

Abstract
The effective treatment of brain tumors is a considerable challenge in part because of the presence of the blood-brain barrier (BBB) that limits drug delivery. Glioblastoma multiforme (GBM) is an aggressive and infiltrative primary brain tumor with an extremely poor prognosis after standard-of-care therapy with surgery, radiotherapy (RT), and chemotherapy. DNA damage response (DDR) pathways play a critical role in DNA repair in cancer cells, and inhibition of these pathways can potentially augment RT and chemotherapy tumor cell toxicity. The ataxia telangiectasia and Rad3-related protein (ATR) kinase is a key regulator of the DDR network and is potently and selectively inhibited by the ATR inhibitor berzosertib. Although in vitro studies demonstrate a synergistic effect of berzosertib in combination with temozolomide, in vivo efficacy studies have yet to recapitulate this observation using intracranial tumor models. In the current study, we demonstrate that delivery of berzosertib to the brain is restricted by efflux at the BBB. Berzosertib has a high binding affinity to brain tissue compared with plasma, thereby leading to low free drug concentrations in the brain. Berzosertib distribution is heterogenous within the tumor, wherein concentrations are substantially lower in normal brain and invasive tumor rim (wherein the BBB is intact) when compared with those in the tumor core (wherein the BBB is leaky). These results demonstrate that high tissue binding and limited and heterogenous brain distribution of berzosertib may be important factors that influence the efficacy of berzosertib therapy in GBM.
SIGNIFICANCE STATEMENT
This study examined the brain delivery and efficacy of berzosertib in patient-derived xenograft models of glioblastoma multiforme (GBM). Berzosertib is actively effluxed at the blood-brain barrier and is highly bound to brain tissue, leading to low free drug concentrations in the brain. Berzosertib is heterogeneously distributed into different regions of the brain and tumor and, in this study, was not efficacious in vivo when combined with temozolomide. These factors inform the future clinical utility of berzosertib for GBM.
Introduction
Glioblastoma multiforme (GBM) is the most common adult primary brain tumor and has a dismal prognosis and limited treatment options. GBMs are highly infiltrative and therefore not surgically curable. Tumor cells invade surrounding brain regions in a diffuse nature, making complete surgical resection impossible (Parrish et al., 2015). The current standard approach to GBM therapy includes surgery followed by concurrent radiotherapy (RT) with temozolomide (TMZ), a DNA alkylating agent (Tan et al., 2020). A major limitation in the treatment of all brain tumors is the presence of the blood-brain barrier (BBB), which acts as both a physical and functional barrier to central nervous system drug delivery. Because of the infiltrative nature of GBM, tumor cells can penetrate deep into normal brain tissue and reside in an environment that is “protected” from effective drug delivery by the BBB.
DNA damage response (DDR) is a collective term that describes a complex signaling network responsible for surveillance, recruitment, and completion of DNA lesions and associated coordination of cell cycle arrest and induction of cell death (Bindra et al., 2017). The cytotoxic effects of both RT and chemotherapy in brain tumors ultimately stem from unrepaired DNA damage; thus, inhibitors of the DDR can potentially enhance RT and chemotherapy cytotoxic effects significantly. One of the key regulators of DDR is the ataxia telangiectasia mutated and Rad3-related protein (ATR) kinase, which is activated by single-stranded DNA breaks and is essential for recovery from these lesions commonly formed at stalled replication forks or other forms of replication stress. In this context, ATR inhibition can increase the sensitivity of cells to DNA-damaging chemotherapies or radiotherapy, which suggests that ATR inhibitors may be effective sensitizing agents for a variety of cancers (Bradbury et al., 2020).
Tumor cell recovery from RT and TMZ is dictated by an effective DDR, and many GBM have endogenous elevation of DDR signaling because of endogenous replication stress and elevation of ATR signaling (Morgan and Canman, 2018). Therefore, there is a strong rationale to study ATR inhibitors for the treatment of GBM. Additionally, Ahmed et al. (2015) recently demonstrated the importance of ATR signaling in GBM stem-like cell survival after DNA-damaging agent exposure (Carruthers, 2018). Importantly, GBMs commonly have dysregulation of DDR pathways, including inactivation of p53 and O6-methylguanine-methyltransferase (MGMT) through mutation or deletion and promoter methylation, respectively (Majd et al., 2021). ATR acts as a cell cycle checkpoint regulator and activates p53 in the case of single-strand DNA breaks, leading to DNA repair. Loss of p53 signaling can further drive reliance on ATR in response to DNA damage, which can be exploited by the use of ATR inhibitors to prevent DNA repair (Reaper et al., 2011) (Fig. 1A). DNA damage induced by TMZ is repaired by MGMT. Methylation of the promoter region of the gene encoding MGMT leads to its inactivation and thereby confers an increased sensitivity to TMZ. MGMT methylation is therefore directly linked to enhanced efficacy of TMZ in patients with GBM (Hegi et al., 2005) (Fig. 1B). Additionally, TMZ-induced DNA damage robustly activates the ATR pathway (Aasland et al., 2019; Ferri et al., 2020) and sensitizes MGMT-deficient tumor cells to ATR inhibitors (Caporali et al., 2004; Jackson, 2019). Therefore, understanding the impact of p53 and MGMT status in GBM with respect to the combination of TMZ and ATR inhibitors is an important focus.
Fig. 1.

Importance of p53 and MGMT status for berzosertib and TMZ combination in GBM. (A) Increased sensitivity to berzosertib in the case of p53 mutation downstream of the ATR pathway and (B) increased sensitivity to TMZ in the case of MGMT methylation and inactivation, both contributing to prevention of DNA repair and thereby contributing to tumor cell death.
Berzosertib (formerly M6620, VX-970) is a highly potent and selective, first-in-class ATR inhibitor currently in clinical trials for a variety of cancers in combination with DNA-damaging agents (Fokas et al., 2012; Gorecki et al., 2020; Konstantinopoulos et al., 2020; Middleton et al., 2021). Given the importance of ATR signaling for recovery from RT and TMZ, berzosertib may be a useful drug in GBM. However, a wide range of small-molecule anticancer-targeted agents tested for treatment of brain tumors have been found to be substrates of active efflux transporters at the BBB, which results in severely limited brain delivery of these agents. In GBM specifically, heterogeneous disruption of the BBB within the tumor and the genetic heterogeneity of tumors pose significant challenges in identifying drugs that may be effective in treating brain tumors (Van Tellingen et al., 2015; Sarkaria et al., 2018; Becker et al., 2021). Therefore, this study evaluated the brain delivery of berzosertib and examined the role of major efflux transporters, P-glycoprotein (P-gp) and breast cancer resistance protein (Bcrp), on its brain distribution. Additionally, we performed a preclinical evaluation of berzosertib in combination with TMZ in patient-derived xenograft (PDX) models for patients with GBM to evaluate free versus total drug distribution of berzosertib in different tumor regions. This study identified factors limiting brain delivery and thereby berzosertib efficacy in an orthotopic xenograft model of patient-derived GBM. These data provide critical insights into the factors to be considered for the clinical development of ATR inhibitors for the treatment of GBM.
Materials and Methods
Chemicals and Reagents
Berzosertib (3-[3-[4-(methylaminomethyl)phenyl]-1,2-oxazol-5-yl]-5-(4-propan-2-ylsulfonylphenyl)pyrazin-2-amine) and TMZ (3-methyl-4-oxoimidazo[5,1-day][1,2,3,5]tetrazine-8-carboxamide) were provided by the National Cancer Institute (Bethesda, MD). Dasatinib (N-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)piperazin-1-yl]-2-methylpyrimidin-4-yl]amino]-1,3-thiazole-5-carboxamide) was purchased from LC Laboratories (Woburn, MA). Elacridar (N-[4-[2-(6,7-dimethoxy-3,4-dihydro-1H-isoquinolin-2-yl)ethyl]phenyl]-5-methoxy-9-oxo-10H-acridine-4-carboxamide) was purchased from Toronto Research Chemicals (Toronto, ON, Canada). All other chemicals and reagents were high-performance liquid chromatography–grade and purchased from Thermo Fisher Scientific (Waltham, MA). Cell culture reagents were purchased from Invitrogen (Carlsbad, CA). A rapid equilibrium dialysis (RED) device consisting of a 96-well base plate and RED membrane inserts (8-kDa molecular mass cutoff cellulose membrane) were purchased from Thermo Fisher Scientific.
Lentiviral Vector and Cell Transduction
MGMT was overexpressed using a lentivirus vector, pGIPZ-MGMT-Puro, in which the turbo green fluorescent protein tag of pGIPZ (Open Biosystems) was replaced with human MGMT cDNA. Lentiviral particles were packaged in human embryonic kidney 293T cells that were cotransfected with lentiviral vector and helper plasmids (psPAX2 and pMD2.G encoding Gag/Pol and vesicular stomatitis virus glycoprotein, respectively). Transduction to U251 cells was performed in the presence of 5 µg/ml polybrene (MilliporeSigma, Jaffrey, NH) as previously described (Gupta et al., 2014), and stably transduced U251 cells expressing MGMT or empty control vector (U251-CV) were selected in 5 µg/ml puromycin.
Cell Culture and Cell Viability Experiments
U251, U87, GBM 12, and GBM 22 cells were maintained as previously described (Nadkarni et al., 2012; Gupta et al., 2014). Five hundred to 2000 cells/well were seeded in 96-well plates and exposed to 0–300 µM TMZ and 0–1000 nM berzosertib for 7 or 9 (GBM22) days until confluent. At endpoint, media were discarded, and plates were rinsed with 1× PBS and stored at −80°C until analysis. CyQUANT cell proliferation assay was performed in accordance with the manufacturer’s instructions (Thermo Fisher Scientific, Waltham, MA). Background CyQUANT values were subtracted from all cell viability data.
Synergy Assessment
CyQUANT cell proliferation assay results were uploaded into the MacSynergy II software, and synergy was calculated without adjustment (Prichard and Shipman, 1990; Smee and Prichard, 2017). Three-dimensional interactions that fell above or below the baseline were graphically plotted using this software. Interpretable values referred to as the volume of synergy were generated at 95% confidence limits for berzosertib (concentrations in nanomolar) and TMZ (concentrations in micromolar) data. The guidelines for the volumes of the synergy determinations expressed as μM × nM unit % at a 95% confidence level were as follows: 0–25 unit %, insignificant; 25–50 unit %, minor but significant synergy; 50–100 unit %, moderate synergy; and >100 unit %, strong synergy with possibly significant effects in vivo (Ilyushina et al., 2008).
Tumor-Bearing Animals and In Vivo Efficacy Studies
All studies using animals were approved by the Institutional Animal Care and Use Committee, Mayo Clinic (Rochester, MN). All animals were housed in a standard 12-hour light/dark cycle with unlimited access to food and water. Studies involving tumor implantation used female athymic nude mice (Hsd:athymic Nude-Foxn1nu; Envigo, Indianapolis, IN) at age 4–5 weeks.
Mice with intracranial tumors were enrolled into vehicle, TMZ (50 mg/kg daily for 5 days), or the TMZ/berzosertib combination (50 mg/kg TMZ + 60 mg/kg berzosertib daily for 5 days) arms. Treatment began on day 14 after injection of GBM 22 cells, and mice were monitored daily until moribund.
Non–Tumor-Bearing Animals
In vivo pharmacokinetic studies were conducted using an equal number of male and female Friend leukemia virus strain B (FVB) wild-type, Bcrp1–/–, Mdr1a/b–/–, and Mdr1a/b–/–Bcrp1–/– mice (Taconic Biosciences, Inc., Germantown, NY) at the age of 8–12 weeks. Breeder pairs were sourced from Taconic Biosciences, Inc., and animal colonies were maintained following an established breeding protocol in the Research Animal Resources animal housing facility at the Academic Health Center, University of Minnesota, Minneapolis, MN, USA. Animal genotypes were routinely verified by conducting tail snip for gene expression (TransnetYX, Cordova, TN). All animal experiments were approved by the University of Minnesota Institutional Animal Care and Use Committee and conducted in accordance with the Guide for the Care and Use of Laboratory Animals established by the US National Institutes of Health (Bethesda, MD).
In Vitro Binding Assays for Determination of Free (Unbound) Fractions of Berzosertib
The free fractions in plasma from wild-type, Bcrp1–/–, Mdr1a/b–/–, and Mdr1a/b–/–Bcrp1–/– mice; brain homogenates from wild-type, Bcrp1–/–, Mdr1a/b–/–, and Mdr1a/b–/–Bcrp1–/– mice; and serum-containing medium (Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum) were determined using RED as per the manufacturer’s protocol. Briefly, brain homogenates were prepared by adding three volumes of PBS (pH 7.4), which were followed by homogenization (PowerGen 125; Thermo Fisher Scientific). Each matrix was spiked with berzosertib to a final concentration of 5 µM containing 0.475% DMSO. Three hundred microliters of the spiked matrix was loaded into the sample chamber, and then 500 µl of blank (drug-free) PBS (pH 7.4) with 0.475% DMSO was loaded into the corresponding receiver chamber. The base plate was sealed with an adhesive covering and incubated at 37°C for 24 hours on an orbital shaker (600 rpm; ShelLab, Cornelius, OR). After the 24-hour incubation, samples were collected from both chambers and stored at −80°C until liquid chromatography–tandem mass spectrometry (LC-MS/MS) analysis.
Brain Distribution of Berzosertib after Intravenous Bolus or Single Oral Administration
A single intravenous bolus dose (tail vein injection) or oral dose (oral gavage) of 20 mg/kg berzosetib (vehicle: 5% w/v Captisol, 3% w/v mannitol, pH adjusted to 5.5 using acetate buffer) was administered to FVB wild-type, Bcrp1–/–, Mdr1a/b–/–, and Mdr1a/b–/–Bcrp1–/– mice. After euthanasia using CO2, brain and blood samples were collected from 0.167 to 16 hours; n = 4 at each time point (additional 24- and 48-hour time points were added for oral dose in Mdr1a/b–/–Bcrp1–/– mice). Blood was collected using heparinized syringes via cardiac puncture and stored in heparinized tubes, which was followed by centrifugation at 3500 rpm at 4°C for 15 minutes to separate plasma. Whole brain was removed and dipped in ice-cold saline, which was followed by removal of superficial meninges by blotting with tissue. Plasma and brain samples were stored at −80°C until LC-MS/MS analysis.
Steady-State Organ Distribution of Berzosertib
Alzet osmotic pumps (1003D; Durect Corporation, Cupertino, CA) were implanted into the intraperitoneal cavity of FVB wild-type and Mdr1a/b–/–Bcrp1–/– mice to release berzosertib at a constant rate of 10 µg/h for 48 hours (n = 5) as described previously (Agarwal et al., 2010). Briefly, berzosertib was loaded into the pumps at a concentration of 10 mg/ml in DMSO and primed at 37°C overnight in sterile saline. Mice were anesthetized using isoflurane, and after surgical implantation, the osmotic pumps were inserted into the intraperitoneal cavity, which was followed by suturing the membrane and closing the skin incision using surgical clips. After 48 hours, the mice were sacrificed, and blood, brain, heart, kidney, liver, and muscle were isolated. Samples were stored at −80°C until LC-MS/MS analysis.
Pharmacological Inhibition of P-gp and Bcrp Using Elacridar
Elacridar, a dual inhibitor of P-gp and Bcrp, was formulated in a microemulasion as described previously (Sane et al., 2013). Cremophor EL, Carbitol, and Captex 355 were combined in a 6:3:1 w/w ratio. Elacridar was added to this premix and sonicated to formulate a 3-mg/ml solution, which was diluted with water to form a 1-mg/ml microemulsion for dosing. Elacridar was administered at a dose of 10 mg/kg intraperitoneally, and berzosertib was administered simultaneously at a dose of 20 mg/kg via oral gavage. Brain and plasma samples were harvested at 2 and 8 hours (n = 5 at each time point) after administration of elacridar and berzosertib and were stored at −80°C until LC-MS/MS analysis.
Regional Distribution of Berzosertib in Tumor-Bearing Mouse Brains
Mice with established GBM 12—enhanced GFP (eGFP)—firefly luciferase 2 (fLuc2) intracranial tumors (18 days after implantation) were treated with a single oral dose of 20 mg/kg or 60 mg/kg berzosertib, which was followed by euthanasia at 2 and 8 hours (n = 5 per time point). Blood- and tumor-bearing brain samples were collected, and whole brains were immediately flash-frozen. Plasma was separated by centrifuging the blood samples at 3500 rpm at 4°C for 15 minutes. A fluorescence-guided punch biopsy technique (Gampa et al., 2020) was employed for isolation of tumor core, tumor rim (brain adjacent to tumor), and normal brain (non–tumor-bearing) regions from brain samples. An acrylic adult mouse brain matrix was used to obtain 1-mm thick coronal brain sections of the eGFP-FLUC2–labeled tumor-bearing brains. The tumor regions were identified by relative fluorescence signal (Nikon AZ100M microscope, Nikon, Japan), and biopsy punches were used to isolate tumor core (tumor region with fluorescence signal 5-fold or higher relative to background signal) and rim (region adjacent to tumor core with fluorescence signal 3- to 5-fold higher relative to background). Both the plasma and individual brain region samples were stored at −80°C until LC-MS/MS analysis.
LC-MS/MS Analysis of Berzosertib
The samples from different regions of the brain isolated as described previously were homogenized with three volumes of 5% bovine serum albumin using a homogenizer (PowerGen 125; Thermo Fisher Scientific). Drug concentration of berzosertib in these plasma and homogenized-brain samples was determined using an LC-MS/MS assay. Twenty-five microliters of plasma and 50 μl of brain homogenate samples were spiked with 50 ng of the internal standard (dasatinib), which was followed by liquid-liquid extraction using 1× volume of cold pH 11 buffer and 5× volumes of ice-cold ethyl acetate. The sample tubes were then shaken vigorously for 5 minutes and centrifuged at 7500 rpm and 4°C for 10 minutes. After centrifugation, the organic supernatant layer was collected and dried under nitrogen flow, which was followed by reconstitution of dried powder residue in 100 µl of freshly prepared mobile phase. An ACQUITY ultra-performance liquid chromatography (UPLC) system (Waters Corporation, Milford, MA) with an ACQUITY UPLC BEH-C18 column (1.7 μm, 2.1 × 50 mm; Waters Corporation, Milford, MA) was used for the chromatographic analysis of samples. An isocratic method with a runtime of 4 minutes was used with the mobile phase consisting of 70% distilled and filtered water with 0.1% formic acid (A) and 30% acetonitrile with 0.1% formic acid (B) supplied at a constant flow rate of 0.2 ml/min. The column effluent from the UPLC system was monitored by a Micromass Quattro Ultima mass spectrometer (Waters, Milford, MA). The electrospray probe in positive-ionization mode functioning at a collision energy of 18 V and cone voltage of 35 V was used in this analytical method. The mass-to-charge transitions were 464.24 > 433.03 for berzosertib and 488.21 > 400.99 for dasatinib (internal standard). The retention time was 1.71 minutes for berzosertib and 1.41 minutes for dasatinib. The calibration curve was sensitive and linear over the range of 1–2000 ng/ml (weighting factor of 1/ Y 2) with a coefficient of variation of <20%. All measured concentrations fell within the range of the calibration curve for each sample analysis.
Pharmacokinetic Modeling and Simulation
A five-compartment pharmacokinetic model with a central compartment, normal brain compartment, tumor core compartment, tumor rim compartment, and peripheral (other tissues) compartment was created using STELLA (iSEEE systems, Lebanon, NH). The model parameters were obtained from the in vivo studies in FVB wild-type mice after a single oral dose and the brain regional distribution studies in tumor-bearing mice. The brain and tumor compartments were linked to the plasma by distributional clearances (bidirectional passive clearance and unidirectional active efflux by P-gp and Bcrp). The peripheral compartment was linked to the plasma compartment by a bidirectional passive clearance. Simulations were performed to obtain plasma, brain, tumor core, and tumor rim concentrations for two dose levels given as single oral dose—20 mg/kg and 60 mg/kg.
Calculations
Unbound fraction (fu) values of berzosertib in plasma and media were calculated using the ratio of buffer concentration to matrix concentration. For the brain homogenates, the calculation of free fraction accounted for dilution resulting from homogenate preparation (dilution factor, D = 4) as shown here (Kalvass and Maurer, 2002):
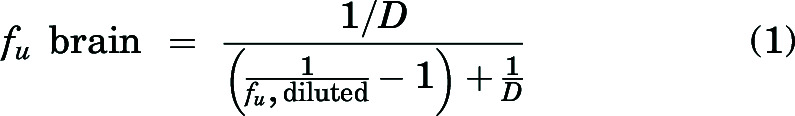 |
A tissue partition coefficient (e.g., brain-, organ-, or tumor-to-plasma ratio), or Kp, was quantified as the ratio of total tissue concentration to total plasma concentration at steady state for tumor core and rim (at two time points) or the ratio of area under the curve from time 0 to infinity (AUC0-∞) for brain (AUC0-∞,brain) to AUC0-∞ for plasma (AUC0-∞,plasma) for single oral and intravenous bolus doses.
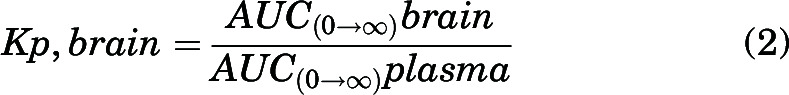 |
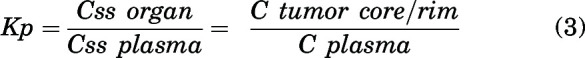 |
The unbound (free) derivative of Kp (Kpuu) was determined by:
 |
Relative drug exposure in the brain between wild-type and knockout (Mdr1a/b−/−, Bcrp1−/−, and Mdr1a/b−/− Bcrp1−/−) mice was compared using the free distribution advantage (DAfree).
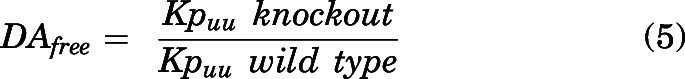 |
Total drug concentrations measured from in vivo pharmacokinetic studies using LC-MS/MS were multiplied by the corresponding matrix (plasma, brain homogenate, and media) fu values that were determined using in vitro binding assay to obtain free drug concentrations (Liu et al., 2008).
Pharmacokinetic Data Analysis
Noncompartmental Analysis
Noncompartmental analysis (NCA) was performed using Phoenix WinNonlin version 8.3 (Certara USA, Inc., Princeton, NJ) to obtain pharmacokinetic parameters from the concentration-time profiles in plasma and brain. Linear trapezoidal integration method was used to calculate the area under the concentration-time curves (AUCs) for plasma and brain. The AUC extrapolation from last time point to infinity was calculated by dividing the last concentration measured by the terminal elimination rate constant, which was determined by the last three to four points in the concentration-time profiles. The percent AUC extrapolation from last time point to infinity was <10%, which indicated that our designed time course was able to capture the majority of the exposure. Other pharmacokinetic parameters, including half-life (t1/2), systemic clearance (CL), and volume of distribution (Vd) for intravenous dose and apparent volume of distribution for oral dose, were calculated using NCA. The standard errors around the means of AUC0-∞ were determined as described previously (Yuan, 1993). The oral bioavailability was calculated using:
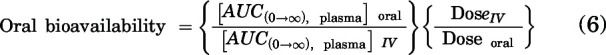 |
Statistical Analysis
The mean estimated free fraction was compared between specimens using unpaired t tests. Median survival was estimated by the Kaplan-Meier method and compared using the log-rank test. Data presentation and statistical tests were completed using GraphPad Prism (Version 8; GraphPad Software, La Jolla, California). Comparisons between two groups were made using an unpaired t test. Comparisons between multiple groups were made using a one-way ANOVA, which was followed by Bonferroni’s multiple comparison test. All experimental data are presented as mean ± S.D. In all cases, P < 0.05 was considered statistically significant.
Results
In Vitro Synergy of TMZ and Berzosertib in GBM Cell Lines
Combination studies of berzosertib (0–1000 nM) and TMZ (0–300 μM) in established GBM cell lines U87 (p53 wild-type, MGMT methylated) and U251 (p53 mutant, MGMT methylated) both showed marked synergy (>100), with a stronger synergy in the U251 cells compared with that in U87 cells (Fig. 2, A and B). In the GBM PDX cell line GBM22 (p53 mutant, MGMT methylated), the combination also showed marked synergistic activity (>100) (Fig. 2C). Transduced U251 cells that overexpress MGMT showed moderate synergy (Fig. 2D) compared with that in U251-CV, which showed strong synergy (Fig. 2E). Across all cell lines, berzosertib concentrations from 100–300 nM show potent synergistic activity in combination with TMZ. In contrast, concentrations <100 nM demonstrate additivity or antagonism, and concentrations above 300 nM show decreased cell viability because of concentration-related toxicity. Therefore, 100–300 nM was deemed to be a reasonable target in vitro concentration for effective berzosertib combinations with TMZ.
Fig. 2.

In vitro synergy studies of berzosertib and TMZ combination in established GBM cell lines. Increasing concentrations of berzosertib (0–1000 nM) and TMZ (0–300 μM) combinations tested for in vitro synergy in (A) U87, (B) U251, (C) GBM22, (D) U251-CV, and (E) U251 MGMT overexpressing cell lines (U251-MGMT). (F) MGMT status, p53 status, and synergy scores for all the tested cell lines.
In Vivo Efficacy of TMZ and Berzosertib in Orthotopic GBM 22 Xenografts
Based on the strong in vitro synergy results for GBM 22 cells, in vivo efficacy of the berzosertib/TMZ combination was evaluated using intracranial models. We found that the addition of berzosertib (60 mg/kg per day via oral gavage for 5 days) to TMZ (50 mg/kg per day via oral gavage for 5 days) did not improve median survival (61 vs. 59 days, P = 0.82; Fig. 3). These negative in vivo results contrast with our in vitro data, which prompted us to speculate that the location of the tumor in the brain may affect the berzosertib/TMZ combination efficacy and the effect of delivery of berzosertib across the BBB into the brain and that the intracranial tumor is a critical factor that influences its lack of in vivo intracranial efficacy.
Fig. 3.

In vivo efficacy studies in GBM 22 orthotopic [intracranial (IC)] tumors treated with 50 mg/kg TMZ for 5 days with and without 60 mg/kg berzosertib. Kaplan-Meier curves for GBM 22 intracranial xenografts (n = 7–8) show no difference in survival with the addition of berzosertib to TMZ (P = 0.82).
Binding of Berzosertib in Plasma, Brain Homogenate, and Media
In vitro binding studies were conducted using RED after a 24-hour (time to reach equilibrium determined from pilot studies) incubation to determine the fu of berzosertib in different matrices. Binding of berzosertib was studied in plasma and brain homogenate matrices from FVB wild-type, Bcrp1–/–, Mdr1a/b–/–, and Mdr1a/b–/–Bcrp1–/– mice to determine the effect of genotype on drug binding. The fu values of berzosertib in plasma and brain homogenate are summarized in Table 1. The fu of berzosertib in the plasma of FVB wild-type mice was 8.1% ± 0.4% as compared to the extremely low fu in the brain homogenate of 0.14% ± 0.12%. The free fraction of bersozertib was 58-fold higher in plasma compared with that in brain. This indicates that berzosertib is highly bound in the brain compared with plasma, and this will have an impact on its free concentration in the brain. Since we used brain homogenate to determine brain free fractions in RED, we cannot determine the exact composition of drug binding sites within the brain. No significant difference was observed in either plasma or brain homogenate binding across different genotypes (P > 0.05). Binding was also calculated in 10% FBS containing media that was used in the in vitro synergy studies, and was found to be 36.4% ± 3.5%. The free fraction of berzosertib in media is 4.5-fold higher compared with that in plasma and 260-fold higher compared with that in brain. These binding results will be critical in determining “free or unbound” concentrations from the in vitro and in vivo studies and relating them to the observed efficacy.
TABLE 1.
The pharmacokinetic parameters of berzosertib in FVB wild-type, Bcrp1–/–, Mdr1a/b–/–, Mdr1a/b–/– Bcrp1–/– mice after administration of a single intravenous bolus dose of 20 mg/kg
| Parameter | Units | Wild-type | Bcrp1 –/– | Mdr1a/b–/– | Mdr1a/b Bcrp1–/– | ||||
|---|---|---|---|---|---|---|---|---|---|
| Plasma | Brain | Plasma | Brain | Plasma | Brain | Plasma | Brain | ||
| t 1/2 | hour | 4.16 | 3.51 | 5.27 | 4.4 | 4.31 | 3.80 | 4.88 | 5.47 |
| AUC0-∞ | hr*ng/ml | 4860 ± 121 | 3097 ± 78 | 6524 ± 487 | 5500 ± 434 | 5562 ± 756 | 13,167 ± 1354 | 5372 ± 358 | 92,068 ± 11,145 |
| Vd | L/kg | 22.6 | — | 22 | — | 21.7 | — | 20.2 | — |
| CL | L/hr/kg | 4.1 | — | 3.0 | — | 3.6 | — | 3.7 | — |
| fu | 0.081 ± 0.004 | 0.0014 ± 0.0002 | 0.072 ± 0.004 | 0.0016 ± 0.0002 | 0.08 ± 0.01 | 0.0015 ± 0.0002 | 0.07 ± 0.008 | 0.0014 ± 0.0002 | |
Brain Distribution of Berzosertib after a Single Intravenous Dose
The plasma and brain concentration-time profiles and the brain-to-plasma ratios after administration of 20 mg/kg of berzosertib via a single intravenous bolus dose are depicted in Fig. 4. Plasma concentrations in FVB wild-type, Bcrp1–/–, Mdr1a/b–/–, and Mdr1a/b–/–Bcrp1–/– mice were similar to each other (Fig. 4A), whereas the brain concentrations vary hugely among the four genotypes, with the wild-type mice having the lowest brain concentrations at all time points, which were followed by Bcrp1–/–, Mdr1a/b–/–, and Mdr1a/b–/–Bcrp1–/– mice (Fig. 4B). The brain-to-plasma ratios at each time point are the highest in the Mdr1a/b–/–Bcrp1–/– mice, which were followed by the Mdr1a/b–/–, Bcrp1–/–, and wild-type mice (Fig. 4C). The pharmacokinetic parameters after NCA are summarized in Table 1. The was similar across all genotypes (P > 0.05), whereas the was the highest in Mdr1a/b–/–Bcrp1–/– mice, which were followed by the Mdr1a/b–/–, Bcrp1–/–, and wild-type mice. The was significantly different in the Mdr1a/b–/– and Mdr1a/b–/–Bcrp1–/– mice compared with the wild-type mice (P < 0.0001); however, the was similar in the Bcrp1–/– mice compared with that in the wild-type mice (P > 0.05). Therefore, the brain-to-plasma AUC ratios in the wild-type, Bcrp1–/–, Mdr1a/b–/–, and Mdr1a/b–/–Bcrp1–/– mice were 0.64, 0.84, 2.36, and 17.13, respectively (Table 3). These values indicate that efflux by P-gp is the major restrictive factor in delivery of berzosertib across the BBB. Although Bcrp has an insignificant efflux contribution in the presence of P-gp, in the absence of both P-gp and Bcrp, the Kp in Mdr1a/b–/–Bcrp1–/– mice is markedly higher than the Kps of Bcrp1–/– and Mdr1a/b–/– mice alone, which indicates a compensatory functional interaction between P-gp and Bcrp in restricting the brain delivery of berzosertib. The wild-type, Bcrp1–/–, Mdr1a/b–/–, and Mdr1a/b–/–Bcrp1–/– mice had similar terminal elimination t1/2, Vd, and CL (Table 1), which indicates that there were no differences in the systemic disposition of berzosertib across all four genotypes.
Fig. 4.

Pharmacokinetic profiles of berzosertib in FVB wild-type, Bcrp1−/−, Mdr1a/b−/−, and Mdr1a/b−/− Bcrp1−/− mice after intravenous administration. Plasma concentrations (A), brain concentrations (B), and brain-to-plasma concentration ratios (C) of berzosertib in FVB wild-type, Bcrp1−/−, Mdr1a/b−/−, and Mdr1a/b−/− Bcrp1−/− mice after administration of a single intravenous bolus dose of 20 mg/kg. Data represent mean ± S.D., n = 4.
TABLE 3.
Total and free brain partition coefficients summarizing the pharmacokinetic studies of berzosertib in FVB wild-type, Bcrp1−/−, Mdr1a/b−/−, and Mdr1a/b−/− Bcrp1−/− mice
| I.V. | P.O. | Steady State | ||||||
|---|---|---|---|---|---|---|---|---|
| Wild-Type | Bcrp1−/− | Mdr1a/b−/− | Mdr1a/b−/− Bcrp1−/− | Wild-Type | Mdr1a/b −/− Bcrp1−/− | Wild-Type | Mdr1a/b−/− Bcrp1−/− | |
| Kp,brain | 0.64 | 0.84 | 2.36 | 17.13 | 0.70 | 13.9 | 0.8 | 14.0 |
| Kpuu,brain | 0.011 | 0.019 | 0.044 | 0.3426 | 0.012 | 0.278 | 0.014 | 0.28 |
| DAfree | 1 | 1.73 | 4 | 31.15 | 1 | 23.16 | 1 | 20 |
P.O., oral administration
Brain Distribution of Berzosertib after a Single Oral Dose
The plasma and brain concentration-time profiles and the brain-to-plasma ratios (Kp,brain) after administration of 20 mg/kg of berzosertib via a single oral dose are depicted in Fig. 5. Plasma concentrations in FVB wild-type and Mdr1a/b–/–Bcrp1–/– mice were similar to each other (Fig. 5A), whereas the brain concentrations and the Kp,brain in the Mdr1a/b–/–Bcrp1–/– mice were significantly higher than those in wild-type mice at each time point (Fig. 5, B and C). The was similar in FVB wild-type and Mdr1a/b–/–Bcrp1–/– mice (P > 0.05), whereas the was significantly higher in Mdr1a/b–/–Bcrp1–/– mice compared with that in wild-type mice (P < 0.0001). Therefore, the brain-to-plasma AUC ratios in the wild-type and Mdr1a/b–/–Bcrp1–/– mice were 0.80 and 13.9, respectively (Table 3). The systemic oral bioavailability was calculated in both wild-type and Mdr1a/b–/–Bcrp1–/– mice as 67% and 74%, respectively (Table 2). Similarity in the oral bioavailabilities across genotypes indicates that P-gp and Bcrp both do not have a significant influence on the systemic bioavailability of berzosertib, even though P-gp– and Bcrp-mediated efflux at the BBB is critical in limiting its brain delivery.
Fig. 5.

Pharmacokinetic profiles of berzosertib in FVB wild-type and Mdr1a/b−/− Bcrp1−/− mice after oral administration. Plasma concentrations (A), brain concentrations (B), and brain-to-plasma concentration ratios (C) of berzosertib in FVB wild-type and Mdr1a/b−/− Bcrp1−/− mice after administration of a single oral dose of 20 mg/kg. Data represent mean ± S.D., n = 4.
TABLE 2.
The pharmacokinetic parameters of berzosertib in FVB wild-type and Mdr1a/b−/− Bcrp1−/− mice after administration of a single oral dose of 20 mg/kg
| Parameter | Units | Wild-Type | Mdr1a/b−/− Bcrp1−/− | ||
|---|---|---|---|---|---|
| Plasma | Brain | Plasma | Brain | ||
| t 1/2 | h | 4.1 | 5.73 | 6.22 | 7.93 |
| Tmax | h | 1 | 2 | 1 | 8 |
| C max | ng/ml | 403.8 | 200.5 | 373.7 | 2375.8 |
| AUC0-∞ | h*ng/ml | 3252 ± 245 | 2294 ± 140 | 4017 ± 211 | 55,909 ± 3359 |
| CL/F | l/h/kg | 6.14 | — | 4.98 | — |
| Vd/F | l/kg | 36.4 | — | 44.7 | — |
| Oral bioavailability | 0.67 | 0.74 | |||
CL/F, apparent clearance; Tmax, time at the maximum drug concentration; Vd/F, apparent volume of distribution.
Steady-State Distribution of Berzosertib
The organ distribution of berzosertib at steady state was determined after a constant rate infusion at 10 μg/h for 48 hours. Figure 6A depicts the concentrations at steady state in plasma, brain, heart, kidney, liver, and muscle in wild-type and Mdr1a/b–/–Bcrp1–/– mice. The only organ showing a significant concentration difference was the brain (P < 0.01). Similarly, the organ-to-plasma ratios in Fig. 6B indicate that the only statistically significant increase in the ratio was seen in the brain (P < 0.0001) between wild-type and Mdr1a/b–/–Bcrp1–/– mice. The Kp,brain obtained using the ratio of concentration at steady state was 0.8 and 14.0, respectively, in wild-type and Mdr1a/b–/–Bcrp1–/– mice. This study indicates that the distribution of berzosertib is unaffected in the absence of P-gp– and Bcrp-mediated efflux to other critical organs except the brain.
Fig. 6.
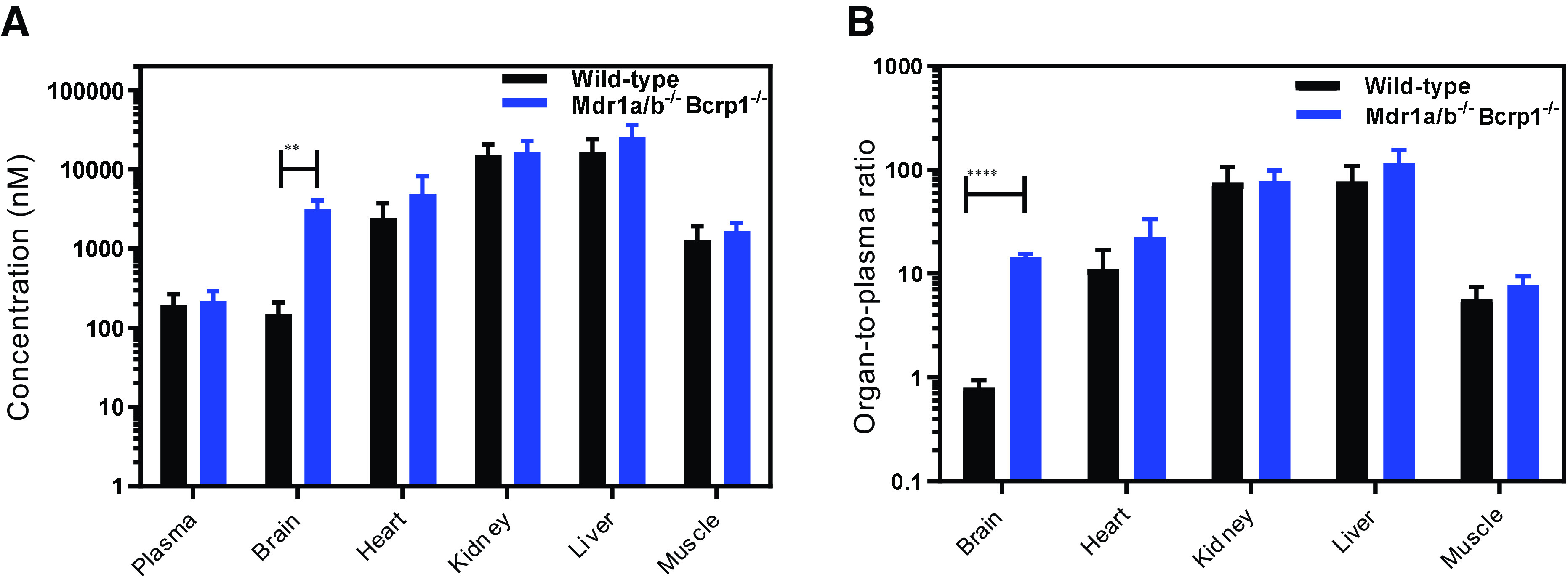
Pharmacokinetics of berzosertib in FVB wild-type and Mdr1a/b−/− Bcrp1−/− mice after intraperitoneal infusion to steady state. (A) Concentrations and (B) organ-to-plasma partition coefficients of berzosertib after an intraperitoneal infusion to steady state at a rate of 10 μg/h in FVB wild-type and Mdr1a/b−/− Bcrp1−/− mice. Data are presented as mean ± S.D., n = 5. **P < 0.01 and ****P < 0.0001.
Impact of Pharmacological Inhibition of P-gp and Bcrp on the Brain Distribution of Berzosertib
The effect of inhibition of the efflux transporters on enhancing the brain disposition of berzosertib was studied by administering 10 mg/kg elacridar (dual inhibitor of P-gp and Bcrp) intraperitoneally with 20 mg/kg berzosertib given orally. Plasma concentrations were similar at both 2 and 8 hours (P > 0.05; Fig. 7A). Brain concentration was significantly higher at 2 hours (P < 0.01; Fig. 7B) but was not different at 8 hours (P > 0.05; Fig. 7B). This is because the half-life of elacridar is 4.3 hours after intraperitoneal administration (Sane et al., 2012), and therefore, at 8 hours almost 75% of the administered elacridar was cleared from systemic circulation, thereby reducing its extent of efflux inhibition. Kp,brain was significantly higher at both 2 (P < 0.005; Fig. 7C) and 8 hours (P < 0.01; Fig. 7C). A moderate 2-fold increase in the brain accumulation of berzosertib was observed at both 2 and 8 hours after coadministration of elacridar.
Fig. 7.

The effect of a pharmacological inhibitor of efflux transport, elacridar, on the plasma and brain concentration of berzosertib. (A) Concentrations in plasma and brain at 2 and 8 hours postdose with and without coadministration of elacridar. (B) Brain-to-plasma ratio at 2 and 8 hours postdose with and without coadministration of elacridar. Data are presented as mean ± S.D., and n = 5 for each group. **P < 0.01 and ***P < 0.005.
Total versus Free Brain Partitioning of Berzosertib
Comparison of brain partitioning of berzosertib in FVB wild-type, Bcrp1–/–, Mdr1a/b–/–, and Mdr1a/b–/–Bcrp1–/– mice after intravenous administration and in FVB wild-type and Mdr1a/b–/–Bcrp1–/– mice after oral and steady-state administration has been summarized in Table 3. The Kp,brain estimates were consistent and robust regardless of the route of administration and method of calculation (AUC ratio or steady-state concentration ratio). Additionally, unbound brain-to-plasma ratio (Kpuu,brain) values calculated by eq. 3 are also summarized in Table 3. Factoring the extent of berzosertib binding in the brain and plasma and determining the free brain partitioning clearly indicate a significant reduction in the Kpuu,brain values. The Kpuu,brain in the wild-type, Bcrp1–/–, Mdr1a/b–/–, and Mdr1a/b–/–Bcrp1–/– mice were 0.011, 0.019, 0.044, and 0.3426, respectively, after an intravenous dose. Similar Kpuu,brain values were observed after oral and steady-state administration of berzosertib (Table 3). The Kpuu,brain in the Mdr1a/b–/–Bcrp1–/–, Mdr1a/b–/–, and Bcrp1–/– mice were 31.15-fold, 4-fold, and 1.73-fold higher than that in the wild-type mice, as indicated by the DAfree in Table 3. These values clearly indicate that, although P-gp–mediated efflux is dominant in the brain delivery of berzosertib, the absence of both P-gp and Bcrp shows an additional increase in the brain accumulation of berzosertib, which indicates a functional cooperativity of these transporters at the BBB. These results also indicate that in addition to efflux considerations mediated by P-gp and Bcrp, the high extent of berzosertib binding in brain and plasma severely restricts the availability of the free concentration in the brain.
Heterogenous Regional Distribution of Berzosertib in Orthotopic GBM 12 PDX Tumors
Determination of orally administered berzosertib distribution within the brain of an orthotopic GBM PDX mouse model is an important factor to consider. To reliably isolate brain tumor from brains of mice, we used a PDX with similar molecular characteristics as GBM22 (GBM12, TP53 mutant, MGMT methylated) but had also been stably transduced with a lentiviral construct for expression of fLuc2-eGFP. Using this fluorescent tumor line, mice with established intracranial tumors were dosed with 20 mg/kg and 60 mg/kg berzosertib orally, and plasma and brain were harvested at 2 and 8 hours postdose. Doses of 20 mg/kg and 60 mg/kg were selected to match the in vivo pharmacokinetic study and efficacy study dosing. Fluorescence-guided punch biopsy was used to separate different brain regions—normal brain, tumor rim, and tumor core (Fig. 8A). At both dose levels, the results indicate a heterogenous distribution of berzosertib, with higher drug concentrations in the tumor core compared with those in normal brain. Additionally, the delivery of berzosertib is restricted in the invasive tumor-rim region compared with that in the tumor core. The rank order of brain concentrations and brain region–to-plasma concentration ratios for the 20-mg/kg dose at both 2 and 8 hours is tumor core > tumor rim = normal brain (Fig. 8, B and C). Similarly at the 60-mg/kg dose, the concentration and the region-to-plasma concentration ratio were the highest in the tumor core, which was followed by similar concentrations in the tumor rim and normal brain (Fig. 8, D and E). At both dose levels, the total concentration of berzosertib was over 100 nM, an efficacious concentration of berzosertib determined in the in vitro synergy studies with TMZ. This picture, however, changed drastically when the total concentrations were converted to free concentrations (Fig. 9). Plasma and brain free fractions from wild-type mice were multiplied to each respective concentration to obtain free concentrations. The unbound fraction in the tumor core and rim was not calculated separately, but the free fraction determined in the brain was used for these regions. The total effective concentration of 100 nM determined from in vitro synergy study was multiplied by the free fraction determined in the media, and therefore, the free effective target concentration in vitro was 36 nM. At both 20 mg/kg and 60 mg/kg, all the normal-brain, tumor-core, and tumor-rim concentrations were below this target free effective concentration. Although we recognize the limitation of the analysis performed in different PDXs for efficacy and delivery, similar suboptimal and severely restricted delivery of berzosertib into orthotopic GBM22 tumors could contribute to the lack of observed efficacy in vivo.
Fig. 8.

Spatial distribution of berzosertib in different regions of the brain after oral administration in mice with GBM 12 intracranial tumors. Data represent mean ± S.D., n = 4–5. *P < 0.05 and **P < 0.01. (A) Representative image of a mouse brain slice marked with tumor core and rim regions. (B) Total concentrations in plasma, normal brain, tumor rim, and tumor core and (C) concentration ratios in brain, tumor rim, and tumor core with respect to plasma after a dose of 20 mg/kg. (D) Total concentrations in plasma, normal brain, tumor rim, and tumor core and (E) concentration ratios in brain, tumor rim, and tumor core with respect to plasma after a dose of 60 mg/kg.
Fig. 9.
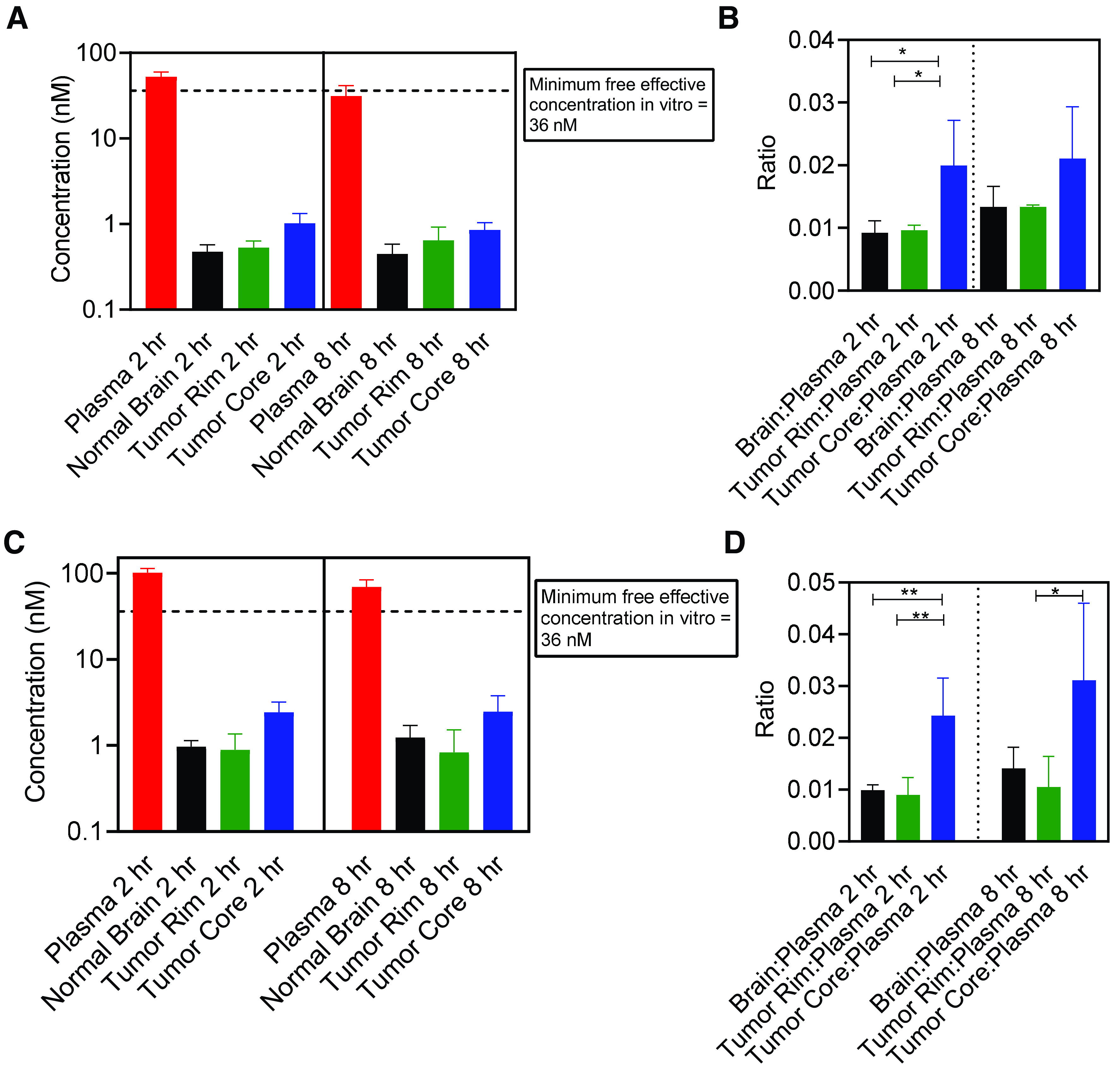
Free/unbound spatial distribution of berzosertib in different regions of the brain after oral administration in mice with GBM 12 intracranial tumors. Data represent mean ± S.D., n = 4–5. *P < 0.05 and **P < 0.01. (A) Free/unbound concentrations in plasma, normal brain, tumor rim, and tumor core and (B) free/unbound concentration ratios in brain, tumor rim, and tumor core with respect to plasma after a dose of 20 mg/kg. (C) Free/unbound concentrations in plasma, normal brain, tumor rim, and tumor core and (D) free/unbound concentration ratios in brain, tumor rim, and tumor core with respect to plasma after a dose of 60 mg/kg.
Pharmacokinetic Modeling and Simulations Explaining Lack of Efficacy of Berzosertib/TMZ Combination in GBM PDX Models
Concentrations and pharmacokinetic parameters obtained from in vivo pharmacokinetic studies and regional distributional studies in tumor-bearing mice were used to develop a model to predict the concentration of berzosertib in plasma, normal brain, tumor rim, and tumor core (Fig. 10A). Systemic pharmacokinetic parameters like elimination rate constant, t1/2, Vd, and CL were obtained from intravenous and oral studies discussed earlier (Tables 1 and 2). The partitioning into normal brain, tumor rim, and tumor core was obtained from the regional tumor distribution study (Fig. 8). Predictivity of the model simulation was confirmed by overlaying plasma and brain concentration data obtained from the oral administration of berzosertib in FVB wild-type mice. The overlay of the experimental data with the simulated concentrations matched visually (Fig. 10B). This model was then used to simulate total concentrations in the plasma, normal brain, tumor rim, and tumor core at 20 mg/kg and 60 mg/kg (Fig. 10, C and D). The simulated profiles at the 60-mg/kg dose showed that the total berzosertib concentration in all these regions was above the total effective concentration for about 16 hours. However, upon converting the total concentration to free concentration, the concentrations in the normal brain, tumor rim, and tumor core did not reach the free effective target concentration of 36 nM at the dose of 60 mg/kg (Fig. 10E). These simulations verify our experimental observations, and this model can therefore be a valuable tool to simulate concentration-time profiles for a variety of doses and dosing regimens to determine an effective dosing regimen to maintain an effective free concentration in vivo for a prolonged duration for the berzosertib/TMZ combination.
Fig. 10.

Pharmacokinetic modeling to predict exposure of berzosertib in plasma and different regions of the brain. (A) Schematic of the pharmacokinetic model. (B) Simulated plasma and brain concentrations overlayed with observed pharmacokinetic data for 20-mg/kg dose. (C) Simulated plasma, brain, tumor rim, and tumor core total concentrations for 20-mg/kg dose. (D) Simulated plasma, brain, tumor rim, and tumor core total concentrations for 60-mg/kg dose. (E) Simulated plasma, brain, tumor rim, and tumor core free concentrations for 60-mg/kg dose.
Discussion
Despite the current multimodal treatment approach of surgery, TMZ, and RT for GBM, the median survival of patients is dismal, with most patients succumbing to their disease in <24 months (Stupp et al., 2018). Therefore, development of effective radio- or chemo-sensitizing strategies is a significant unmet medical need. DNA repair pathways have been widely studied in the last 2 decades with multiple molecules now in clinical trials. Among these pathways, ATR is a key orchestrator in the DDR after therapy-induced replication stress. In this study, we evaluated the brain delivery of berzosertib, a potent ATR inhibitor, and its potential chemosensitization with TMZ for the treatment of GBM.
In vitro synergy studies performed in multiple GBM cell lines demonstrated synergistic activity for the berzosertib/TMZ combination with marked synergy when berzosertib concentrations were between 100 and 300 nM. Thus, 100 nM was established as the minimum effective concentration that would be necessary for effective chemosensitization in vivo with TMZ. Combination synergy scores were enhanced in p53 mutant and MGMT methylated cell lines, which suggests the possibility of increased sensitivity of berzosertib with TMZ in these subsets of GBM PDX cell lines. These results align with previous studies demonstrating that the absence of wild-type p53 confers greater sensitivity to berzosertib in a variety of cancers, and MGMT-deficient glioma cells showed enhanced sensitivity to the berzosertib/TMZ combination (Middleton et al., 2018; Jackson, 2019; Gorecki et al., 2020).
In vivo efficacy studies conducted in orthotopic (intracranial) GBM 22 PDX models demonstrated a lack of survival improvement after berzosertib/TMZ treatment. These in vivo data contrasted with our in vitro synergy data in these same models, and Jackson et al. have previously demonstrated the efficacy of an ATR inhibitor and TMZ combination in flank tumor models derived from both LN229 and GBM22 cell lines in vivo (Jackson, 2019). Collectively, these findings suggest the possibility that drug delivery may be impaired in intracranial tumors. Subsequent brain distribution studies were conducted to investigate this possibility.
In vivo pharmacokinetic studies were conducted after single intravenous, oral, and steady-state infusion dosing. An intravenous dose of berzosertib was administered to wild-type and transporter knockout mice to determine systemic pharmacokinetic parameters and the role of the efflux transporters P-gp and Bcrp in limiting its brain delivery. Oral-dose pharmacokinetics were studied because the route of administration in the in vivo efficacy studies was oral. Our in vivo studies clearly indicate that P-gp plays a major role in restricting delivery of berzosertib into brain. Although Bcrp does not play a significant role on its own (in the presence of P-gp) in limiting the delivery of berzosertib, Mdr1a/b–/–Bcrp1–/– mice had a total Kp,brain value that was greater than the additive Kp from Bcrp1–/– and Mdr1a/b–/– mice, which indicates a functional compensation in the role of these two major efflux transporters, P-gp and Bcrp, at the BBB. This compensation of P-gp and Bcrp at the BBB has been described for a variety of compounds (Polli et al., 2008; Chen et al., 2009; Kodaira et al., 2010; Agarwal et al., 2012; Laramy et al., 2018). The oral bioavailability in wild-type mice was 67%, indicating that oral dosing was feasible for in vivo efficacy studies. The oral bioavailability was similar in Mdr1a/b–/–Bcrp1–/– mice (74%), indicating that the efflux transporters in the intestine did not affect drug absorption, as opposed to their restrictive effect in the brain. A possible reason for this might be the high intestinal lumen concentrations of berzosertib at the current dose compared with those in the plasma, which may have led to the saturation of the intestinal transporters (Lin and Yamazaki, 2003). Steady-state infusion studies indicated that the efflux transporters significantly restricted the delivery of berzosertib to the brain and not to other organs tested like the heart, kidney, liver, and muscle. This is an important finding considering that understanding delivery to these organs is critical in toxicological considerations for the clinical translation of berzosertib.
Although the delivery of berzosertib was restricted by P-gp and Bcrp, the Kp in brain in wild-type mice was 0.7 (after a single oral dose). This, however, is an incomplete picture, since a Kp value close to unity does not necessarily mean that drug in plasma effectively permeates across the BBB. These “high” Kp values (close to unity) can be misinterpreted as efficient brain delivery of a drug and therefore may not be an accurate predictor of its efficacious concentration to the brain. Differences in relative binding affinity of a drug to plasma and brain that can lead to change in free drug partitioning across the BBB is an additional factor that needs to be considered. According to the free drug hypothesis, only free drug is available to move across cell membranes, and evaluation of BBB penetration requires a critical understanding of parameters that influence the movement of free/unbound drug across the BBB, including Kpuu (Hammarlund-Udenaes et al., 2008). We found that berzosertib is highly bound to brain tissues compared with the plasma (∼58-fold higher). We also found that the there was no difference in binding in the plasma and brain of wild-type and knockout mice of all genotypes. Extremely high binding of berzosertib in brain compared with that in the plasma led to a low Kpuu,brain value of 0.011 in wild-type mice. The importance of measuring the relative drug binding in plasma and brain is critical to explain this disagreement between Kp and Kpuu.
The use of elacridar, a dual inhibitor of P-gp and Bcrp, in combination with berzosertib showed a modest 2-fold increase in brain accumulation. This indicates that coadministration of a transporter inhibitor might not be an effective option to improve the brain accumulation of berzosertib.
We also determined the concentrations of berzosertib within different regions of intracranial tumor—tumor core, tumor rim, and normal brain—to determine whether our purported effective concentration was achieved from the dosage regimen employed in the in vivo efficacy study. The drug distribution around the tumor was heterogeneous, with the rank order of tumor core > tumor rim = normal brain. Although the total drug concentrations achieved in all the regions were above 100 nM for the 60-mg/kg dose as used in the efficacy study (Fig. 8), when the total concentration was converted to free concentration by incorporating the brain and plasma binding data, none of the brain regions had achieved the efficacious in vitro free concentration of 36 nM (adjusted for binding in the media used for in vitro synergy studies) (Fig. 9). The concentrations in the tumor core, tumor rim, and normal brain were all multiplied by the free fraction obtained from the normal brain in this case since we were not able to determine binding in tumor core and rim separately. However, an earlier published study for ponatinib, an epidermal growth factor receptor inhibitor, indicated that the binding of the drug in tumor core was lower than that in the tumor rim, which was similar to the normal brain (Laramy et al., 2017). This indicates that our assumption might not be ideal; however, in the case of GBM, the tumor core is usually surgically resected, and therefore, drugs must reach the invasive tumor rim and normal brain to prevent tumor regrowth. Similarity in the binding between tumor rim and the normal brain, therefore, might be a reasonable conservative assumption for berzosertib.
Utilizing data from our in vivo pharmacokinetic studies and the tumor drug distribution study, we developed a pharmacokinetic model for berzosertib that would be able to simulate concentration-time profiles in the plasma, normal brain, tumor rim, and tumor core. In simulated concentration-time profiles for the in vivo efficacy study dose of 60 mg/kg, the free concentration-time profiles in all the brain regions were well below the free efficacious target concentration of 36 nM throughout the entire time course of the dose (Fig. 10E). Assuming linearity, this model can therefore be used to predict concentration-time profiles for different dosing regimens and to aid in determining an efficacious dosing regimen of berzosertib in combination with TMZ.
In conclusion, the berzosertib/TMZ combination showed synergy in vitro in GBM cell lines and PDXs but not in orthotopic tumors. Key factors driving this observation include restricted brain delivery of berzosertib due to efflux at the BBB, a high extent of brain binding, and heterogeneous drug distribution within the tumor; however, further exploration is warranted since mechanisms impacting efficacy are diverse and BBB penetration is one aspect of drug delivery. Other factors include the maturity of the vascular bed, expression of efflux transporters, integrity of the intercellular junctions, and perfusion within the tumor. As noted earlier, a previous study showed a significant improvement in efficacy with the berzosertib/TMZ combination in a flank tumor model of GBM 22 (Jackson, 2019). However, in this study, a 5-mg/kg dose of TMZ was used compared with our TMZ dose of 50 mg/kg, and the berzosertib/TMZ combination was not administered concurrently. This suggests that dose, duration, and sequencing of the combination are key parameters that must be considered when testing ATR inhibitors like berzosertib in combination with TMZ (Fig. 11). Determining efficacious synergistic doses of TMZ is a key factor for the efficacy of this combination. In the case of berzosertib, dosing regimens should be designed to ensure that the efficacious free concentrations cannot only be achieved in vivo but also can be maintained over a prolonged period to ensure inhibition of ATR. Determining the effective dose of a combination therapy from in vitro synergy studies, understanding the brain delivery of ATR inhibitors, and using in vivo pharmacokinetic data to guide dosing regimens in the in vivo efficacy studies in GBM PDXs form a framework for the future preclinical testing of ATR inhibitors in combination with DNA-damaging agents for the treatment of GBM.
Fig. 11.

Factors involved in the observed lack of efficacy of berzosertib for GBM.
Acknowledgments
The authors thank Jim Fisher, Clinical Pharmacology Analytical Laboratory, University of Minnesota, for his support in the development of the LC-MS/MS assay.
Abbreviations
- AUC
area under the concentration-time curve
- AUC0-∞
area under the curve from time 0 to infinity
- AUC0-∞,brain
AUC0-∞ for brain
- AUC0-∞,plasma
AUC0-∞ for plasma
- ATR
ataxia telangiectasia and Rad3-related protein
- BBB
blood-brain barrier
- Bcrp
breast cancer resistance protein
- CL
clearance
- DAfree
free distribution advantage
- DDR
DNA damage response
- eGFP
enhanced GFP
- fLuc2
firefly luciferase 2
- fu
unbound fraction
- FVB
Friend leukemia virus strain B
- GBM
glioblastoma multiforme
- Kp
tissue-to-plasma ratio
- Kp
brain, brain-plasma ratio
- Kpuu
unbound (free) Kp
- Kpuu
brain, unbound Kp, brain
- LC-MS/MS
liquid chromatography–tandem mass spectrometry
- MGMT
O6-methylguanine methyltransferase
- NCA
noncompartmental analysis
- PDX
patient-derived xenograft
- P-gp
P-glycoprotein
- RED
rapid equilibrium dialysis
- RT
radiotherapy
- t 1/2
half-life
- TMZ
temozolomide
- U251-CV
U251 empty control vector
- UPLC
ultra-performance liquid chromatography
- Vd
volume of distribution
Authorship Contributions
Participated in research design: Talele, Sarkaria, Elmquist.
Conducted experiments: Talele, Zhang, Burgenske, Kim, Mohammad, Dragojevic, Gupta.
Performed data analysis: Talele, Burgenske, Sarkaria, Elmquist.
Wrote or contributed to the writing of the manuscript: Talele, Burgenske, Bindra, Sarkaria, Elmquist.
Footnotes
This work was supported by National Institutes of Health National Cancer Institute [Grants RO1 CA138437, RO1 NS077921, U54 CA210180, U01 CA227954, and P50 CA108960]. S.T. was supported by the Rory P. Remmel and Cheryl L. Zimmerman Fellowship in Drug Metabolism and Pharmacokinetics, Edward G. Rippie Fellowship, Bighley Graduate Fellowship, Ronald J. Sawchuk Fellowship in Pharmacokinetics, and Doctoral Dissertation Fellowship.
No author has an actual or perceived conflict of interest with the contents of this article.
References
- Aasland DGötzinger LHauck LBerte NMeyer JEffenberger MSchneider SReuber EERoos WPTomicic MT, et al. (2019) Temozolomide induces senescence and repression of DNA repair pathways in glioblastoma cells via activation of ATR-CHK1, p21, and NF-κB. Cancer Res 79:99–113. [DOI] [PubMed] [Google Scholar]
- Agarwal S, Hartz AMS, Elmquist WF, Bauer B (2012) Breast cancer resistance protein and P-glycoprotein in brain cancer: two gatekeepers team up. Curr Pharm Des 17:2793–2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal S, Sane R, Gallardo JL, Ohlfest JR, Elmquist WF (2010) Distribution of gefitinib to the brain is limited by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2)-mediated active efflux. J Pharmacol Exp Ther 334:147–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed, U, Carruthers R, Gilmour L, Yildirim S, Watts C, Chalmers AJ (2015) Selective inhibition of parallel DNA damage response pathways optimizes radiosensitization of glioblastoma stem-like cells. Cancer Res 75:4416–4428. [DOI] [PubMed] [Google Scholar]
- Becker AP, Sells BE, Haque SJ, Chakravarti A (2021) Tumor heterogeneity in glioblastomas: from light microscopy to molecular pathology. Cancers 13:761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindra RS, Chalmers AJ, Evans S, Dewhirst M (2017) GBM radiosensitizers: dead in the water…or just the beginning? J Neurooncol 134:513–521. [DOI] [PubMed] [Google Scholar]
- Bradbury A, Hall S, Curtin N, Drew Y (2020) Targeting ATR as cancer therapy: a new era for synthetic lethality and synergistic combinations? Pharmacol Ther 207:107450. [DOI] [PubMed] [Google Scholar]
- Caporali S, Falcinelli S, Starace G, Russo MT, Bonmassar E, Jiricny J, D’Atri S (2004) DNA damage induced by temozolomide signals to both ATM and ATR: role of the mismatch repair system. Molecular Pharmacology 66:478–491. [DOI] [PubMed] [Google Scholar]
- Carruthers RDAhmed SURamachandran SStrathdee KKurian KMHedley AGomez-Roman NKalna GNeilson MGilmour L, et al. (2018) Replication stress drives constitutive activation of the DNA damage response and radioresistance in glioblastoma stem-like cells. Cancer Res 78:5060–5071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Agarwal S, Shaik NM, Chen C, Yang Z, Elmquist WF (2009) P-glycoprotein and breast cancer resistance protein influence brain distribution of dasatinib. J Pharmacol Exp Ther 330:956–963. [DOI] [PubMed] [Google Scholar]
- Ferri A, Stagni V, Barilà D (2020) Targeting the DNA damage response to overcome cancer drug resistance in glioblastoma. Int J Mol Sci 21:4910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fokas EPrevo RPollard JRReaper PMCharlton PACornelissen BVallis KAHammond EMOlcina MMGillies McKenna W, et al. (2012) Targeting ATR in vivo using the novel inhibitor VE-822 results in selective sensitization of pancreatic tumors to radiation. Cell Death Dis 3:e441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gampa GKenchappa RSMohammad ASParrish KEKim MCrish JFLuu AWest RHinojosa AQSarkaria JN, et al. (2020) Enhancing brain retention of a KIF11 inhibitor significantly improves its efficacy in a mouse model of glioblastoma. Sci Rep 10:6524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorecki L, Andrs M, Rezacova M, Korabecny J (2020) Discovery of ATR kinase inhibitor berzosertib (VX-970, M6620): clinical candidate for cancer therapy. Pharmacol Ther 210:107518. [DOI] [PubMed] [Google Scholar]
- Gupta SK, Mladek AC, Carlson BL, Boakye-Agyeman F, Bakken KK, Kizilbash SH, Schroeder MA, Reid J, Sarkaria JN (2014) Discordant in vitro and in vivo chemopotentiating effects of the PARP inhibitor veliparib in temozolomide-sensitive versus -resistant glioblastoma multiforme xenografts. Clin Cancer Res 20:3730–3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarlund-Udenaes M, Fridén M, Syvänen S, Gupta A (2008) On the rate and extent of drug delivery to the brain. Pharm Res 25:1737–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegi MEDiserens A-CGorlia THamou M-Fde Tribolet NWeller MKros JMHainfellner JAMason WMariani L, et al. (2005) MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 352:997–1003. [DOI] [PubMed] [Google Scholar]
- Ilyushina NA, Hay A, Yilmaz N, Boon ACM, Webster RG, Govorkova EA (2008) Oseltamivir-ribavirin combination therapy for highly pathogenic H5N1 influenza virus infection in mice. Antimicrob Agents Chemother 52:3889–3897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson CBNoorbakhsh SISundaram RKKalathil ANGanesa SJia LBreslin HBurgenske DMGilad OSarkaria JN, et al. (2019) Temozolomide sensitizes MGMT-deficient tumor cells to ATR inhibitors. Cancer Res 79:4331–4338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalvass JC, Maurer TS (2002) Influence of nonspecific brain and plasma binding on CNS exposure: implications for rational drug discovery. Biopharm Drug Dispos 23:327–338. [DOI] [PubMed] [Google Scholar]
- Kodaira H, Kusuhara H, Ushiki J, Fuse E, Sugiyama Y (2010) Kinetic analysis of the cooperation of P-glycoprotein (P-gp/Abcb1) and breast cancer resistance protein (Bcrp/Abcg2) in limiting the brain and testis penetration of erlotinib, flavopiridol, and mitoxantrone. J Pharmacol Exp Ther 333:788–796. [DOI] [PubMed] [Google Scholar]
- Konstantinopoulos PACheng SCWahner Hendrickson AEPenson RTSchumer STDoyle LALee EKKohn ECDuska LRCrispens MA, et al. (2020) Berzosertib plus gemcitabine versus gemcitabine alone in platinum-resistant high-grade serous ovarian cancer: a multicentre, open-label, randomised, phase 2 trial. Lancet Oncol 21:957–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laramy JK, Kim M, Parrish KE, Sarkaria JN, Elmquist WF (2018) Pharmacokinetic assessment of cooperative efflux of the multitargeted kinase inhibitor ponatinib across the blood-brain barrier. J Pharmacol Exp Ther 365:249–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laramy JKKim MGupta SKParrish KEZhang SBakken KKCarlson BLMladek ACMa DJSarkaria JN, et al. (2017) Heterogeneous binding and central nervous system distribution of the multitargeted kinase inhibitor ponatinib restrict orthotopic efficacy in a patient-derived xenograft model of glioblastoma. J Pharmacol Exp Ther 363:136–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JH, Yamazaki M (2003) Role of P-glycoprotein in pharmacokinetics: clinical implications. Clin Pharmacokinet 42:59–98. [DOI] [PubMed] [Google Scholar]
- Liu X, Chen C, Smith BJ (2008) Progress in brain penetration evaluation in drug discovery and development. J Pharmacol Exp Ther 325:349–356. [DOI] [PubMed] [Google Scholar]
- Majd NK, Yap TA, Koul D, Balasubramaniyan V, Li X, Khan S, Gandy KS, Yung WKA, de Groot JF (2021) The promise of DNA damage response inhibitors for the treatment of glioblastoma. Neurooncol Adv 3:b015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton FK, Pollard JR, Curtin NJ (2018) The Impact of p53 dysfunction in ATR inhibitor cytotoxicity and chemo- and radiosensitisation. Cancers (Basel) 10:275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton MR, Dean E, Evans TRJ, Shapiro GI, Pollard J, Hendriks BS, Falk M, Diaz-Padilla I, Plummer R (2021) Phase 1 study of the ATR inhibitor berzosertib (formerly M6620, VX-970) combined with gemcitabine ± cisplatin in patients with advanced solid tumours. Br J Cancer 125:510–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan MA, Canman CE (2018) Replication stress: an Achilles’ heel of glioma cancer stem–like cells. Cancer Res 78:6713–6716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadkarni A, Shrivastav M, Mladek AC, Schwingler PM, Grogan PT, Chen J, Sarkaria JN (2012) ATM inhibitor KU-55933 increases the TMZ responsiveness of only inherently TMZ sensitive GBM cells. J Neurooncol 110:349–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish KE, Sarkaria JN, Elmquist WF (2015) Improving drug delivery to primary and metastatic brain tumors: strategies to overcome the blood-brain barrier. Clin Pharmacol Ther 97:336–346. [DOI] [PubMed] [Google Scholar]
- Polli JW, Humphreys JE, Harmon KA, Castellino S, O’Mara MJ, Olson KL, St. John-Williams L, Koch KM, Serabjit-Singh CJ (2008) The role of efflux and uptake transporters in N-{3-chloro-4-[(3- fluorobenzyl)oxy]phenyl}-6-[5-({[2-(methylsulfonyl)ethyl]amino}methyl)-2-furyl] -4-quinazolinamine (GW572016, lapatinib) disposition and drug interactions. Drug Metab Dispos 36:695–701. [DOI] [PubMed] [Google Scholar]
- Prichard MN, Shipman C Jr (1990) A three-dimensional model to analyze drug-drug interactions. Antiviral Res 14:181–205. [DOI] [PubMed] [Google Scholar]
- Reaper PM, Griffiths MR, Long JM, Charrier JD, Maccormick S, Charlton PA, Golec JMC, Pollard JR (2011) Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat Chem Biol 7:428–430. [DOI] [PubMed] [Google Scholar]
- Sane R, Agarwal S, Elmquist WF (2012) Brain distribution and bioavailability of elacridar after different routes of administration in the mouse. Drug Metab Dispos 40:1612–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sane R, Agarwal S, Mittapalli RK, Elmquist WF (2013) Saturable active efflux by p-glycoprotein and breast cancer resistance protein at the blood-brain barrier leads to nonlinear distribution of elacridar to the central nervous system. J Pharmacol Exp Ther 345:111–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkaria JNHu LSParney IFPafundi DHBrinkmann DHLaack NNGiannini CBurns TCKizilbash SHLaramy JK, et al. (2018) Is the blood-brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro-oncol 20:184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smee DF, Prichard MN (2017) Comparison of three dimensional synergistic analyses of percentage versus logarithmic data in antiviral studies. Antiviral Res 145:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stupp RTaillibert SKanner ARead WSteinberg DMLhermitte BToms SIdbaih AAhluwalia MSFink K, et al. . 2018. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: a randomized clinical trial. JAMA 318:2306–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan AC, Ashley DM, López GY, Malinzak M, Friedman HS, Khasraw M (2020) Management of glioblastoma: state of the art and future directions. CA Cancer J Clin 70:299–312. [DOI] [PubMed] [Google Scholar]
- van Tellingen O, Yetkin-Arik B, de Gooijer MC, Wesseling P, Wurdinger T, de Vries HE (2015) Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist Updat 19:1–12. [DOI] [PubMed] [Google Scholar]
- Yuan J (1993) Estimation of variance for AUC in animal studies. J Pharm Sci 82:761–763. [DOI] [PubMed] [Google Scholar]