated beads.
Post-blocking, wash the beads once with washing buffer (PBSB).
Thaw antibody solution and centrifuge at maximum speed for 5 min in a table-top centrifuge. Move the supernatant to a fresh tube and measure the concentration.
Incubate beads with antibody at desired concentrations. It is recommended that a broad range of antibody concentrations, such as from 0.1 to 300 nM, be tested during affinity maturation. Examined concentrations should display a range of signals, from minimal to no detectable binding at the lowest concentrations to saturated binding signal at the highest concentrations (see Fig. 2C). If a sigmoidal curve cannot be obtained with this range of antibody concentrations, it is recommended the mass of antigen on the beads be reduced.
From this point, sample preparation and flow cytometry analysis is performed as described in Section 3.2.3, beginning with Step 4.
4. NOTES
Cells can be induced in either SDGCAA at 20 °C for ~ 36–40 h or SGCAA at 30 °C for ~ 20–24 h. SGCAA contains the same composition as SDGCAA without glucose. We find that induction in SDGCAA increases the percentage of yeast cells in the library which express antibodies in some cases.
It is important to use freshly prepared milk solution for each sort. Centrifugation of the milk solution prior to use removes insoluble aggregates, which improves experimental reproducibility.
The anti-Myc antibody is used to detect antibody expression on the yeast surface. It is preferred that antibody expression be measured via detection of a peptide tag that is on the opposite terminus of the antibody from the linker (i.e., linker-antibody-tag or tag-antibody-linker). This ensures that only full-length antibodies are selected.
Neutralization should be performed only for IgGs. For scFv-Fc fusion proteins, it is recommended to buffer exchange the protein rather than neutralize immediately after elution from the Protein A beads. The isoelectric points of scFv-Fc fusion proteins are typically lower than those for IgGs, and neutralization may cause precipitation of the fusion proteins.
Filtered electroporation buffer and DI water should be prepared fresh on the day that the libraries are prepared. These solutions can be prepared immediately after starting the EBY100 culture and kept on ice until use. It is important that these buffers are cold at the time of use.
It is important lithium acetate/DTT buffer is prepared immediately before use to prevent the degradation of DTT.
It is best to perform a titration to evaluate the concentration of fibrils immobilized on magnetic beads. For initial antibody discovery, it is preferred to saturate the beads with the antigen to avoid enriching for antibodies binding to streptavidin or the magnetic bead surface.
It is important to perform multiple positive and negative selections for selection of conformational antibodies. However, the order in which they are performed is flexible and we find that both orders of operation are useful. For example, we performed three negative selections against the disaggregated monomer followed by a positive selection against fibrils in one round. This was repeated 2–5 times in succession. For initial antibody discovery, we recommend emphasizing negative selections to isolate lead clones with highest conformational specificity.
It is recommended to always centrifuge antibody solutions after thawing to remove aggregates formed during freezing and/or thawing. This improves reproducibility in antibody binding assays.
We consider HCDR1 to be a combination of the Kabat and Chothia definitions, H26 to H35B. We also define HCDR3 as H93 to H102, which includes two additional residues on the N-terminus of the CDR. All other CDRs follow the Kabat scheme.
A simple diversification strategy would be to mutate positions with WT frequency < 50%.
The maximum number of encoded amino acids, in this case six, can be altered to fit other design criteria.
In PCRs with degenerate oligos, it is recommended that the template does not contain the WT sequence complimentary to the degenerate sequence on the primer. Removing the WT sequence during this PCR step minimizes biasing of the PCR product to WT codons.
If digested backbone is prepared in advance of ethanol precipitation, it should be kept frozen to decrease the level of background WT antibody present in the library.
It is crucial to perform negative selections during affinity maturation as trade-offs in antibody properties (e.g., affinity-conformational specificity tradeoffs) are possible. The type and stringency of negative selections depend on the levels of conformational specificity of library compared to WT antibody.
ACKNOWLEDGMENTS
We thank the Tessier lab for their feedback on the manuscript. This work was supported by the National Institutes of Health (RF1AG059723, R01AG050598 and R35GM136300 to P.M.T.) and National Science Foundation (CBET 1159943, 1605266 and 1813963 to P.M.T., Graduate Research Fellowship to M.D.S.), the Albert M. Mattocks Chair (to P.M.T).
Footnotes
CONFLICTS OF INTEREST
There are no conflicts of interest.
REFERENCES
- 1.Chiti F and Dobson CM (2006) Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem 75:333–366 [DOI] [PubMed] [Google Scholar]
- 2.Forloni G, Terreni L, Bertani I, et al. (2002) Protein misfolding in Alzheimer’s and Parkinson’s disease: Genetics and molecular mechanisms. Neurobiol Aging 23:957–976 [DOI] [PubMed] [Google Scholar]
- 3.Onoue S, Ohshima K, Debari K, et al. (2004) Peptide Glucagon Generates. 21:1274–1283 [DOI] [PubMed] [Google Scholar]
- 4.Tiller KE and Tessier PM (2015) Advances in Antibody Design. Annu Rev Biomed Eng 17:191–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Munke A, Persson J, Weiffert T, et al. (2017) Phage display and kinetic selection of antibodies that specifically inhibit amyloid self-replication. 1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Julian MC, Rabia LA, Desai AA, et al. (2019) Nature-inspired design and evolution of anti-amyloid antibodies. J Biol Chem 294:8438–8451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stimple SD, Kalyoncu S, Desai AA, et al. (2019) Sensitive detection of glucagon aggregation using amyloid fibril-specific antibodies. Biotechnol Bioeng 116:1868–1877 [DOI] [PubMed] [Google Scholar]
- 8.Lou W, Stimple SD, Desai AA, et al. (2021) Directed evolution of conformation‐specific antibodies for sensitive detection of polypeptide aggregates in therapeutic drug formulations. Biotechnol Bioeng 118:797–808 [DOI] [PubMed] [Google Scholar]
- 9.Desai AA, Smith MD, Zhang Y, et al. (2021) Rational affinity maturation of anti-amyloid antibodies with high conformational and sequence specificity. J Biol Chem 296:100508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jain T, Sun T, Durand S, et al. (2017) Biophysical properties of the clinical-stage antibody landscape. Proc Natl Acad Sci U S A 114:944–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tiller KE, Chowdhury R, Li T, et al. (2017) Facile affinity maturation of antibody variable domains using natural diversity mutagenesis. Front Immunol 8:986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dunbar J and Deane CM (2015) ANARCI: antigen receptor numbering and receptor classification. 32:btv552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Swindells MB, Porter CT, Couch M, et al. (2017) abYsis: Integrated Antibody Sequence and Structure—Management, Analysis, and Prediction. J Mol Biol 429:356–364 [DOI] [PubMed] [Google Scholar]
- 14.Benatuil L, Perez JM, Belk J, et al. (2010) An improved yeast transformation method for the generation of very large human antibody libraries. Protein Eng Des Sel 23:155–159 [DOI] [PubMed] [Google Scholar]