

Abstract
We report that exposing the dipyrrin complex (EMindL)Cu(N2) to air affords rapid, quantitative uptake of O2 in either solution or the solid-state to yield (EMindL)Cu(O2). The air and thermal stability of (EMindL)Cu(O2) is unparalleled in molecular copper-dioxygen coordination chemistry, attributable to the ligand flanking groups which preclude the [Cu(O2)]1+ core from degradation. Despite the apparent stability of (EMindL)Cu(O2), dioxygen binding is reversible over multiple cycles with competitive solvent exchange, thermal cycling, and redox manipulations. Additionally, rapid, catalytic oxidation of 1,2-diphenylhydrazine to azoarene with generation of hydrogen peroxide is observed, through the intermittency of an observable (EMindL)Cu(H2O2) adduct. The design principles gleaned from this study can provide insight for the formation of new materials competent capable of reversible scavenging of O2 from air under ambient conditions with low-coordinate CuI sorbents.
Graphical Abstract
Authors are required to submit a graphic entry for the Table of Contents (TOC) that, in conjunction with the manuscript title, should give the reader a representative idea of one of the following: A key structure, reaction, equation, concept, or theorem, etc., that is discussed in the manuscript. Consult the journal’s Instructions for Authors for TOC graphic specifications.
1. INTRODUCTION
The industrial separation of high-purity dioxygen (O2) from air is of paramount importance for chemical synthesis1,2 with myriad applications in oxy-fuel combustion, medical treatments, and steel manufacturing.3 The current methodology for O2 purification involves desiccating and filtration of air, followed by fractional distillation at both cryogenic temperatures and elevated pressures to remove undesirable components, primarily dinitrogen (N2). Distillation purification of O2 is currently conducted on scales exceeding 100 Mton annually.4 The direct separation of O2 from humid, unpurified air without the requisite desiccation, particulate filtration, and intermittent cryogenic distillation would represent an advancement in O2 separations. Various N2-selective cation-exchanged zeolites5 and O2-selective metal-organic frameworks with coordinately unsaturated metal ions6–14 have demonstrated promise for O2 separation from binary O2/N2 mixtures under milder conditions; however, the water-sensitivity of these activated frameworks and their diminished capacity over multiple cycles at ambient temperatures hinders wide-scale implementation in O2 separation. Fine tuning the O2 binding site at the molecular level may yet enable new strategies to achieve selective O2 separation from air.
Nature employs selective O2 binding in metalloenzymes for O2 transport.15,16 In particular, the CuI sites in hemocyanin17 reversibly binds O2, inspiring a number of biomimetic Cu-based model complexes.18–20 In contrast to metalloenzymes, synthetic copper-dioxygen complexes suffer from poor thermal stability and poor control of nuclearity, attributable to the absence of the protein superstructure which modulates substrate access to avoid framework degradation.21 In general, the binding and activation of O2 by molecular complexes is well-precedented;22–26 yet synthetic complexes often suffer from facile O2 displacement without excess O2,27 low O2 affinity in the solid-state,28,29 ligand oxidative degradation,30 or irreversible O2 activation.31 Thus, an ideal transition metal-ligand combination would preserve low-coordination numbers for the bound transition metal, lack activatable functionalities for oxidative robustness, and facilitate O2 binding reversibly.
Recently, we described an isolable, triplet copper-nitrene supported by a dipyrrin scaffold featuring sterically encumbered peralkylated hydridacene EMind32 substituents (EMind: 1,1,7,7-tetraethyl-1,2,3,5,6,7-hexahydro-3,3,5,5-tetramethyl-s-indacene).33 The Cu resides in a hydrophobic, sterically protected pocket ideal for stabilizing traditionally reactive species. Herein, we report the preparation of an air-stable, side-bound cupric superoxide (EMindL)Cu(O2) which displays remarkable aqueous, thermal, and vacuum stability. Nonetheless, O2 displacement can be promoted using a variety of stimuli. The bound O2 can also be converted to hydrogen peroxide catalytically via arylhydrazine oxidation under ambient air. These observations of using reversible O2 binding to a low-coordinate CuI site provides new design insights for the generation of new O2 separation materials.
2. RESULTS AND DISCUSSION
2.1. Cu Oxygenation.
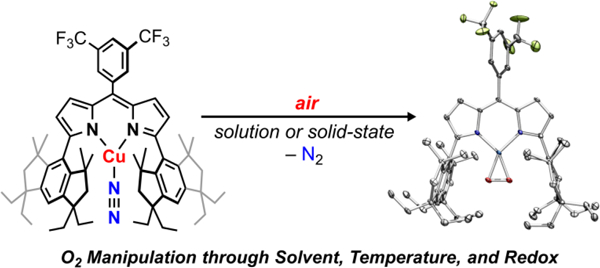
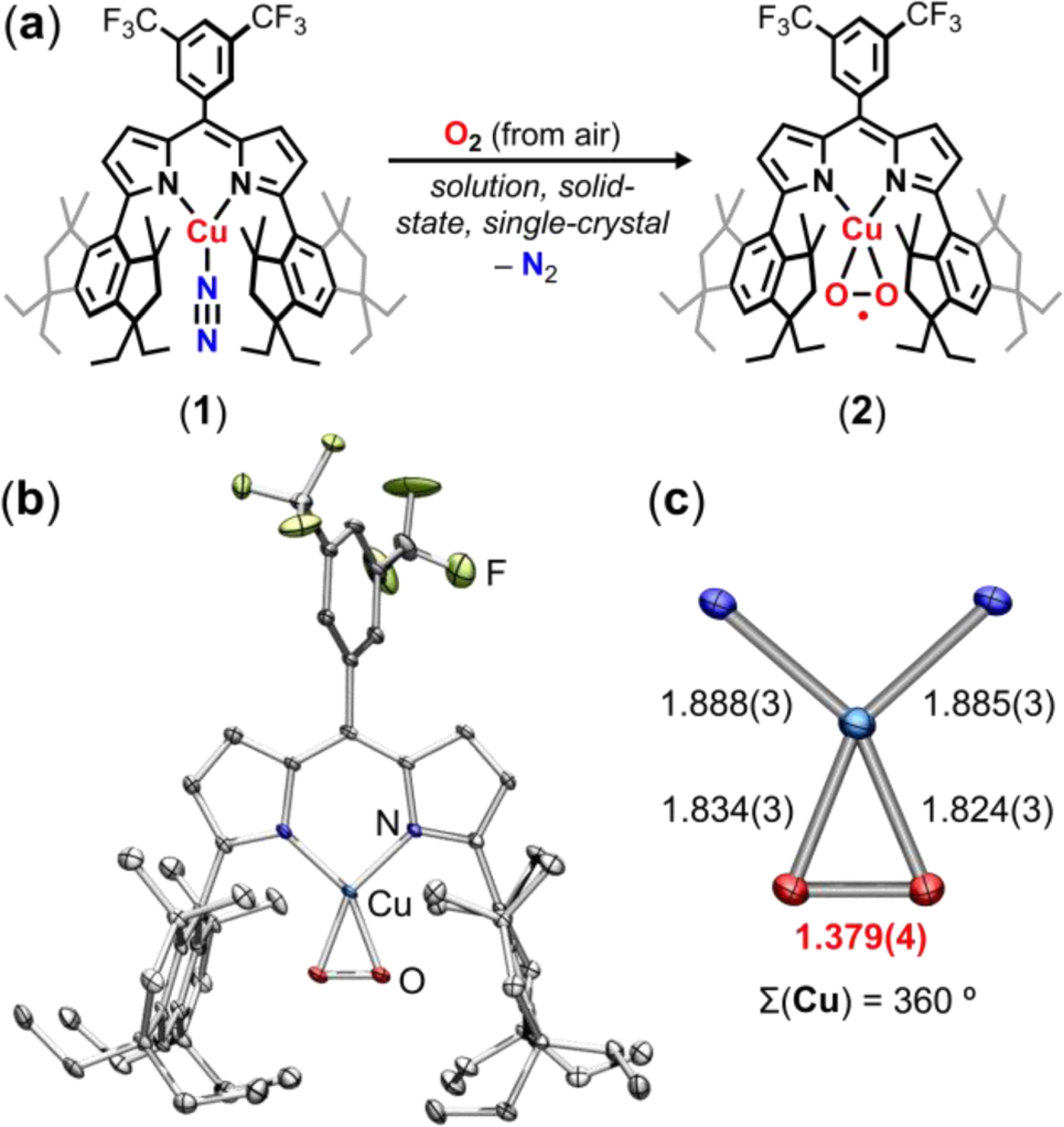
During routine manipulations of (EMindL)Cu(N2) (1),33 we observed a rapid color change from orange to red upon exposure of 1 to ambient air to yield 2 (Figure 1a, Figure S23). This color change from 1 to 2 was distinct from the yellow-orange hue attributed to free ligand (EMindL)H, arguing against ligand protonolysis from atmospheric water, which is prevalent in 3d transition metal dipyrrin complexes. In hexanes, the color change upon air exposure was accompanied by notable effervescence, consistent with loss of the coordinated dinitrogen ligand from 1. Analysis by UV/vis spectroscopy revealed a subtle red-shift in the Soret band (λ1 = 515 cm–1, ε1 = 65,000 M–1 cm–1; λ2 = 520 cm–1, ε2 = 59,000 M–1 cm–1) as well as a substantial blue-shift in a less-intense absorbance feature (λ1 = 400 cm–1, ε1 = 5,200 M–1 cm–1; λ2 = 360 cm–1, ε2 = 7,000 M–1 cm–1; Figure 2a). Multinuclear (1H/19F/13C{1H}) NMR spectroscopy revealed a single diamagnetic species (Figure 2b, see supporting information for assignments of spectra resonances), accessed in quantitative yield from 1 (Figures S1–S3). A high-resolution mass spectrum of air-exposed 1 was satisfactorily modelled as [(EMindL)Cu(N2) + 2O – 2N]+, suggesting exchange of the coordinated N2 for O2 (Figure S4). In accord, single crystal X-ray diffraction at 100 K on crystals produced from a concentrated pentane solution of 1 under air at –10 ºC overnight revealed the corresponding side-bound dioxygen-adduct (EMindL)Cu(O2) (2) (Figures 1b, 1c).
Figure 1.
(a) Air-exposure of (EMindL)Cu(N2) (1) affords thermally robust (EMindL)Cu(O2) (2) in quantitative yield in either solution of the solid state. (b) Solid state structure of 2 at 100 K depicted at 50 % displacement ellipsoid probability. (c) Pertinent bond metrics in [Cu(O2)]1+ core. Color scheme: Cu (cobalt blue), F (yellow-green), N (blue), and O (red).
Figure 2.
(a) UV/visible spectroscopy depicting shifts in absorbances from (EMindL)Cu(N2) (1) upon air exposure to yield (EMindL)Cu(O2) (2). Inset depicts shift in Soret band. (b) 1H NMR spectrum of 2 in C6D6, displaying diamagnetic resonances. Inset depicts a single resonance by 19F NMR spectroscopy. (c) Resonance Raman measurements on 2-16O2, displaying an isotoptically sensitive resonance at 1003 cm–1 (blue) with a shift to 953 cm–1 for 18O2 labelling (red) most consistent with a superoxide motif. The difference map (green) indicates changes between 2-16O2 and 2-18O2.
Interestingly, high-quality single crystals of 2 could be prepared through a single-crystal-to-single-crystal transformation by exposure of 1 to air over one week, reflecting the minimal changes in lattice parameters between 1 and 2 as well as the low volatility of the encapsulated pentane solvent molecule within the crystalline lattice (Figure S47). Analysis of the solid-state molecular structure reveals a planar geometry about the Cu ion and relatively short Cu–O bond lengths (1.824(3), 1.834(3) Å), (Figure 2C). The O–O bond length (1.379(4) Å) is comparable to that observed in our previously reported (ArL)Cu(O2) (1.383(2) Å; ArL: 5-mesityl-1,9-(2,4,6-Ph3C6H2)dipyrrin), assigned as a predominant CuII(O2•–) electronic structure based on the observed bonds lengths and multiconfigurational calculations.34 By analogy, we tentatively assign 2 as a cupric superoxide as a more appropriate descriptor than a high-valent cupryl peroxide CuIII(O22–) formulation. The relatively short Ndipyrrin–Cu bond parameters (1.888(3), 1.885(3) Å) are similarly comparable to an authentic cupric species in the same dipyrrin platform (EMindL)CuCl, (1.901(3), 1.894(3) Å).33 Resonance Raman studies on 2-16O2 and isotopically labelled (EMindL)Cu(18O2) (2-18O2) reveal an isotopically sensitive feature at 1003 cm–1 (shifted 50 cm–1 to 953 cm–1 for 2-18O2 with an expected shift of 57 cm–1 to 946 cm–1 for an ideal harmonic oscillator; see supporting information for synthesis details), in agreement with mononuclear β-diketiminate-supported (968 cm–1) and tris(pyrazolyl)hydroborate-supported (1043 cm–1) CuO2 species, both of which display an electronic structure most consistent with a predominant cupric superoxide formulation (Figure 2c).19,35
2.2. Stability Assessment.
The steric protection afforded by the hydrindacene units engenders both kinetic persistence and thermodynamic stability, preventing protolytic decomposition products and oligomerization. No noticeable decomposition was observed upon aqueous workup of 2 in aliphatic solvents. Thermogravimetric analysis (TGA) measurements on crystalline 2 illustrate no apparent decomposition below 120 ºC under active flow of Ar, pure O2, or air. Allowing crystalline 2 to stand at 100 ºC under dynamic vacuum for 36 h afforded no decomposition as evident by multinuclear NMR spectroscopy, conducted by dissolution in C6D6 under Ar following thermolysis without intermittent air exposure. Similarly, crystalline samples of 2 left under air for one year show no signs of degradation or substantial changes in crystallinity based on periodically collected single crystal X-ray diffraction data sets. Prolonged thermolysis of a solution of 2 in C6D6 (at 10–30 mM concentrations) at 60 ºC over multiple days reveals no starting material consumption by 1H/19F NMR spectroscopy. Nonetheless, thermolysis at elevated temperatures under air (80 ºC in solution, 150 ºC in the solid-state) afforded partial, albeit incomplete, decomposition of 2 to (EMindL)H over a 24 h period (Figures S41, S42), accompanied by deposition of a precipitate.
2.3. Displacement of O2.
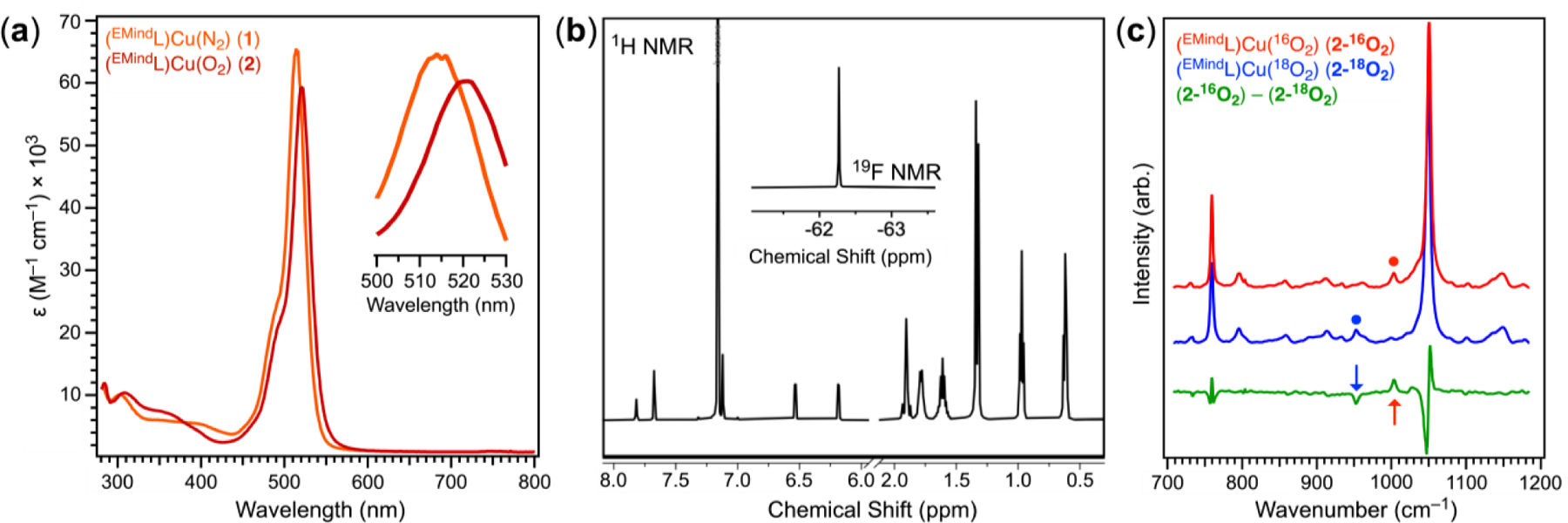
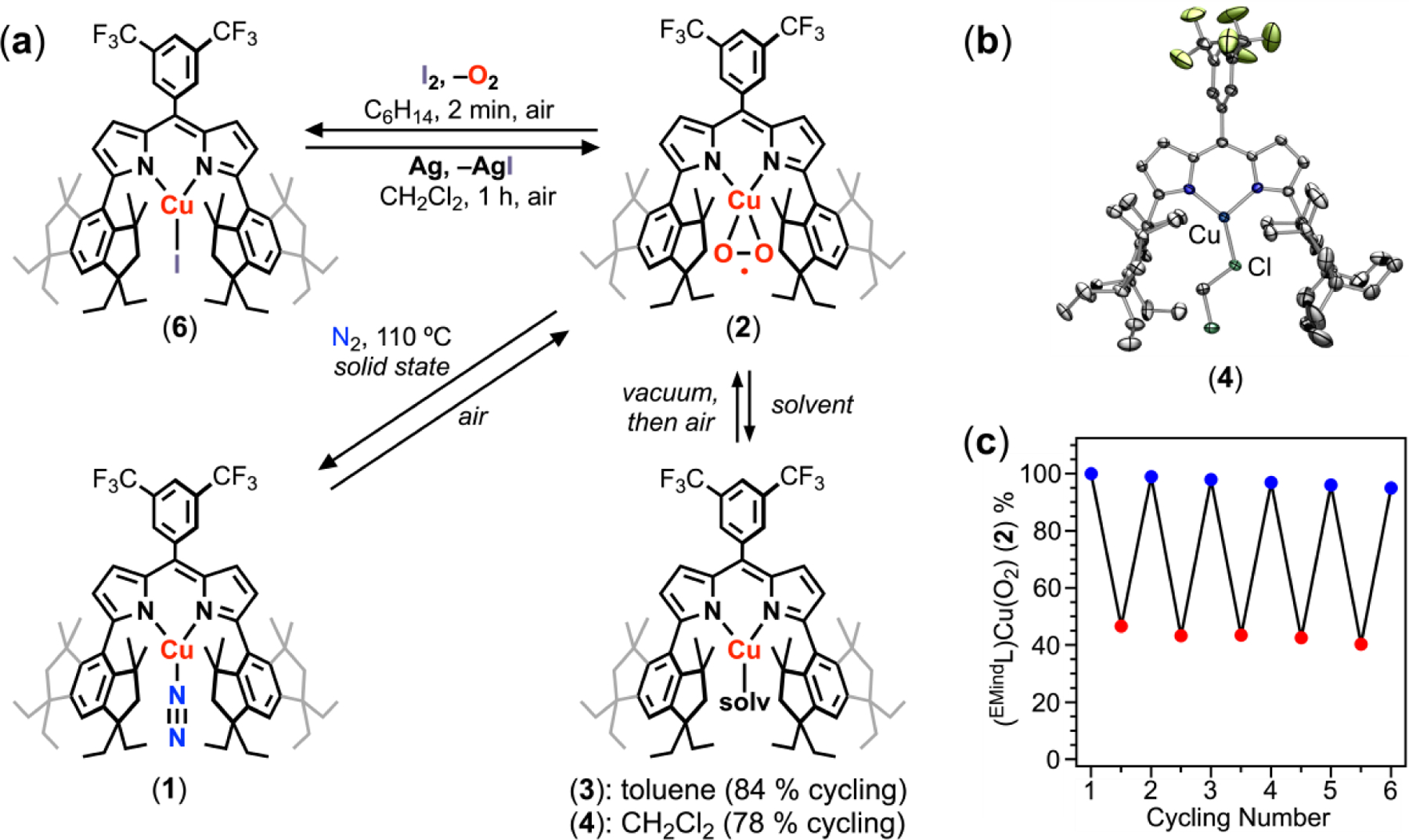
Despite the apparent stability of 2, removal of O2 could be accomplished through deliberate solvation, redox processes, or thermolysis. Addition of IMes2Pd in C6D6 to solid 2 under N2 afforded (EMindL)Cu(η2-C6H6) and IMes2Pd(O2), consistent with O2 transfer (Figure S23).36 Whereas dissolution of 2 in C6D6 under air failed to elicit O2 release, dissolution of 2 in neat d8-toluene (C7D8) afforded rapid effervescence with a color change from red to orange-red with formation of a single diamagnetic species by multinuclear NMR spectroscopy assigned as (EMindL)Cu(η2-C7H8) (3) (Figure 3a; Figures S26, S27). The same spectroscopic features were accessed upon dissolution of crystalline 1 in toluene under N2, accompanied by release of N2. Mixtures of 2 and 3 could be observed from titration of 2 with varying quantities of toluene. Although single crystals of 3 have remained elusive, crystallization of 1 under Ar in the presence of benzene affords (EMindL)Cu(η2-C6H6), featuring an η2-interaction between the Cu center and rotationally disordered arene (Figure S55).37 Interestingly, removal of solvent from 3 under vacuum at room temperature afforded 2 (84 % yield, 1H/19F NMR, C6D6) with partial remaining 3 (16 % yield, 1H/19F NMR), producing 0.84 equivalents of O2 per cycle (Figures S28, S29). These cycling experiments could be performed at least five times with no apparent degradation and consistent ratios of 2:3 upon removal of excess toluene. Similarly, dissolution of 2 in neat dichloromethane rapidly furnished a new diamagnetic species (EMindL)Cu(CH2Cl2) (4) (4, 95 %; 2, 5 % remaining) with reformation of 2 (2, 83 % yield; 4, 17 % remaining) upon evacuation and air exposure, producing 0.78 equivalents of O2 per cycle (Figure 3c; Figures S30–S33). Single crystals of the dichloromethane adduct 4 were isolated under Ar, demonstrating that solvent binding facilitates O2 displacement (Figure 3b). An extensive screen of additional solvents including cyclic alkenes, ketones, amides, sulfoxides, and sulfur-containing solvents revealed partial or irreversible O2 displacement with diminished efficacies compared to those of toluene for generating 3 and dichloromethane for generating 4 (Table S1). Strongly coordinating additives such as pyridine and acetonitrile irreversibly displaced O2 to yield air-stable CuI species.
Figure 3.
(a) Displacement of O2 from (EMindL)Cu(O2) (2) through chemical redox, thermolysis under N2, or solvent ligation. (b) solid state structure of (EMindL)Cu(CH2Cl2) (4) at 100 K depicted at 50 % displacement ellipsoid probability. Hydrogen atoms of the ligand scaffold are omitted for clarity, excluding those on the hydrazine motif. Color scheme: Cu (cobalt blue), Cl (green), F (yellow-green), N (blue). (c) Thermal gravimetric analysis (TGA), illustrating reversible coordination of O2 under active N2 flow at 110 ºC. Color scheme denotes 2 under N2 (red) and under O2 (blue).
Although d6-benzene solutions of 2 display indefinite stability in air, dissolution of 2 in C6D6 under an N2 atmosphere afforded detectable quantities (ca. 10 %) of (EMindL)Cu(η2-C6D6) through intermittency of 1, suggesting O2 displacement could be affected through N2 binding (Figures S39, S40). Monitoring the conversion between 2 and 1 by mass changes through TGA, a sample of finely divided microcrystalline 2 (ca. 10 mg, 0.010 mmol) in the solid state at 110 ºC under flowing N2 (10.0 mL/min) for 24 h showed ca. 50–60 % conversion to 1, with rapid conversion back to 2 upon O2 or air exposure (Figures 3a, 3c). Indeed, exposure of 1 to a stoichiometric quantity of O2 in air afforded 2 in quantitative yield upon mixing. Performing TGA cycling measurements between N2 and O2 atmospheres revealed partially conversion of 2 to 1 (see supporting information for experimental details) with approximately 95% 2 upon cycling completion by multinuclear NMR studies (Figures S36, S37). The incomplete conversion of 2 to 1 under N2 is attributed to kinetic limitations, consequent of poor or incomplete gas penetration in the bulk sample.
Release of coordinated O2 could be similarly affected through redox mediation. Addition of iodine to 2 induced rapid effervescence from O2 dissociation, accompanied by a color change from red to dark pink (Figure 3a). Analysis by multinuclear NMR revealed formation of a single paramagnetic species identified as (EMindL)CuI (6) by electron paramagnetic resonance for a doublet species and single crystal X-ray diffraction (Figures S17, S58). A quasi-reversible reduction was observed by cyclic voltammetry (E1/2 = –0.46 vs. Fc/Fc1+) for 6, attributed to reduction to the corresponding CuI species (Figure S19). Accordingly, treatment of 6 with excess metallic silver (3.0 equiv.) in dichloromethane under air (1 h) afforded full consumption of 6, with reformation of a 2/4 mixture in combined quantitative yield upon filtration and workup (Figure S34, S35). Exposure of this mixture to I2 rapidly re-afforded 6 in quantitative yield with effervescence from O2 dissociation, with no diminished capacity over multiple cycles.
Previously reported copper–dioxygen adducts typically exhibit thermal sensitivity and commonly require cryogenic temperature (e.g., –125 ºC,38 –95 ºC,39 –80 ºC30), excess O2, and strictly anhydrous reaction conditions to prepare. Additionally, dimeric and trimeric oligomers are observed as consequence of insufficient steric protection30 or pre-arrangement of the CuI centers in a multinucleating scaffold.40 Two notable exceptions of thermally robust copper-dioxygen adducts include [((tBu3tacn)Cu)2(μ–η2:η2–O2)][OTf]2 (tacn: 1,4,7-triazacyclononane)41 and (ArL)Cu(O2).34 The former complex, prepared by aerobic oxidation of [(tBu3tacn)Cu][OTf], exhibits gradual decay over prolonged solvation in polar solvents at room temperature (the thermal stability at elevated temperatures was not reported). The latter complex exhibits a reversible, intramolecular η2-interaction with one of the ortho-phenyl groups from the dipyrrin aryl flanking unit, necessitating excess dioxygen (1 atm) and mild cooling (–15 ºC) with dilute samples to promote full conversion to the dioxygen adduct and displace the proximal aryl ring. Furthermore, (ArL)Cu(O2) reverts to (ArL)Cu under reduced pressure, reflecting the entropically favored O2 displacement. Exposure of (ArL)Cu to ambient air afforded minor formation of (ArL)Cu(O2) (1.0:6.2 by 1H NMR for Cu(O2): Cu), accompanied by gradual decomposition over hours to unidentified paramagnetic species and free ligand, attributed to diminished water stability relative to that of 2 (Figure S24).
2.4. Catalytic O2 Transfer.
The reversible binding of O2 from 2 prompted us to explore chemical transformations in which 2 could act as a molecular O2 source. We targeted the catalytic synthesis of hydrogen peroxide from O2, noting the industrial anthraquinone auto-oxidation process suffers from side-reactions in the palladium-catalyzed hydrogenation of the 2-alkylanthraquinone species.42,43 Direct addition of stoichiometric hydrazine to 2 afforded immediate effervescence to yield the air-stable hydrazine adduct (EMindL)Cu(NH2NH2) (5) without generation of hydrogen peroxide (Figure S58). However, addition of 1,2-diphenylhydrazine in dichloromethane to 2 (1 mol%) under air at ambient conditions afforded the corresponding azoarene (94 % yield) after 2 h (Figure 4; Figure S48–S51). The azoarene product could be selectively extracted with ethanol to remove dipyrrin-containing species and converted back quantitively into 1,2-diphenylhydrazine through previously reported chemical reduction protocols.45 Analysis of the reaction mixture by 1H NMR spectroscopy reveals a diagnostic singlet resonance at δ 9.13 ppm attributable to hydrogen peroxide which could be similarly observed in stock solutions of aqueous H2O2 in C6D6 (Figure S53). Interestingly, repeating the analogous reaction in hexanes afforded 62 % azoarene over 2 h with observation of (EMindL)H and full consumption of 2 by 1H NMR spectroscopy, suggesting the partial equilibrium between 2 and 4 in CH2Cl2 may prevent ligand hydrolysis from the acidic hydrogen peroxide. Consistent with this hypothesis, (EMindL)H was rapidly observed upon exposure of 2 to silica in neat hexanes, whereas we observed no free ligand (EMindL)H upon flash column chromatography (silica) in neat dichloromethane of 4. For comparison, the autooxidation of 1,2-diphenylhydrazine to azoarene and hydrogen peroxide under an O2 atmosphere in the absence of 2 required ca. 48 h to observe the corresponding azoarene (92% yield). By contrast, trace formation (13% yield) of azoarene was formed under ambient air after 2 h in the absence of 2 due to background auto-oxidation (Figure S50). Similarly low yields of the diazene product were obtained with free ligand (EMindL)H and related CuI species (e.g., CuCl, (ArL)Cu, (tBuL)2Cu2),44 suggesting the sequestration and stabilization of Cu within the (EMindL) ligand scaffold is necessary for the reaction.
Figure 4.
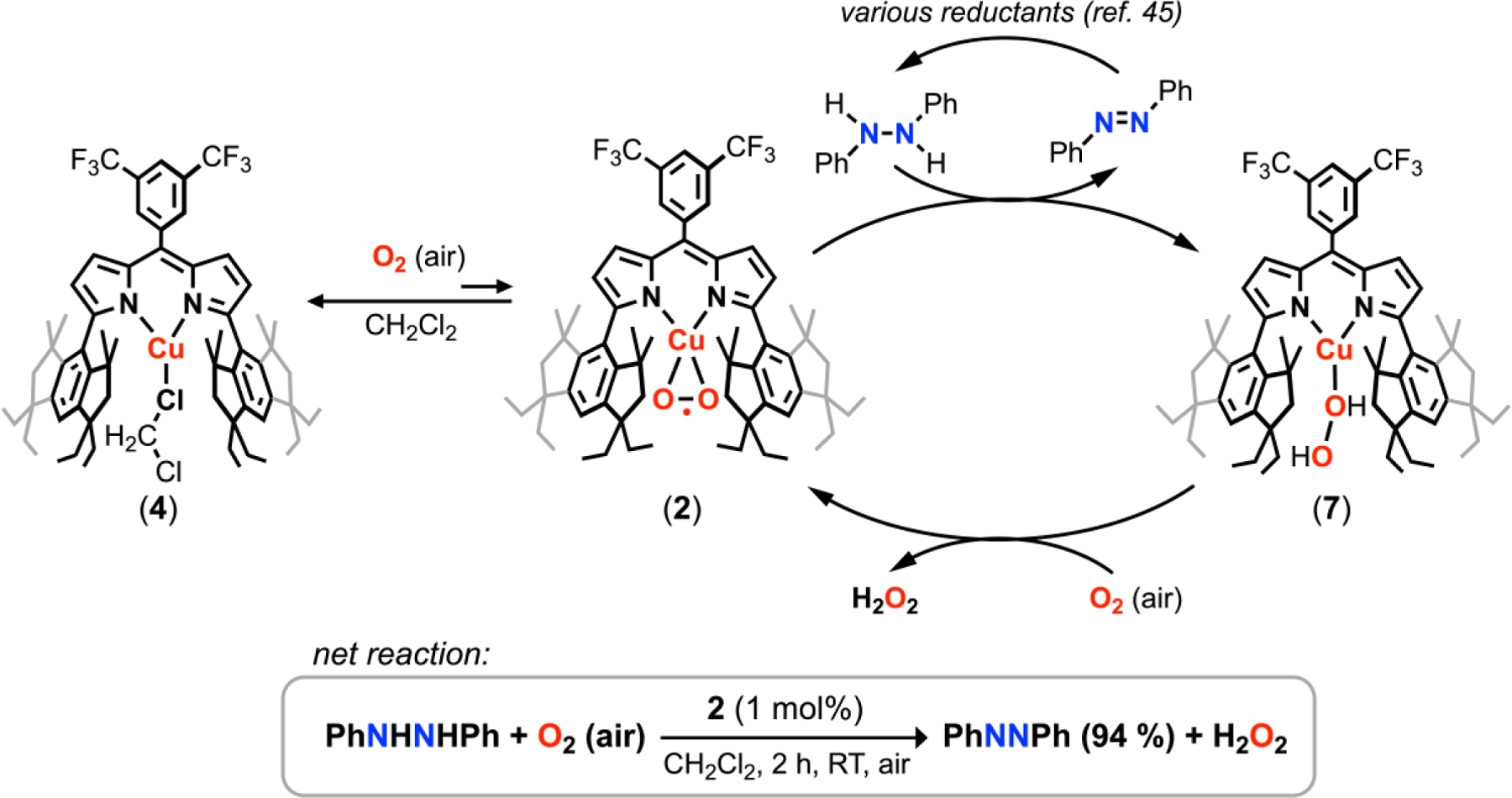
Proposed catalytic cycle for hydrogen peroxide formation through 1,2-diphenylhydrazine oxidation, accompanied by azoarene formation. The off-cycle formation of (EMindL)Cu(CH2Cl2) (4) is proposed to augment stability of the catalyst toward H2O2.
Whereas treatment of 2 under air with stoichiometric 1,2-diphenylhydrazine afforded a combination of azoarene, H2O2 and 2, repeating the analogous experiment under N2 or Ar afforded azoarene and a new diamagnetic Cu-containing species. Control experiments of 1 with 1,2-diphenylhydrazine and azoarene afforded no changes in 1H NMR resonances. To rule out H2O generation and coordination following H2O2 disproportionation, addition of H2O to 1 was assessed, affording a separate, air-sensitive diamagnetic species consistent with an aquo complex. Based on these observations, we assign the Cu-containing species resulting from hydrazine oxidation under inert atmosphere as the air-sensitive peroxide adduct (EMindL)Cu(H2O2) (7). Accordingly, treatment of 1 with anhydrous hydrogen peroxide in the form of (Ph3PO)2(H2O2)46 afforded the same multinuclear NMR resonances, corroborating our assignment of the previous product as 7 (Figure S22). We note treatment of 1 with triphenylphosphine oxide elicited no changes by 1H NMR spectroscopy. Although isolable adducts of hydrogen peroxide are known,47–50 prior reports feature hydrogen bond acceptors in the secondary coordination sphere to overcome the poor σ-donating properties of H2O2.50 Density functional theory computations reveal coordination of H2O2 is accompanied by no apparent perturbation in H2O2 bond metrics, indicative of minimal activation (Figure S54). By contrast, the highly electrophilic Cu center and hydrindancene flanking units promote coordination and thermal stability of the ligated hydrogen peroxide despite the lack of hydrogen bond acceptors in 7.
3. CONCLUSION.
The foregoing data demonstrates a biomimetic approach to molecular oxygen scavenging using a dipyrrin-supported copper complex. The side-bound, superoxide adduct is remarkably robust and can form from ambient air via electron transfer from CuI. Release of O2 could be achieved through solvent displacement, molecular redox processes, and thermolysis under N2. Rapid oxidation of arylhydrazines with 2 was observed under ambient conditions, generating the corresponding azoarene and hydrogen peroxide. These results suggest the use of low-coordinate, electrophilic CuI frameworks may provide promising platforms for O2 purification from air. The low oxophilicity of CuI makes it ideal for O2 capture and release, occurring via facile electron transfer to O2 to form superoxide or peroxide adducts that may be subsequently displaced. Further evidence for this proposal was recently reported that a CuI metal-organic framework reversibly changes color upon air exposure14 with isotherm measurements indicating a strong Cu–O2 heat of adsorption. Detailed spectroscopic and computational studies of 2 as well as related dipyrrin-supported O2 adducts are underway.
Supplementary Material
ACKNOWLEDGMENT
T.A.B. gratefully acknowledges support by grants from NIH (GM-115815), NSF (CHE-1954690), the Dreyfus Foundation in the form of a Teacher-Scholar Award, and Harvard University. K.M.C. acknowledges the Fannie & John Hertz Foundation and the National Science Foundation Graduate Research Fellowship for financial support of this research. A.I. acknowledges Harvard College Research Program and the Harvard University Center for Environment Summer Undergraduate Research Fund for financial support of this research. Collection of Raman spectra were obtained at the Center for Nanoscale Systems (CNS, Harvard University), a member of the National Nanotechnology Coordinated Infrastructure Network (NNCI), which is supported by the National Science Foundation under NSF award no. 1541959.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The data supporting the finding of this study are available in this article and the Supplementary Information. The crystallographic datasets for the structures reported in this study have been deposited at the Cambridge Crystallographic Data Centre, under deposition numbers CCDC 2049910 (1-benzene), 2049906 (2), 2049909 (4), 2049908 (5), and 2049907 (6). Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/. All other data supporting the findings of this study and detailed experimental procedures and characterization of compounds are available in the Supplementary Information files, and in the depository.
The authors declare no competing financial interest.
REFERENCES
- (1).Smith AR; Klosek J, A Review of Air Separation Technologies and Their Integration with Energy Conversion Processes. Fuel Proc. Technol 2001, 70, 115. [Google Scholar]
- (2).Kirschner MJ; Alekseev A; Dowy S; Grahl M; Jansson L; Keil P; Lauermann G; Meilinger M; Schmehl W; Weckler H, Oxygen. Ullmann's Encyclopedia of Industrial Chemistry 2000, 1.
- (3).Buhre BJ; Elliott LK; Sheng C; Gupta RP; Wall TF, Oxy-Fuel Combustion Technology for Coal-Fired Power Generation. Prog. Energy Combust. Sci 2005, 31, 283. [Google Scholar]
- (4).Greenwood NN; Earnshaw A Oxygen, In Chemistry of the Elements; Elsevier, 2012. [Google Scholar]
- (5).Nandi SP; Walker PL, Separation of Oxygen and Nitrogen Using 5A Zeolite and Carbon Molecular Sieves. Sep. Sci 1976, 11, 441. [Google Scholar]
- (6).Jaffe A; Ziebel ME; Halat DM; Biggins N; Murphy RA; Chakarawet K; Reimer JA; Long JR, Selective, High-Temperature O2 Adsorption in Chemically Reduced, Redox-Active Iron-Pyrazolate Metal–Organic Frameworks. J. Am. Chem. Soc 2020, 142, 14627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Xiao DJ; Gonzalez MI; Darago LE; Vogiatzis KD; Haldoupis E; Gagliardi L; Long JR, Selective, Tunable O2 Binding in Cobalt(II)–Triazolate/Pyrazolate Metal–Organic Frameworks. J. Am. Chem. Soc 2016, 138, 7161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Murray LJ; Dinca M; Yano J; Chavan S; Bordiga S; Brown CM; Long JR, Highly-Selective and Reversible O2 Binding in Cr3(1,3,5-Benzenetricarboxylate)2. J. Am. Chem. Soc 2010, 132, 7856. [DOI] [PubMed] [Google Scholar]
- (9).Bloch ED; Murray LJ; Queen WL; Chavan S; Maximoff SN; Bigi JP; Krishna R; Peterson VK; Grandjean F; Long GJ; Smit B; Bordiga S; Brown CM; Long JR, Selective Binding of O2 over N2 in a Redox–Active Metal–Organic Framework with Open Iron(II) Coordination Sites. J. Am. Chem. Soc 2011, 133, 14814. [DOI] [PubMed] [Google Scholar]
- (10).Reed DA; Xiao DJ; Jiang HZH; Chakarawet K; Oktawiec J; Long JR, Biomimetic O2 Adsorption in an Iron Metal–Organic Framework for Air Separation. Chem. Sci 2020, 11, 1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Bloch ED; Queen WL; Hudson MR; Mason JA; Xiao DJ; Murray LJ; Flacau R; Brown CM; Long JR, Hydrogen Storage and Selective, Reversible O2 Adsorption in a Metal–Organic Framework with Open Chromium(II) Sites. Angew. Chem. Int. Ed 2016, 55, 8605. [DOI] [PubMed] [Google Scholar]
- (12).Rosen AS; Mian MR; Islamoglu T; Chen H; Farha OK; Notestein JM; Snurr RQ, Tuning the Redox Activity of Metal–Organic Frameworks for Enhanced, Selective O2 Binding: Design Rules and Ambient Temperature O2 Chemisorption in a Cobalt–Triazolate Framework. J. Am. Chem. Soc 2020, 142, 4317. [DOI] [PubMed] [Google Scholar]
- (13).Oktawiec J; Jiang HZ; Vitillo JG; Reed DA; Darago LE; Trump BA; Bernales V; Li H; Colwell KA; Furukawa H, Negative Cooperativity Upon Hydrogen Bond-Stabilized O2 Adsorption in a Redox-Active Metal–Organic Framework. Nat. Commun 2020, 11, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Denysenko D; Grzywa M; Jelic J; Reuter K; Volkmer D, Scorpionate-Type Coordination in MFU-4l Metal–Organic Frameworks: Small-Molecule Binding and Activation Upon the Thermally Activated Formation of Open Metal Sites. Angew. Chem. Int. Ed 2014, 53, 5832. [DOI] [PubMed] [Google Scholar]
- (15).Solomon EI; Heppner DE; Johnston EM; Ginsbach JW; Cirera J; Qayyum M; Kieber-Emmons MT; Kjaergaard CH; Hadt RG; Tian L, Copper Active Sites in Biology. Chem. Rev 2014, 114, 3659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Sono M; Roach MP; Coulter ED; Dawson JH, Heme-Containing Oxygenases. Chem. Rev 1996, 96, 2841. [DOI] [PubMed] [Google Scholar]
- (17).Van Holde KE; Miller KI In Advances in Protein Chemistry; Elsevier: 1995; Vol. 47, p 1. [DOI] [PubMed] [Google Scholar]
- (18).Cramer CJ; Tolman WB, Mononuclear Cu–O2 Complexes: Geometries, Spectroscopic Properties, Electronic Structures, and Reactivity. Acc. Chem. Res 2007, 40, 601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Elwell CE; Gagnon NL; Neisen BD; Dhar D; Spaeth AD; Yee GM; Tolman WB, Copper–Oxygen Complexes Revisited: Structures, Spectroscopy, and Reactivity. Chem. Rev 2017, 117, 2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Trammell R; Rajabimoghadam K; Garcia-Bosch I, Copper-Promoted Functionalization of Organic Molecules: from Biologically Relevant Cu/O2 Model Systems to Organometallic Transformations. Chem. Rev 2019, 119, 2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Shook RL; Borovik AS, Role of the Secondary Coordination Sphere in Metal-Mediated Dioxygen Activation. Inorg. Chem 2010, 49, 3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Li GQ; Govind R, Separation of Oxygen from Air Using Coordination Complexes: A Review. Ind. Eng. Chem. Res 1994, 33, 755. [Google Scholar]
- (23).Southon PD; Price DJ; Nielsen PK; McKenzie CJ; Kepert CJ, Reversible and Selective O2 Chemisorption in a Porous Metal–Organic Host Material. J. Am. Chem. Soc 2011, 133, 10885. [DOI] [PubMed] [Google Scholar]
- (24).Niederhoffer EC; Timmons JH; Martell AE, Thermodynamics of Oxygen Binding in Natural and Synthetic Dioxygen Complexes. Chem. Rev 1984, 84, 137. [Google Scholar]
- (25).Sundberg J; Cameron LJ; Southon PD; Kepert CJ; McKenzie CJ, Oxygen Chemisorption/Desorption in a Reversible Single-Crystal-to-Single-Crystal Transformation. Chem. Sci 2014, 5, 4017. [Google Scholar]
- (26).Henrici-Olivé G; Olivé S, Activation of Molecular Oxygen. Angew. Chem. Int. Ed 1974, 13, 29. [Google Scholar]
- (27).Karlin KD; Cruse RW; Gultneh Y; Farooq A; Hayes JC; Zubieta J, Dioxygen-Copper Reactivity. Reversible Binding of O2 and Co to a Phenoxo-Bridged Dicopper(I) Complex. J. Am. Chem. Soc 1987, 109, 2668. [Google Scholar]
- (28).Vaska L, Oxygen-Carrying Properties of a Simple Synthetic System. Science 1963, 140, 809. [DOI] [PubMed] [Google Scholar]
- (29).Holt SL; DeIasi R; Post B, Crystal Structure of the Oxygen-Inactive Form of Bis(Salicylaldehyde) ethylenediiminecobalt(II). Inorg. Chem 1971, 10, 1498. [Google Scholar]
- (30).Cole AP; Root DE; Mukherjee P; Solomon EI; Stack TDP, A Trinuclear Intermediate in the Copper-Mediated Reduction of O2: Four Electrons from Three Coppers. Science 1996, 273, 1848. [DOI] [PubMed] [Google Scholar]
- (31).Hong S; Lee Y-M; Ray K; Nam W, Dioxygen Activation Chemistry by Synthetic Mononuclear Nonheme Iron, Copper and Chromium Complexes. Coord. Chem. Rev 2017, 334, 25. [Google Scholar]
- (32).Matsuo T; Suzuki K; Fukawa T; Li B; Ito M; Shoji Y; Otani T; Li L; Kobayashi M; Hachiya M; Tahara Y; Hashizume D; Fukunaga T; Fukazawa A; Li Y; Tsuji H; Tamao K, Synthesis and Structures of a Series of Bulky “Rind-Br” Based on a Rigid Fused-Ring S-Hydrindacene Skeleton. Bull. Chem. Soc. Jpn 2011, 84, 1178. [Google Scholar]
- (33).Carsch KM; DiMucci IM; Iovan DA; Li A; Zheng S-L; Titus CJ; Lee SJ; Irwin KD; Nordlund D; Lancaster KM; Betley TA, Syntheses of a Copper-Supported Triplet Nitrene Complex Pertinent to Copper-Catalyzed Amination. Science 2019, 365, 1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Iovan DA; Wrobel AT; McClelland AA; Scharf AB; Edouard GA; Betley TA, Reactivity of a Stable Copper-Dioxygen Complex. Chem. Commun 2017, 53, 10306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Tomson NC; Williams KD; Dai X; Sproules S; DeBeer S; Warren TH; Wieghardt K, Re-Evaluating the Cu K Pre-Edge Xas Transition in Complexes with Covalent Metal–Ligand Interactions. Chem. Sci 2015, 6, 2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Konnick MM; Guzei IA; Stahl SS, Characterization of Peroxo and Hydroperoxo Intermediates in the Aerobic Oxidation of N-Heterocyclic-Carbene-Coordinated Palladium(0). J. Am. Chem. Soc 2004, 126, 10212. [DOI] [PubMed] [Google Scholar]
- (37). Although the synthesis of 1 is conducted in benzene, the species isolation from workup and crystallization is identified as (EMindL)Cu(N2) (1). Nonetheless, dissolution of 1 in neat benzene yields (EMindL)Cu(η2-C6H6) (1-C6H6), which reverts to 1 upon removal of solvent and exposure to N2, as asertained by infrared spectroscopy and combustion analysis. for clarity, we refer to 1 either as the crystalline solid or as solutions of (EMindL)Cu(N2) in aliphatic solvents.
- (38).Citek C; Gary JB; Wasinger EC; Stack TDP, Chemical Plausibility of Cu(III) with Biological Ligation in pMMO. J. Am. Chem. Soc 2015, 137, 6991. [DOI] [PubMed] [Google Scholar]
- (39).Gary JB; Citek C; Brown TA; Zare RN; Wasinger EC; Stack TDP, Direct Copper(III) Formation from O2 and Copper(I) with Histamine Ligation. J. Am. Chem. Soc 2016, 138, 9986. [DOI] [PubMed] [Google Scholar]
- (40).Lionetti D; Day MW; Agapie T, Metal-Templated Ligand Architectures for Trinuclear Chemistry: Tricopper Complexes and Their O2 Reactivity. Chem. Sci 2013, 4, 785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Karahalis GJ; Thangavel A; Chica B; Bacsa J; Dyer RB; Scarborough CC, Synthesis and Catalytic Reactivity of a Dicopper(II) μ-η2:η2-Peroxo Species Supported by 1,4,7-Tri-Tert-Butyl-1,4,7-Triazacyclononane. Inorg. Chem 2016, 55, 1102. [DOI] [PubMed] [Google Scholar]
- (42).Campos-Martin JM; Blanco-Brieva G; Fierro JL, Hydrogen Peroxide Synthesis: An Outlook Beyond the Anthraquinone Process. Angew. Chem. Int. Ed 2006, 45, 6962. [DOI] [PubMed] [Google Scholar]
- (43).Goor G; Glenneberg J; Jacobi S, Hydrogen Peroxide. Ullmann's Encyclopedia of Industrial Chemistry, 2000, 1.
- (44).Carsch KM; Lukens JT; DiMucci IM; Iovan DA; Zheng S-L; Lancaster KM; Betley TA, Electronic Structures and Reactivity Profiles of Aryl Nitrenoid-Bridged Dicopper Complexes. J. Am. Chem. Soc 2020, 142, 2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Shi X, Diazene, 1,2-Diphenyl-. In Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, Ltd, 2012. [Google Scholar]
- (46).Kadassery KJ; Dey SK; Cannella AF; Surendhran R; Lacy DC, Photochemical Water-Splitting with Organomanganese Complexes. Inorg. Chem 2017, 56, 9954. [DOI] [PubMed] [Google Scholar]
- (47).Wallen CM; Bacsa J; Scarborough CC, Hydrogen Peroxide Complex of Zinc. J. Am. Chem. Soc 2015, 137, 14606. [DOI] [PubMed] [Google Scholar]
- (48).Wallen CM; Palatinus L; Bacsa J; Scarborough CC, Hydrogen Peroxide Coordination to Cobalt(II) Facilitated by Second-Sphere Hydrogen Bonding. Angew. Chem. Int. Ed 2016, 55, 11902. [DOI] [PubMed] [Google Scholar]
- (49).Wallen CM; Bacsa J; Scarborough CC, Coordination of Hydrogen Peroxide with Late-Transition-Metal Sulfonamido Complexes. Inorg. Chem 2018, 57, 4841. [DOI] [PubMed] [Google Scholar]
- (50).DiPasquale AG; Mayer JM, Hydrogen Peroxide: A Poor Ligand to Gallium Tetraphenylporphyrin. J. Am. Chem. Soc 2008, 130, 1812. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.