

Abstract
Conversion plateaus rapidly in radical photopolymerizations (RPPs) following discontinuation of irradiation due to rapid termination of reactive radicals, which restricts the wider use of RPPs in applications that involve nonuniform light access including those with attenuated light transmission or irregular surfaces. Based on our recent report of a radical dark-curing photoinitiator (DCPI) that continues polymerization beyond the cessation of irradiation by enabling latent redox initiation with photo-released amine in the presence of a suitable oxidant, we developed a new DCPI with an absorption spectrum that extends well into the visible range. Our design process involved a series of computational investigations of candidate molecules, including a systematic study of substituents and their position-dependent effects on absorption characteristics, electronic transitions, and the photochemical mechanism and its associated energetics. Our quantum chemical computations identified the target compound 5,7-dimethoxy-6-bromo-3-aroylcoumarin-DMPT/BPh4 and predicted that it would facilitate the dark-curing mechanism by concurrent photo-radical generation and photo-induced release of an efficient redox reductant under visible irradiation. This reductant-tethered chromophore was then synthesized and optically characterized with UV–vis spectroscopy that revealed its strong visible-light absorption with a molar absorptivity of 5710 M−1 cm−1 at 405 nm and 50 M−1 cm−1 at 455 nm. We then demonstrated extensive dark-curing of >35% additional conversion over 25 min following brief activation of the shelf-stable one-part system by irradiation with a 455 nm LED that was ceased at 20% conversion. In contrast, shuttering irradiation of the control formulation at that same point resulted in immediate cessation of conversion, which plateaued at 20%. We determined a remarkable initiator efficiency of 2.82 that results from the additional redox-generated radicals with a 77% photo-reductant generation quantum yield. The combination of superior photo- and dark-curing efficiencies of this new visible DCPI is expected to open new application opportunities in RPP, especially those involving resins that are highly light attenuating, surfaces that possess irregular features that produce uneven irradiance, and production lines where continued dark-curing downstream of the light activation step enhances line efficiencies.
Graphical Abstract

INTRODUCTION
Photopolymerization (PP) is one of the most environmentally friendly methods to produce materials due to its ambient temperature processing, the absence of volatile organic compounds, low energy consumption, and increased productivity with a well-controlled onset and rate of polymerization dependent on the formulation and photocuring conditions.1 This has driven the development of a photopolymer market currently estimated to be $4.8 billion and projected to grow at a rate of 9.4% through 2025 to $8.3 billion.2 Over 90% of all PP is radical based, which uniquely exhibits nearly immediate termination of polymerization upon discontinuation of irradiation due to its exceptionally reactive radical intermediates that lead to efficient biradical termination. Such termination behavior increases the likelihood of producing highly heterogeneous or undercured regions, even with careful tailoring of the PP process to its specific application. Therefore, solving this issue would broaden PP for a multitude of polymeric material applications that involve dimensionally thick (>100 μm) or otherwise light-attenuating systems, including highly filled or pigmented materials, and surface coatings on curved or irregular substrates.3,4
In response to such limitations, we recently reported radical-mediated UV PP that exhibits extensive post-polymerization after cessation of irradiation.5 This demonstration was enabled by the innovative design of a dark-curing photoinitiator (DCPI) that incorporated photo-reductant generation concurrent with a direct photo-radical generation process during irradiation. Photolysis released radicals that initiated PP in a similar manner to conventional photoinitiators while also photo-releasing amines that function as reductants in reactions with a formulated peroxide oxidant.5 Such a redox reaction leads to additional initiating radicals via the amine–peroxide redox mechanism, which effectively extends polymerization well beyond the irradiation interval.5–7 A demonstration of this DCPI included effectively achieving a vitrification-limited final conversion of 80%, although PP irradiation was discontinued at only 20% conversion. Furthermore, the mechanical performances of a dark-cured polymer, including its storage modulus and shrinkage stress, were equal to or exceeded those of the continuously photocured polymer.
This prior DCPI with a benzophenone chromophore (BP-DMPT/BPh4) absorbs light with wavelengths shorter than 390 nm and hence is activated only by UV, which has a few disadvantages in comparison to visible-light activation. In particular, shorter-wavelength UV photons are significantly more attenuated as a function of depth, which are detrimental to many industrial and biomedical applications that involve thick, pigmented, or filled resins. Although our UV-based DCPI can chemically mitigate such depth-of-curing issues by raising the conversion at the deepest depth reached by photons, the greater light attenuation of UV light physically limits the curing depth in comparison to longer wavelength visible light. Additionally, highly energetic UV photons present significant occupational safety and health issues (particularly to vision) along with the higher cost of UV LEDs.7 With the potential of DCPIs being particularly suitable for difficult-to-cure resins or applications that present irregular light access, an analogous photoinitiating system with visible-light activation would be exceptionally useful in enabling or improving existing and future high-value PP applications. For example, such a visible-light DCPI may allow autonomous post-cure in photo-activated 3D printing, as the dark-curing mechanism continues curing of the previously irradiated areas in 3D-printed structures under ambient conditions without additional light or heat input. Most 3D-printed parts require time-consuming and costly post-processing of “green parts”, such as by thermal and/or light treatment, as these green parts are only cured to varying stages beyond the gel point that holds its shape but not sufficiently to ultimately possess viable mechanical properties. As such, the development of a visible-light-activated DCPI would enable many applications of light-attenuated resins with greater productivity, safety, and energy efficiency.
Over the past 5 years, three examples of visible-light-initiated radical dark-curing have been demonstrated, but each with practical limitations. Our group reported a methylene blue-based initiating system of bulk acrylate resins that exhibited delayed polymerization following irradiation and produced heavily colored products.8 The Boyer group also reported a dye-based initiating system based on rose bengal and eosin Y that photoinitiated polymerization of a solution of acrylamide in water and also resulted in similar problems as our methylene blue system.9 Last, the Lalevée group reported a Cu(I)/iodonium salt/tin(II)-based photoinitiating system that produced ROOH in situ.10 Despite the impressive capability exhibited by this ternary initiating system, its incorporation of metallic components introduces application limitations.
Herein, we report the first demonstration of visible-light activation of solvent-free radical dark-curing that does not rely on a reaction exotherm or photo-induced thermal assistance. The design of this new visible-light-absorbing DCPI was enabled by an extensive computational quantum chemical study that identified compounds with the desired photoinitiator properties before synthesis. We chose 3-aroylcoumarin (ARC) as the chromophore scaffold for candidate DCPIs because of its potential to accommodate synthetic diversity as well as the dark-curing mechanism. Our work is distinctively different from previous studies that examined coumarin species as type II photoinitiators,11–13 although these photoinitiators were also activated by low-intensity visible light. Our novel photoinitiator system enables continued radical production that extends significantly beyond the irradiation interval. A systematic study was conducted of its substituted derivatives in which we evaluated their computationally predicted absorption characteristics and identified 5,7-dimethoxy-6-bromo-3-aroyl-coumarin as a target compound. We synthesized this chromophore and conducted a mechanistic study to confirm that its energetics allow concurrent photo-reductant and photo-radical generation as a visible-light DCPI. This new DCPI was demonstrated to be activated by 365, 405, and 455 nm LEDs, with absorption up to 470 nm, which resulted in rapid visible-light photocuring during irradiation and substantial dark-curing following irradiation when the initiator package included a peroxide oxidant to enable redox radical production pathways. The new development we report here provides a valuable tool to polymer scientists that enables the use visible light to synthesize industrial and biomedical materials that commonly involve light-attenuating resins or applications comprising irregular surfaces or otherwise poorly controlled access to the activating light source.
RESULTS AND DISCUSSION
Dark-Curing Mechanism.
This study aims to enable visible-light absorption that facilitates the reaction cascade shown in Figure 1. The aryl groups allow tuning of absorption characteristics while the methylene ammonium groups release a tertiary amine upon photoexcitation. The borate salt is involved in both photo-radical and photo-reductant generation. The photo-released amine reacts with formulated peroxide to amplify radical production during photoactivation but more critically serves as a source of latent radical formation after irradiation is ceased. Our previous UV-absorbing DCPI is based on a benzophenone chromophore and undergoes a complex mechanism composed of three homolysis (HM), three electron-transfer (ET; two outer spheres and one inner sphere), and one proton-transfer (PT) reactions, whereby direct photolysis and a latent redox reaction produce initiating radicals over twoime scales (Figure 1).5 Here, we seek to exploit our understanding of the DCPI mechanism to enable an analogous set of reactions with a new DCPI that is efficiently activated by visible light. As with all photoinitiation, the dark-curing mechanism also begins with photoexcitation by irradiation. Following photoexcitation, the chromophore, which is covalently bound to a photo-releasable amine, is reduced by ET from the tetraphenyl borate (BPh4) anion (ET1). The resultant oxidized borate radical homolyzes into triphenyl borane and a phenyl radical that efficiently initiates monomers (HM1), while the reduced chromophore–ammonium radical undergoes HM on its own into the chromophore anion and the amine radical cation (HM2). The homolyzed chromophore anion donates an electron back to the amine radical cation (ET2), producing chromophore radical and amine with the restored lone pair. The chromophore radical adds to a monomer with efficiency similar to a styrenic radical. Such dissociative ETs to generate initiating radicals are an effective strategy here14 because visible-light photons typically are not sufficiently energetic to directly cleave C–C bonds to create carbon-centered radicals.15
Figure 1.

Light- and dark-curing mechanism. The proposed DCPI undergoes a complex mechanism composed of three HM, three ET, and one PT reactions, whereby direct photolysis and latent redox reaction produce four initiating radicals over two time scales.
The dark-curing mechanism proceeds with the photo-released amine and then attacking of formulated benzoyl peroxide (BPO). It should be noted that by design, the photo-protected amine species in the form of chromophore–ammonium cation does not contain any reactive lone pair, which consequently prevents ground-state interactions with BPO, resulting in a stable one-component formulation only limited by the thermal degradation potential of BPO, whereby its thermolysis exceeds the inhibitor capacity in the formulation (Figure S30).5 Attack of BPO by the photo-released amine results in ammonium species and the benzoate anion, where the amine is singly oxidized and the benzoate anion is singly reduced via inner-sphere ET (ET3).6,7 The ammonium species homolyzes into another amine radical cation and benzoyloxy initiating radical (HM3). When the benzoate anion abstracts a proton from the amine radical cation, an initiating aminoalkyl radical is generated along with benzoic acid. Essentially, a single photon can generate two radicals via photolysis during irradiation and two more radicals via redox reaction, which occurs either during irradiation or post-irradiation. Therefore, a design of a new DCPI that enables this complex dark-curing mechanism subsequent to visible-light (>400 nm) absorption involves the development of a visible-light-absorbing photobase generator that releases a tertiary aromatic amine, which is a weak base but an effective reductant, in addition to the initiating radicals formed directly by photoexcitation. In this approach, there is no potential for undesired reactions with any component of the formulation including peroxide prior to irradiation (Figure S30). We designed such a photoinitiator that fulfills all the requirements by conducting an extensive quantum chemical study prior to undertaking the initiator synthesis.
Chromophore Scaffold.
We first considered various aryl ketone chromophores to enable an analogous photobase/radical generation, as the notable feature of benzophenone-based DCPI is diaryl ketone.32 None of these molecules have their maximum absorption (λmax) within the visible-light range. For example, the λmax of benzophenone (BP), xanthone, thioxanthone, and anthraquinone occur at 335, 340, 360, and 326 nm, respectively. Furthermore, modification with various functional groups does not necessarily red shift λmax. BP derivatives with several functional groups, including dianhydride, biphenyl, dihydroxy, and (di)alkyl, essentially exhibit a similar λmax (~335 nm), although such functionalization is often accompanied by higher molar absorptivity (ε) relative to the unfunctionalized BP. On the other hand, some functionalization was shown to achieve an ~30 nm red shift (λmax = ~370 nm) as in Michler’s ketone that is a BP derivative with para-bisdimethyl-amino functionality. Last, a few of these chromophores with λmax located in the UV are currently used in industry as visible photoinitiators (e.g., thioxanthones) due to their low but extended absorption in the visible-light range. As such, our targeted chromophore need not have λmax in the visible-light range to act as a visible photoinitiator and judicious selection of functional groups could lead to desirable absorption characteristics (vide infra). Besides, the photobase generation mechanism requires that the desired chromophore contains a para-tolyl group to furnish a tertiary amine via benzylic bromination. Based on such restrictions, we decided to pursue ARCs as our chromophore scaffold among many possible aryl ketone chromophore candidates due to the synthetic accessibility of the coumarin moiety for tuning absorption characteristics and of the tolyl group for accommodating a tertiary amine (Figure 2).16
Figure 2.

ARC structural moiety and its potential synthetic route that facilitates facile functionalization to tune absorption. The design components of visible-light-absorbing DCPIs include diaryl ketone that mediates photochemistry, the coumarin moiety with functionalization opportunities that modify light absorption characteristics, and the methylene phenyl group that attaches to a tertiary amine as ammonium salt.
Benzophenone versus ARC.
We then computed the photoabsorption of ARC and its derivatives to identify promising new visible-light-absorbing chromophores. Primarily, time-dependent density functional theory (TD-DFT) was used at the M06–2X/6–311++g(d,p)/SMD-acetonitrile level of theory with geometries optimized with DFT at the M06–2X/6–31+g(d,p)/SMD-acetonitrile level of theory. TD-DFT predicted the photophysical properties of λmax, its corresponding molar absorptivity (εmax), and changes in the electron occupation of active orbitals upon photoexcitation of each new chromophore structure. To understand how ARC functionalization affects these photophysical properties and to narrow the set of potential target compounds, we truncated the size of the studied molecules to only consider the paratolyl-functionalization, excluding ammonium functional groups to reap the reduction in computational cost.
In addition to the absorption of visible light (≥400 nm), we sought ideal molar absorptivity of the candidate visible-light-absorbing DCPI to be ≥1000 M−1 cm−1 at 405 nm for a balance of mild coloration and fast reaction for implementation in thin-film applications such as coatings, inks, adhesives, and photolithography,17 as well as ≤ 100 M−1 cm−1 at 450 nm to reduce optical density and increase light transmission for increased depth of cure for thick-film and bulk applications.18 For instance, in the case of dental composite applications, ~50 M−1 cm−1 at 470 nm would be highly desirable to replace the camphorquinone/amine system that remains the state-of-the-art for commercial dental resin composite applications due to its appropriate molar absorptivity level for deep curing.19,20 The difference in the desirable molar absorptivity criteria for the given application results from the intrinsic competitive effects where higher ε provides an increased rate of initiation and faster rates of polymerization, yet lower ε results in higher light transmission through the material due to lower photon absorption, which reduces filtering of the active wavelengths.18 Last, rapid photo-bleaching is also desired in order to further facilitate increased depth of cure.18
The benzophenone chromophore utilized for our UV-activated DCPI is computed to have λmax at 329 and 250 nm, which closely agree with the observed values of 335 and 248 nm (Table 1).21,22 Additionally, the εmax values predicted by our quantum chemical calculations are 19,029 M−1 cm−1 for 250 nm absorption and 69 M−1 cm−1 for 329 nm absorption, also in close agreement with the experimentally observed values of 19,400 and 121 M−1 cm−1, respectively (Figure S1).21 The close agreement validates our computational method for reliably computing absorption characteristics of various chromophores in search of a chromophore that enables a visible DCPI. The unfunctionalized ARC possesses a computed λmax,1 at 331 nm with εmax,1 of 733 M−1 cm−1 and λmax,2 at 295 nm with εmax,2 of 13361 M−1 cm−1 (Table 1). The εmax,1 of ARC was greater by a factor of ~10 with a similar λmax,1 to BP, while the λmax,2 of ARC was red-shifted to approximately 300 nm, whereas the λmax,2 of BP lies at 250 nm, indicating that ARC is a more strongly absorbing chromophore scaffold than benzophenone. In addition to λmax and its associated εmax, UV–vis spectra were simulated using Gaussian profiles that take collision and proximity broadening into account.23,24 As a result of its relatively high εmax,1 of 733 M−1 cm−1 at 331 nm, the unfunctionalized ARC had some visible range absorption with an ε400nm of 47 M−1 cm−1 (Figure 3a). On the other hand, the simulated absorption spectrum of benzophenone is such that the absorptivity of 47 M−1 cm−1 is found only at 350 nm while its ε400nm is barely 3 M−1 cm−1. Although BP and ARC have a similar λmax,1, the significant difference in their εmax,1 makes ARC a more suitable scaffold for a visible DCPI (733 vs 69 M−1 cm−1). Ultimately, the unfunctionalized ARC does not fulfill the photophysical property requirements with a λmax,1 of ~1000 M−1 cm−1 or higher at 405 nm and thus is predicted to not be a high-performing visible-light chromophore for our purposes. Therefore, we proceeded by instead investigating the absorption of various substituted ARCs.
Table 1.
Computed Photophysical Properties of Benzophenone and ARC Derivatives Including Their Orbital Energies and Dipole Momentsa
| λmax,1 (nm) | εmax,1 (M−1 cm−1) | λmax,2 (nm) | εmax,2 (M−1 cm−1) | n orbital (eV) | π orbital (eV) | π* orbital (eV) | dipole (D) | |
|---|---|---|---|---|---|---|---|---|
| BP | 329 | 69 | 250 | 19,030 | −9.19 | −8.26 | −1.07 | 4.94 |
| ARC | 331 | 733 | 295 | 21,362 | −9.36 | −8.07 | −1.59 | 6.38 |
| 5-MeO | 332 | 2729 | 312 | 17,289 | −9.33 | −7.79 | −1.59 | 8.40 |
| 6-MeO | 331 | 2020 | 321 | 6482 | −9.36 | −7.57 | −1.59 | 7.49 |
| 7-MeO | 331 | 4741 | 311 | 30,002 | −9.29 | −7.61 | −1.50 | 8.60 |
| 8-MeO | 331 | 632 | 307 | 1777 | −9.35 | −7.76 | −1.56 | 7.72 |
| 5-CI | 332 | 741 | 295 | 21,208 | −9.42 | −8.24 | −1.77 | 5.92 |
| 6-CI | 331 | 688 | 300 | 15,677 | −9.41 | −8.07 | −1.70 | 4.56 |
| 7-CI | 331 | 1008 | 297 | 27,755 | −9.38 | −8.08 | −1.68 | 5.33 |
| 8-CI | 331 | 563 | 292 | 18,333 | −9.41 | −8.23 | −1.70 | 6.89 |
| 5,7-diOMe | 338 | 18,422 | 322 | 13,361 | −9.18 | −7.36 | −1.43 | 10.99 |
| 5,7-diOMe-6-Br | 332 | 1506 | 303 | 21,718 | −9.38 | −7.95 | −1.62 | 8.26 |
TD-DFT calculations were conducted on truncated chromophores without ammonium groups at the M06–2X/6–311++g(d,p)/SMD-acetonitrile level of theory with geometries computed with the M06–2X/6–31+g(d,p)/SMD-acetonitrile level of theory.
Figure 3.

(a) Simulated UV–vis spectra of benzophenone (BP) and ARC. Despite their λmax,1 being similar at ~330 nm, the εmax,1 of ARC is much greater than that of BP (733 vs 69 M−1 cm−1), making ARC a superior scaffold for visible-light-absorbing DCPIs. (b) Simulated UV–vis spectra of ARC and its methoxy-substituted derivatives. Methoxy substitutions at the C5 and C7 positions increased molar absorptivity more efficiently than those at the C6 and C8 positions. The substitution positions are described in Figure 2. Relevant photophysical constants were computed by TD-DFT.
Functionalization of ARC.
With the predicted weak absorption in the visible-light range for the unfunctionalized ARC, we computationally screened various functional groups to enhance visible-light absorption. Synthetic access of functionalization at the C5, C6, C7, and C8 positions of ARC is easily achieved with many commercially available salicylaldehyde precursors (Figure 2). We first examined how the nature of functionalization and the location of such functionalization affected the absorption characteristics. ARC was subjected to single substitution with methoxy and chlorine groups that respectively represent an electron-donating and an electron-withdrawing group (EDG and EWG), which were chosen based on the commercially available salicylic aldehydes. It should be noted that amino-functional groups as an EDG were excluded due to their potential ground-state interactions with BPO in the absence of light via a spontaneous amine–peroxide redox mechanism while the nitro group as an EWG was also excluded because of its potential to act as a radical polymerization inhibitor.25,26
Our selected EDG and EWG single substitution at all positions retain the same λmax,1 at 331 nm as the unsubstituted ARC. However, other absorption characteristics such as εmax,1, λmax,2, and εmax,2 all improved to cause further red-shifting (Table 1). Compared to the εmax,1 of 733 M−1 cm−1 of ARC, functionalization with an electron-donating methoxy group at the C5, C6, and C7 positions increased εmax,1 to 2729, 2020, and 4741 M−1 cm−1, respectively. In contrast, the methoxy group substitution at the C8 position decreased εmax,1 to 632 M−1 cm−1. Furthermore, these substitutions significantly change λmax,2 and εmax,2. While the unfunctionalized ARC has a λmax,2 of 295 nm with a εmax,2 of 21,362 M−1 cm−1, methoxy substitution at the C5 and C7 positions both increased λmax,2 to 311 nm with a εmax,2 of 17,289 and 30,002 M−1 cm−1, respectively. Although λmax,2 is still 20 nm shorter than λmax,1, such second absorptions with exceptionally large molar absorptivities may contribute to visible-light absorption. The methoxy substitution at the C6 position increased λmax,2 to an even longer wavelength of 321 nm, but with a greatly reduced absorptivity of 6482 M−1 cm−1. On the other hand, the methoxy substitution at the C8 position performed poorly with a λmax,2 of 307 nm and εmax,2 of a mere 1777 M−1 cm−1.
The methoxy substituents are predicted to affect the light-absorbing characteristics through their effect on orbital energies and dipole strengths. The λmax,1 remained unchanged because the corresponding electronic transition is mostly n → π* in nature, where both the n and π* orbitals are not significantly affected by the methoxy substitution. The unsubstituted ARC possesses n and π* orbitals at −9.36 and −1.59 eV, respectively. The resultant energy changes in these orbitals from methoxy substitution are less than 0.1 eV for all positions, and most are less than 0.05 eV (~1 kcal/mol) due to the orthogonally oriented n lone pair orbital that prevents overlap with the lone pairs of the methoxy groups (Figure 4). In contrast, λmax,2 was bathochromically shifted from 295 nm to ~310 nm, where the corresponding electronic excitation is a π → π* transition. Unlike the unaffected n and π* orbitals, the energy of the π orbital increased by 0.28 to 0.50 eV (6.5 to 11.5 kcal/mol). This is because electron donation increases electron density in the coumarin moiety and raises its π orbital energy. The three relevant orbitals of chromophores that result from chlorine substitutions are less affected than those from the methoxy substitutions. The energetic change in the n orbitals is less than 0.1 eV, similar to those produced by methoxy substitutions, while energies of the π and π* orbitals decreased slightly and to a comparable degree so that its calculated effect on λmax,2 is marginal.
Figure 4.

Molecular orbitals of ARC and 7-methoxy-3-aroylcoumarin (ARC-7-MeO). λmax,1 remained unchanged because the participating orbitals (n and π*) are only marginally affected by the methoxy substituent, while λmax,2 was bathochromically shifted from 295 to ~310 nm due to the shift of the π orbital to higher energies caused by the methoxy substituents.
We attribute the change of molar absorptivity predicted by our computational study to the push–pull effect that mediates intramolecular charge transfer. Absorption of electromagnetic (EM) radiation by organic molecules occurs along particular directions within the molecular framework that correspond to the transition dipole moments, where each electronic transition involves an oscillating electric dipole that couples the two states to each other. Increases in the dipole moments caused by a substituent increase the interaction of the electric field of the photon with the electron cloud of the molecule. Therefore, molecules with higher dipole moments possess higher molar absorptivities.27–30 Based on these principles and literature reports on coumarin as a chromophore for dye-based solar cell applications,31–34 we anticipated that ARC would also absorb EM by polarization along its long molecular axis to excite the S0 → S1 transition that consists of both n → π* (HOMO – 4 → LUMO) and π → π* (HOMO → LUMO) characteristics (Figures 4 and 5). When ARC is substituted with either an EWG or an EDG, the substitution changes the dipole moment along the long axis. The methoxy EDG substituent increases the polarizability by establishing a push–pull effect jointly with the lactone and ketone moieties, which function as EWGs. In contrast, the chlorine EWG decreases the polarizability relative to the unsubstituted ARC. To confirm this push–pull hypothesis, we computed the dipole moments—a proxy for polarizability—for variously substituted ARC to show that indeed the dipole moments are correlated with higher molar absorptivities in agreement with the findings by Sutradhar et al.35 The highly absorbing ARC-5-OMe and ARC-7-OMe possess dipole moments of 8.40 and 8.60 D, while the dipole moments of all chlorine-substituted ARCs are less than 7 D. Substitutions that increase the polarizability parallel to the transition dipole moment corresponding to the S0 → S1 transition intensified absorptivity to a greater degree by increasing the effective absorption cross section. This is why methoxy substitutions at the C5 and C7 positions modulate the absorption characteristic most effectively, while the methoxy substitution at the C6 position has a less dramatic effect due to resonance interference that hinders the push–pull effect (Figure S2, resonance structures).
Figure 5.

Illustration of the push–pull effect in ARC used to modulate light absorption in this study. Polarizability along the long molecular axis enhances the desired light absorption and increasing the push–pull characteristic of ARC increases the dipole moment, resultant polarizability, and thus the intensity of light absorption.
When these predictions are incorporated into the UV–vis spectra simulated with peak broadening effects, ARC with methoxy substitutions at the C5 and C7 positions led to predicted ε405nm of 185 and 286 M−1 cm−1, higher than 172 M−1 cm−1 for C6, 29 M−1 cm−1 for C8, and 33 M−1 cm−1 for the unsubstituted ARC (Figure 3b). This suggests that substitutions with EDGs at the C5 and C7 positions improve light absorption and that the double methoxy substitution at C5 and C7 may yield even larger red shifts and stronger absorption in the visible-light range. In contrast, Cl substitutions at all positions causes unfavorable orbital energy changes that blue-shifts λmax,1. It also failed to increase the molar absorptivity as EWGs because Cl does not produce a push–pull effect with the lactone or ketone groups. As a result, the methoxy-substituted ARC was predicted to significantly outperform the chlorine-substituted ARC.
Using the insights gained from the single-substitution study, we proposed that 5,7-dimethoxy-3-aroylcoumarin (ARC-5,7-diOMe) would be an optimal chromophore derivative of ARC to access the visible-light range with appropriate absorption characteristics. ARC-5,7-diOMe is predicted to exhibit a λmax at 338 nm with a εmax,1 of 18,422 M−1 cm−1 and a εmax,2 at 322 nm of 13361 M−1 cm−1 (Table 1). The combination of large εmax,1, εmax,2 and the red shift of λmax,2 to lie near λmax,1 produced a strong visible-light absorption of 2371 M−1 cm−1 at 400 nm according to the simulated UV–vis spectra (Figure 6). In particular, 5,7-dimethoxy-3-aroylcoumarin was computed to effectively absorb the common wavelength output by commercial LEDs,36 exhibiting absorptivities of 1731 M−1 cm−1 at 405 nm, 635 M−1 cm−1 at 420 nm, 48 M−1 cm−1 at 455 nm, and 15 M−1 cm−1 at 470 nm (Figure 6). Such photophysical properties of ARC-5,7-diOMe exceed our desired molar absorptivity target of ≥1000 M−1 cm−1 at 405 nm and ≤100 M−1 cm−1 at 450 nm.
Figure 6.
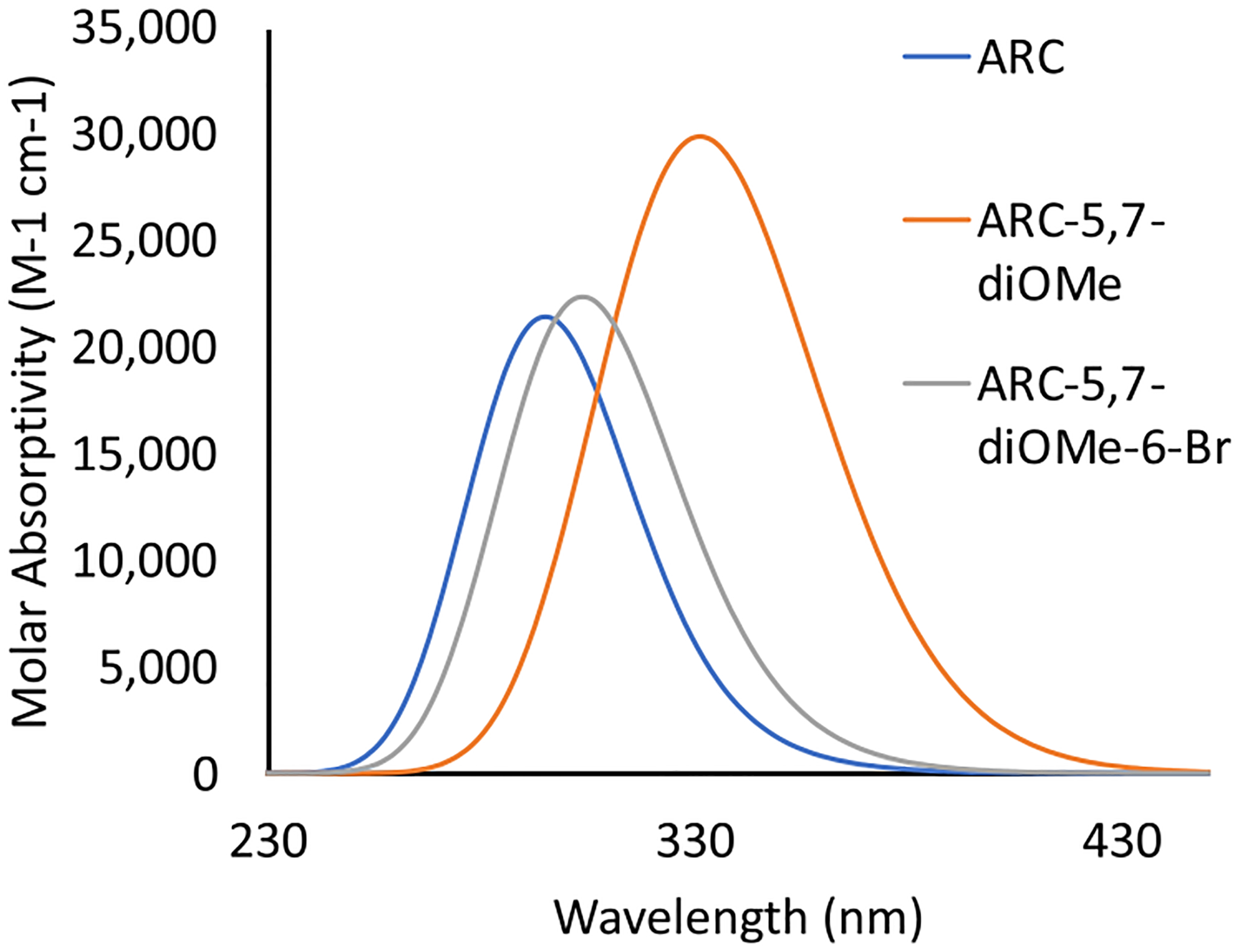
Simulated UV–vis of ARC, ARC-5,7-diOMe, and ARC-5,7-diOMe-6-Br. ARC-diOMe and ARC-diOMe-Br exhibit better visible-light absorption than unsubstituted ARC. When para-tolyl bromination of ARC-diOMe was attempted, bromination at the sixth position between two methoxy substituents continuously occurred; therefore, ARC-diOMe-Br was instead targeted as a potential chromophore for visible DCPI after computationally confirming its viability as a visible-light DCPI.
5,7-Dimethoxy-6-bromo-3-aroylcoumarin.
A deeper analysis of the synthesis of 5,7-dimethoxy-3-aroylcoumarin suggested that the dimethoxy substitutions at the C5 and C7 positions result in a considerable increase in the electron density at the C6 position from −0.242 e in ARC to −0.675 in ARC-diOMe. Such density changes may lead to electrophilic aromatic bromination at the C6 position, producing 5,7-dimethoxy-6-bromo-3-aroylcoumarin (5,7-diOMe-6-Br) when attempting to brominate the benzylic methyl group (Figure 2).37,38 Our speculation was confirmed by NMR studies that we indeed synthesized the brominated chromophore 5,7-diOMe-6-Br with a benzylic brominated site (SI2, synthesis of visible photobase). Therefore, we changed our target compound and evaluated 5,7-diOMe-6-Br as the new prospective chromophore to confirm its potential as a visible DCPI. The presence of a bromine group affected both max absorption and its absorptivity. Br is considerably larger than H, which led to a slight rotation of both methoxy groups. Therefore, the favorable electron donation to the π orbital was reduced but not eliminated (Figure S22), which decreased λmax,2 from 322 nm in ARC-5,7-diOMe to 303 nm in ARC-5,7-diOMe-6-Br. Additionally, the higher electronegativity of Br relative to H (2.8 vs 2.1) reduced the push–pull characteristic slightly, which led to a lower dipole moment of 10.99 versus 8.26 D (Figure S22). However, we ultimately found that the computationally predicted absorption characteristics of ARC-5,7-diOMe-6-Br led to sufficient visible-light absorption (Figure 6) for the DCPI. Therefore, we continued the computational study to confirm that the energetics of the photochemical reaction are feasible. For the calculation of photochemistry energetics, the entire chromophore–ammonium structures were modeled including the photo-releasable amine [dimethyl-p-toluidine (DMPT)] without tetraphenyl borate at the M06–2X/6–31+g(d,p)/SMD-ethyl acetate level of theory. Free energies were computed within the bulk reaction approximation used in our previous publications,39,40 where the contributions from the entropies of the solvent-hindered molecular motions including rotation and translation were excluded to reflect the highly viscous nature of the photocuring resin phase.
We next examined several aspects of the photochemical mechanism of ARC-5,7-diOMe-6-Br-DMPT. First, we established that a visible-light photon could promote the C–N bond scission. The computed C–N bond dissociation energies (BDE) of BP, ARC, ARC-5,6-diOMe, and ARC-5,6-diOMe-6-Br were nearly identical (52.4, 52.5, 53.1, and 53.2 kcal/mol, respectively). These BDEs indicate that photons with wavelengths up to ~540 nm possess sufficient energy to dissociate the C–N bond if no energy is dissipated into other modes. Given that the simulated absorption of ARC-5,7-diOMe-6-Br has ample absorptivity below 500 nm, such a low BDE allows nearly the entire absorption spectrum of ARC-5,7-diOMe-Br to be useful. Second, we confirmed that an unintended ET reaction between the ground-state chromophore–ammonium and tetraphenyl borate does not occur. We calculated ground-state redox potentials of BP-DMPT+, ARC-DMPT+, ARC-5,7-diOMe-DMPT+, and ARC-5,7-diOMe-6-Br-DMPT+ of −1.81, −1.39, −1.60, and −1.30 V versus SCE in ethyl acetate, while the redox potential of the tetraphenylborate anion is reported to range from −0.78 to −0.98 V versus SCE in various organic solvents.41 The more negative redox potential of the ground-state photoinitiator relative to of the tetraphenylborate anion indicates that the photo-reductant generator is stable in the formulation in the absence of photoexcitation of the chromophore, where we previously showed that a formulation of BP-DMPT+/BPh4 is stable with and without BPO.5 Third, we determined that the photoexcitation of the chromophore leads to the desired reactions. The excited-state redox potentials of all four chromophores are significantly larger than of the ground state, making them electron acceptors in ET reactions with tetraphenyl borate (Figure 7). The redox potentials of BP-DMPT+, ARC-DMPT+, ARC-5,7-diOMe-DMPT+, and ARC-5,7-diOMe-6-Br-DMPT+ singlet excited states were calculated to be 1.53, 1.92, 1.66, and 1.98 V versus SCE, which facilitates ET from tetraphenyl borate (ET1) with redox potentials of −0.78 to −0.98 V and instigates the resultant reaction cascade of the mechanism described above (Figures 1 and 7). If triplet excited states instead of singlet excited states mediate the ET, their reduction potentials are still sufficient (1.10 V for BP, 1.27 V for ARC, 0.90 V for ARC-5,7-diOMe, and 1.09 V for ARC-5,7-diOMe-6-Br vs SCE).
Figure 7.

Changes in the electronic configurations of chromophore–ammonium/BPh4 leading up to the generation of two radicals and an amine. The positions of the orbitals qualitatively represent energies of the orbitals for all chromophores studied herein.
After ET1, two HM reactions ensue (Figures 1 and 7). The HM of tetraphenylborate (HM1) is well-known and discussed in our previous publication and many others.42–46 On the other hand, the HM of the chromophore–DMPT radical (HM2) is a novel aspect of the new visible DCPI design that enables the photo-release of an amine, where HM2 of the zwitterionic radical leads to an amine radical cation and chromophore anion that form an ionic complex (SI8, ionic complexation). We computed that the HM2 of the BP-DMPT radical was exergonic with a reaction free energy of −16.8 kcal/mol, while the HM2 of the ARC-DMPT radical, ARC-5,7-diOMe-DMPT radical, and ARC-5,7-diOMe-6-Br-DMPT radical was calculated to be exergonic with free energy changes of −7.7, −11.5, and −6.5 kcal/mol, respectively. The exergonicity indicates that C–N HM reactions in these four reduced chromophores after ET1 are spontaneous and rapid; this is supported by the observed irreversibility after reduction of BP-DMPT/BPh4 in cyclic voltammetry.5
The last reaction of interest is ET from the chromophore anion to the amine radical cation (ET2) to generate an amine with a restored lone pair that facilitates a ground-state redox reaction with the peroxide and hence extended dark-curing. We calculated ET2 from the BP anion to the DMPT radical cation to be 12.6 kcal/mol and the analogous reaction for ARC, ARC-5,7-diOMe, and ARC-5,7-diOMe-6-Br to be 13.2, 12.9, and 14.8 kcal/mol, respectively. The energetics of the DCPI mechanism activated by the new chromophores are nearly identical to that of the preceding BP-based DCPI, which we contend validates the viability of photo-generating the tertiary amine (Table 2). Consequently, our systematic study led to ARC-5,7-diOMe-6-Br that we proposed to be a desirable DCPI structure after computing the absorption wavelength, corresponding molar absorptivity, and favorable photochemical energetics. Therefore, we synthesized ARC-5,7-diOMe-6-Br-DMPT/BPh4 and report its photophysical and photochemical properties that ultimately culminated in the demonstration of visible-light-activated dark-curing PP in the following section.
Table 2.
Computed Photochemical Properties of Benzophenone and ARC Derivativesa
| C-N BDE (kcal/mol) | (V) | (V) | (V) | HM2 (kcal/mol) | ET2 (kcal/mol) | |
|---|---|---|---|---|---|---|
| BP-DMPT | 52.4 | −1.81 | 1.53 | 1.10 | −16.8 | 12.6 |
| ARC-DMPT | 52.5 | −1.39 | 1.92 | 1.27 | −7.7 | 13.2 |
| ARC-5,7-diOMe-DMPT | 53.1 | −1.60 | 1.66 | 0.90 | −11.5 | 12.9 |
| ARC-5,7-diOMe-6-Br-DMPT | 53.2 | −1.30 | 1.98 | 1.09 | −6.5 | 14.8 |
DFT and TD-DFT calculations were conducted on chromophore–ammonium species without tetraphenyl borate at the M06–2X/6–31+g(d,p)/SMD-ethyl acetate level of theory. The of BPh4 ranges from −0.78 to −0.98 V. Reduction potentials are reported referenced to SCE.
Synthesis of 5,7-Dimethoxy-6-bromo-3-aroylcoumarin.
Based on the proposed synthetic method (Figure 2), we synthesized ARC-5,7-diOMe-6-Br-DMPT/BPh4. First, ARC-5,7-diOMe was prepared with a high yield of ~98% from the condensation of ethyl(4-methylbenzoyl)acetate with 2,4-dimethoxy-6-hydroxybenzaldehyde. ARC-5,7-diOMe was then subjected to bromination using 3-bromosuccinimide and BPO. The bromination occurred at the desired tolyl group and the hypothesized C6 position that is evidenced by the disappearance of the C6 hydrogen NMR peak at 6.33 ppm (SI2, synthetic details).37,38 The resultant brominated chromophore was prepared with a 50% yield and the addition of DMPT and then sodium tetraphenyl borate to this compound produced the target quaternary ammonium borate salt PBG with a 25% yield of the last step.
Optical Measurement of 5,7-Dimethoxy-6-bromo-3-aroylcoumarin.
We measured an optical spectrum of the synthesized ARC-5,7-diOMe-6-Br-DMPT/BPh4 and compared it to the spectrum of BP-DMPT/BPh4 in N,N-dimethylformamide. Using a series of spectra, a plot of molar absorptivity versus wavelength was generated for a direct comparison. BP-DMPT/BPh4 has a maximum absorption of 134 M−1 cm−1 at 342 nm with a tail-end absorption of 80 M−1 cm−1 at the commonly used 365 nm wavelength. In comparison, ARC-5,7-diOMe-6-Br-DMPT/BPh4 has drastically improved light-absorbing characteristics over the useful range of wavelengths including the UV (Figure 8). The maximum absorption of ARC-5,7-diOMe-6-Br-DMPT/BPh4 red-shifted to 371 nm with a molar absorptivity of 13066 M−1 cm−1. At the near-UV wavelength of 365 nm, its molar absorptivity is 4 orders of magnitude higher than BP-DMPT/BPh4 (12924 vs 80 M−1 cm−1), indicating that this new DCPI likely initiates at dramatically higher rates in UV-initiated PP and subsequent dark-curing assuming a similar efficiency for each photoinitiator. More importantly, ARC-5,7-diOMe-6-Br-DMPT/BPh4 strongly absorbs in the visible light range; the violet absorption rates at 405 and 420 nm are 5710 and 1620 M−1 cm−1, respectively, while its blue absorption rate is much weaker with 50 M−1 cm−1 at 455 nm and 25 M−1 cm−1 at 470 nm, respectively, which favorably compares to our desired molar absorptivity target of ≥1000 M−1 cm−1 at 405 nm and ≤100 M−1 cm−1 at 450 nm. Interestingly, its experimentally measured UV–vis spectrum can be deconvoluted into two major peaks that correspond to the computationally predicted n → π* and π → π* transitions (Figure S25).
Figure 8.

Experimentally measured optical spectra of BP-DMPT/BPh4 and ARC-5,7-diOMe-6-Br-DMPT/BPh4 in N,N-dimethylformamide. ARC-based PBG exhibits strong visible-light absorption while BP-based PBG is limited to near-UV absorption. Two y-axes were used to accommodate the large molar absorptivity difference between two DCPI structures. The concentration of BP-PBG is 7.50 mM and that of ARC-PBG is 0.16 mM.
We then calculated the wavelength-dependent uniform depth of curing for a given initiator concentration, where 90% transmission is defined as meeting the thin-film approximation that guarantees simultaneous through-cure of a sample without the need to overirradiate the exposed surface (Table S2 and Figure S26). Due to the extreme molar absorptivity, the effective thin-film approximation depth of curing at 365 nm is only 4.6 μm with this photoinitiator in UV polymerizations. This would ostensibly limit its use in UV cure thin coating applications except that the dark cure mode of radical production promotes latent redox polymerization even with significantly attenuated photon access. However, optically uniform depths of 10.5 and 36.9 μm were achieved at 405 and 420 nm, respectively, which matches the requirements of most thin-film coating applications (~5 to 50 μm). More importantly, the use of 455 and 470 nm light enables the thin-film approximation to extend to 1195 and 2389 μm, respectively, allowing photopolymer resins more than a millimeter thick to be uniformly photocured at these wavelengths even before considering the DCPI’s latent radical production capability.
In addition to the blue absorption, ARC-5,7-diOMe-6-Br-DMPT/BPh4 exhibited photobleaching that increases the depth of curing (Figure S27).18 With the increased light dose from 0.6 to 201.5 kJ/m2, the maximum absorption of 371 nm progressively disappeared, which reduced the violet and blue absorptions and hence the potential for yellow coloration in the photocured resin leading to a clear solution. Concurrently, the deep UV peak at 270 nm gradually increased in response, likely due to the product of type II-like reactions between photoproducts.16 Last, the UV–vis spectrum of the complete formulation does not differ from the linear addition of two spectra that respectively contain BPO and ARC-5,7-diOMe-6-Br-DMPT/BPh4 (Figure S28). This result indicates that the formulation is stable where BPO and ARC-5,7-diOMe-6-Br-DMPT/BPh4 do not engage in ground-state reactions and only react with each other following irradiation due to the intentional design of the PBG without the reactive lone pair. With the confirmation of visible-light absorption, we next investigated its photochemistry leading to reductant release and polymerization.
Photochemistry of 5,7-Dimethoxy-6-bromo-3-aroylcoumarin.
To probe whether photogeneration of the redox initiator DMPT from ARC-5,7-OMe-6-Br-DMPT/BPh4 is indeed occurring, we studied the evolution of 1H NMR peaks specific to ARC-5,7-OMe-6-Br-DMPT and DMPT (Figure 9) in deuterated DMSO with ethylene carbonate as an internal standard. Following irradiation with a 405 nm LED, the peak associated with the methylene hydrogens of ARC-5,7-OMe-6-Br-DMPT at 5.09 ppm diminishes while methyl hydrogen peaks of DMPT (N–CH3 at 2.83 ppm and Ph-CH3 at 2.19 ppm) emerge, demonstrating that DMPT is absent before irradiation and is photo-generated from ARC-5,7-OMe-6-Br-DMPT/BPh4. This indicates this new chromophore’s potential to induce dark-curing with BPO within the amine–peroxide redox polymerization framework. Furthermore, we conducted quantitative NMR to study the dose dependence of photobase-release from ARC-5,7-OMe-6-Br-DMPT/BPh4 by varying irradiation time at a fixed irradiance of 2.0 mW/cm2. The PBG degradation and amine generation linearly responded to increasing time, leading to pseudo-zero-order reaction kinetics within the low conversion of the reaction (~20%). Under our experimental conditions, ~77% of ARC-5,7-diOMe-6-Br-DMPT/BPh4 degradation yielded DMPT amine generation. The high efficiency of the new DCPI is similar to that of our previous UV-based BP-DMPT/BPh4 that was found to be the most photon-efficient photoinitiator.5 These results directly validate our computational study where the mechanism energetics between BP-DMPT and ARC-5,7-OMe-6-Br-DMPT were essentially identical.
Figure 9.

NMR evidence of a photobase generator from ARC-5,7-diOMe-6-Br-DMPT/BPh4. The peaks of DMPT emerge after irradiation of the PBG, where the efficiency of photo-reductant generation is 77%. Ethylene carbonate was used as an internal standard for quantitative NMR. The concentration was 14.7 mM in deuterated dimethyl sulfoxide with 2.0 mW/cm2 405 nm LED irradiation.
Last, we studied photo- and dark polymerization enabled by the new visible DCPI initiated by 365, 405, and 455 nm LEDs in a methacrylate resin of 80 wt % triethylene glycol dimethacrylate and 20 wt % α-methylene-γ-butyrolactone, a cyclic analogue of methyl methacrylate. As in our previous study of DCPI, in addition to complete PP, we used a partial curing protocol where 20% of functional group conversion is achieved by irradiation to allow observation of the subsequent post-irradiation dark-curing behavior. PBG without the BPO oxidant is used as a control. With continuous irradiation by a 455 nm LED at 50 mW/cm2, both DCPI (PBG + BPO) and the control PBG rapidly photopolymerized in the methacrylate resin (Figure 10). Interestingly, final conversions observed after 10 min of irradiation are noticeably different. The DCPI sample reached 75% conversion while the PBG-only sample reached 65%. Such a difference is commonly attributed to increased chain mobility enabled by the exothermicity of polymerization where faster rates of polymerization lead to a larger exothermicity that surpasses the sample’s ability to dissipate the released heat.47 However, the temperature evolution was insignificant (<3 °C), does not differ between the two samples, and thus likely has a minimal effect on chain mobility in glassy polymers.48 We hypothesized that the final conversion difference is likely due to the difference in excess free volume generated from the delayed shrinkage with respect to conversion.47,49 The faster polymerization rates can “freeze” the developing polymer into a kinetically trapped structure further from the thermodynamic equilibrium, creating excess free volume that allows higher vitrification-limited conversion to be achieved.
Figure 10.

Methacrylate PP and dark-curing with DCPI and the control photoinitiator. The control formulation only contains PBG (ARC-5,7-diOMe-6-Br-DMPT/BPh4) that generates amine and radicals during irradiation, excluding the dark-curing potential due to the lack of BPO peroxide. The DCPI formulation contains PBG and BPO that enables an extended conversion post-irradiation. The experimental conditions include the initiator of 0.6 wt % ARC-PBG and 1.8 wt % BPO, the resin of 80 wt % TEGDMA and 20 wt % MBL, and a 50 mW/cm2 455 nm LED.
A steady-state analysis of the PP kinetics based on Figure 10 indicated that the rates of polymerization during irradiation are 0.24 s−1 for the PBG without peroxide and 0.50 s−1 for full DCPI composed of the PBG and peroxide, the difference of which must originate from the occurrence of ground-state redox initiation between amine and peroxide during irradiation. This yields initiator efficiencies (Φ) for the respective photoinitiators of 0.65 and 2.82 as calculated from the free radical PP kinetic equation (SI10, kinetic property calculation).50 The ARC-based PBG activated with a 455 nm LED is more efficient than the previous BP-based PBG activated with a 365 nm LED (ΦARC-455nm = 0.65 vs ΦBP-365nm = 0.43). This is likely due to a greater redox potential of the ARC-based PBG relative to the BP-based PBG (1.98 V for ARC-PBG vs 1.54 V for BP-PBG vs SCE assuming excitations into their lowest energy singlet excited states). The larger redox potential difference with respect to that of tetraphenyl borate results in a larger thermodynamic driving force that often leads to faster ET kinetics. This also implies that the photochemical mechanism is likely operative through the singlet excited state, facilitated by the proximity of chromophore–ammonium and tetraphenyl borate in the ion pair complex. This is supported by the very similar redox potentials of the first triplet excited states of the ARC- and BP-based PBGs at 1.09 and 1.10 V versus SCE, respectively, which are too small to significantly affect the ET kinetics. On the other hand, we found the initiator efficiency of DCPI to be larger than 1, whose theoretical maximum value is 1 by definition.50 This apparent anomaly results from the additional radicals generated from the redox reactions initiated by photo-released amine reductants, which occurs concurrently with photolysis during irradiation. The higher efficiency relative to our previous report is primarily due to our use of a higher concentration of the formulated peroxide in this study (1.82 wt % here vs 0.5 wt % previously). The ability to modulate PP rates by changing the concentration of visible-light-transparent peroxide is exceptionally useful, as it enables increasing PP rates while maintaining the relatively low overall optical density for depth of curing. Otherwise, with conventional photoinitiators, increasing photoinitiator concentrations to increase PP rates reduces the depth of cure due to the internal filter effect.
More importantly, when irradiation was ceased at the respective 20% conversion level, DCPI and the control PBG exhibited different post-irradiation behaviors (Figure 10). The conversion with the control photoinitiator plateaued essentially immediately following irradiation, which indicates the rapid biradical termination behavior expected of all conventional photoinitiators in the low-conversion regime where relatively high mobility persists. Contrarily, DCPI continued its polymerization to 55% conversion within 25 min post-irradiation. A steady-state analysis of dark-polymerization between 21% and 25% resulted in a polymerization rate of 0.16 s−1, which approximately corresponds to the difference of polymerization rates between the PBG and DCPI observed in our PP experiments. Generation of these additional initiating radicals did not require photons and initiated radical polymerization with an initiator efficiency of 0.3. The efficiencies for the PP of the PBG and the dark-curing of the DCPI do not linearly add up to the efficiency found for the PP initiated by DCPI because termination-dependent initiator efficiency is not an additive property. We also demonstrated a similar dark-curing behavior with 365 nm and 405 nm LEDs while the absence of irradiation resulted in the lack of any spontaneous polymerization over 1 h (Figures S31 and S32).
Last, because of the lack of ground-state interaction between DCPI and BPO, the stability of the one-part system was only limited by the thermal stability of BPO. The formulations used in the polymerization studies were examined for a week at room temperature and no polymerization occurred, showing that light is necessary to initiate polymerization. Our analogous system involving a benzophenone-based DCPI was found to be so stable that two resins respectively containing a benzophenone type II photoinitiator and the DCPI thermally degraded at the same temperature.5 BPO appears to be the only stability-limiting component of our DCPI system and it has sufficient thermal stability to be included in many two-part redox-curable commercial resins.51 Under refrigeration, the storage time of BPO-containing methacrylate solutions exceeds 12 months,52 which is also expected to be the case here.
CONCLUSIONS
In this work, we used quantum chemical methods, namely, DFT and TD-DFT, to search for a novel DCPI with a chromophore that offers significant and well-characterized visible-light absorption. The theoretical insights into the photochemical mechanism drawn from our previous UV-absorbing DCPI guided the approach taken here. We identified a ARC chromophore that proved to be a superior scaffold for photon absorption by a factor of ~10 relative to the benzophenone chromophore used for UV absorption. We then functionalized ARC at several positions with either methoxy or chlorine groups to red-shift the absorption into the visible-light range. Overall, the methoxy substitution, particularly at the C5 and C7 positions, allowed moderate visible-light absorption while chlorine substitution universally failed to increase the absorption wavelength. We found that the bathochromic shift from methoxy substitutions at certain positions is primarily due to increased molar absorptivity rather than maximum absorption changes. Such high molar absorptivity results from increased polarizability that originates from the push–pull characteristic through the long axis of the molecule, evidenced by the ground-state dipole moments. A substitution study indicated that 5,7-dimethoxy-3-aroylcourmain would be a promising visible-light PBG chromophore with optimal absorption characteristics, which we showed using a simulated UV–vis spectrum. However, due to a synthetic limitation, 5,7-dimethoxy-6-bromo-3-aroylcoumarin with slightly poorer absorption was instead targeted and the computed energetics of the proposed photochemical mechanism predicted that the ARC-5,7-diOMe-6-Br-based DCPI would perform as efficiently as the benzophenone-based predecessor.
ARC-5,7-diOMe-6-Br-DMPT/BPh4 was optically examined and found to possess molar absorptivities of 5710 M−1 cm−1 at 405 nm and 1620 M−1 cm−1 at 420 nm with weaker blue absorption with 50 M−1 cm−1 at 455 nm and 25 M−1 cm−1 at 470 nm. We also observed significant photo-bleaching, which reduces the internal filter effect and increases the depth of cure potential before considering the redox component of the mechanistic cascade. The NMR spectra confirmed that the DMPT amine is indeed generated from ARC-5,7-diOMe-6-Br-DMPT/BPh4 with 77% efficiency. Last, we demonstrated this DCPI’s dark-curing capability in a methacrylate resin with 365, 405, and 455 nm light. Based on a steady-state analysis, we found that the initiator efficiency of ARC-5,7-diOMe-6-Br-DMPT/BPh4 without peroxide is 0.65 while its initiator efficiency with BPO increased to 2.82 due to additional radicals produced by redox initiation that do not require further light activation. More importantly, we demonstrated extensive dark-curing that can rescue any undercured regions post-irradiation. The new DCPI with visible absorption will improve many current applications and enable new ones that conventional visible-light photoinitiators could not achieve due to nonideal photocuring conditions, making radical PP an even more useful technology.
EXPERIMENTAL AND COMPUTATIONAL DETAILS
Computational Methods.
All computations were conducted using the GAUSSIAN 16 (Revision A.03) software package53 and density functional theory with the M06–2X exchange correlation functional with the 6–31+g(d,p) and 6–311++g(d,p) basis sets.54 The M06–2X functional outperformed other functionals for calculation of the thermochemistry and kinetics of ground-state and excited-state organic molecules.54 Vibrational frequencies were computed to confirm that the stationary states were optimized to the correct structures and to compute vibrational entropies, zero-point energies, and thermal corrections to enthalpies at 298 K. Solvent effects were applied using the universal solvent model of acetonitrile and ethyl acetate.55 Gaussian broadening of the UV–vis spectra was conducted using a Python code.23 The M06–2X-computed molecular orbitals were viewed using Avogadro. Reductional potential calculations were performed based on the approach developed by Tossell.56,57 Electron densities were computed using atomic polar tensor population analysis.58,59
The photophysical properties of λmax, its corresponding molar absorptivity (εmax), and changes in the electron occupation of active orbitals upon photoexcitation of chromophores were computed using TD-DFT at the M06–2X/6–311++g(d,p)/SMD-acetonitrile level of theory with geometries optimized with DFT at the M06–2X/6–31+g(d,p)/SMD-acetonitrile level of theory. To limit the computational expense of TD-DFT calculations, the studied molecules were truncated to the para-tolyl-functionalization, excluding ammonium functional groups. On the other hand, the energetics of the photochemical reaction were computed at the M06–2X/6–31+g-(d,p)/SMD-ethyl acetate level of theory using the entire chromophore–ammonium structures including the photo-releasable amine (DMPT) without tetraphenyl borate. Free energies were computed within the bulk reaction approximation used in our previous publications,39,40 where the entropic contributions from the solvent-hindered molecular rotations and translations were excluded to reflect the highly viscous nature of the photocuring resin phase.
Instrumentation and Methods.
1H NMR and 13C NMR spectra were recorded on a Bruker 400 MHz NMR spectrometer. Proton chemical shifts are expressed in parts per million (δ) using TMS as an internal standard. For quantitative NMR, ethylene carbonate was used as an internal standard. HRMS analysis was performed on a Waters Synapt G2 HDMS Q-TOF Mass Spectrometer. UV absorption spectra were recorded by a Thermo-Fisher Scientific UV/vis spectrophotometer, corrected for baseline with a solvent-filled cuvette. The real-time polymerization kinetics was recorded using a Nicolet Magna-IR Series II FT-IR spectrophotometer. A detailed description of the polymerization kinetic study and analysis is included in the Supporting Information (Section 3, FTIR characterization).
Materials.
The desired quaternary-ammonium-salt-based visible photobase generator (5,7-dimethoxy-6-bromo-3-aroylcoumarin-DMPT/BPh4) was synthesized starting from ARC by the condensation of ethyl (4-methylbenzoyl)acetate with 2,4-dimethoxy-6-hydroxybenzaldehyde. The ARC derivative hence obtained was subjected to bromination using N-bromosuccinimide and BPO in chlorobenzene as a solvent, resulting in a mixture of aromatic brominated and tolyl brominated compounds. The mixture was further treated with DMPT in a toluene/DCM solvent mixture that exclusively resulted in a quaternary ammonium bromide salt. This quaternary ammonium bromide salt was further reacted with sodium tetraphenyl borate to yield the desired quaternary ammonium borate salt as the visible photobase. A more detailed procedure describing the synthesis of each step performed to obtain the desired visible photobase generator is included in the Supporting Information (Section 2, synthesis of a visible photobase generator).
Supplementary Material
ACKNOWLEDGMENTS
Funding was provided by the Industry/University Cooperative Research Center for the Fundamentals and Applications of Photopolymerization as well as by NIH/NIDCR R21DE028017.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.macromol.1c00761.
Instrumental methods and materials, synthesis of a visible photobase generator, FTIR characterization, molar absorptivity, resonance structures, NMR, effect of 6-bromo substitution, ionic complexation, depth cure, and kinetic property calculations (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.macromol.1c00761
The authors declare no competing financial interest.
Contributor Information
Kangmin Kim, Chemistry, University of Colorado, Boulder, Colorado 80309, United States.
Jasmine Sinha, Chemical and Biological Engineering, University of Colorado, Boulder, Colorado 80309, United States.
Jeffrey W. Stansbury, Chemical and Biological Engineering, University of Colorado, Boulder, Colorado 80309, United States; Craniofacial Biology, School of Dental Medicine, Aurora, Colorado 80045, United States.
Charles B. Musgrave, Chemistry, Chemical and Biological Engineering, and Materials Science and Engineering, University of Colorado, Boulder, Colorado 80309, United States; National Renewable Energy Laboratory, Golden, Colorado 80401, United States.
REFERENCES
- (1).Fouassier JP; Allonas X; Burget D Photopolymerization reactions under visible lights: Principle, mechanisms and examples of applications. Prog. Org. Coatings 2003, 47, 16–36. [Google Scholar]
- (2).Marqual IT Solutions Pvt Ltd (KBV Research). Global UV Curable Resins Market Outlook to 2025. https://www.researchandmarkets.com/reports/4848974/global-uv-curable-resins-market-2019-2025?utm_source=BW&utm_medium=PressRelease&utm_code=bgjwxn&utm_campaign=1309806+-+Global+UV+Curable+Resins+Market+Outlook+to+2025&utm_exec=joca220prd, accessed July 21st, 2020
- (3).Palin WM; Leprince JG; Hadis MA Shining a light on high volume photocurable materials. Dent. Mater 2018, 34, 695–710. [DOI] [PubMed] [Google Scholar]
- (4).Garra P; Dietlin C; Morlet-Savary F; et al. Photopolymerization processes of thick films and in shadow areas: a review for the access to composites. Polym. Chem 2017, 8, 7088–7101. [Google Scholar]
- (5).Kim K; Sinha J; Gao G; et al. High-Efficiency Radical Photopolymerization Enhanced by Autonomous Dark Cure. Macromolecules 2020, 53, 5034–5046. [Google Scholar]
- (6).Kim K; Singstock NR; Childress KK; et al. Rational Design of Efficient Amine Reductant Initiators for Amine-Peroxide Redox Polymerization. J. Am. Chem. Soc 2019, 141, 6279–6291. [DOI] [PubMed] [Google Scholar]
- (7).Musgrave CB III; Kim K; Singstock NR; Salazar AM; Stansbury JW; Musgrave CB Computational and experimental evaluation of peroxide oxidants for amine-peroxide redox polymerization. Macromolecules 2020, 53, 9736–9746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Aguirre-Soto A; Lim C-H; Hwang AT; Musgrave CB; Stansbury JW Visible-light organic photocatalysis for latent radical-initiated polymerization via 2e−/1H+ transfers: Initiation with parallels to photosynthesis. J. Am. Chem. Soc 2014, 136, 7418–7427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Shanmugam S; Xu J; Boyer C Photoinduced Oxygen Reduction for Dark Polymerization. Macromolecules 2017, 50, 1832–1846. [Google Scholar]
- (10).Garra P; Dumur F; Gigmes D; Al Mousawi A; Morlet-Savary F; Dietlin C; Fouassier JP; Lalevee J Copper (Photo)redox Catalyst for Radical Photopolymerization in Shadowed Areas and Access to Thick and Filled Samples. Macromolecules 2017, 50 (10), 3761–3771. [Google Scholar]
- (11).Abdallah M; Hijazi A; Graff B; Fouassier J-P; Rodeghiero G; Gualandi A; Dumur F; Cozzi PG; Lalevee J Coumarin derivatives as versatile photoinitiators for 3D printing, polymerization in water and photocomposite synthesis. Polym. Chem 2019, 10, 872. [Google Scholar]
- (12).Abdallah M; Hijazi A; Lin J-T; Graff B; Dumur F; Lalevee J Coumarin Derivatives as Photoinitiators in Photo-Oxidation and Photo-Reduction Processes and a Kinetic Model for Simulations of the Associated Polymerization Profiles. ACS Appl. Polym. Mater 2020, 2, 2769–2780. [Google Scholar]
- (13).Abdallah M; Dumur F; Hijazi A; Rodeghiero G; Gualandi A; Cozzi PG; Lalevee J Keto-coumarin scaffold for photoinitiators for 3D printing and photocomposites. J. Polym. Sci 2020, 58, 1115–1129. [Google Scholar]
- (14).Savéant J-M Electron transfer, bond breaking and bond formation. Adv. Phys. Org. Chem 2000, 35, 117–192. [Google Scholar]
- (15).Shao J; Huang Y; Fan Q Visible light initiating systems for photopolymerization: Status, development and challenges. Polym. Chem 2014, 5, 4195–4210. [Google Scholar]
- (16).Freddi AB; Morone M; Norcini G Design of new 3-ketocoumarins for UV LED curing. UV+EB Technology, 2016. [Google Scholar]
- (17).Eibel A; Fast DE; Gescheidt G Choosing the ideal photoinitiator for free radical photopolymerizations: Predictions based on simulations using established data. Polym. Chem 2018, 9, 5107–5115. [Google Scholar]
- (18).Miller GA; Gou L; Narayanan V; Scranton AB Modeling of photobleaching for the photoinitiation of thick polymerization systems. J. Polym. Sci. Part A Polym. Chem 2002, 40, 793–808. [Google Scholar]
- (19).Guimaräes T; Schneider LF; Braga RR; Pfeifer CS Mapping camphorquinone consumption, conversion and mechanical properties in methacrylates with systematically varied CQ/amine compositions. Dent. Mater 2014, 30, 1274–1279. [DOI] [PubMed] [Google Scholar]
- (20).Schneider LFJ; Cavalcante LM; Prahl SA; Pfeifer CS; Ferracane JL Curing efficiency of dental resin composites formulated with camphorquinone or trimethylbenzoyl-diphenyl-phosphine oxide. Dent. Mater 2012, 28, 392–397. [DOI] [PubMed] [Google Scholar]
- (21).Taniguchi M; Lindsey JS Database of Absorption and Fluorescence Spectra of >300 Common Compounds for use in Photochem CAD. Photochem. Photobiol 2018, 94, 290–327. [DOI] [PubMed] [Google Scholar]
- (22).Dantas JA; Correia JTM; Paixão MW; Corrêa AG Photochemistry of Carbonyl Compounds: Application in Metal-Free Reactions. ChemPhotoChem 2019, 3, 506–520. [Google Scholar]
- (23).Creating UV/Visible Plots from the Results of Excited States Calculations; Gaussian Inc; Pittsburgh PA, https://gaussian.com/uvvisplot/, 2017. Published. [Google Scholar]
- (24).Bradley MS; Bratu C Vibrational line profiles as a probe of molecular interactions. J. Chem. Educ 1997, 74, 553–555. [Google Scholar]
- (25).Hammond GS; Bartlett PD Polymerization of allyl compounds. V. Inhibition by nitro compounds. J. Polym. Sci 1951, 6, 617–624. [Google Scholar]
- (26).Bagdasar’Ian KS; Sinitsina ZA Polymerization inhibition by aromatic compounds. J. Polym. Sci 1961, 52, 31–38. [Google Scholar]
- (27).Pavlopoulos T Prediction of Laser Action Properties of Organic Dyes From their Structure and the Polarization Characteristics of their Electronic Transitions. IEEE J. Quantum Electron 1973, 9, 510–516. [Google Scholar]
- (28).Albota M; Beljonne D; Brédas JL; et al. Design of organic molecules with large two-photon absorption cross sections. Science 1998, 281, 1653–1656. [DOI] [PubMed] [Google Scholar]
- (29).Bayliss NS; Mcrae EG Solvent effects in organic spectra: Dipole forces and the Franck-Condon principle. J. Phys. Chem 1954, 58, 1002–1006. [Google Scholar]
- (30).Nagakura S; Baba H Dipole Moments and Near Ultraviolet Absorption of Some Monosubstituted Benzenes - The Effect of Solvents and Hydrogen Bonding. J. Am. Chem. Soc 1952, 74, 5693–5698. [Google Scholar]
- (31).Liu X; Cole JM; Waddell PG; Lin T-C; Radia J; Zeidler A Molecular origins of optoelectronic properties in coumarin dyes: Toward designer solar cell and laser applications. J. Phys. Chem. A 2012, 116, 727–737. [DOI] [PubMed] [Google Scholar]
- (32).Liu X; Cole JM; Waddell PG; Lin T-C; McKechnie S Molecular origins of optoelectronic properties in coumarins 343, 314T, 445, and 522B. J. Phys. Chem. C 2013, 117, 14130–14141. [DOI] [PubMed] [Google Scholar]
- (33).Liu X; Xu Z; Cole JM Molecular design of UV-vis absorption and emission properties in organic fluorophores: Toward larger bathochromic shifts, enhanced molar extinction coefficients, and greater stokes shifts. J. Phys. Chem. C 2013, 117, 16584–16595. [Google Scholar]
- (34).Yong Lee J; Kim KS; Jin Mhin B Intramolecular charge transfer of π-conjugated push-pull systems in terms of polarizability and electronegativity. J. Chem. Phys 2001, 115, 9484–9489. [Google Scholar]
- (35).Sutradhar T; Misra A Role of Electron-Donating and Electron-Withdrawing Groups in Tuning the Optoelectronic Properties of Difluoroboron-Napthyridine Analogues. J. Phys. Chem. A 2018, 122, 4111–4120. [DOI] [PubMed] [Google Scholar]
- (36).Dietlin C; Schweizer S; Xiao P; et al. Photopolymerization upon LEDs: new photoinitiating systems and strategies. Polym. Chem 2015, 6, 3895–3912. [Google Scholar]
- (37).Carreño MC; Garcia Ruano JL; Sanz G; Toledo MA; Urbano A N-Bromosuccinimide in Acetonitrile: A Mild and Regiospecific Nuclear Brominating Reagent for Methoxybenzenes and Naphthalenes. J. Org. Chem 1995, 60, 5328–5331. [Google Scholar]
- (38).Bloomer JL; Zheng W Using NBS as a mild bromination reagent for polyalkoxyaromatic systems. Synth. Commun 1998, 28, 2087–2095. [Google Scholar]
- (39).Love DM; Kim K; Goodrich JT; et al. Amine Induced Retardation of the Radical-Mediated Thiol-Ene Reaction via the Formation of Metastable Disulfide Radical Anions. J. Org. Chem 2018, 83, 2912. [DOI] [PubMed] [Google Scholar]
- (40).Lim C-H; Holder AM; Hynes JT; Musgrave CB Reduction of CO2 to Methanol Catalyzed by a Biomimetic Organo-Hydride Produced from Pyridine. J. Am. Chem. Soc 2014, 136, 16081–16095. [DOI] [PubMed] [Google Scholar]
- (41).Pal PK; Chowdhury S; Drew MGB; Datta D The electrooxidation of the tetraphenylborate ion revisited. New J. Chem 2002, 26, 367–371. [Google Scholar]
- (42).Grinevich O; Serguievski P; Sarker AM; Zhang W; Mejiritski A; Neckers DC Relative Activity of Possible Initiating Species Produced from Photolysis of Tetraphenyl and Triphenylbutyl Borates As Measured by Fluorescence Probe Techniques. Macromolecules 1999, 32, 328–330. [Google Scholar]
- (43).Schuster GB; Yang X; Zou C; Sauerwein B Photo-initiated electron transfer reactions in dye-borate ion pairs: energy, distance and solvent dependence. J. Photochem. Photobiol. A Chem 1992, 65, 191–196. [Google Scholar]
- (44).Zhang W; Feng K; Wu X; Martin D; Neckers DC Photochemical Properties Of 4-Benzoylbenzylammonium Borates. J. Org. Chem 1999, 64, 458–463. [Google Scholar]
- (45).Kaneko Y; Sarker AM; Neckers DC Mechanistic Studies of Photobase Generation from Ammonium Tetraorganyl Borate Salts1. Chem. Mater 1999, 11, 170–176. [Google Scholar]
- (46).Hu S; Sarker AM; Kaneko Y; Neckers DC Reactivities of Chromophore-Containing Methyl Tri-n-butylammonium Organoborate Salts as Free Radical Photoinitiators: Dependence on the Chromophore and Borate Counterion1. Macromolecules 1998, 31, 6476–6480. [Google Scholar]
- (47).Truffier-Boutry D; Demoustier-Champagne S; Devaux J; et al. A physico-chemical explanation of the post-polymerization shrinkage in dental resins. Dent. Mater 2006, 22, 405–412. [DOI] [PubMed] [Google Scholar]
- (48).Stansbury JW Dimethacrylate network formation and polymer property evolution as determined by the selection of monomers and curing conditions. Dent. Mater 2012, 28, 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Abu-Elenain DA; Lewis SH; Stansbury JW Property evolution during vitrification of dimethacrylate photopolymer networks. Dent. Mater 2013, 29, 1173–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Odian G Principles of Polymerization, 4th ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2004. [Google Scholar]
- (51).Kwon T-Y; Bagheri R; Kim YK; Kim K-H; Burrow MF Cure mechanisms in materials for use in esthetic dentistry. J. Investig. Clin. Dent 2012, 3, 3–16. [DOI] [PubMed] [Google Scholar]
- (52).Shim JB; Warner SJ; Hasenwinkel JM; Gilbert JL Analysis of the shelf life of a two-solution bone cement. Biomaterials 2005, 26, 4181–4187. [DOI] [PubMed] [Google Scholar]
- (53).Frisch MJ; Trucks GW; Schlegel HB; et al. Gaussian, 2016; Vol. 16. [Google Scholar]
- (54).Zhao Y; Truhlar DG; Truhlar DG The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc 2008, 120, 215. [Google Scholar]
- (55).Marenich AV; Cramer CJ; Truhlar DG Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [DOI] [PubMed] [Google Scholar]
- (56).Tossell JA Calculation of the properties of molecules in the pyridine catalyst system for the photochemical conversion of CO2 to methanol. Comput. Theor. Chem 2011, 977, 123–127. [Google Scholar]
- (57).Alherz A; Lim C-H; Kuo Y-C; et al. Renewable Hydride Donors for the Catalytic Reduction of CO2: A Thermodynamic and Kinetic Study. J. Phys. Chem. B 2018, 122, 10179–10189. [DOI] [PubMed] [Google Scholar]
- (58).Cioslowski J A new population analysis based on atomic polar tensors. J. Am. Chem. Soc 1989, 111 (22), 8333–8336. [Google Scholar]
- (59).Ferreira MMC xx. J. Mol. Struct 1993, 294, 75–78. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.