

Abstract
Cognitive decline is a debilitating aspect of aging and neurodegenerative diseases such as Alzheimer's disease are closely associated with mitochondrial dysfunction, increased reactive oxygen species, neuroinflammation, and astrogliosis. This study investigated the effects of decreased mitochondrial antioxidant response specifically in astrocytes on cognitive performance and neuronal function in C57BL/6J mice using a tamoxifen-inducible astrocyte-specific knockout of manganese superoxide dismutase (aSOD2-KO), a mitochondrial matrix antioxidant that detoxifies superoxide generated during mitochondrial respiration. We reduced astrocyte SOD2 levels in male and female mice at 11–12 months of age and tested in an automated home cage (PhenoTyper) apparatus for diurnal patterns, spatial learning, and memory function at 15 months of age. aSOD2-KO impaired hippocampal-dependent spatial working memory and decreased cognitive flexibility in the reversal phase of the testing paradigm in males. Female aSOD2-KO showed no learning and memory deficits compared with age-matched controls despite significant reduction in hippocampal SOD2 expression. aSOD2-KO males further showed decreased hippocampal long-term potentiation, but paired-pulse facilitation was unaffected. Levels of d-serine, an NMDA receptor coagonist, were also reduced in aSOD2-KO mice, but female knockouts showed a compensatory increase in serine racemase expression. Furthermore, aSOD2-KO mice demonstrated increased density of astrocytes, indicative of astrogliosis, in the hippocampus compared with age-matched controls. These data demonstrate that reduction in mitochondrial antioxidant stress response in astrocytes recapitulates age-related deficits in cognitive function, d-serine availability, and astrogliosis. Therefore, improving astrocyte mitochondrial homeostasis may provide a therapeutic target for intervention for cognitive impairment in aging.
SIGNIFICANCE STATEMENT Diminished antioxidant response is associated with increased astrogliosis in aging and in Alzheimer's disease. Manganese superoxide dismutase (SOD2) is an antioxidant in the mitochondrial matrix that detoxifies superoxide and maintains mitochondrial homeostasis. We show that astrocytic ablation of SOD2 impairs hippocampal-dependent plasticity in spatial working memory, reduces long-term potentiation of hippocampal neurons and levels of the neuromodulator d-serine, and increases astrogliosis, consistent with defects in advanced aging and Alzheimer's disease. Our data provide strong evidence for sex-specific effects of astrocytic SOD2 functions in age-related cognitive dysfunction.
Keywords: aging, astrogliosis, cognitive function, d-serine, long-term potentiation, SOD2
Introduction
Aging is associated with reductions in mitochondrially derived energy production (Vančová et al., 2010), global increases in oxidative load (Poon et al., 2004a,b), and a concomitant decline in correlates of cognitive function (Gibbs et al., 2009; Gibbs and Bowser, 2009; Kubik and Philbert, 2015), with cell-specific effects on neuronal and astroglial function (Parihar and Brewer, 2007a,b; Parihar et al., 2008; Kubik and Philbert, 2015). Dysregulation of mitochondrial function and neuroinflammation are part of the etiology of many age-related human diseases, including neurodegenerative disorders such as Alzheimer's disease (AD; Parihar and Brewer, 2007a,b; Flynn and Melov, 2013; Guo et al., 2013). Within the brain, astrocytes have a key role in maintaining cognitive function and provide energy substrates to neurons, respond to injury, and regulate the synaptic milieu for optimal neuronal function. Enhancing astrocyte mitochondrial metabolism has been shown to have neuroprotective effects against the pathologic outcomes of neuroinflammation (Zheng et al., 2010; Diekman et al., 2013), suggesting a key link between astrocyte mitochondrial function and age-related diseases (Rose et al., 2020).
Oxidative stress in the brain is critically regulated by the astrocyte antioxidant system, which, when dysregulated, contributes to oxidative stress and pathology in the brain (Gibbs et al., 2009; Gibbs and Bowser, 2009; Kubik and Philbert, 2015; Rizor et al., 2019; Habib et al., 2020). Astrocytes produce an order of magnitude more mitochondrial-derived reactive oxygen species (ROS) than neurons (Lopez-Fabuel et al., 2016). Additionally, astrocytes release glutathione and superoxide dismutases (SODs) that facilitate the breakdown and detoxification of ROS (Birben et al., 2012; Bolanos, 2016; McBean, 2018). Recent studies, using single-cell RNA sequencing, have shown a downregulation of antioxidant response gene expression in reactive astrocytes in aged mice (Clarke et al., 2018), suggesting a key link between astrocyte redox capacity and pathologic phenotype of astrocytes in aging and Alzheimer's disease (Habib et al., 2020). However, whether increased mitochondrial-derived ROS in astrocytes alone drive induction of an inflammatory astrocyte phenotype and cognitive dysfunction in the aging brain is unclear.
Mitochondria generate ROS, such as superoxide anions, during energy metabolism and ATP synthesis by the electron transport chain. Superoxide is detoxified by conversion to hydrogen peroxide via manganese superoxide dismutase (SOD2) located in the mitochondrial matrix. Ablation of SOD2 in the brain results in pathologic gliosis, encephalopathy, and perinatal death (Izuo et al., 2015). Reduction in SOD2 levels impairs mitochondrial respiration, increasing oxidative stress, resulting in tissue disfunction (Williams et al., 1998; Van Remmen et al., 2001; Song et al., 2020). SOD2 deficiency is also associated with accelerated Alzheimer's pathology in transgenic mice (Esposito et al., 2006; Flynn and Melov, 2013) and age-related decline in NMDA receptor (NMDAR)-driven activity (Carvajal et al., 2018). These reports suggest a critical role for SOD2 in the brain, albeit cell-specific mechanisms of SOD2 in the aging brain are not clearly understood.
In this study, we investigated the effects of astrocyte-specific induction of mitochondrial oxidative stress on cognitive function and neuroinflammation. We show that Sod2 levels are reduced in the hippocampus of aged mice and that astrocyte-specific knockout of SOD2 (aSOD2-KO) impaired hippocampal-dependent working memory specifically in male mice, while no specific deficits were seen in the female mice. These behavioral impairments correlated with decreased hippocampal long-term potentiation (LTP) and reduced levels of d-serine. aSOD2-KO also increased the density of astrocytes within the CA1 region of the hippocampus, indicative of astrogliosis, and decreased the branch length of astrocyte projections, specifically in males. The preservation of cognitive function in females despite the reduction of SOD2 suggests the presence of protective compensatory mechanisms and possibly for estrogen-mediated effects in females. These data suggest that astrocytic mitochondrial redox homeostasis plays a critical role in d-serine availability and cognitive function in males, and that improving astrocytic mitochondrial function may be a potential therapy for age-related cognitive impairment. Understanding sex differences in redox homeostasis may provide insights into protective mechanisms that can be harnessed for therapeutic intervention.
Materials and Methods
Animals.
All procedures were approved by and followed the guidelines of the Institutional Animal Care and Use Committee of the University of Oklahoma Health Sciences Center (OUHSC). Mice were housed (3–4/cage) in Allentown XJ cages with Anderson's Enrich-o'Cobs bedding in the specific pathogen-free (including Helicobacter and parvovirus) Rodent Barrier Facility at OUHSC. Mice were bred on a 14/10 h light/dark cycle, and weaned mice were maintained on a 12 h light/dark cycle at 21°C with ad libitum access to standard irradiated bacteria-free rodent chow (5053 Pico Lab, Purina Mills) and reverse osmosis filtered water. Both male and female mice were used in all experiments.
Astrocyte-specific ablation of SOD2.
Sod2 floxed (Sod2f/f) mice were generated by Takahiko Shimizu (National Center for Geriatrics and Gerontology; Aichi, Japan) (Ikegami et al., 2002) and provided to H.V.R. as described previously (Ikegami et al., 2002; Lustgarten et al., 2009). GFAP-CreERT2 male mice (stock #012849, The Jackson Laboratory) were bred with Sod2f/f female mice to obtain the founder colony of Cre+/Sod2 heterozygous floxed mice. These mice were allowed to breed with Sod2f/f female mice to generate experimental cohorts of Cre–/(floxed and C57 age-matched controls) and Cre+/Sod2f/f mice. Astrocyte-specific knockout of Sod2 (referred to as SOD2) was induced via intraperitoneal injections of tamoxifen (TAM) dissolved in corn oil (75 mg tamoxifen/kg body weight) for 5 d at 11–12 months of age. Mice were allowed to recover for 4 months before behavioral assessments. Mice were genotyped for GFAP-CreERT2 and Sod2f/f using primers and PCR conditions specified by The Jackson Laboratory (stock #012849). Primer pairs for GFAP-CreERT2 transgene (forward, 5′-GCCAGTCTAGCCCACTCCTT-3′; and reverse, 5′-TCCCTGAACATGTCCATCAG-3′) yielded a 200 bp PCR product. Sod2f/f was genotyped using the three primers in the same reaction, 5′-TTAGGGCTCAGGTTTGTCCATAA-3′, 5′-CGAGGGGCATCTAGTGGAGAAG-3′, and 5′-AGCTTGGCTGGACGTAA-3′, as previously described (Ikegami et al., 2002; Izuo et al., 2015).
Automated home-cage testing (PhenoTyper).
The PhenoTyper (model 3000, Noldus) automated home-cage testing apparatus was used to assess the spontaneous activity, initial discrimination learning, and reversal learning of individual mice in our study, as previously described (Maroteaux et al., 2012; Loos et al., 2014; Logan et al., 2018b, 2019). The PhenoTyper apparatus consists of a transparent plastic cage (length, 30 cm; width, 30 cm; height, 35 cm) that includes a shelter and CognitionWall in opposing corners of the cage. Water was available ad libitum. Following a 6 h adaptation period in the apparatus, the activity of animals was continuously recorded for 90 h beginning at 4:00 P.M. on day 1 using EthoVision version 14 (Noldus) software (Maroteaux et al., 2012). During cognitive testing, diurnal spontaneous activity was tracked for the 90 h of the study with data points collected every 15th of a second and used to calculate the total distance traveled by each mouse per hour. Mice were required to pass through the left entrance of the CognitionWall during the initial discrimination (acquisition) phase, and the right entrance during the reversal phase to obtain a food reward (Dustless Precision Rodent Pellets, catalog #F05684, Bio-Serv). Mice were rewarded with a food pellet using an FR5 (fixed ratio 5) after five successful entries into the correct hole during testing, as previously described (Logan et al., 2018a). Following acquisition (49 h), the reversal phase modified the task to require entry into the right hole as the correct response, requiring the animal to extinguish previously learned behavior and acquire a new response. Following testing, data were exported from EthoVision and processed using Python scripts. Success rates for both initial discrimination and reversal learning were calculated post hoc and defined as the percentage (80% criterion) of correct entries of the trailing 30 entries through the appropriate entrances of the CognitionWall and plotted as survival graphs with the number of entries plotted against the percentage of mice that met the criterion for the group. The following dependent variables to reach a specific success rate were calculated for both initial discrimination and reversal learning: the percentage of animals reaching criterion, entries to criterion, errors to criterion, and time (hours) to criterion (Logan et al., 2018b). The Learning Index was calculated as the correct entries minus the incorrect entries divided by the total number of entries and plotted as a cumulative value (Cumulative Learning Index; Logan et al., 2018b, 2019). Cognitive flexibility was calculated as correct entries minus the incorrect entries divided by the total number of entries during the first dark phase of the reversal between the 51 and 61 h after initiation of the experiment. Young (6-month-old) C57BL/6J male and female mice were included as a reference group for optimal spontaneous activity and cognitive function in the PhenoTyper.
Radial arm water maze.
Mice (Control and aSOD2-KO; 15–16 months; n = 10/group) were tested for spatial learning using an eight-arm radial arm water maze (RAWM) during the morning hours (9:00 A.M. to 12:00 P.M.) only as previously described (Logan et al., 2018b). Briefly, the eight-arm water maze is 67 cm in diameter and filled approximately two-thirds with an opaque liquid (water with white food coloring) containing a hidden platform just beneath the surface of the liquid in one arm. Each mouse was placed on the hidden platform in the target arm of the maze for a few seconds before beginning the actual test. During testing, each mouse was given four 60 s trials a day (beginning at 9:00 A.M.) for 2 d to find a hidden platform in one specific target arm. Animals were randomly placed in an arm other than the target arm for each trial. Mice were guided to the platform if they failed to find it in the target arm at the end of each 60 s trial. Movement of the mice within the maze was recorded by video monitoring and analyzed using EthoVision version 14 software. The number of errors (number of entries into incorrect arms) and path length (in centimeters; total distance traveled) to target were recorded, and nonmoving duration (in seconds) and latency (time to reach platform) were recorded. Data were exported from EthoVision and analyzed using GraphPad Prism 6.0.2.3.
Electrophysiology.
Following behavioral testing, acute brain slices (350 µm thick) from both male and female mice at 15 months were used for electrophysiological assessments. Field EPSPs (fEPSPs) were generated in the CA1 region of the hippocampus by stimulating electrodes placed in the CA1 and CA3 regions of the hippocampus along the Schaffer collateral pathway. Input/output (I/O) curves were generated by applying increasing stimulus currents to the pathway from 0 to 100 µA and recording the responses as previously described (Orock et al., 2018, 2020; Nagaraja et al., 2021). For LTP experiments, the stimulus strength for generating fEPSPs was determined as 40–50% of the stimulus strength needed to generate the maximum fEPSP amplitude during the I/O curve measurement. The slice was stimulated once every 30 s until a stable baseline lasting at least 10 min was observed. LTP was induced using 100 high-frequency stimulation pulses at 100 Hz applied three times with 30 s intervals. Baseline stimulation was resumed and fEPSPs recorded for at least 60 more minutes. Finally, we recorded another I/O curve generated as described above. For all recordings, we used the MED-64 system and Mobius software (Alpha MED Scientific). Potentiation was calculated as the percentage increase of the mean fEPSP descending slope (10–90 section) after high-frequency stimulation and normalized to the mean fEPSP descending slope of baseline recordings during 3 min prior tetanus. Statistical significance was calculated on the averaged values obtained from the last 10 min of recordings. We also examined the paired-pulse facilitation (PPF) ratio by applying two stimuli at an intensity of 40–50% of the maximum fEPSP in rapid succession. We applied the second identical stimulus 10, 20, 30, 40, 50, 75, 100, and 200 ms after the first stimulus. PPF was measured by examining the ratio of the fEPSP slope of second response to the first. Data are presented as multiple recordings (N as listed per experiment) from a minimum of two to three brain slices from n = 5 mice/group.
Brain sectioning and immunohistochemistry.
Mice that were not used for electrophysiology were decapitated and the brains were rapidly extracted from the skull and processed as previously described (Logan et al., 2018a). Half the brain was fixed in 4% paraformaldehyde in 0.1 m PBS overnight at 4°C, cryoprotected in 30% sucrose in PBS, embedded in tissue blocks using Cryo-Gel embedding medium (Leica Biosystems), and frozen at −80°C. Sagittal sections (30 µm) were obtained using a cryostat with adjacent sections collected in each well of a 12-well plate. Thus, sections in one well are in increments 360 µm apart. Corresponding wells with sections from young and old brains were rinsed in PBS and processed for immunohistochemistry. Sections were incubated in sodium citrate buffer for antigen retrieval and subsequently blocked in goat blocking buffer (10% normal goat serum, 5% BSA, 1% Triton X in HBSS). Sections were then incubated with primary antibodies against glial fibrillary acidic protein (GFAP; 1:1000; catalog #G3893; Sigma-Aldrich) and Iba1 (1:50; catalog #019–19 741 FUJIFILM Wako) overnight followed by incubation with goat anti-mouse and goat anti-rabbit secondary antibodies (1:2000 dilution). The immunostained sections were mounted using Prolong Gold with DAPI and imaged using a Leica THUNDER large specimen imaging microscope using Leica LasX software (Leica Biosystems) to stitch together image montages of immunostained hippocampi for analysis. For Sholl and Delaunay analysis of astrocyte morphology and spacing, respectively, images encompassing the CA1 region of the hippocampus were captured using a Ti2 microscope (Nikon Instruments) 40× objective lens using the extended depth of focus and image stitching functions of Nikon Elements software. Image montages were captured from several sections from each specimen for analysis.
Image processing and analysis.
High-magnification (40× objective) images were taken from the comparable CA1 region of the hippocampus for all mice (using DAPI channel for selection). Individual GFAP-labeled astrocytes were processed using despeckled maximum intensity projections and binarized. Sholl (semi-log) analysis was performed using the Neuroanatomy Plugin (Legacy: Sholl analysis) in Fiji-ImageJ 2 (Schindelin et al., 2012; Rueden et al., 2017) with the first concentric circles set at 3 µm for astrocytes, and the subsequent concentric circles set with a radius step of 1.5 µm. The maximal branch length (rmax), critical value (rcrit), and maximal number of intersections (Nmax) were calculated for each astrocyte as previously described (Hösli et al., 2022), and group distributions are plotted as violin plots using Prism software. For all analyses, 45 individual CA1 astrocytes imaged from at least two to four sections per animal (n = 3–4/group) were analyzed. The experimenter was blinded to the genotypes during analysis.
Delaunay analysis was performed using the Delaunay-Voronoi plug-in of Fiji-ImageJ 2 (Schindelin et al., 2012; Rueden et al., 2017) to determine the mean distance between neighboring astrocytes in the stratum radiatum/lacunosum and stratum oriens of the CA1 region in control and aSOD2-KO mice (n = 5 control mice per sex; n = 4 male, n = 5 female aSOD2-KO mice). High-magnification montages of GFAP-immunolabeled astrocytes in the CA1 region of the hippocampus were imported into Fiji/ImageJ, and spatial scale was calibrated. GFAP-labeled astrocytes were visually identified and marked using the Point tool. The Delaunay triangulation was generated to calculate the mean distance between neighboring astrocytes and map the triangular domains formed by three neighboring astrocyte cell bodies. The overall mean distance between astrocytes for each specimen was then determined by averaging the distance measurement across all images from a given specimen.
Primary astrocyte cultures.
Primary astrocytes from mouse cortical/hippocampal mixed cultures were established from postnatal day 1–3 pups from C57BL/6J and Sod2f/f mice as previously described (Logan et al., 2018a). Briefly, following papain enzymatic digestion and trituration, cells were resuspended in growth media [DMEM containing 2% NuSerum, 10% fetal bovine serum, penicillin (10 U/ml), streptomycin (10 µg/ml), and l-glutamine (29.2 µg/ml)] and seeded on 50 µg/ml poly-d-lysine-coated plates. Astrocytes were split on day 7 and seeded at a density of 1.5 × 106/10 cm2 plate. Sod2f/f astrocyte cultures (∼90% confluence) were treated with AAV1-CMV-GFP (GFP) or AAV1-CMV-GFP-CRE (CRE) at 8 × 103 gene copies/cell (Addgene) on day 2 after splitting. Cells were fed every 3–4 d, with half of the media being replaced with fresh growth media. Following washes with 1× PBS on day 5 post-viral treatment, astrocytes were scraped and cell pellets were flash frozen for further analysis. Human hippocampal astrocytes (catalog #1830, ScienCell Research Laboratories) were cultured using the same methodology for mouse astrocytes. Astrocytes (C57BL/6J mouse and human) were treated with 5 µm MitoPQ (catalog #18808, Cayman Chemicals) in serum-free media for 24 h and analyzed for d-serine biosynthesis.
Western blotting.
Lysates (hippocampal and cortical) were prepared in RIPA buffer (catalog #R0278, Sigma-Aldrich) containing protease inhibitors (cOmplete Protease Inhibitor Cocktail, Roche). Equal amounts of protein (10 µg/lane) were subjected to SDS/PAGE and subsequently transferred to a nitrocellulose membrane. The membrane was blocked in a solution containing 5% bovine serine albumin (catalog #A7030, Sigma-Aldrich) in TBST (20 mm Tris, 0.9% NaCl, 0.1% Tween-20, pH 7.4; catalog #T5912, Sigma-Aldrich), and probed with primary antibodies for SOD2 (1:5000; catalog #ab13533, Abcam), GADPH (1:2000; catalog #97166, Cell Signaling Technology), and H2B (1:1000; catalog #ab52484, Abcam). Additionally, membranes were probed for GluA1 (1:1000; catalog #75–327), GluN2A (1:500; catalog #74–325), and GluN2B (1:1000; catalog #75–097, University of California, Davis, Davis, California); GluN2C (1:1000; catalog #ab110, Abcam); GluN1 (1:500; catalog #5704S), and GluA2 (1:1000; catalog #13607S, Cell Signaling Technology); and serine racemase (SRR; 1:1000; catalog #17955–1-AP, Proteintech). For 3-nitrotyrosine (3-NT; 1 µg/ml; catalog #MAB3248, Biotechne) immunoblotting, protein subjected to SDS/PAGE was transferred to a PVDF membrane, blocked with 2% nonfat dry milk in TBST and incubated with primary antibody overnight. Following secondary incubation with IRDye donkey anti-mouse and donkey anti-rabbit antibodies (1:5000; catalog #926–68072 and #926–32213, LI-COR), images were captured using an Odyssey CLX imaging system (LI-COR). Band densitometry was quantified using ImageStudio software (LI-COR), with all signals normalized to GAPDH or H2B (as indicated) as a loading control.
RNA/cDNA preparation and quantitative real-time PCR.
Total RNA from control and aSOD2-KO male and female hippocampus was extracted using the RNeasy Mini Kit (Qiagen). cDNA was prepared from equal concentrations of total RNA (2.0 µg) using the High-Capacity RNA-to-cDNA Kit (Thermo Fisher Scientific). Quantitative RT-PCR was performed using the following gene-specific TaqMan probes: Sod2 (spanning exons 2 and 3; Mm00449726_m1), Gfap (Mm01253033_m1), and Aldh1l1 (Mm03048957_m1). Housekeeping genes Ywhaz (Mm03950126_s1), Hprt (Mm03024075_m1), and B2M (Mm00437762) were used for normalization. Quantitative PCR and melt–curve analyses were performed using TaqMan Universal PCR Master Mix with UNG (Thermo Fisher Scientific) and the QuantStudio 12K Flex Real-Time PCR System (Thermo Fisher Scientific). Expression data were calculated from six independent samples unless otherwise stated, each with two replicates, and are presented (mean ± SEM) relative to the expression of the geometric mean of the housekeeping genes.
Quantification of d-serine concentrations.
Equal protein (200 µg) from hippocampal lysates was resuspended in serine assay buffer, deproteinized through 10 kDa spin columns (Corning) and processed using the DL-Serine Assay Kit (Sigma-Aldrich) according to manufacturer instructions (controls: n = 7 male, n = 9 female; aSOD2-KO: n = 5 male, n = 7 female). Control, MitoPQ-treated (C57BL/6J and human hippocampal), and SOD2 knock-out astrocytes (∼1.5 × 106 cells/n; n = 5/group) were pelleted, frozen, and later homogenized directly in assay buffer. d-serine only and total serine reaction wells were performed in duplicate, while sample background control wells were done in singlicate. The assay and data analysis were completed according to the manufacturer instructions to calculate d-serine and total serine concentrations of samples.
Targeted quantitative proteomics.
Lysates (100 µg protein; n = 4–5/group) were run on an SDS gel for analysis as previously described (Kinter et al., 2012). Each gel lane was cut as a complete sample, then divided into smaller pieces and washed/destained. The proteins were reduced with DTT and alkylated with iodoacetamide. Samples were then washed with ethanol and bicarbonate and digested with 1 µg of trypsin overnight at room temperature. The peptides produced were extracted from the gel, the extract evaporated to dryness, and reconstituted in 1% acetic acid for analysis. The digest samples were injected in 5 µl aliquots and quantified using QEx orbitrap and TSQ systems. A BSA internal standard was added for quantification, and the mass spectrometer was operated in selected reaction monitoring mode to analyze two peptides per protein. Data were analyzed using the program SkyLine, and the response for each protein was calculated as the geometric mean of the two-peptide area normalized to the response for the BSA standard. The principal component analysis (PCA) plot was generated using ClustVis with default settings (row scaling = unit variance scaling; PCA method = singular value decomposition with imputation; clustering distance for rows = correlation; clustering method for rows = average; tree ordering for rows = tightest cluster first; Metsalu and Vilo, 2015).
Statistical analyses.
All experiments were performed in multiple independent replicates per group as described for each experiment. Behavioral data in the PhenoTyper was analyzed using repeated-measures ANOVA as previously reported using JMP (version 15.2.0; SAS; Logan et al., 2018b). Longitudinal measurements of interest for each mouse and their dependence on covariates were analyzed by fitting linear mixed-effects models as implemented by the “lme” function in the R package “nlme.” The mixed-effects model extends the classical linear model (regression/ANOVA) by including random effects to accommodate for longitudinal intracorrelation and complex nested multilayer experimental design. Statistical differences between experimental groups for RAWM experiments were analyzed using a multivariate repeated-measures ANOVA followed by Dunnett's post hoc test. Statistical analyses on electrophysiology data were performed using Student's t test for LTP time course (on averaged values of last 10 min) and two-way ANOVA followed by Bonferroni's post hoc test for I/O curves and PPF. Sholl data were analyzed using a two-way ANOVA with mixed-effects model as previously described (Hösli et al., 2022). All other statistical methods were performed using GraphPad Prism version 9.1 for Mac OS X. Statistical significance for all other experiments with two groups was determined using unpaired two-tailed (unless otherwise noted) Student's t test with p < 0.05 was considered significant. For all data, outliers were determined using ROUT (Q = 2%) method. Data are represented as the mean ± SEM with p values identified in figure legends. Male and female data histograms are represented on a shared axis but are divided using a dotted line to denote sex-specific analyses. Significance is indicated by p value measurements with p < 0.05 considered to be significant (*p < 0.05; **p < 0.01; ***p < 0.001).
Results
Astrocyte-specific SOD2 knockout increases antioxidant response in males
We have previously reported that an increase in the reactive astrogliosis marker GFAP correlates with the loss of hippocampal-dependent spatial working memory in aged male mice (Logan et al., 2018a). Mitochondrial redox imbalance is associated with cognitive decline in aging, prompting our exploration of the role of SOD2 in astrocyte redox function. We show that Sod2 transcript expression was significantly reduced in the hippocampus of aged (22 months; n = 11) male mice compared with young (6 months; n = 7) controls (Fig. 1A; p = 0.02), mice that we have previously shown to have increased GFAP expression compared with young controls (Logan et al., 2018a).
Figure 1.
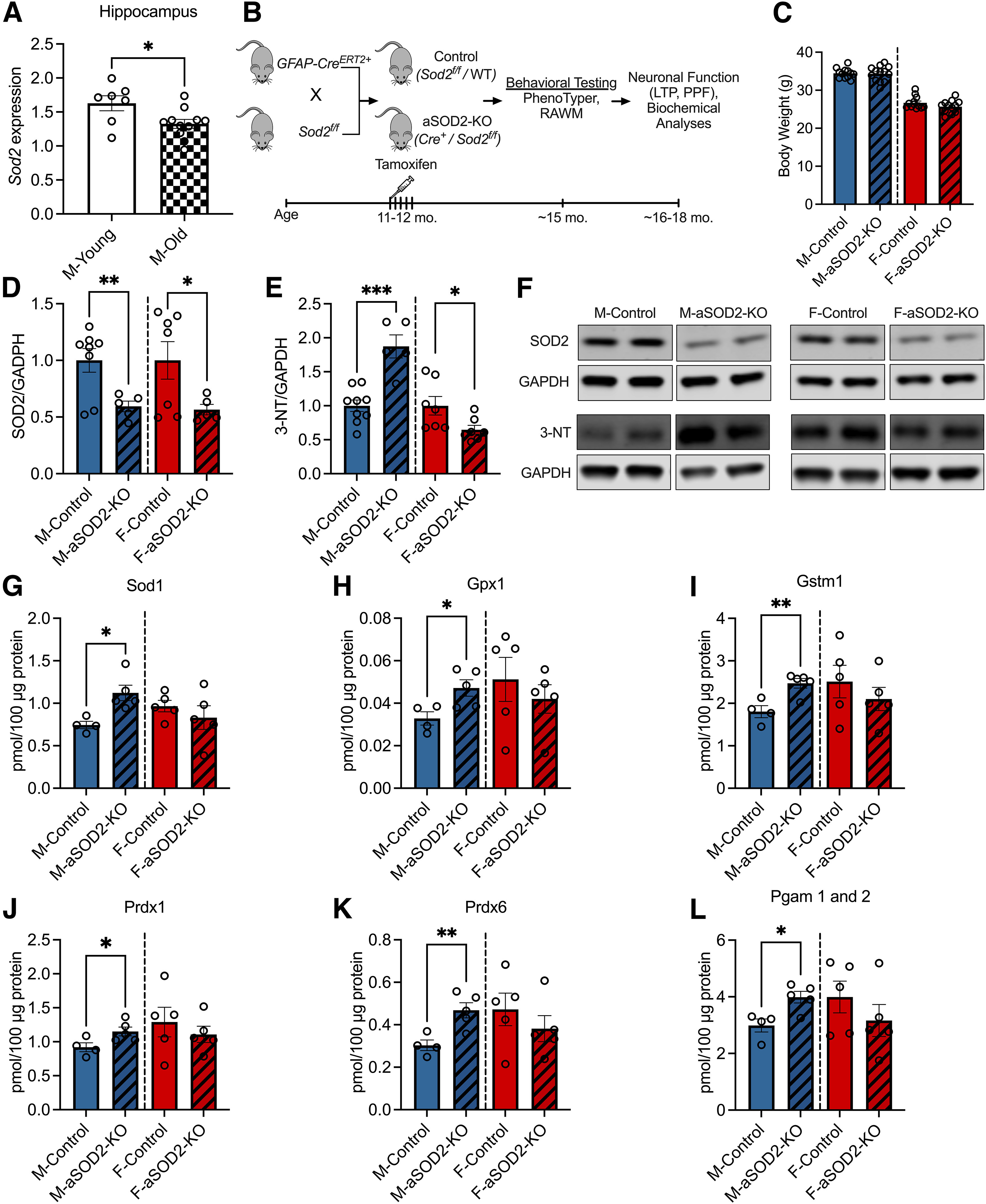
Effect of astrocyte-specific knockout of SOD2 on antioxidant response in male and female mice. A, Bar plots depicting decreased Sod2 mRNA expression in the hippocampus of aged (22 months of age; n = 11; shaded bar) male mice compared with young (6 months of age; n = 7; white bar) C57 controls (p = 0.02, two-tailed t test). B, Experimental schematic displaying the timeline of tamoxifen-induced aSOD2-KO in mice and subsequent behavioral testing, neuronal functional studies, and biochemical analyses. C, Bar plots depicting the weight of animals in our experimental cohort at the time of behavioral testing. There was no difference detected between the weights of aSOD2-KO animals compared with controls in male or female animals. D, Representative blot depicting reduced expression of SOD2 in the hippocampus of aSOD2-KO in male (n = 5; p = 0.006) and female (n = 5; p = 0.029) mice compared with aged-matched male (n = 8) and female (n = 7) controls, respectively (one-tailed t test). F, Blot was normalized to GAPDH. E, Western blot quantification of the oxidative stress marker 3-NT is significantly increased in aSOD2-KO males (n = 9/control, n = 5/KO; p = 0.001) and decreased in aSOD2-KO females (n = 6/controls, n = 7/KO; p = 0.018) compared with age-matched controls. F, Blot was normalized to GAPDH. G–L, Bar plots depicting quantification of mitochondrial antioxidant proteins Sod1 (p = 0.011), Gpx1 (p = 0.027), Gstm1 (p = 0.007), Prdx1 (p = 0.042), Prdx6 (p = 0.0087), and Pgam 1 and 2 (p = 0.014) in aSOD2-KO mice via targeted proteomic mass spectrometry analysis (n = 4–5 animals/group). For graphs in C–J, colored bars represent the following: males (blue), females (red), and aSOD2-KO (shaded). Error bars depict the mean ± SEM. Significance was tested using unpaired Student's t test (*p < 0.05, **p < 0.01, ***p < 0.001). Please see Extended Data Tables 1-1, 1-2, and 1-3.
Mass spectrometry analysis listing abundance of targeted proteins in glycolysis and gluconeogenesis. Download Table 1-1, DOCX file (17.6KB, docx) .
Mass spectrometry analysis listing abundance of targeted proteins in β-oxidation. Download Table 1-2, DOCX file (17.4KB, docx) .
Mass spectrometry analysis listing abundance of targeted proteins in Kreb's cycle. Download Table 1-3, DOCX file (20.6KB, docx) .
To understand the effects of astrocyte SOD2 on cognitive function and neuroinflammation, we generated astrocyte-specific SOD2 knockout in cohorts of both male and female mice. Female Sod2f/f mice were bred with male mice expressing CreERT2 under the control of the astrocyte-specific GFAP promoter. Astrocyte-specific knockout was induced via intraperitoneal TAM injection (11–12 months of age) to generate age-matched controls (Cre–/floxed and C57BL6/J) and aSOD2-KO (Cre+/Sod2f/f; aSOD2-KO) mice that were behaviorally tested at ∼15 months. Timeline of end points are depicted in the schematic (Fig. 1B). No differences in the body weights of control and aSOD2-KO mice (Fig. 1C) in either sex were noted at the time of behavioral testing.
Following behavioral testing, brain tissue was harvested at 16–18 months for molecular analyses. We analyzed the expression of SOD2 via Western blotting along with targeted proteomics of mitochondrial protein expression in brain lysates. Mice with aSOD2-KO showed a significant reduction in SOD2 expression compared with age-matched controls in male (41% decline; p = 0.006) and female (44% decline; p = 0.029) mice, respectively (Fig. 1D,F; n = 5–8 animals/group, single-tailed t test). To assess the effects of aSOD2-KO on oxidative stress, we measured levels of 3-NT via immunoblotting on male and female cortical lysates. Nitrotyrosine is a marker of protein damage resulting from the reaction between superoxide anion and nitric oxide, and is elevated with neuroinflammation (Bourgognon et al., 2021) and neurodegenerative conditions such as AD (Guivernau et al., 2016). Male aSOD2-KO mice showed a significant increase in 3-NT levels (Fig. 1E,F; n = 5–9 animals/group; p = 0.002), while 3-NT levels were significantly decreased in females (n = 7 animals/group; p = 0.036) compared with their respective age-matched controls.
Targeted proteomic analysis of mitochondrial antioxidant proteins by mass spectrometry revealed significant increases in Sod1 (Fig. 1G; p = 0.011), glutathione peroxidase (gpx1; Fig. 1H; p = 0.027), glutathione S-transferase Mu-1 (gstm1; Fig. 1I; p = 0.007), and peroxiredoxins (prdx1 and prdx6; Fig. 1J,K; p = 0.0429 and p = 0.0087, respectively) in male aSOD2-KO mice, but not in females (n = 4–5 mice/group). Expression of glycolytic enzyme phosphoglycerate mutase (Pgam1 and 2) protein was significantly elevated in males but not in females (Fig. 1L; p = 0.014). Other important proteins in the glycolysis/gluconeogenesis pathway that were elevated in male aSOD2-KO were enolase (Eno2; p = 0.046) and triosephosphate isomerase 1 (Tpi1; p = 0.036; Extended Data Table 1-1). Mass spectrometry analysis showed no differences in the tissue levels of other glycolysis/gluconeogenesis (Extended Data Table 1-1), β-oxidation (Extended Data Table 1-2), and Kreb's cycle (Extended Data Table 1-3) targeted protein expression in either sex. Overall, these data indicate that both male and female aSOD2-KO mice exhibit decreased tissue expression of SOD2, but only males showed a compensatory antioxidant response.
Impaired spatial working memory in aged aSOD2-KO male but not female mice
To understand the role of astrocytic SOD2 knockout on cognitive function, we used an automated home-cage behavioral testing apparatus, the PhenoTyper (model 3000, Noldus) as described previously (Logan et al., 2018b, 2019; Grieco et al., 2021). In this testing paradigm, male and female mice (control and aSOD2-KO, 15 months of age; n = 7–11 mice/group) learned to enter one of three holes to receive a food pellet reward with performance recorded over a 90 h period through multiple 12 h day/night cycles. Young (6 months) male and female C57BL/6 mice were used as reference controls for optimal sex-specific performance (n = 12 animals/sex).
Circadian activity (distance moved) was comparable among young (n = 12), aSOD2-KO (n = 11), and aged-matched control (n = 7) males (Fig. 2A). No differences in circadian activity were observed in female aSOD2-KO (n = 11) mice compared with control littermates (n = 9) despite an increase in distance moved with age compared with the young female (n = 12) reference group (Fig. 2B). Entry choice into the CognitionWall was used to calculate the learning index (correct – incorrect/total entries) and plotted as cumulative values per hour across the 90 h of testing for male (Fig. 2C) and female (Fig. 2D) mice. During acquisition (initial discrimination; 1–49 h), no differences were observed in either the learning index or maximum learning between aSOD2-KO mice and controls in males or females, as depicted in the cumulative learning index plots. In the reversal phase, male aSOD2-KO mice showed reduced reversal learning compared with controls (Fig. 2C), but no differences were seen between experimental groups in females (Fig. 2D).
Figure 2.
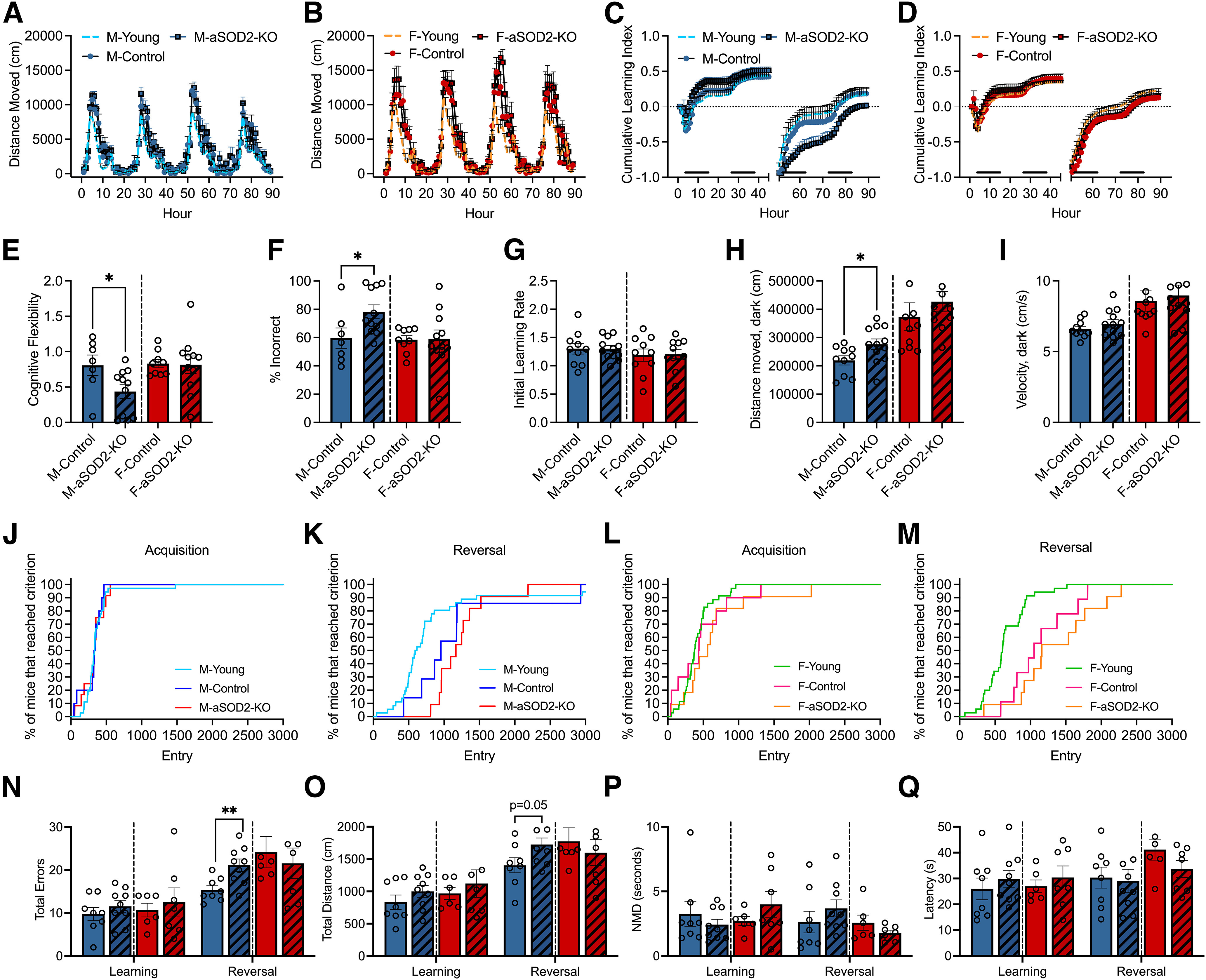
Astrocyte-specific SOD2-KO impairs cognitive performance in males. A–Q, Behavioral data in the PhenoTyper (A–M) and RAWM (N–Q) are represented below. A, B, Circadian activity (distance moved) of aSOD2-KO (males: n = 11, blue square; females: n = 11, red square) and age-matched control (males: n = 7, blue circle; females: n = 9, red circle) males (A) and females (B) plotted per hour over a 90 h period in the PhenoTyper. Young (6 months) C57BL/6J (males: n = 12, blue dashed line; females: n = 12, red dashed line) mice served as reference controls for optimal activity and cognitive performance. C, D, Cumulative learning index during initial discrimination (acquisition; 0–49 h) and reversal (50–89 h) phases show a decline in aSOD2-KO males (C), but not in females (D), over the 90 h testing period. Black bars near the x-axis indicate dark periods of the light/dark cycle. E, Cognitive flexibility measured during hours 51–61 of the reversal phase is reduced in aSOD2-KO males (p = 0.040) but not in females. F, Percentage (%) of incorrect entries (left + middle entries/total entries) calculated during hours 51–61 of the reversal phase is increased in males (p = 0.040) but not in females. G, Bar plot depicting no changes in the initial learning rate of males and females assessed using the PhenoTyper between aSOD2-KO and age-matched controls. H, Bar plots depicting the distanced moved of males (p = 0.033) and females during the dark phase of the reversal phase in the PhenoTyper. I, Bar plots depicting differences between groups in the velocity of males and females during the dark phase of the PhenoTyper. J–M, Survival graphs depicting the number of entries to reach 80% criteria in the acquisition phase and reversal phase for males (E, F; n = 7–11/group) and females (G, H; n = 9–11/group). Young controls (6 months of age; n = 12/sex) were used as reference for optimal performance. N, Total errors (number of entries into incorrect arms) plotted during acquisition (learning) and reversal learning in the radial arm water maze in males (n = 8-9/group) and females (n = 6-7/group). Male aSOD2-KO mice showed a significant decline in reversal learning (p = 0.0058) compared with controls. O, Distance moved (path length) plotted during acquisition (learning) and reversal learning in the radial arm water maze in males and females. Male aSOD2-KO showed an increase in reversal learning path length compared with controls (p = 0.051). P, Bar plots depicting the nonmoving duration (NMD) in the radial arm water maze showed no differences between aSOD2-KO and control in males (H; n = 8–9/group) and females (I; n = 6–7/group). Q, Bar plots depicting the latency to target showed no differences in the initial and reversal phases in either groups for males and females in the radial arm water maze (n = 8–9/group). For graphs I–Q, colored bars represent the following: males (blue), females (red), aSOD2-KO (shaded). Error bars depict the mean ± SEM. Significance was tested using unpaired Student's t test (*p < 0.05; **p < 0.01).
We further analyzed the cognitive flexibility and percentage of incorrect entries during hours 51–61 of the reversal dark phase. Cognitive flexibility (Fig. 2E) and the percentage of incorrect entries (Fig. 2F) were derived based on total entries during the first dark segment of the reversal phase (51–61 h). Male aSOD2-KO mice showed significantly reduced cognitive flexibility (Fig. 2E, left; p = 0.04) and increased the number of incorrect entries (Fig. 2F, left; p = 0.04) compared with age-matched controls. There were no differences between groups in cognitive flexibility (Fig. 2E, right) or the percentage of incorrect entries (Fig. 2F, right) for female mice. No differences were observed in the initial learning rate (Fig. 2G), total distance moved (Fig. 2H), or velocity (Fig. 2I) during the dark phase in either male or female aSOD2-KO mice and age-matched controls.
Finally, we analyzed the proportion of male and female mice that reached 80% criteria in both the acquisition and reversal phases in males (Fig. 2J,K) and females (Fig. 2L,M) as previously described (Logan et al., 2018b). Survival graphs showed an age-related increase in the number of entries to 80% criterion in the reversal phase for both male (Fig. 2K) and female (Fig. 2M) mice, but no differences were observed during acquisition between aSOD2-KO and age-matched control mice in either sex (Fig. 2J,L).
To further assess effects on spatial learning and memory, we tested the same cohort of mice using the radial arm water maze, as previously described (Logan et al., 2018a). Male and female aSOD2-KO and control littermates were tested over 2 d with four trials per day to reach a hidden platform in one arm of the eight-arm maze. No differences were observed between groups in either total distance moved or errors during acquisition (Learning; day 1) for either sex. During reversal (day 2) when the hidden platform was moved to a different arm, male aSOD2-KO mice showed a significant increase in total number of errors (Fig. 2N; p = 0.0058) to find the platform as well as a trend toward increased total distance moved (Fig. 2O; p = 0.051), indicating impaired cognitive performance in males. No differences were noted for female mice during reversal in either total errors or distance moved (Fig. 2N,O). Effects on cognitive performance in the maze were not because of either lack of movement (Fig. 2P) or increased latency (Fig. 2Q) in either group or sex, suggesting a central mechanism for cognitive dysfunction.
Together, aSOD2-KO mice show a deficit in spatial working memory in the reversal phase that is specific to males, while females showed no differences despite the significant reduction in SOD2 expression in knock-out mice.
aSOD2-KO selectively reduces hippocampal LTP in male mice
LTP at excitatory synapses on of hippocampal CA1 neurons is widely accepted as the mechanism that underlies hippocampal-mediated learning and memory (Whitlock et al., 2006; Citri and Malenka, 2008). To assess whether loss of cognitive function in male aSOD2-KO mice was associated with LTP deficits, we measured LTP at the Schaffer collateral–CA1 synapses in acute hippocampal slices using field recordings recorded with a MED64 multielectrode array stimulation system in male (Fig. 3A–C) and female (Fig. 3D–F) aSOD2-KO mice and aged-matched controls (n = 5 animals/group) following previously established protocols (Oka et al., 1999). LTP was successfully induced in both aSOD2-KO and control mice; however 1 h after induction, the level of potentiation was lower in male aSOD2-KO mice (Fig. 3A; 132.17 ± 7.26%; N = 12, p = 0.012) than in male controls (164.5 ± 8.39%; N = 11). Interestingly, there was no difference in the level of potentiation in female aSOD2-KO mice (Fig. 3D; 132.28 ± 9.17%; N = 12) compared with female controls (142.59 ± 9.16%; N = 14) 1 h after induction. This indicates that LTP was impaired in male aSOD2-KO mice, but not in female aSOD2-KO mice.
Figure 3.
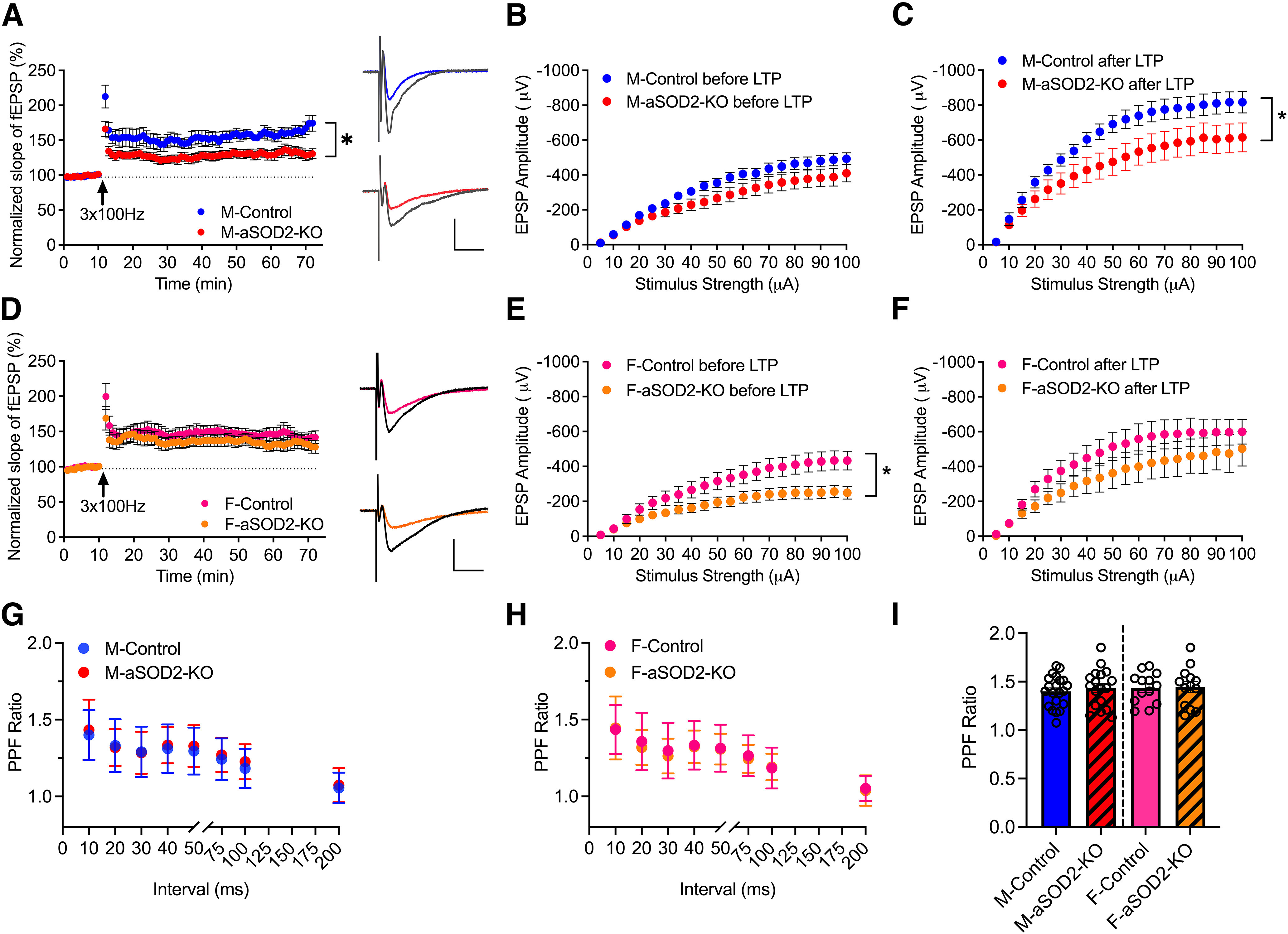
LTP at hippocampal CA1 synapses is impaired in aSOD2-KO male mice. A, Normalized fEPSP showing the induction of LTP in male aSOD2-KO mice (N = 12 recordings, n = 5; p = 0.012) compared with control mice (N = 11 recordings, n = 5). Representative traces show EPSPs before (color line) and after (black line) 3× 100 Hz (arrow) stimulation to induce LTP. Each data point represents the average of two successive test responses. Calibration: y, 500 µV; x = 50 ms. B, C, Graphs depicting the input–output curve amplitude of fEPSP evoked before (B) and after (C; p = 0.020) LTP in male mice. LTP from control (N = 9 recordings) and aSOD2-KO (n = 8 recordings) slices by stepwise increase in the stimulus from 5 to 100 µA. Both groups showed an increase in the fEPSP amplitudes after 1 h of LTP induction. Two-way ANOVA genotype × stimulation intensity interaction: F(19,300) = 1.819, p = 0.0204; genotype: F(1,300)= 284.9, p < 0.0001; stimulation intensity: F(19,300) = 26.54, p < 0.0001. D, Normalized fEPSP showing induction of LTP in female aSOD2-KO mice (N = 12 recordings, n = 5 compared with control mice; N = 14 recordings, n = 5). Representative traces show EPSPs before (color line) and after (black line) 3× 100 Hz (arrow) stimulation to induce LTP. Each data point represents the average of two successive test responses. Calibration: y = 500 µV; x = 50 ms. E, F, Graphs depicting the input–output curve amplitude of fEPSP evoked before (E; p = 0.0162) and after (F) LTP in female mice. LTP from control and aSOD2-KO slices by stepwise increase in the stimulus from 5 to 100 µA. Both groups showed an increase in the fEPSP amplitudes after 1 h of LTP induction. Two-way ANOVA genotype × stimulation intensity interaction: F(19,380) = 1.856, p = 0.0162; genotype: F(1,380)= 158.1, p < 0.0001; stimulation intensity: F(19,380) = 16.41, p < 0.0001. G, H, Paired-pulse facilitation in male (G; N = 18–21 recordings/group) and female (H; N = 13–14 recordings/group) control and aSOD2-KO mice with step-wise increase in interval in milliseconds between two pulses. I, The highest PPF values obtained at the 10 ms interval are plotted. For all graphs, colored points and bars represent the following: male-control, blue; male-aSOD2-KO, red; female-control, pink; female-aSOD2-KO, orange. Error bars depict the mean ± SEM. Significance was tested using unpaired Student's t test or two-way ANOVA (*p < 0.05). n = 5 animals/group, with N as listed per respective experiment.
To further assess the properties of synaptic transmission in these hippocampal slices, we measured the amplitude of the fEPSPs in response to a range of stepwise increased stimuli (5–100 µA) before and after LTP induction. In male mice, there was no significant difference in fEPSP amplitude between controls and the aSOD2-KO group at any stimulation strength of the I/O curve obtained before induction of LTP (Fig. 3B). However, after LTP induction, both groups showed significantly increased fEPSP amplitude, but aSOD2-KO mice showed a reduced response relative to controls (Fig. 3C; N = 8–9/group, p = 0.020), again demonstrating that LTP was impaired in slices from male aSOD2-KO mice in particular.
Female mice showed a modest decrease in fEPSP amplitude in the aSOD2-KO group compared with controls in the I/O curve obtained before LTP (Fig. 3E; N = 10–11 recordings/group, p = 0.016). However, in alignment with the LTP time course data (Fig. 3B), there was no significant difference in fEPSP amplitude between female control and aSOD2-KO groups at any stimulation strength along the I/O curve obtained after LTP induction, although the response for both groups increased significantly (Fig. 3F). These data suggest that LTP and basal synaptic transmission are differentially affected by aSOD2-KO in males and females.
We also examined the PPF ratio in these slices before inducing LTP, measuring the ratio of the response between two pulses at 10, 20, 30, 40, 50, 75, 100, and 200 ms apart (Fig. 3G–I). Both control and aSOD2-KO mice showed PPF with the highest PPF values at the 10 ms interval (Fig. 3I; males: N = 18–21/group; females: N = 13–14/group). There was no difference in the PPF ratio between male and female control or aSOD2-KO mice at any duration between 10 and 200 ms intervals (Fig. 3G,H), indicating that this measure of short-term plasticity was not affected in aSOD2-KO mice of either sex.
As LTP responses are initiated by the postsynaptic activation of AMPA and NMDA receptors, we assessed the expression of AMPA and NMDA receptor subunits by Western blotting (Fig. 4A–G). No differences were observed in AMPA or NMDA receptor expression in males between groups. Females showed a trend of enhanced AMPA receptor expression (GluA1 and GluA2; Fig. 4E,F; p = 0.054 and p = 0.067, respectively) and significant increases in NMDA receptor subunit expression (GluN2B and GluN2C; Fig. 4C,D; p = 0.006 and p = 0.012, respectively), suggesting a compensatory increase in females that may be protective of cognitive function.
Figure 4.

AMPA receptor and NMDA receptor expression in aSOD2-KO brain tissue of both males and females. A, Blots representing the expression of AMPA (GluA1 and GluA2) and NMDA (GluN1, GluN2A, GluN2B, and GluN2C) receptor subunits in brain homogenates of male and female aSOD2-KO mice compared with controls (n = 4/group). Blots were normalized to H2B. B–G, Bar plots depicting quantification of GluN2A (B), GluN2B (C; p = 0.006), GluN2C (D; p = 0.012), GluA1 (E; p = 0.054), GluA2 (F; p = 0.067), and GluN1 (G) for male and female aSOD2-KO mice compared with controls. Blots were normalized to H2B. Colored bars represent the following: males, blue; females, red; aSOD2-KO, shaded. Error bars depict the mean ± SEM. Significance was tested using unpaired Student's t test (*p < 0.05; **p < 0.01).
d-serine levels are reduced by mitochondrial oxidant stress in astrocytes
Induction of LTP in postsynaptic neurons is facilitated by d-serine, levels of which decline in the hippocampus with age (Dememes et al., 2006; Panatier et al., 2006; Papouin et al., 2012; Billard, 2015). d-serine is an NMDAR coagonist that is synthesized and released in part by astrocytes into the synaptic cleft to enable LTP induction (Panatier et al., 2006). We therefore quantified whether mitochondrial oxidative stress affected levels of d-serine in vivo. We measured d-serine, total serine, and the rate-limiting enzyme in its biosynthesis, SRR in aSOD2-KO male and female hippocampal lysates. Male aSOD2-KO mice had a 30% reduction (Fig. 5A, left; n = 5–7/group, p = 0.041) in hippocampal d-serine levels, while aSOD2-KO females had a 17% reduction in levels relative to controls (Fig. 5A, right; n = 7–9/group, p = 0.047). Total levels of serine were comparable between groups of either sex (Fig. 5B). SRR protein expression was significantly decreased in male aSOD2-KO hippocampi (Fig. 5C, left; n = 6–8/group, p = 0.040), while females aSOD2-KO hippocampi showed a significant increase in hippocampal SRR expression (Fig. 5C, right; n = 7/group, p = 0.0036) relative to controls. These data suggest that astrocytic redox imbalance has a negative impact on the tissue levels of d-serine in the hippocampus but has a sex-specific effect on SRR expression that may contribute to cognitive protection in females.
Figure 5.
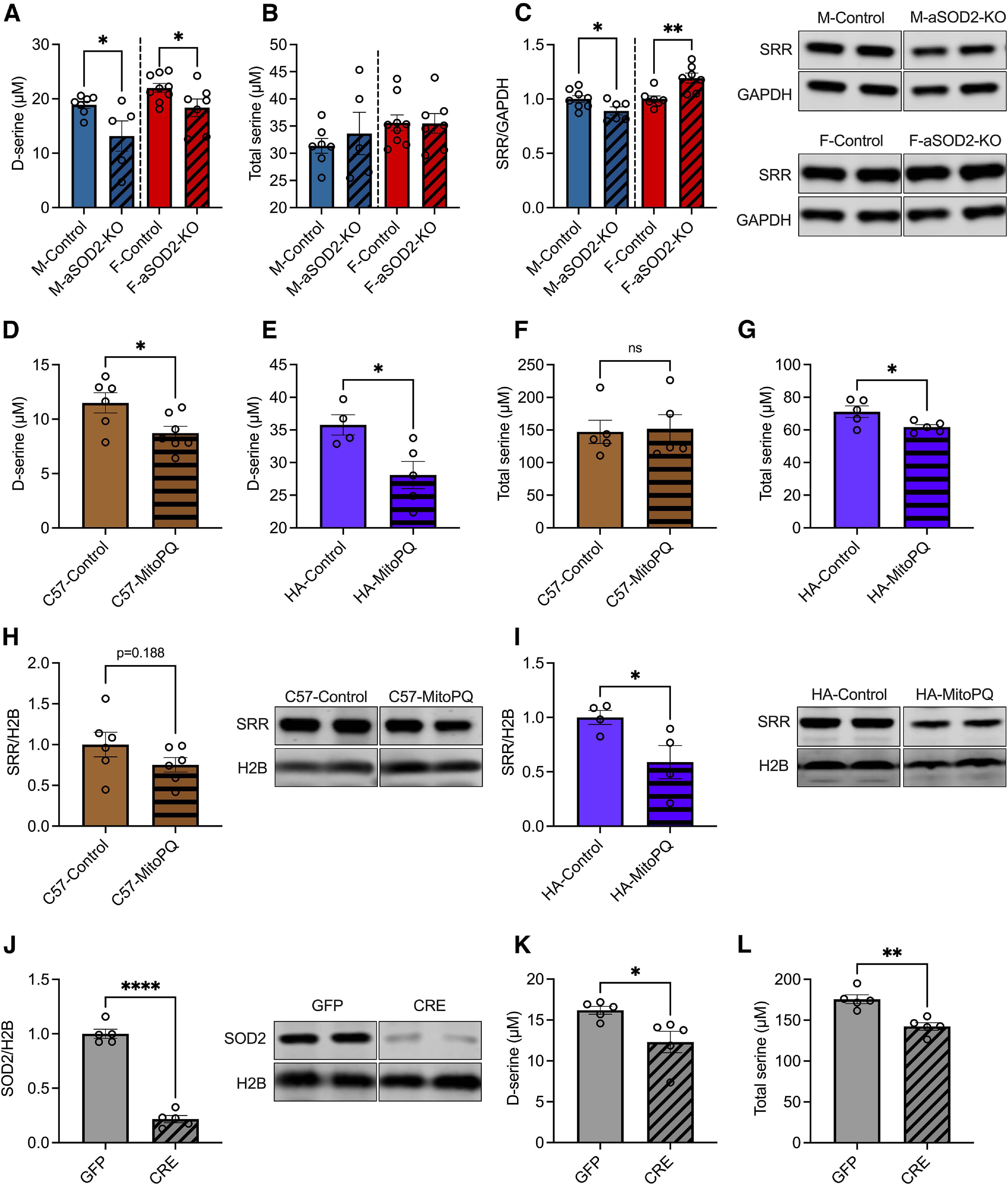
Mitochondrial oxidative stress results in impaired d-serine availability in aSOD2-KO and cultured astrocytes. A, Bar plots depicting significant declines in d-serine levels in male and female aSOD2-KO hippocampi (male: n = 5; p = 0.041; female: n = 7; p = 0.047) relative to controls (male, n = 7; female, n = 9). B, No differences in total serine were detected in aSOD2-KO male or female hippocampi relative to controls (n = 3–5 animals/group). C, Bar plots and representative blot of SRR expression in hippocampal lysates show a significant reduction in SRR expression in aSOD2-KO males (n = 5–7/group; p = 0.041) and a significant increase in SRR expression in aSOD2-KO females relative to controls (n = 7–9/group; p = 0.047). Blot was normalized to GAPDH. D, E, Cultured primary mouse astrocytes (D; n = 6–7/group; p = 0.026) and human (E; n = 4–5/group; p = 0.025) astrocytes treated with MitoPQ (5 µm for 24 h) show a significant decline in d-serine levels compared with controls. F, G, Total serine of MitoPQ-treated (5 µm for 24 h) astrocytes cultured from C57BL/6J mice and human hippocampus. Total serine levels were unchanged in mouse astrocytes with MitoPQ treatment but were significantly reduced in human astrocytes compared with controls. H, I, Quantification and representatives blot of SRR protein expression normalized to H2B from mouse and human primary astrocytes. SRR expression showed a trending decline in mouse (n = 6/group, p = 0.188) and a significant decrease in human (n = 4/group; p = 0.048) astrocytes treated with MitoPQ compared with controls. J, Primary astrocytes cultures from Sod2f/f mice treated with AAV-Cre or -GFP showed significant reduction (n = 5/group; p = 0.023) in SOD2 levels in the knockout (CRE) relative to controls (GFP). Blot was normalized to H2B. K, L, Levels of d-serine and total serine were significantly reduced in SOD2 knock-out astrocytes (n = 5/group; p = 0.023 and p = 0.001). Colored bars represent the following: males, blue; females, red; primary mouse astrocytes, brown; human astrocytes, purple; Sod2f/f astrocyte cultures, gray. All shaded bars represent either SOD2-KO or MitoPQ-treated groups. Error bars depict the mean ± SEM. Significance was tested using unpaired two-tailed Student's t test (***p < 0.001; **p < 0.01; *p < 0.05). n as listed per respective experiment.
To investigate whether mitochondrial redox imbalance directly affects d-serine synthesis in astrocytes, we cultured primary mouse and human astrocytes treated with the mitochondrial redox-cycler MitoParaquat (MitoPQ; 5 µm for 24 h) that induces superoxide production specifically in mitochondria (Robb et al., 2015). Levels of d-serine in cell lysates were significantly reduced in MitoPQ-treated primary astrocytes compared with untreated controls in both mouse (Fig. 5D; n = 6–7/group, p = 0.026) and human hippocampal astrocytes (Fig. 5E; n = 4–5/group, p = 0.025). Levels of total serine were unaffected in mouse astrocytes (Fig. 5F) treated with MitoPQ but were reduced in human hippocampal astrocytes (Fig. 5G; p = 0.039) relative to their respective controls. Human hippocampal astrocytes showed a significant reduction in SRR (Fig. 5H; n = 4/group, p = 0.048) relative to controls, while mouse astrocytes showed a trending decline (25%) in SRR expression (Fig. 5I; n = 6/group, p = 0.188). The lack of significance in the latter may be due to the lack of separation of sexes from postnatal brain cultures.
We further determined whether SOD2 levels in astrocytes directly influenced d-serine synthesis. We cultured primary astrocytes from Sod2f/f mice and reduced SOD2 expression using CRE-mediated knockout. Controls were treated with GFP. We assessed levels of SOD2 via Western blotting that showed a significant reduction in SOD2 expression in CRE compared with GFP astrocytes (Fig. 5J; n = 5/group, p = 0.0001). We then measured d-serine in cell pellets from CRE and GFP astrocytes and found that SOD2 knock-out astrocytes showed a significant decrease in d-serine levels (Fig. 5K; n = 5/group, p = 0.023) and total serine levels (Fig. 5L; p = 0.001) compared with GFP controls. These results recapitulate our findings in human astrocytes treated with MitoPQ, suggesting that mitochondrial redox imbalance induced either chemically (via MitoPQ) or genetically (via SOD2-knockout) impairs astrocytic biosynthesis of d-serine. Together, the decline in hippocampal d-serine levels in aSOD2-KO mice suggests that astrocytic contributions to d-serine biosynthesis in the hippocampus is critical for neuronal modulation of learning and memory.
aSOD2 knockout increases protoplasmic astrocyte density in the hippocampus
Mitochondrial dysfunction and increased ROS have been shown to induce astrogliosis and neuroinflammation in mice with brain-specific knockout of SOD2 (Izuo et al., 2015). To understand the effect of increased mitochondrial ROS specifically in astrocytes, we assessed the morphology and density of astrocytes by Delaunay and Sholl analyses on high-magnification image montages of GFAP-immunolabeled cells in the CA1 region of the hippocampus in male and female aSOD2-KO mice. Representative image montages are of GFAP-labeled astrocytes from control and aSOD2-KO male and females are depicted (Fig. 6A).
Figure 6.
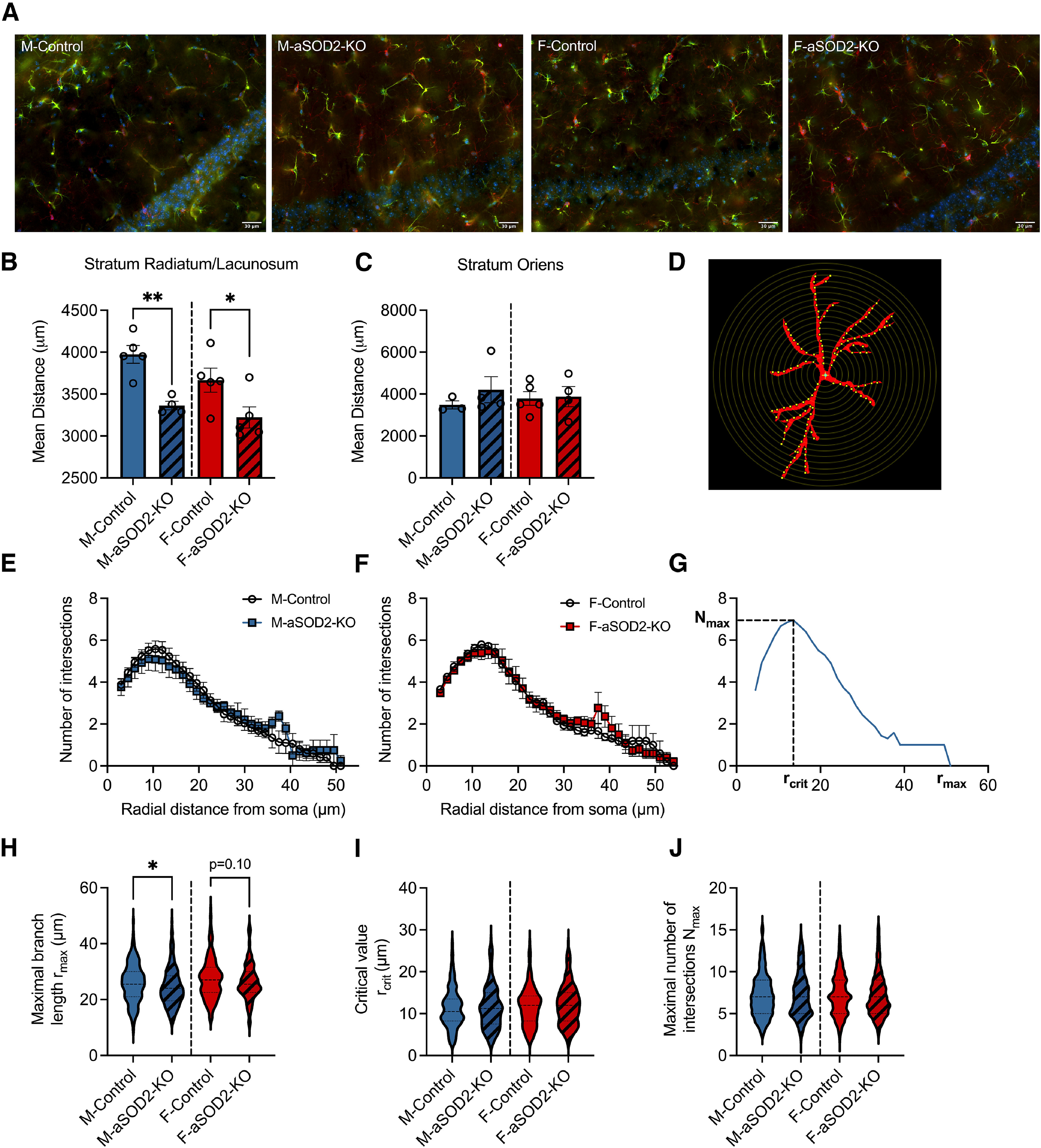
Astrocyte-specific SOD2-KO increases gliosis in the hippocampus of aSOD2-KO mice. A, Representative image montages of merged immunostained images labeled for GFAP (green; astrocytes), Iba1 (red; microglia), and DAPI (blue; nuclei) within the CA1 region of the hippocampus of male and female control and aSOD2-KO mice. Scale bar, 30 µm. B, Knockout of SOD2 in astrocytes produced a decrease in astrocyte spacing in stratum radiatum and stratum lacunosum (n = 4-5/group; p = 0.002 and p = 0.047) compared with control mice. C, No differences were detected in the mean distance between neighboring astrocytes in stratum oriens. D, Example of a GFAP-labeled astrocyte with superimposed concentric circles (step size, 1.5 µm) used for Sholl analysis. E, F, Quantification of the number of projection intersections plotted against radial distance from the soma shows no difference between aSOD2-KO and controls in males (E; N = 45 astrocytes/n, n = 4–5/group) and females (F; N = 45 astrocytes/n, n = 5/group). Clear circles represent controls, while colored squares represent aSOD2-KO for each sex, respectively. G, Representative Sholl plot of one astrocyte displaying the parameters of rmax, rcrit, and Nmax quantified in H–J. H, The rmax of astrocytic projections was decreased in aSOD2-KO males (N = 180–225 astrocytes, n = 4–5/group; p = 0.031) compared with controls while aSOD2-KO females had a trending decrease (N = 225 astrocytes, n = 5/group; p = 0.10) compared with controls. I, J, No changes were detected in the rcrit or the Nmax between aSOD2-KO males and females compared with respective controls. Colored bars represent the following: males, blue; females, red; aSOD2-KO, shaded. Error bars depict the mean ± SEM. Significance was tested using unpaired Student's t test (*p < 0.05; **p < 0.01).
To assess whether knockout of aSOD2-KO altered the numbers and spatial arrangement of astrocytes, we used the Delaunay tessellation to determine the mean distance between astrocyte cell bodies in stratum radiatum/lacunosum and in stratum oriens of CA1 (Fig. 6B,C). The mean distance between astrocyte cell bodies in stratum radiatum/lacunosum (Fig. 6B; n = 4–5/group; p = 0.002 and p = 0.047) was smaller in both male and female aSOD2-KO mice than in age-matched controls, indicating that astrocytes in the aSOD2-KO mice were more tightly spaced than in control mice. No differences in the spacing of astrocytes were noted in stratum oriens (Fig. 6C).
To assess whether aSOD2-KO alters astrocyte morphology and projections, we performed Sholl analysis on individual astrocytes (Fig. 6D) from control and aSOD2-KO male and female CA1 region of the hippocampus (N = 45 astrocytes/n, n = 4–5/group). Sholl analysis showed that the number of intersections of astrocyte processes along the radial distance from the soma were unchanged in control and aSOD2-KO males (Fig. 6E) or females (Fig. 6F). We then determined the rmax, the rcrit, and the Nmax for each astrocyte as depicted (Fig. 6G) and previously described (Hösli et al., 2022). Astrocytes from male aSOD2-KO showed reduced maximal branch length compared with controls (Fig. 6H; p = 0.031), while no differences were noted in female aSOD2-KO mice relative to controls. No differences were seen in either the critical value (Fig. 6I) or maximal number of intersections (Fig. 6J) in either group of each sex.
Together, these data suggest that aSOD2-KO induced astrocyte proliferation regardless of sex, with limited morphologic alterations that were specific to male aSOD2-KO.
Discussion
Mitochondrial dysfunction, neuroinflammation, and synaptic defects have been reported as early changes in brain aging and neurodegenerative disorders (Reddy et al., 2012). Moreover, decrements in astrocyte mitochondrial function and oxidative stress are associated with memory impairments (Gibbs et al., 2009; Gibbs and Bowser, 2009; Kubik and Philbert, 2015). Using an inducible astrocyte-specific SOD2 knock-out model in middle-aged mice (15–18 months of age), we show that redox dysregulation in astrocytes impairs spatial working memory and LTP selectively in male mice with no specific differences observed in females, despite the comparable reduction in mitochondrial SOD2 expression in the hippocampus. Additionally, we show that the astrocyte-specific loss of SOD2 results in a decline in hippocampal levels of d-serine, a critical NMDA coagonist, that may contribute to the loss of cognitive function in males. Furthermore, astrocytic redox imbalance in males, indicated by increases in protein nitrosylation and antioxidant response protein expression, resulted in an increase in astrocyte density within the CA1 region of the hippocampus indicative of astrogliosis. Our results indicate a fundamental role for astrocytic redox regulation in preserving cognitive function in the hippocampus in the aged brain.
While steady-state levels of ROS regulate intracellular signaling, apoptosis, and other cellular functions, aberrant ROS production can adversely affect tissue function with age and in diseased states. Additionally, hippocampal astrocytes from aged mice show downregulation of genes involved in mitochondrial oxidative phosphorylation and redox regulation (Clarke et al., 2018). Here, we show that Sod2 levels decline in the hippocampus of aged mice that we have previously shown to exhibit learning and memory impairments and increased astrogliosis (Logan et al., 2018a,b). We show that aSOD2 knockout in male mice results in deficits in spatial working memory and reduced cognitive flexibility in the reversal phase as assessed using the high-resolution PhenoTyper and RAWM. Our data are in alignment with reports showing protection from oxidative stress with astrocyte-specific overexpression of SOD2 (Xu et al., 2010). Importantly, despite the reduction in SOD2 expression, female aSOD2-KO hippocampus was protected from protein nitrosylation and cognitive deficits. Our data suggest a role for sex-specific factors that mediate the response to mitochondrial oxidative stress in astrocytes. Furthermore, while our data are based on 15- to 18-month-old mice, whether female mice are more susceptible to oxidative stress in advanced age (24 months of age) needs further investigation. Overall, these data indicate that the compensatory antioxidant response of aSOD2-KO in males may be maladaptive and that inherent biological mechanisms (e.g., estrogen signaling) in females may protect against oxidative stress-induced cognitive dysfunction in females. This notion requires further exploration.
LTP is a well documented process of sustained synaptic strengthening occurring during hippocampal learning and memory processing, which is impaired in aging and neurodegenerative diseases. LTP at the Schaffer collateral–CA1 synapses is induced by the activation of NMDA receptors in response to neuronal firing and synaptic glutamate release. Importantly, ROS (e.g., hydrogen peroxide) impairs LTP (Kamsler and Segal, 2003a,b). Both LTP and NMDA receptor function are impaired in mice with whole-body knockout of SOD2, which also display mitochondrial dysfunction and increased oxidative stress (Carvajal et al., 2018). Our data support and augment these findings with astrocyte-specific knockout of SOD2 impairing LTP in CA1 neurons in males, suggesting a critical role of cell-specific age-related redox imbalance on learning and memory processing in the hippocampus. Interestingly, we found that I/O of basal synaptic transmission was reduced selectively in female aSOD2-KO mice, pointing to either a decrease in the number of functional synapses or reduced AMPA receptor number per synapse. The compensatory increase in NMDA receptor expression in females may provide protection from aSOD2-KO-induced LTP deficits in females. Overall, our data suggest the presence of sex-dependent mechanisms in the regulation of synaptic AMPA and NMDA receptors by redox balance in astrocytes.
Protoplasmic astrocytes ensheath synapses and express the rate-limiting enzyme in d-serine biosynthesis, SRR, that isomerizes l-serine to d-serine, which serves as a coagonist at NMDA receptors (Wolosker et al., 1999). Reducing the age-associated increase in oxidative stress has been shown to rescue hippocampal LTP deficits via d-serine-mediated NMDA receptor activation (Haxaire et al., 2012). In support, we show that oxidative stress induced specifically in astrocytes impairs LTP and reduces d-serine levels in aSOD2-KO hippocampus and that reduced d-serine levels are a direct effect of mitochondrial oxidative stress in cultured primary mouse and human astrocytes. Furthermore, we show that the decline in hippocampal d-serine levels correlates with the reduced cognitive flexibility in males. These data are supported by recent reports of astrocytic d-serine modulating flexibility in memory processing (Koh et al., 2022). Decreased SRR expression has been shown to induce deficits in hippocampal-mediated cognitive function in aging (Turpin et al., 2011). In agreement, our data show a decline in SRR expression in male aSOD2-KO mice with cognitive deficits, while SRR expression was increased in female aSOD2-KO mice, which showed protected cognitive function. SRR expression in the subiculum of the hippocampus modulates the glycolytic pathway and d-serine synthesis to provide a critical energy source for learning and memory function (Suzuki et al., 2015). Additionally, our data show an upregulation of Pgam in aSOD2-KO male mice, suggesting a shift in glycolytic metabolism (Hitosugi et al., 2012) by regulating the concentrations of the glycolytic intermediates, phosphoglycerate (3-PG and 2-PG), that feed into the d-serine synthetic pathway (Oslund et al., 2017). These data suggest a critical role for astrocytic oxidative stress in age-related loss of cognitive function via modulation of glycolytic flux and d-serine metabolism.
Altered mitochondrial function and serine metabolism is linked to many of the age-related diseases, including Alzheimer's disease (Orzylowski et al., 2021), diabetes (Holm and Buschard, 2019), and cancer (Yang and Vousden, 2016). Synthesis of d-serine by astrocytes, however, has been contentious, with reports suggesting that d-serine synthesis mainly occurs in neurons, which subsequently augments NMDAR function on synaptic release (Wolosker et al., 2016). We show a direct effect of aSOD2-KO on d-serine levels in cultured astrocytes, supported by previous studies on astrocytic release of d-serine as a gliotransmitter (Yang et al., 2003; Mothet et al., 2005; Martineau et al., 2014). The reduction in d-serine in the hippocampus from aSOD2-KO suggests a direct link between astrocytic redox status and tissue levels of d-serine, whether synthesized by astrocytes or neurons. Future experiments will determine whether redox imbalance in astrocytes differentially regulates l-serine shuttle to neurons and consequently d-serine synthesis in a sex-specific manner.
The prevalence of inflammatory astrocytes increases with age and is closely associated with cognitive dysfunction in Alzheimer's disease (Habib et al., 2020). Brain-specific SOD2 knock-out mice show increased astrogliosis and a compensatory antioxidant response (Izuo et al., 2015). Furthermore, reduction in d-serine metabolism is linked to a reactive astrocyte phenotype (Li et al., 2018) that exhibits increased GFAP expression in the aged hippocampus (Clarke et al., 2018). Morphologic changes and loss of fine processes in astrocytes is a hallmark of reactive astrogliosis and is indicative of the early onset of neurodegeneration (Zhou et al., 2019). Sholl analysis of astrocyte morphology revealed a decrease in maximal branch length in aSOD2-KO males that is consistent with reactive astrogliosis. Furthermore, Delaunay analysis of astrocyte spacing suggests that knockout of SOD2 in astrocytes induces changes in astrocyte density, at least in synaptic layers, although the closer spacing of astrocytes was observed selectively in stratum radiatum/lacunosum and not in stratum oriens. This potentially could reflect differences in the magnitude of the effects of aSOD2-KO metabolism between astrocytes in the two synaptic layers, leading to a small increase in the astrocyte population in stratum radiatum/lacunosum. Alternatively, knockout of SOD2 in astrocytes may have induced a selective increase in GFAP expression by astrocytes in stratum radiatum/lacunosum, leading to increased detection of astrocytes. Differences in GFAP expression by astrocytes induced by SOD2 knockout could reflect diversity in neurochemical responses among astrocytes (Escartin et al., 2021). Other reports have shown proliferation of SOD2-KO astrocytes under hypoxic conditions in culture through a c-Myc-dependent activation of cell cycle (Liu et al., 2006). This suggests that astrocyte-specific loss of SOD2 in mice may create a hypoxic environment, potentially through neurovascular unit dysfunction, which is a known component of cognitive aging and neurodegeneration (Duncombe et al., 2017; Tarantini et al., 2017; Kugler et al., 2021), to promote astrocyte proliferation and gliosis in the brain. Whether the loss of SOD2 in astrocytes promotes proliferation of existing inflammatory GFAP-positive astrocytes or recruitment of other astrocytic or microglial populations to a more inflammatory phenotype requires further investigation.
In summary, our behavioral and molecular assessments show that astrocyte-specific SOD2 knock-out mice recapitulate many of the aging phenotypes that manifest in cognitive deficits, altered synaptic plasticity, d-serine availability, and astrogliosis. Importantly, our findings also indicate that these effects are sex specific. Future studies could focus on whether aging interventions, such as nonfeminizing estrogens, may improve cognitive function through the modulation of d-serine metabolism in astrocytic subpopulations. Finally, by delineating the impact of astrocytic SOD2 ablation, we provide evidence for astrocyte mitochondrial redox enzymes as potential therapeutic targets for cognitive deficits in aging.
Footnotes
This work was supported by the National Institutes of Health (NIH; National Institute on Aging Grant R00-AG-056662 to S.L.). Additional support came from the Nathan Shock Center of Oklahoma Integrative Redox Biology and Multiplexing Protein Quantification Cores (NIH Grant P30-AG-050911. This research was also supported in part by the Molecular Analysis Cellular Imaging (MACI) and Animal Model Development and Behavioral Assessment (AMD-BA) Cores of the University of Oklahoma Health Sciences Center and “Cellular and Molecular GeroScience CoBRE” Grant 1P20-GM-125528 from NIH | National Institute of General Medical Sciences.
The authors declare no competing financial interests.
References
- Billard JM (2015) D-Serine in the aging hippocampus. J Pharm Biomed Anal 116:18–24. 10.1016/j.jpba.2015.02.013 [DOI] [PubMed] [Google Scholar]
- Birben E, Sahiner UM, Sackesen C, Erzurum S, Kalayci O (2012) Oxidative stress and antioxidant defense. World Allergy Organ J 5:9–19. 10.1097/WOX.0b013e3182439613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolanos JP (2016) Bioenergetics and redox adaptations of astrocytes to neuronal activity. J Neurochem 139 [Suppl 2]:115–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgognon JM, Spiers JG, Robinson SW, Scheiblich H, Glynn P, Ortori C, Bradley SJ, Tobin AB, Steinert JR (2021) Inhibition of neuroinflammatory nitric oxide signaling suppresses glycation and prevents neuronal dysfunction in mouse prion disease. Proc Natl Acad Sci U|S|A 118:e2009579118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvajal FJ, Mira RG, Rovegno M, Minniti AN, Cerpa W (2018) Age-related NMDA signaling alterations in SOD2 deficient mice. Biochim Biophys Acta Mol Basis Dis 1864:2010–2020. 10.1016/j.bbadis.2018.03.019 [DOI] [PubMed] [Google Scholar]
- Citri A, Malenka RC (2008) Synaptic plasticity: multiple forms, functions, and mechanisms. Neuropsychopharmacology 33:18–41. 10.1038/sj.npp.1301559 [DOI] [PubMed] [Google Scholar]
- Clarke LE, Liddelow SA, Chakraborty C, Münch AE, Heiman M, Barres BA (2018) Normal aging induces A1-like astrocyte reactivity. Proc Natl Acad Sci U|S|A 115:E1896–E1905. 10.1073/pnas.1800165115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dememes D, Mothet JP, Nicolas MT (2006) Cellular distribution of D-serine, serine racemase and D-amino acid oxidase in the rat vestibular sensory epithelia. Neuroscience 137:991–997. 10.1016/j.neuroscience.2005.09.020 [DOI] [PubMed] [Google Scholar]
- Diekman CO, Fall CP, Lechleiter JD, Terman D (2013) Modeling the neuroprotective role of enhanced astrocyte mitochondrial metabolism during stroke. Biophys J 104:1752–1763. 10.1016/j.bpj.2013.02.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncombe J, Lennen RJ, Jansen MA, Marshall I, Wardlaw JM, Horsburgh K (2017) Ageing causes prominent neurovascular dysfunction associated with loss of astrocytic contacts and gliosis. Neuropathol Appl Neurobiol 43:477–491. 10.1111/nan.12375 [DOI] [PubMed] [Google Scholar]
- Escartin C, et al. (2021) Reactive astrocyte nomenclature, definitions, and future directions. Nat Neurosci 24:312–325. 10.1038/s41593-020-00783-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito L, Raber J, Kekonius L, Yan F, Yu GQ, Bien-Ly N, Puoliväli J, Scearce-Levie K, Masliah E, Mucke L (2006) Reduction in mitochondrial superoxide dismutase modulates Alzheimer's disease-like pathology and accelerates the onset of behavioral changes in human amyloid precursor protein transgenic mice. J Neurosci 26:5167–5179. 10.1523/JNEUROSCI.0482-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn JM, Melov S (2013) SOD2 in mitochondrial dysfunction and neurodegeneration. Free Radic Biol Med 62:4–12. 10.1016/j.freeradbiomed.2013.05.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs ME, Bowser DN (2009) Astrocytes and interneurons in memory processing in the chick hippocampus: roles for G-coupled protein receptors, GABA(B) and mGluR1. Neurochem Res 34:1712–1720. 10.1007/s11064-009-9980-1 [DOI] [PubMed] [Google Scholar]
- Gibbs ME, Gibbs Z, Hertz L (2009) Rescue of Abeta(1-42)-induced memory impairment in day-old chick by facilitation of astrocytic oxidative metabolism: implications for Alzheimer's disease. J Neurochem 109 [Suppl 1]:230–236. 10.1111/j.1471-4159.2009.05800.x [DOI] [PubMed] [Google Scholar]
- Grieco F, et al. (2021) Measuring behavior in the home cage: study design, applications, challenges, and perspectives. Front Behav Neurosci 15:735387. 10.3389/fnbeh.2021.735387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guivernau B, Bonet J, Valls-Comamala V, Bosch-Morató M, Godoy JA, Inestrosa NC, Perálvarez-Marín A, Fernandez-Busquets X (2016) Amyloid-beta peptide nitrotyrosination stabilizes oligomers and enhances NMDAR-mediated toxicity. J Neurosci 36:11693–11703. 10.1523/JNEUROSCI.1081-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C, Sun L, Chen X, Zhang D (2013) Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen Res 8:2003–2014. 10.3969/j.issn.1673-5374.2013.21.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habib N, McCabe C, Medina S, Varshavsky M, Kitsberg D, Dvir-Szternfeld R, Green G, Dionne D, Nguyen L, Marshall JL, Chen F, Zhang F, Kaplan T, Regev A, Schwartz M (2020) Disease-associated astrocytes in Alzheimer's disease and aging. Nat Neurosci 23:701–706. 10.1038/s41593-020-0624-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haxaire C, Turpin FR, Potier B, Kervern M, Sinet PM, Barbanel G, Mothet JP, Dutar P, Billard J-M (2012) Reversal of age-related oxidative stress prevents hippocampal synaptic plasticity deficits by protecting D-serine-dependent NMDA receptor activation. Aging Cell 11:336–344. 10.1111/j.1474-9726.2012.00792.x [DOI] [PubMed] [Google Scholar]
- Hitosugi T, et al. (2012) Phosphoglycerate mutase 1 coordinates glycolysis and biosynthesis to promote tumor growth. Cancer Cell 22:585–600. 10.1016/j.ccr.2012.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm LJ, Buschard K (2019) L-serine: a neglected amino acid with a potential therapeutic role in diabetes. APMIS 127:655–659. 10.1111/apm.12987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hösli L, Binini N, Ferrari KD, Thieren L, Looser ZJ, Zuend M, Zanker HS, Berry S, Holub M, Möbius W, Ruhwedel T, Nave K-A, Giaume C, Weber B, Saab AS (2022) Decoupling astrocytes in adult mice impairs synaptic plasticity and spatial learning. Cell Rep 38:110484. 10.1016/j.celrep.2022.110484 [DOI] [PubMed] [Google Scholar]
- Ikegami T, Suzuki Y, Shimizu T, Isono K, Koseki H, Shirasawa T (2002) Model mice for tissue-specific deletion of the manganese superoxide dismutase (MnSOD) gene. Biochem Biophys Res Commun 296:729–736. 10.1016/s0006-291x(02)00933-6 [DOI] [PubMed] [Google Scholar]
- Izuo N, Nojiri H, Uchiyama S, Noda Y, Kawakami S, Kojima S, Sasaki T, Shirasawa T, Shimizu T (2015) Brain-specific superoxide dismutase 2 deficiency causes perinatal death with spongiform encephalopathy in mice. Oxid Med Cell Longev 2015:238914. 10.1155/2015/238914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamsler A, Segal M (2003a) Hydrogen peroxide modulation of synaptic plasticity. J Neurosci 23:269–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamsler A, Segal M (2003b) Paradoxical actions of hydrogen peroxide on long-term potentiation in transgenic superoxide dismutase-1 mice. J Neurosci 23:10359–10367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinter CS, Lundie JM, Patel H, Rindler PM, Szweda LI, Kinter M (2012) A quantitative proteomic profile of the Nrf2-mediated antioxidant response of macrophages to oxidized LDL determined by multiplexed selected reaction monitoring. PLoS One 7:e50016. 10.1371/journal.pone.0050016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh W, Park M, Chun YE, Lee J, Shim HS, Park MG, Kim S, Sa M, Joo J, Kang H, Oh S-J, Woo J, Chun H, Lee SE, Hong J, Feng J, Li Y, Ryu H, Cho J, Lee CJ (2022) Astrocytes render memory flexible by releasing D-serine and regulating NMDA receptor tone in the hippocampus. Biol Psychiatry 91:740–752. 10.1016/j.biopsych.2021.10.012 [DOI] [PubMed] [Google Scholar]
- Kubik LL, Philbert MA (2015) The role of astrocyte mitochondria in differential regional susceptibility to environmental neurotoxicants: tools for understanding neurodegeneration. Toxicol Sci 144:7–16. 10.1093/toxsci/kfu254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kugler EC, Greenwood J, MacDonald RB (2021) The “neuro-glial-vascular” unit: the role of glia in neurovascular unit formation and dysfunction. Front Cell Dev Biol 9:732820. 10.3389/fcell.2021.732820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Uno Y, Rudolph U, Cobb J, Liu J, Anderson T, Levy D, Balu DT, Coyle JT (2018) Astrocytes in primary cultures express serine racemase, synthesize d-serine and acquire A1 reactive astrocyte features. Biochem Pharmacol 151:245–251. 10.1016/j.bcp.2017.12.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Narasimhan P, Lee YS, Song YS, Endo H, Yu F, Chan PH (2006) Mild hypoxia promotes survival and proliferation of SOD2-deficient astrocytes via c-Myc activation. J Neurosci 26:4329–4337. 10.1523/JNEUROSCI.0382-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan S, Pharaoh GA, Marlin MC, Masser DR, Matsuzaki S, Wronowski B, Yeganeh A, Parks EE, Premkumar P, Farley JA, Owen DB, Humphries KM, Kinter M, Freeman WM, Szweda LI, Van Remmen H, Sonntag WE (2018a) Insulin-like growth factor receptor signaling regulates working memory, mitochondrial metabolism, and amyloid-β uptake in astrocytes. Mol Metab 9:141–155. 10.1016/j.molmet.2018.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan S, Owen D, Chen S, Chen WJ, Ungvari Z, Farley J, Csiszar A, Sharpe A, Loos M, Koopmans B, Richardson A, Sonntag WE (2018b) Simultaneous assessment of cognitive function, circadian rhythm, and spontaneous activity in aging mice. Geroscience 40:123–137. 10.1007/s11357-018-0019-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan S, Royce GH, Owen D, Farley J, Ranjo-Bishop M, Sonntag WE, Deepa SS (2019) Accelerated decline in cognition in a mouse model of increased oxidative stress. Geroscience 41:591–607. 10.1007/s11357-019-00105-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loos M, Koopmans B, Aarts E, Maroteaux G, van der Sluis S, Neuro BMPC, Verhage M, Smit AB (2014) Sheltering behavior and locomotor activity in 11 genetically diverse common inbred mouse strains using home-cage monitoring. PLoS One 9:e108563. 10.1371/journal.pone.0108563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Fabuel I, Le Douce J, Logan A, James AM, Bonvento G, Murphy MP, Almeida A, Bolaños JP (2016) Complex I assembly into supercomplexes determines differential mitochondrial ROS production in neurons and astrocytes. Proc Natl Acad Sci U|S|A 113:13063–13068. 10.1073/pnas.1613701113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lustgarten MS, Jang YC, Liu Y, Muller FL, Qi W, Steinhelper M, Brooks SV, Larkin L, Shimizu T, Shirasawa T, McManus LM, Bhattacharya A, Richardson A, Van Remmen H (2009) Conditional knockout of Mn-SOD targeted to type IIB skeletal muscle fibers increases oxidative stress and is sufficient to alter aerobic exercise capacity. Am J Physiol Cell Physiol 297:C1520–C1532. 10.1152/ajpcell.00372.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroteaux G, Loos M, van der Sluis S, Koopmans B, Aarts E, van Gassen K, Geurts A, Largaespada DA, Spruijt BM, Stiedl O, Smit AB, Verhage M (2012) High-throughput phenotyping of avoidance learning in mice discriminates different genotypes and identifies a novel gene. Genes Brain Behav 11:772–784. 10.1111/j.1601-183X.2012.00820.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martineau M, Parpura V, Mothet JP (2014) Cell-type specific mechanisms of D-serine uptake and release in the brain. Front Synaptic Neurosci 6:12. 10.3389/fnsyn.2014.00012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBean GJ (2018) Astrocyte antioxidant systems. Antioxidants (Basel) 7:112. 10.3390/antiox7090112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metsalu T, Vilo J (2015) ClustVis: a web tool for visualizing clustering of multivariate data using principal component analysis and heatmap. Nucleic Acids Res 43:W566–W570. 10.1093/nar/gkv468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mothet JP, Pollegioni L, Ouanounou G, Martineau M, Fossier P, Baux G (2005) Glutamate receptor activation triggers a calcium-dependent and SNARE protein-dependent release of the gliotransmitter D-serine. Proc Natl Acad Sci U|S|A 102:5606–5611. 10.1073/pnas.0408483102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraja RY, Sherry DM, Fessler JL, Stiles MA, Li F, Multani K, Orock A, Ahmad M, Brush RS, Anderson RE, Agbaga M-P, Deák F (2021) W246G mutant ELOVL4 impairs synaptic plasticity in parallel and climbing fibers and causes motor defects in a rat model of SCA34. Mol Neurobiol 58:4921–4943. 10.1007/s12035-021-02439-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka H, Shimono K, Ogawa R, Sugihara H, Taketani M (1999) A new planar multielectrode array for extracellular recording: application to hippocampal acute slice. J Neurosci Methods 93:61–67. 10.1016/s0165-0270(99)00113-2 [DOI] [PubMed] [Google Scholar]
- Orock A, Logan S, Deak F (2018) Munc18-1 haploinsufficiency impairs learning and memory by reduced synaptic vesicular release in a model of Ohtahara syndrome. Mol Cell Neurosci 88:33–42. 10.1016/j.mcn.2017.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orock A, Logan S, Deak F (2020) Age-related cognitive impairment: role of reduced synaptobrevin-2 levels in deficits of memory and synaptic plasticity. J Gerontol A Biol Sci Med Sci 75:1624–1632. 10.1093/gerona/glz013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orzylowski M, Fujiwara E, Mousseau DD, Baker GB (2021) An overview of the involvement of d-serine in cognitive impairment in normal aging and dementia. Front Psychiatry 12:754032. 10.3389/fpsyt.2021.754032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oslund RC, Su X, Haugbro M, Kee JM, Esposito M, David Y, Wang B, Ge E, Perlman DH, Kang Y, Muir TW (2017) Bisphosphoglycerate mutase controls serine pathway flux via 3-phosphoglycerate. Nat Chem Biol 13:1081–1087. 10.1038/nchembio.2453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panatier A, Theodosis DT, Mothet JP, Touquet B, Pollegioni L, Poulain DA, Oliet SH (2006) Glia-derived D-serine controls NMDA receptor activity and synaptic memory. Cell 125:775–784. 10.1016/j.cell.2006.02.051 [DOI] [PubMed] [Google Scholar]
- Papouin T, Ladépêche L, Ruel J, Sacchi S, Labasque M, Hanini M, Groc L, Pollegioni L, Mothet J-P, Oliet SHR (2012) Synaptic and extrasynaptic NMDA receptors are gated by different endogenous coagonists. Cell 150:633–646. 10.1016/j.cell.2012.06.029 [DOI] [PubMed] [Google Scholar]
- Parihar MS, Brewer GJ (2007a) Mitoenergetic failure in Alzheimer disease. Am J Physiol Cell Physiol 292:C8–C23. 10.1152/ajpcell.00232.2006 [DOI] [PubMed] [Google Scholar]
- Parihar MS, Brewer GJ (2007b) Simultaneous age-related depolarization of mitochondrial membrane potential and increased mitochondrial reactive oxygen species production correlate with age-related glutamate excitotoxicity in rat hippocampal neurons. J Neurosci Res 85:1018–1032. 10.1002/jnr.21218 [DOI] [PubMed] [Google Scholar]
- Parihar MS, Kunz EA, Brewer GJ (2008) Age-related decreases in NAD(P)H and glutathione cause redox declines before ATP loss during glutamate treatment of hippocampal neurons. J Neurosci Res 86:2339–2352. 10.1002/jnr.21679 [DOI] [PubMed] [Google Scholar]
- Poon HF, Calabrese V, Scapagnini G, Butterfield DA (2004a) Free radicals and brain aging. Clin Geriatr Med 20:329–359. 10.1016/j.cger.2004.02.005 [DOI] [PubMed] [Google Scholar]
- Poon HF, Calabrese V, Scapagnini G, Butterfield DA (2004b) Free radicals: key to brain aging and heme oxygenase as a cellular response to oxidative stress. J Gerontol A Biol Sci Med Sci 59:478–493. 10.1093/gerona/59.5.m478 [DOI] [PubMed] [Google Scholar]
- Reddy PH, Tripathi R, Troung Q, Tirumala K, Reddy TP, Anekonda V, Shirendeb UP, Calkins MJ, Reddy AP, Mao P, Manczak M (2012) Abnormal mitochondrial dynamics and synaptic degeneration as early events in Alzheimer's disease: implications to mitochondria-targeted antioxidant therapeutics. Biochim Biophys Acta 1822:639–649. 10.1016/j.bbadis.2011.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizor A, Pajarillo E, Johnson J, Aschner M, Lee E (2019) Astrocytic oxidative/nitrosative stress contributes to Parkinson's disease pathogenesis: the dual role of reactive astrocytes. Antioxidants (Basel) 8:265. 10.3390/antiox8080265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robb EL, Gawel JM, Aksentijević D, Cochemé HM, Stewart TS, Shchepinova MM, Qiang H, Prime TA, Bright TP, James AM, Shattock MJ, Senn HM, Hartley RC, Murphy MP (2015) Selective superoxide generation within mitochondria by the targeted redox cycler MitoParaquat. Free Radic Biol Med 89:883–894. 10.1016/j.freeradbiomed.2015.08.021 [DOI] [PubMed] [Google Scholar]
- Rose J, Brian C, Pappa A, Panayiotidis MI, Franco R (2020) Mitochondrial metabolism in astrocytes regulates brain bioenergetics, neurotransmission and redox balance. Front Neurosci 14:536682. 10.3389/fnins.2020.536682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueden CT, Schindelin J, Hiner MC, DeZonia BE, Walter AE, Arena ET, Eliceiri KW (2017) ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinformatics 18:529. 10.1186/s12859-017-1934-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez J-Y, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song JQ, Jiang LY, Fu CP, Wu X, Liu ZL, Xie L, Wu XD, Hao SY, Li S-Q (2020) Heterozygous SOD2 deletion deteriorated chronic intermittent hypoxia-induced lung inflammation and vascular remodeling through mtROS-NLRP3 signaling pathway. Acta Pharmacol Sin 41:1197–1207. 10.1038/s41401-019-0349-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, Sasabe J, Miyoshi Y, Kuwasako K, Muto Y, Hamase K, Matsuoka M, Imanishi N, Aiso S (2015) Glycolytic flux controls D-serine synthesis through glyceraldehyde-3-phosphate dehydrogenase in astrocytes. Proc Natl Acad Sci U|S|A 112:E2217–E2224. 10.1073/pnas.1416117112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarantini S, Tran CHT, Gordon GR, Ungvari Z, Csiszar A (2017) Impaired neurovascular coupling in aging and Alzheimer's disease: contribution of astrocyte dysfunction and endothelial impairment to cognitive decline. Exp Gerontol 94:52–58. 10.1016/j.exger.2016.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turpin FR, Potier B, Dulong JR, Sinet PM, Alliot J, Oliet SH, Dutar P, Epelbaum J, Mothet J-P, Billard J-M (2011) Reduced serine racemase expression contributes to age-related deficits in hippocampal cognitive function. Neurobiol Aging 32:1495–1504. 10.1016/j.neurobiolaging.2009.09.001 [DOI] [PubMed] [Google Scholar]
- Van Remmen H, Williams MD, Guo Z, Estlack L, Yang H, Carlson EJ, Epstein CJ, Huang TT, Richardson A (2001) Knockout mice heterozygous for Sod2 show alterations in cardiac mitochondrial function and apoptosis. Am J Physiol Heart Circ Physiol 281:H1422–1432. 10.1152/ajpheart.2001.281.3.H1422 [DOI] [PubMed] [Google Scholar]
- Vančová O, Bačiak L, Kašparová S, Kucharská J, Palacios HH, Horecký J, Aliev G (2010) In vivo and in vitro assessment of brain bioenergetics in aging rats. J Cell Mol Med 14:2667–2674. 10.1111/j.1582-4934.2009.00879.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlock JR, Heynen AJ, Shuler MG, Bear MF (2006) Learning induces long-term potentiation in the hippocampus. Science 313:1093–1097. 10.1126/science.1128134 [DOI] [PubMed] [Google Scholar]
- Williams MD, Van Remmen H, Conrad CC, Huang TT, Epstein CJ, Richardson A (1998) Increased oxidative damage is correlated to altered mitochondrial function in heterozygous manganese superoxide dismutase knockout mice. J Biol Chem 273:28510–28515. 10.1074/jbc.273.43.28510 [DOI] [PubMed] [Google Scholar]
- Wolosker H, Blackshaw S, Snyder SH (1999) Serine racemase: a glial enzyme synthesizing D-serine to regulate glutamate-N-methyl-D-aspartate neurotransmission. Proc Natl Acad Sci U|S|A 96:13409–13414. 10.1073/pnas.96.23.13409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolosker H, Balu DT, Coyle JT (2016) The rise and fall of the d-serine-mediated gliotransmission hypothesis. Trends Neurosci 39:712–721. 10.1016/j.tins.2016.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Emery JF, Ouyang YB, Voloboueva LA, Giffard RG (2010) Astrocyte targeted overexpression of Hsp72 or SOD2 reduces neuronal vulnerability to forebrain ischemia. Glia 58:1042–1049. 10.1002/glia.20985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Vousden KH (2016) Serine and one-carbon metabolism in cancer. Nat Rev Cancer 16:650–662. 10.1038/nrc.2016.81 [DOI] [PubMed] [Google Scholar]
- Yang Y, Ge W, Chen Y, Zhang Z, Shen W, Wu C, Poo M, Duan S (2003) Contribution of astrocytes to hippocampal long-term potentiation through release of D-serine. Proc Natl Acad Sci U|S|A 100:15194–15199. 10.1073/pnas.2431073100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W, Watts LT, Holstein DM, Prajapati SI, Keller C, Grass EH, Walter CA, Lechleiter JD (2010) Purinergic receptor stimulation reduces cytotoxic edema and brain infarcts in mouse induced by photothrombosis by energizing glial mitochondria. PLoS One 5:e14401. 10.1371/journal.pone.0014401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B, Zuo YX, Jiang RT (2019) Astrocyte morphology: diversity, plasticity, and role in neurological diseases. CNS Neurosci Ther 25:665–673. 10.1111/cns.13123 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Mass spectrometry analysis listing abundance of targeted proteins in glycolysis and gluconeogenesis. Download Table 1-1, DOCX file (17.6KB, docx) .
Mass spectrometry analysis listing abundance of targeted proteins in β-oxidation. Download Table 1-2, DOCX file (17.4KB, docx) .
Mass spectrometry analysis listing abundance of targeted proteins in Kreb's cycle. Download Table 1-3, DOCX file (20.6KB, docx) .