

Abstract
Patient derived xenograft models are critical in defining how different cancers respond to drug treatment in an in vivo system. Mouse models are the standard in the field, but zebrafish have emerged as an alternative model with several advantages, including the ability for high-throughput and low-cost drug screening. Zebrafish also allow for in vivo drug screening with large replicate numbers that were previously only obtainable with in vitro systems. The ability to rapidly perform large scale drug screens may open up the possibility for personalized medicine with rapid translation of results back to clinic. Zebrafish xenograft models could also be used to rapidly screen for actionable mutations based on tumor response to targeted therapies or to identify new anti-cancer compounds from large libraries. The current major limitation in the field has been quantifying and automating the process so that drug screens can be done on a larger scale and be less labor-intensive. We have developed a workflow for xenografting primary patient samples into zebrafish larvae and performing large scale drug screens using a fluorescence microscope equipped imaging unit and automated sampler unit. This method allows for standardization and quantification of engrafted tumor area and response to drug treatment across large numbers of zebrafish larvae. Overall, this method is advantageous over traditional cell culture drug screening as it allows for growth of tumor cells in an in vivo environment throughout drug treatment, and is more practical and cost-effective than mice for large scale in vivo drug screens.
Keywords: zebrafish, patient derived xenograft, high-throughput, drug screen, leukemia, automated
SUMMARY:
Zebrafish xenograft models allow for high-throughput drug screening and fluorescent imaging of human cancer cells in an in vivo microenvironment. We developed a workflow for large scale, automated drug screening on patient-derived leukemia samples in zebrafish using an automated fluorescence microscope equipped imaging unit.
INTRODUCTION:
Xenografting of primary patient cancers or human cancer cell lines into model organisms is a widely used technique to study tumor progression and behavior in vivo, tumor response to drug treatment, and cancer cell interaction with the microenvironment, among others. Traditionally, cells are xenografted into immune-compromised mice, and this remains the standard in the field. However, this model system has several limitations, such as high cost, low replicate numbers, difficulties in accurately quantifying tumor burden in vivo, and the extended time that it takes for tumors to engraft and drug testing to be completed. In recent years, zebrafish have emerged as an alternate xenograft model, with the first being reported in 2005, with green fluorescent protein (GFP)-labeled human melanoma cell lines transplanted into blastula-stage embryos1,2. More recently, 2 day post-fertilization (dpf) zebrafish larvae have been used as xenograft recipients to allow for control of anatomic location of injection and for use in high resolution in vivo imaging of tumor interaction with the surrounding microenvironment3,4.
Zebrafish offer many advantages as a xenograft model. First, adult zebrafish can be housed and rapidly bred in large quantities at a relatively low cost. Each mating pair of adult zebrafish can produce hundreds of larval fish per week. Due to their small size, these larval zebrafish can be maintained in 96-well plates for high-throughput drug screening. Larvae do not have to be fed during the course of a typical xenograft experiment, as their yolk-sac provides the nutrients to sustain them for their first week of life. Furthermore, zebrafish do not have a fully functional immune system until 7 dpf, meaning that they do not require irradiation or immunosuppressive regimens prior to xenograft injection. Finally, optically clear zebrafish lines allow for high-resolution imaging of tumor-microenvironment interactions.
Perhaps the most promising application of zebrafish as a xenograft model is the ability to perform high-throughput drug screening on human cancer samples in a way that is not possible using any other model organism. Larvae absorb drugs from the water through the skin, enhancing the ease of drug administration5. Because animals are maintained in 96-well plates, typically in 100–300 μL of water, screens require smaller drug quantities compared to mice. Currently, there are several different methods for standardization and quantification of the effect of drugs on human tumor burden in zebrafish, some of which are more practical than others for scaling-up single drug testing to high-throughput screening. For example, some groups dissociate fish into single cell suspensions, and quantify fluorescently labeled or stained tumor cells by imaging individual droplets of the suspension and quantifying fluorescence using a semi-automated ImageJ macro4. A semi-automated whole-larvae imaging method was developed in which larval fish were fixed in 96-well plates and imaged using an inverted fluorescent microscope before realignment of composite images and quantification of tumor cell foci6. Both of these assays are fairly labor-intensive methods for quantification, which has made truly high-throughput drug screening in zebrafish xenograft models impractical.
This issue has been addressed by the development of the Vertebrate Automated Screening Technology (VAST) Bioimager and Large Particle (LP) Sampler, a fluorescence microscope equipped imaging unit and automated sampler unit (Figure 1 and Table of Materials), which is a truly automated method for high-throughput imaging of zebrafish larvae7–9. With this unit, fish are anesthetized, sampled automatically from a 96-well plate, positioned in a capillary and rotated into the set orientation based on a preset user preference, imaged, and then either placed back into the same well of a new 96-well plate for further studies or discarded. Combining this imaging technology with zebrafish xenografts may allow for the possibility of personalized medicine that uses high-throughput drug screening of large drug compound libraries against individual patient tumors. Zebrafish xenografts also offer a large-scale and low-cost method for testing both toxicity and efficacy of novel compounds in vivo. Zebrafish can be used as a preliminary screening step before proceeding to mouse xenograft models.
Figure 1: Xenograft drug screen and imaging workflow.
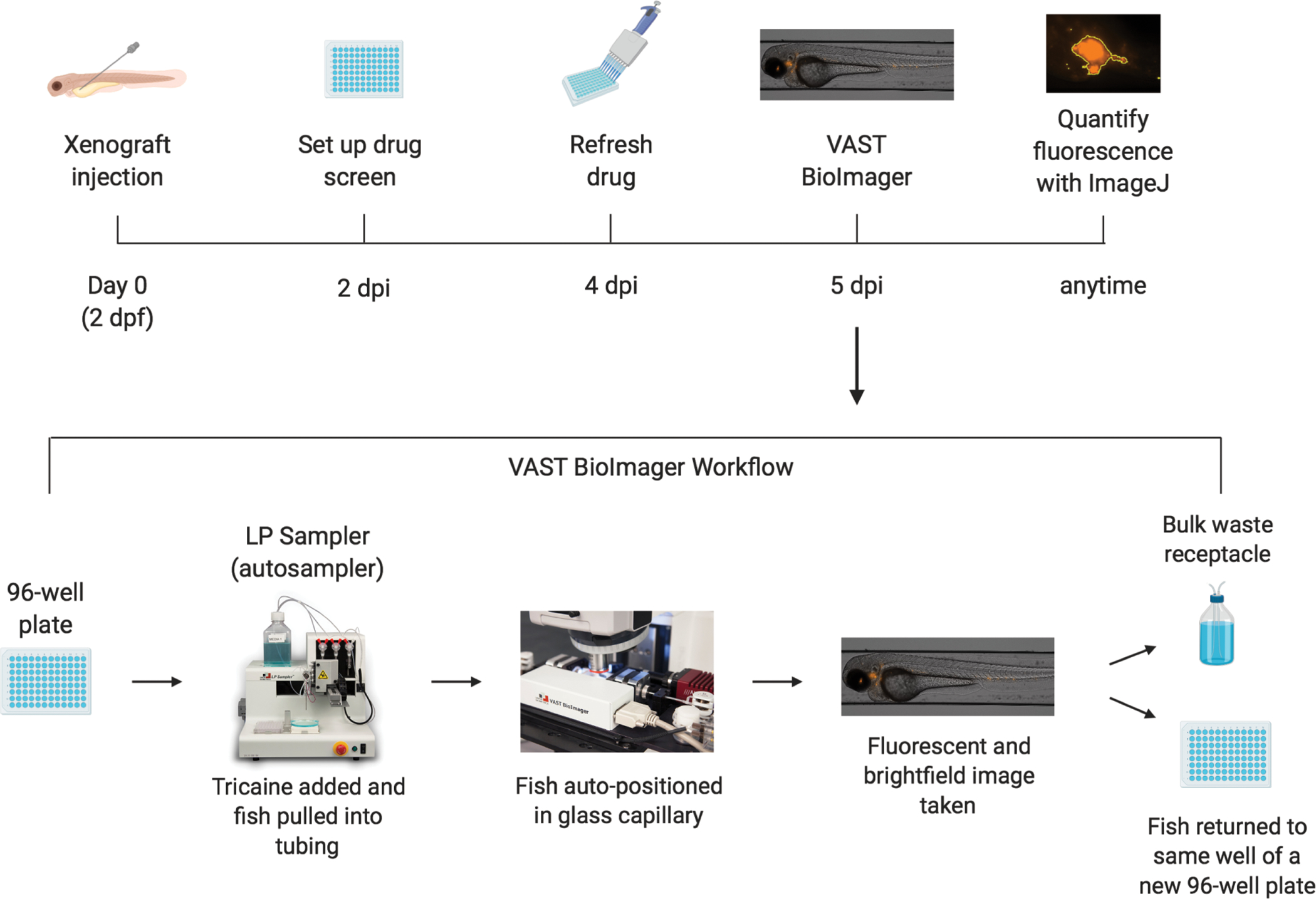
Schematic of workflow of xenografting zebrafish larvae and performing drug screen, including imaging on a fluorescence microscope equipped imaging unit and automated sampler unit.
Table of Materials.
| Name of Material/Equipment | Company | Catalog Number | Comments/Description |
|---|---|---|---|
| 10× TBE Liquid Concentrate | VWR | 0658-5L | |
| 96-well plate, flat bottom | CELLTREAT | 229195 | VAST is compatible with a variety of standard or deep well 24, 48, or 96 well plates |
| Agarose | Fisher Scientific | BP160-500 | |
| Borosilicate Glass Capillary without Filament | Sutter Instrument Company | B100-50-10 | |
| Dexamethasone | Enzo Life Sciences | BML-EI126-0001 | |
| DMSO | Sigma-Aldrich | D2438-5X10ML | |
| E3 media | N/A | 5 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl2, 0.33 mM MgSO4 | |
| Femtotips Microloader Tips | Eppendorf | 930001007 | |
| Fetal Bovine Serum (Premium Heat Inactivated) | Atlanta Biologicals | S11150H | |
| ImageJ | FIJI | N/A | https://imagej.net/Fiji |
| Iscove’s Modified Dulbecco’s Medium | STEMCELL Technologies | 36150 | |
| Large Particle (LP) Sampler | Union Biometrica | N/A | http://www.unionbio.com/copas/features.aspx?id=8 |
| Methotrexate | Sigma-Aldrich | A6770-10MG | |
| Mineral Oil | Fisher Scientific | BP26291 | |
| Phosphate Buffered Saline (1×) | Caisson labs | PBL06-6X500ML | |
| Stage Micrometer (400-Stage) | Hausser Scientific | 400-S | |
| Tricaine-S | Pentair Aquatic | TRS1 | |
| Trypan Blue | Thermo Fisher | T10282 | |
| VAST Bioimager | Union Biometrica | N/A | https://www.unionbio.com/vast/ |
| Vincristine Sulfate | Enzo Life Sciences | BML-T117-0005 | |
| Vybrant DiI Stain | Thermo Fisher | V22885 |
We have developed a streamlined workflow for xenografting primary patient leukemia cells into zebrafish and performing high-throughput drug screens with automated imaging and quantification, which can be applied to any other primary patient tumor cells or cancer cell line. This workflow utilized a fluorescence microscope equipped imaging unit and automated sampler unit to improve upon current standardization and quantification methods and offers an automated alternative to previous, more labor-intensive methods of quantifying tumor mass in vivo.
PROTOCOL:
All procedures described in this protocol have been approved by the University of Kentucky’s Institutional Animal Care and Use Committee (protocol 2015–2225). Patient samples were collected under University of Kentucky’s Institutional Review Board (protocol 44672). All animal experiments performed following this protocol must be approved by the user’s Institutional Animal Care and Use Committee.
1. Thawing primary patient acute lymphoblastic leukemia cells
1.1. Thaw primary patient peripheral blood mononuclear cells (PBMCs) from frozen stock in a 37 °C water bath. Immediately after cells have thawed, transfer cells in their freezing media (90% FBS + 10% dimethyl sulfoxide [DMSO]) to a 15 mL conical tube with slow pipetting, avoiding air bubbles. Add 10 mL of prewarmed 37 °C thawing media (25% fetal bovine serum [FBS] in Iscove’s modified Dulbecco’s medium [IMDM]) dropwise (approximately 2–3 s per mL) to the cells in the 15 mL conical tube.
NOTE: PBMCs were collected from patient blood samples at the time of diagnosis. The buffy coat was separated by density centrifugation and cells were washed 2× in RPMI 1640 + 10% FBS. Cells were counted and 107 cells were frozen per cryovial in 1 mL of freezing media, and stored at −80 °C.
1.2. Centrifuge cells at 100 × g for 10 min and aspirate media from the cell pellet. Repeat the addition of thaw media, centrifugation, and aspiration one additional time to remove any residual DMSO.
1.3. Resuspend cells in 5 mL of phosphate-buffered saline (PBS) and remove 10 μL for counting on an automated cell counter or hemocytometer. Add 10 μL of trypan blue to 10 μL of cells removed for counting. Count number of cells per mL and record to later calculate the volume to resuspend cells in for xenografting (see step 2.5).
NOTE: Typically, 500 cells are xenografted per larval zebrafish. For example, 5 × 105 cells are needed to inject 1,000 zebrafish. Viability should be >85% to use for xenografting. In this experiment, cell viability was 96% after thawing, assessed by trypan blue staining.
2. Fluorescently labeling cells with DiI
2.1. Centrifuge the desired cell number in 5 mL of PBS at 200 × g for 5 min and aspirate supernatant. Stain a minimum of 2 × 106 cells in case of clumping or problems loading the needle during injection.
2.2. Make 1,1’-dioctadecyl-3,3,3’,3’-tetramethylindocarbocyanine perchlorate (DiI) staining solution (5 mL of PBS containing 4 μL per mL of DiI stain, Table of Materials) and resuspend the cell pellet in the staining solution.
NOTE: Cell density should not exceed 2 × 106 cells/mL when resuspended in the staining solution.
2.3. Incubate cells at 37 °C protected from light for 20 min, vortex gently, then incubate cells on ice for 15 min protected from light.
2.4. Centrifuge cells at 200 × g for 5 min and aspirate supernatant. Wash cells with 5 mL of PBS, centrifuge at 200 × g for 5 min and aspirate supernatant. Repeat wash, centrifugation, and aspiration one additional time.
2.5. Resuspend cells in 1 μL of PBS per 250,000 live cells and transfer to a 1.5 mL microcentrifuge tube. Keep resuspended cells on ice in the dark and immediately continue to microinjections.
NOTE: This ensures that 500 patient cells are injected into the zebrafish with each injection pump of 2 nL volume.
3. Microinjecting zebrafish larvae
NOTE: Microinjections should be completed within 1–3 h of staining to improve viability of cells.
3.1. Prior to staining cells and injecting, make agarose plates for injecting by pouring 25 mL of 3% agarose in 1× Tris/borate/EDTA (TBE) into a Petri dish and allow it to solidify. Store plates at 4 °C for up to 2 weeks.
3.2. Also prior to staining cells and injecting, dechorionate 2 dpf zebrafish using forceps under a dissecting microscope. For manual dechorionation, pull from opposite ends of the protective chorion of the zebrafish with forceps until the chorion tears and the zebrafish becomes unenveloped10.
NOTE: Dechorionation can also be performed by using enzymatic treatment with pronase, as previously described10. Casper (roy−/−;nacre−/−) zebrafish were used for these experiments11. Any zebrafish larval strain can be used for xenografting. If pigment interferes with imaging or visualization, propylthiouracil (PTU) treatment can be used to block melanin synthesis if optically clear zebrafish strains are not available12.
3.3. Prechill needles at 4 °C or on ice to prevent clumping of cells during microinjection. Load 5 μL of stained cells into a chilled nonfilamentous borosilicate glass needle using microloader pipette tips.
NOTE: Microinjector and needle setup methods have been previously published13.
3.4. Load the needle into the microinjector arm. Bevel the needle tip using a sterile razor blade. Measure the droplet size in mineral oil using a stage micrometer, keeping droplet volume consistently at 2 nL (~0.15 mm diameter) throughout.
3.5. Use 350 μL of 4 mg/mL tricaine-S to anesthetize ~30 dechorionated 2 dpf zebrafish in a Petri dish containing 25 mL of E3 media. After ~1 min transfer anesthetized larvae to a flat-surface injection plate (3% agarose in a Petri dish) and inject larvae with one pump of stained cells at the desired injection site (e.g., the yolk or the pericardium; see Figure 2A,B).
Figure 2: Injection site and representative images of screening xenografted fish.
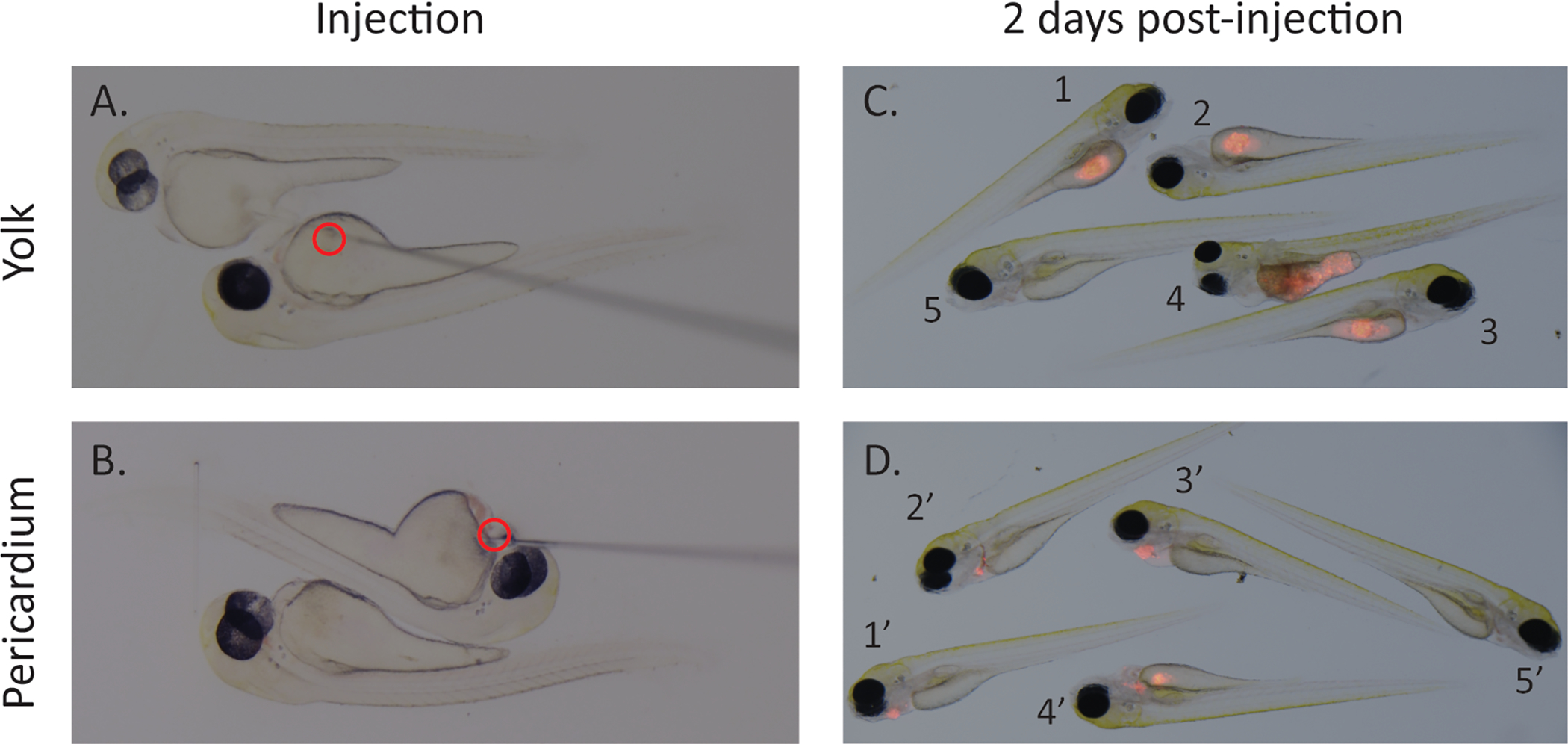
Images of microinjector needle at time of injection into either the yolk (A) or pericardium (B) of 2 dpf zebrafish larvae. Representative images of screening at 2 dpi depict selection of zebrafish for drug screen (C,D). Zebrafish with similar engraftment (1–3 and 1’–3’) should be selected, unengrafted zebrafish (5 and 5’) should be removed. For yolk injected fish, remove fish where borders of the yolk cannot be seen around engrafted cell mass (4) as it makes quantification difficult. For pericardium injected fish, remove fish where injected cell mass encroaches into yolk sac (4’). Scale bar = 0.5 mm.
NOTE: Injection site should be chosen based on the goal of the experiment. The most common injection site is the yolk. To get cells circulating in the bloodstream, the pericardium, duct of Cuvier, perivitelline space, or retro-orbital space can be used as injection sites. Orthotopic injection sites can also be used, such as the brain.
3.6. Wash larvae off the injection plate into a 10 cm2 Petri dish (30 larvae per plate) containing E3 media14 without methylene blue and incubate at 28 °C for a 1 h recovery period. Continue injecting until a desired number of larvae have been injected.
NOTE: Ideally, inject 2–2.5-fold the number of larvae needed for experiments. There will be some die-off of larvae due to stress from injection and the increased incubation temperature. Typically, after practice with the technique, 800–1,500 zebrafish larvae can be injected by a single person within the 1–3 h when stained cells should be injected.
3.7. Move plates of injected larvae to a 34 °C incubator. Do not place the Petri dishes of larvae directly on a metal shelf in the incubator to prevent overheating of the E3 water. For example, place an empty Petri dish between the shelf and Petri dish of larvae to act as a buffer. Remove dead zebrafish larvae after 24 and 48 hours post injection (hpi).
4. Setting up drug screen with xenografted zebrafish
4.1. At 48 hpi, screen zebrafish larvae for fluorescence/tumor engraftment and health (Figure 2C,D). Remove any dead or malformed zebrafish and select zebrafish with similar engraftment (Figure 2C,D, 1–3 and 1’–3’). Remove unengrafted zebrafish (Figure 2C,D, 5 and 5’).
4.1.1. For yolk injected fish, remove fish where borders of the yolk cannot be seen around engrafted cell mass (Figure 2C, 4) as it makes quantification difficult. For pericardium injected fish, remove fish where injected cell mass encroaches into the yolk sac (Figure 2D, 4’).
4.2. Add zebrafish to a 96-well plate. To do this, cut the tip off of a 200 μL pipette tip, just large enough for a 4 dpf zebrafish to fit through. Aspirate 150 μL of E3 media with one zebrafish from the plate using a P200 pipette, and add to an empty well of a flat-bottom 96-well plate.
4.3. Dilute the drug to be tested in the required volume of E3 media, at 150 μL per well. Prepare drug at 2-fold the desired concentration. For example, prepare 20 μM of drug in E3 if the desired final concentration is 10 μM, since half of the total volume in each well is comprised of the drug solution. For the DMSO control group, add DMSO at the same volume as the drug.
4.4. Add 150 μL of 2x diluted drug solution to each well containing zebrafish larvae in 150 μL of E3, for a final volume of 300 μL with 1x drug solution per well.
4.5. Incubate the plate at 34 °C. Check for dead zebrafish daily. After 2 days, if desired, refresh the drug by removing 200 μL of liquid from each well of the 96-well plate and replacing with 200 μL of 1x dilute drug solution or DMSO in E3 media.
NOTE: The best results were found after 3 days of drug treatment for this experiment; however, the length of drug treatment can vary between 2–4 days and may need to be optimized based on the experiment being done or drugs being used.
5. Imaging xenografted zebrafish using a fluorescence microscope equipped imaging unit and automated sampler unit
5.1. Prepare 1 L of fresh 4 mg/mL tricaine and 1.5 L of E3 media. Fill media bottle 1 with E3 media and media bottle 2 with tricaine.
5.2. Remove all unwanted fluorescent channels in the imaging software and add the desired channel (DiI for this experiment). Check the desired fluorescent channel as an image will only be taken for the channels with a check mark. Also, select how the images will be taken (z-stacks, automation, loops in series, etc.).
NOTE: For this experiment, the focus was manually set for each fish imaged to obtain the highest number of images with optimal focus.
5.3. Image the DMSO control fish before the drug-treated fish so that the appropriate exposure time can be set in the imaging software. Once the exposure is set, do not change the exposure time for the duration of the experiment.
NOTE: The focus can either be adjusted manually for each zebrafish to ensure there are no out of focus images, or for fully automated imaging, the same focus can be used between fish with out of focus images being discarded or fish reimaged prior to performing analysis. Furthermore, this experiment could be conducted by using any fluorescent imager followed by quantification of fluorescence using the ImageJ software.
6. Quantifying fluorescence using ImageJ
6.1. Open ImageJ software.
6.2. Go to File | Open and select the desired .czi file. The software will bring up an import options window.
6.2.1. For stack viewing select Hyperstacks, check Open Files Individually, check Autoscale, and check Split Channels. For the color option select Colorized.
6.3. Click Plugins | Macros | Record.
6.4. Click Image | Adjust | Threshold. Select image type as red in the dropdown menu on the right side of the threshold window. Adjust the minimum threshold until the software is only highlighting areas with fluorescence (Figure 3A) and click Apply. The software will convert the photo to black and white, with the selected area in black.
Figure 3: Drug treatment can reduce xenografted tumor area in vivo.

Representative images of zebrafish injected in the pericardium or yolk after 3 days of treatment with DMSO or drugs, either vincristine or dexamethasone. Area of engrafted tumor mass was quantified by setting a fluorescence threshold using ImageJ, selecting all pixels above the set threshold, and measuring the area and mean fluorescence of the selected regions. Pixels above the selected threshold appear in black, while pixels below the threshold appear in white. Pixels were measured in both the yolk and pericardium injected zebrafish images (A). Treatment with vincristine led to a decrease in engrafted tumor area compared to DMSO control with n = 4 fish treated per group (B). SD = standard deviation. Scale bar = 250 μm.
NOTE: Use the same threshold for each image in the drug screen to keep results standardized and comparable.
6.5. Click Analyze | Measure. The software will pull up a results window containing the fluorescent area for that image.
6.6. Click Create on the Macro Recorder window. This will open up a new window with the code for the macro. Highlight all of the desired images for analysis and open as in step 6.2.
6.7. Select Run on the window with the macro. The results window will now contain the area for each image.
NOTE: The image analysis can be done individually without recording and running a macro as well as by repeating the above steps for each image.
6.8. Copy the measured data into a spreadsheet. Average the total fluorescence of all control (DMSO) samples. Calculate the percent difference using the following formula: –[(average DMSO area – experimental area)/average DMSO area] × 100% (Figure 3B).
REPRESENTATIVE RESULTS:
Following the protocol described above, zebrafish were xenografted in the yolk and pericardium with primary patient PBMCs that were originally isolated from a T-cell acute lymphoblastic leukemia (T-ALL) patient at diagnosis and banked as a viable, frozen sample. At 48 hpi, xenografted fish were screened for fluorescently labeled tumor cells (Figure 2C,D) and treated with chemotherapy (dexamethasone or vincristine) or DMSO. Fish were imaged at 7 dpi, after 3 days on drug treatment using a fluorescence microscope equipped imaging unit and automated sampler unit (Figure 3A).
The fluorescent area/tumor burden was measured for each fish imaged using ImageJ and compared between the different drug treatment groups and DMSO (Figure 3B). Overall, xenografted fish treated with vincristine showed the largest and most consistent decrease in xenografted cell mass compared to DMSO treated fish. Dexamethasone treated fish showed about half the reduction in tumor area compared to vincristine, but still showed a reduction in tumor area compared to DMSO (Figure 3). This mimicked what was seen in the patient, as their leukemia rapidly responded to therapy with a combination of dexamethasone and vincristine. These results demonstrate the ability of zebrafish xenograft models to be amenable to drug screening and automated imaging and quantification, providing a platform for testing various patient samples or cell lines with different drugs or drug combinations.
DISCUSSION:
In this study, we demonstrated a standardized method for thawing and injection of primary patient leukemia cells into zebrafish as a xenograft model. We also established a protocol for high-throughput drug screening of xenografted zebrafish using a fluorescence microscope equipped imaging unit and automated sampler unit. Previously, xenografts have been reported with human cell lines, and quantification of xenografted tumors in a high-throughput manner has been a challenge in the field. This method serves as a basis for studies to utilize a fluorescence microscope equipped imaging unit and automated sampler unit as a way to automate imaging of xenografted zebrafish, with the ultimate goal of performing high-throughput drug screens to predict which drugs a specific patient’s cancer may respond to, opening the possibility for more personalized medicine.
Despite the ability to automate much of this protocol, there are still many technical challenges that should not be overlooked. First, it is critical that cells are injected into zebrafish as quickly as possible after staining to prevent cell clumping and cell death. Larvae will need to be dechorionated prior to performing the cell staining protocol. Glass filament needles should also be kept cold prior to loading the needle to reduce needle clogging. Additionally, using embryos produced by healthy adult zebrafish aged 6 months to 1 year is critical to ensure the best viability of xenografted larvae. Finally, xenografted fish should be carefully screened for tumor engraftment, and only those with similar tumor volumes should be used in drug screening to reduce variability in the final results.
Although we used primary patient leukemia PBMCs for our experiments, this protocol can be performed with any tumor type or cancer cell line. For cell lines in culture, adherent cells should be trypsinized and then washed in PBS before proceeding with the staining protocol. It is also important to note that engraftment rates can vary from one sample to the next and between sample types15. For example, in our PBMC sample, >90% of circulating PBMCs were leukemic blasts, but this number can vary significantly from one patient to the next, which may affect engraftment rate. Because results are compared to a DMSO control within the same sample type, there is an internal control for engraftment rate, yet this variation should be taken into consideration when deciding how many cells to inject per zebrafish. We have found success when using 250–1,000 cells injected per animal, with 500 being optimal for our studies. While our experiments concluded when larvae were 7 dpf, we would not expect xenografts to survive in the animals for longer timepoints, as the immune system begins to develop at this point, and would likely cause rejection of human cells. Immune compromised zebrafish lines have been created, with prkdc−/−;il2rga−/− zebrafish capable of engrafting human cancer cells16,17, which may be useful for longer term xenografts or assessing tumor recurrence after drug treatment. However, these immunodeficient lines must be maintained as heterozygotes, so larvae must be genotyped before use. Homozygous fish must also be treated with drugs to deplete macrophages to enable reliable engraftment of human cells, which may complicate drug screening results. Currently, these lines are neither practical nor necessary for large scale drug screening on larvae, which can be completed before the immune system is fully functional at 7 dpf18.
Our representative results focus on injection of cells into the pericardium and yolk for ease and speed of injection and increased viability; however, cancer cells can be injected into many other anatomic locations and we have had success using this workflow at other sites, including the duct of Cuvier, brain, retro-orbital, and the perivitelline sac. Additionally, it is difficult to estimate how much of each drug the larval fish absorb; if few drugs are used, ideally a toxicity screen at a range of doses (usually 0.1 to 25 μM) will be performed prior to the large scale assay to determine the maximum tolerated dose (MTD). We chose to use the MTD for each drug for our assay, however, 10 μM of drug is commonly used in the zebrafish field as a starting concentration for high-throughput drug screening and is generally well tolerated. Combinations of drugs in pools can be used as an initial screen, as well, to increase efficiency of screening through a large-volume compound library19.
Although this approach is more automated and efficient than previously reported workflows, this is still a labor-intensive and technically challenging protocol for anyone without prior experience in microinjecting zebrafish. Drug screening in zebrafish xenografts is unlikely to ever reach the ease and efficacy of in vitro screening of compound libraries and lacks some advantages of mouse xenograft models. For example, one major limitation with zebrafish xenografts is that cancer cells cannot be easily retrieved from fish after xenografting at useful numbers for cell banking or most downstream experiments. Even if this were possible, the human cancer cells would have been growing for several days in non-physiologic temperatures and environments and would not be practical for use in later applications. The effects of a slightly lower than physiological temperature on drug kinetics and tumor cell response is also not known, and may produce confounding results. Despite these caveats, zebrafish xenografts do fill a void in being a more practical and cost-efficient method for performing larger scale in vivo drug screens than is possible in mouse xenograft models. Additionally, zebrafish xenografts require far fewer cells for injection than mouse models, so a small amount of patient sample can be spread amongst hundreds to thousands of zebrafish, allowing for drug screens with large sample numbers. With fluorescent labeling, tumor cells can be monitored from the moment they are xenografted into the larval zebrafish, providing some standardization between the animals used in drug screens. Combining these benefits of zebrafish xenografts with the possibility of automated imaging and quantification of engrafted cells opens up many possibilities for making high-throughput drug screening of patient tumors for personalized medicine a reality.
ACKNOWLEDGMENTS:
This research was supported by a V Foundation V Scholar Award and NIH Grants DP2CA228043, R01CA227656 (to J.S. Blackburn) and NIH Training Grant T32CA165990 (to M.G. Haney).
Footnotes
A complete version of this article that includes the video component is available at http://dx.doi.org/10.3791/60996.
DISCLOSURES:
The authors have nothing to disclose.
REFERENCES:
- 1.Lee LM, Seftor EA, Bonde G, Cornell RA, Hendrix MJ The fate of human malignant melanoma cells transplanted into zebrafish embryos: assessment of migration and cell division in the absence of tumor formation. Developmental Dynamics. 233 (4), 1560–70 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Topczewska JM et al. Embryonic and tumorigenic pathways converge via Nodal signaling: role in melanoma aggressiveness. Nature Medicine. 12 (8), 925–32 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Haldi M, Ton C, Seng WL, McGrath P Human melanoma cells transplanted into zebrafish proliferate, migrate, produce melanin, form masses and stimulate angiogenesis in zebrafish. Angiogenesis. 9 (3), 139–51 (2006). [DOI] [PubMed] [Google Scholar]
- 4.Corkery DP, Dellaire G, Berman JN Leukaemia xenotransplantation in zebrafish--chemotherapy response assay in vivo. British Journal of Haematology. 153 (6), 786–9 (2011). [DOI] [PubMed] [Google Scholar]
- 5.Rennekamp AJ, Peterson RT 15 years of zebrafish chemical screening. Current Opinion in Chemical Biology. 24, 58–70 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ghotra VP et al. Automated whole animal bio-imaging assay for human cancer dissemination. PLoS One. 7 (2), e31281 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chang T-YY, Pardo-Martin C, Allalou A, Wählby C, Yanik MF Fully automated cellular-resolution vertebrate screening platform with parallel animal processing. Lab On a Chip. 12 (4), 711–6 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pardo-Martin C et al. High-throughput in vivo vertebrate screening. Nature Methods. 7 (8), 634–6 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pulak R Tools for automating the imaging of zebrafish larvae. Methods. 96, 118–126 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Henn K, Braunbeck T Dechorionation as a tool to improve the fish embryo toxicity test (FET) with the zebrafish (Danio rerio). Comparative Biochemistry and Physiology Part C: Toxicology & Pharmacology. 153 (1), 91–8 (2011). [DOI] [PubMed] [Google Scholar]
- 11.White RM et al. Transparent adult zebrafish as a tool for in vivo transplantation analysis. Cell Stem Cell. 2 (2), 183–9 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paatero I, Alve S, Gramolelli S, Ivaska J, Ojala P Zebrafish Embryo Xenograft and Metastasis Assay. Bio-Protocol. 8 (18), (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rosen JN, Sweeney MF, Mably JD Microinjection of zebrafish embryos to analyze gene function. Journal of Visualized Experiments. (25), e1115 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.E3 medium (for zebrafish embryos). Cold Spring Harbor Protocols. 2011. (10), pdb.rec66449–pdb.rec66449 (2011). [Google Scholar]
- 15.Wertman J, Veinotte CJ, Dellaire G, Berman JN The Zebrafish Xenograft Platform: Evolution of a Novel Cancer Model and Preclinical Screening Tool. Advances in Experimental Medicine and Biology. 916, 289–314 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Tang Q et al. Optimized cell transplantation using adult rag2 mutant zebrafish. Nature Methods. 11 (8), 821–4 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moore JC et al. Single-cell imaging of normal and malignant cell engraftment into optically clear prkdc-null SCID zebrafish. The Journal of Experimental Medicine. 213 (12), 2575–2589 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yan C et al. Visualizing Engrafted Human Cancer and Therapy Responses in Immunodeficient Zebrafish. Cell. 177 (7), 1903–1914.e14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kawahara G et al. Drug screening in a zebrafish model of Duchenne muscular dystrophy. Proceedings of the National Academy of Sciences of United States of America. 108 (13), 5331–5336 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]