

Abstract
BACKGROUND:
Early administration of unfractionated heparin (UFH) after traumatic brain injury (TBI) reduces early in vivo circulating leukocytes (LEUs) in peri-injury penumbral brain tissue, enhancing cognitive recovery 2 days after injury. It remains unclear how long this effect lasts and if this is related to persistently accumulating LEUs in penumbral brain tissue. We hypothesized that UFH reduces LEU brain tissue sequestration resulting in prolonged cognitive recovery.
METHODS:
CD1 male mice underwent either TBI by controlled cortical impact (CCI) or sham craniotomy. Unfractionated heparin (75 or 225 U/kg) or vehicle was repeatedly administered after TBI. Neurologic function (Garcia Neurological Test [maximum score = 18]) and body weight loss ratios were evaluated at 24 hours to 96 hours after TBI. Brain and lung wet-to-dry ratios, hemoglobin levels, and brain LEU sequestration (Ly6G immunohistochemistry) were evaluated 96 hours postmortem. Analysis of variance with Bonferroni correction determined significance ( p < 0.05).
RESULTS:
Compared with untreated CCI animals (24 hours, 14.7 ± 1.0; 48 hours, 15.5 ± 0.7; 72 hours, 15.0 ± 0.8; 96 hours, 16.5 ± 0.9), UFH75 (24 hours, 16.0 ± 1.0, p < 0.01; 48 hours, 16.5 ± 0.7, p < 0.05; 72 hours, 17.1 ± 0.6, p < 0.01; 96 hours, 17.4 ± 0.7, p < 0.05) increased cognitive recovery throughout the entire observation period after TBI. At 48 hours, UFH225 significantly worsened body weight loss (10.2 ± 4.7%) as compared with uninjured animals (5.5 ± 2.9%, p < 0.05). Both UFH75 (60.8 ± 40.9 PMNs per high-power field [HPF], p < 0.05) and UFH225 (36.0 ± 17.6 PMNs/HPF, p < 0.01) significantly decreased brain neutrophil sequestration found in untreated CCI animals (124.2 ± 44.1 PMNs/HPF) 96 hours after TBI. Compared with untreated CCI animals (78.8 ± 0.8%), UFH75 (77.3 ± 0.6%, p = 0.04) reduced cerebral edema to uninjured levels (77.4 ± 0.6%, p = 0.04 vs. CCI). Only UFH225 (10.6 ± 1.2 g/dL) resulted in lower hemoglobin than in uninjured animals (13.0 ± 1.2 g/dL, p < 0.05).
CONCLUSIONS:
Heparin after TBI reduces tissue LEU sequestration and edema in injured brain for up to 4 days. This is associated with persistent improved cognitive recovery, but only when low-dose UFH is given. Early administration of UFH following TBI may blunt LEU-related cerebral swelling and slow progression of secondary brain injury.
Keywords: Brain edema, heparin, neutrophil, TBI
Traumatic brain injury (TBI) remains the leading cause of death and disability in young Americans, with 1.7 million cases occurring yearly.1 Despite extensive efforts to develop neuroprotective therapies, morbidity and mortality remain high. Following the initial mechanical cerebral damage of severe TBI, ongoing secondary brain injury often persists, driven by cellular, metabolic, and molecular processes including oxidative stress and apoptosis.2–5
Loss of integrity of the blood-brain barrier (BBB) through inappropriate activation of endothelial cells (ECs) and leukocytes (LEUs) is a key manifestation of secondary brain injury.5,6 This maladaptive phenomenon results in uncontrolled release of metalloproteinases, proteases, inflammatory cytokines, and reactive oxygen species, which further promote local BBB destruction and progression to secondary brain injury.5,7,8 Indeed, preventing polymorphonuclear neutrophil (PMN) emigration into injured brain tissue presents an appealing therapeutic intervention in severe TBI.
One family of drugs gaining interest for their effect on activated PMNs in injured tissue is heparinoids.9 A large body of evidence has now demonstrated that heparinoids possess not only anticoagulant activity but also intriguing anti-inflammatory properties.10–15 To date, no study has evaluated the time course of neuromodulating heparin effects on TBI, and it remains unknown how long these effects persist after injury.
In the current study, we sought to determine if unfractionated heparin (UFH) treatment early after TBI was neuroprotective. We hypothesized that UFH reduces cerebral edema and PMN tissue accumulation beyond the early postinjury period and that this is associated with persistent cognitive improvements up to 96 hours after injury.
MATERIALS AND METHODS
Experimental Design and Study Groups
All experimental procedures were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania. Adult male mice (CD1; Charles River Laboratories, Wilmington, MA) weighing 25 g to 30 g were housed for 5 days to 7 days in standard facilities before experiments. Mice underwent severe TBI by controlled cortical impact (CCI) or sham craniotomy. Injured animals were then randomly assigned (by double coin toss) to receive 75 U/kg or 225 U/kg UFH (Medefil, Inc, Glendale Heights, IL) or an equal vehicle volume of 0.9% saline (VEH, 1 mL/kg; Baxter Healthcare Corporation, Deerfield, IL) in repeated subcutaneous doses (at 2, 11, 20, 27, 34 hours after injury). Heparin dosages were chosen to reflect a typical clinical dose used for VTE prophylaxis (75 U/kg) and a triplication of this dose, to reflect equivalent doses utilized in prior publications. Forty-four animals were randomized into four groups: no CCI + VEH (NoCCI, n = 10), CCI + VEH (CCI, n = 11), CCI + 75 U/kg UFH (UFH75, n = 13), and CCI + 225 U/kg UFH (UFH225, n = 10) (Fig. 1). In order to detect differences in immunohistochemistry-sequestered PMNs of at least 7 cells per high-power field (HPF), with an SD of 3, a minimum of five animals in each group was necessary to achieve a significance of less than 0.05 with 80% power. Because all groups’ animal brains were divided equally into those that were oven dried for w-t-d determination and those that were perfused with formalin for immunohistochemistry, this required a minimum of 10 animals per group (Fig. 1).
Figure 1.
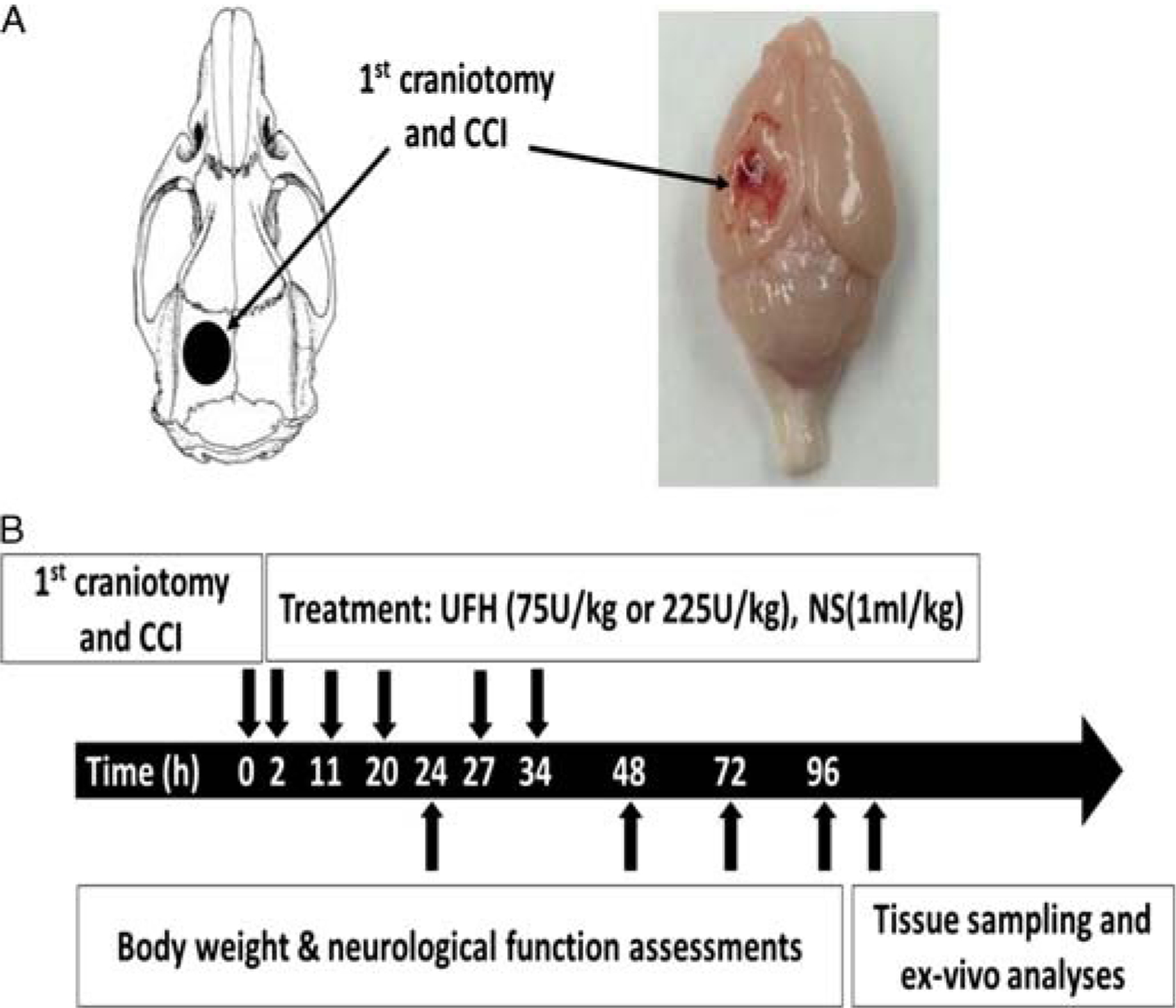
Experimental design and procedures. (A) Schematic diagram showing the location of craniotomies and the CCI-induced cortical contusion area. (B) Timeline of experimental procedures. NS, normal saline.
Severe TBI Model
Controlled cortical impact was used to create severe TBI as described elsewhere.6,16 Briefly, after intraperitoneal anesthesia with ketamine (Hospira, Lake Forest, IL), xylazine (Akorn, Decatur, IL), and acepromazine (Boehringer Ingelheim, St. Joseph, MO) (100/10/1 mg/kg), mice underwent a sagittal incision exposing the skull, and a left-sided, 4-mm craniotomy was created between bregma and lambda sutures using a dental drill (Henry Schein, Melville, NY). The left parietotemporal cortex was then injured using a CCI device (AMS 201; AmScien Instruments, Richmond, VA), resulting in a reproducible injury consistent with severe TBI16 (3-mm-diameter impactor tip, impact velocity of 6 m/s, and cortical deformation of 1.0 mm).
Body Weight Loss, Neurological Recovery, and Systemic Hemodynamics
Animal body weights were measured at 0, 24, 48, 72, and 96 hours after TBI, and extent of weight loss was expressed as weight loss ratio [(W0 h – W24 h or 48 h or 72 h or 96 h)/W0 h × 100%].
Neurologic function was evaluated 24, 48, 72, and 96 hours after TBI using the validated, modified Garcia Neurological Test (GNT),17,18 which scores motor, sensory, reflex, and balance ability to a maximum sum score of 18 points.
Ninety-six hours after injury, the left common carotid artery was cannulated to measure mean arterial pressure. After mean arterial pressure measurement, animals were killed by ketamine overdose and cervical dislocation.
Circulating Hemogoblin Levels
Immediately after the animals were killed, animals underwent cardiac puncture. A 1.0-mL aliquot of fresh blood was evaluated using an i-Stat blood analyzer (Abbott Laboratories, Abbott Park, IL) for serum hemoglobin (Hb) determination per the manufacturer instructions.
Brain and Lung Water Content
After the animals were killed, a subgroup of animals (n = 16) underwent brain excision and left lung excision through sternotomy. Both organs were immediately weighed (wet weight [WW], in grams) and again after 72 hours of drying at 70°C (dry weight [DW]). Tissue water content in each organ was calculated using wet-to-dry ratios (% water content = [WW – DW]/WW × 100%).
Immunohistochemistry to Quantify PMN Sequestration (Ly6G)
At 96 hours from CCI, a subgroup of animals (n = 20) was killed and subjected to cardiac perfusion with 1× PBS (Life Technologies, Carlsbad, CA) and formalin (Sigma-Aldrich Corp., St. Louis, MO). The brain was postfixed in 10% neutral-buffered formalin. Two-millimeter-thick blocks of brain tissue in the coronal plane were processed to paraffin using standard techniques.
For immunohistochemical analysis, 8-μm-thick coronal sections were generated at the midpoint of the contusion. After dewaxing and rehydration, endogenous peroxidase activity was quenched using 3% aqueous hydrogen peroxide. Antigen retrieval was performed via immersion in Tris-EDTA buffer (pH 8.0) within a microwave pressure cooker for 8 minutes. Brain sections were blocked using 2% normal goat serum in Optimax buffer for 1 hour. Incubation with a rat monoclonal antibody Ly6G (clone 1A8; BD Biosciences, San Jose, CA) was then performed (1:15 K for 20 hours at 4°C) in Opimax buffer (BioGenex, San Ramon, CA). Slides were then incubated for 30 minutes at room temperature with biotinylated goat anti–rat secondary antibody followed by 30 minutes with avidin-biotin complex. Visualization was achieved using the DAB peroxidase substrate kit (ABC kit, Vectastain; Vector Labs, Burlingame, CA), as per manufacturer’s instructions. Counterstaining with hematoxylin was also performed.
High-resolution whole-section scans of tissue were generated using the Aperio Scan Scope (Leica Microsystems, Buffalo Grove, IL). ImageScope software (Leica Biosystems, Wetzlar, Germany) was used to visualize the scanned imaged at a magnification of ×20. The number of PMNs was manually counted by an observer unaware of treatment groups and reported as number of cells per HPF.
Statistical Analysis
All data are presented as mean ± SD. Statistical analyses were performed using SPSS software (version 19; SPSS Inc., Chicago, IL). Differences in means between groups were compared using two-tailed analysis of variance (ANOVA) with Bonferroni correction. Six such corrected comparisons were evaluated, and p < 0.05 was considered statistically significant.
RESULTS
Brain and Lung Edema 96 Hours After TBI
As compared with CCI animals (78.8 ± 0.8%), brain water content 96 hours after injury in the injured hemisphere was significantly reduced in UFH75 animals (p = 0.035) approaching NoCCI levels (p = 0.039 vs. CCI; p > 0.05 vs. UFH75) (Fig. 2). There were no significant differences in contralateral cerebral hemisphere and lung water content among the different groups.
Figure 2.
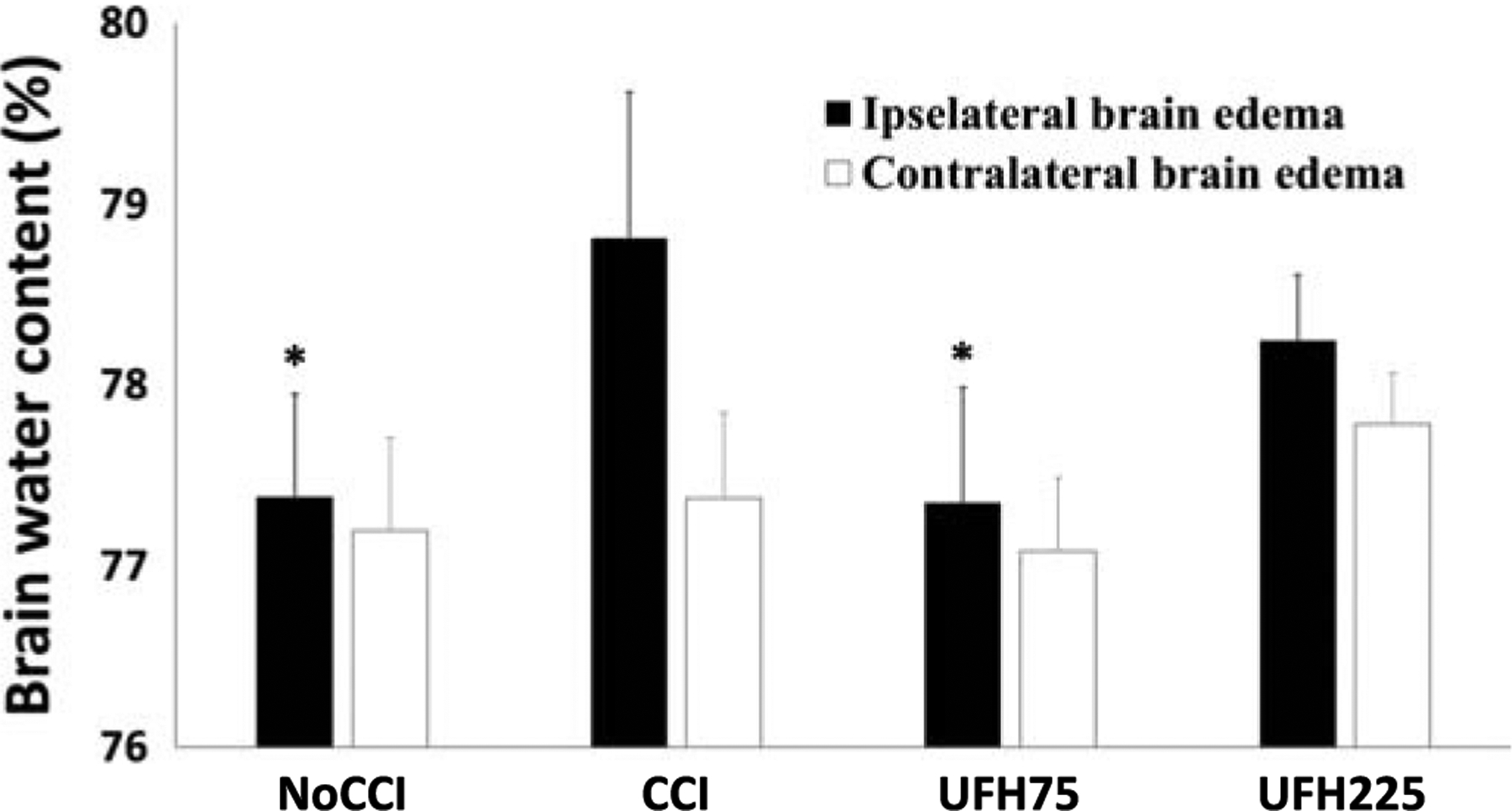
Brain water content by wet-to-dry analyses. Low-dose UFH decreased brain water in the injured (ipsilateral) hemisphere to levels nearing uninjured animals. Ipsilateral hemisphere: *p < 0.05 vs. CCI. There were no significant differences in contralateral cerebral edema among all groups.
Neurological Function, Body Weight Loss
Neurologic functional ability as determined by the GNT was complete during the entire observation period in negative controls (NoCCI). Compared with untreated CCI animals, only UFH75 (24 hours, p = 0.004; 48 hours, p = 0.02; 72 hours, p = 0.001; 96 hours, p = 0.02) demonstrated greater cognitive scores throughout the entire observation period after TBI (Fig. 3A). At 24 hours and 48 hours, UFH75 demonstrated significantly higher neurological scores as compared with UFH225 (24 and 48 hours; p < 0.05 for each). As shown in Figure 3B, body weight loss at 48 hours was greatest in the UFH225 especially when compared with uninjured animals at the same time frame. At 48 hours and 72 hours, there were no significant differences in body weight loss among the different groups.
Figure 3.
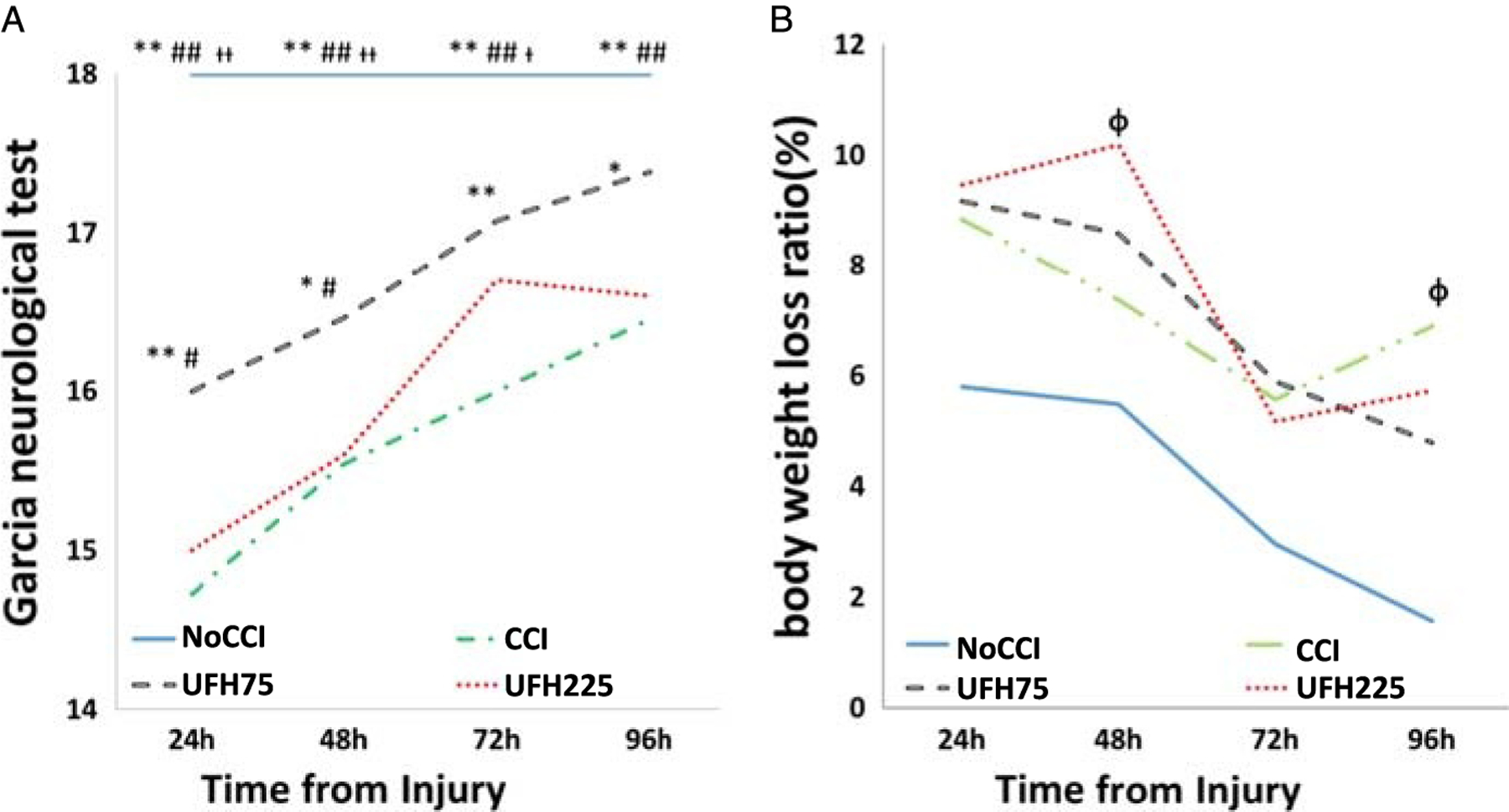
(A) Neurological function was normal in negative controls with significantly higher GNT scores than all other groups at all time frames before and including 72 hours after injury. At 96 hours, uninjured animals (NoCCI) demonstrated similar scores as injured animals treated with 75UFH. Similarly, for the entire study period, 75UFH animals persistently demonstrated better GNT scores than untreated injured animals (CCI). Compared with UFH225, UFH75 significantly improved GNT at both 24 and 48 hours after injury. (B) The greatest weight loss persistence was observed in CCI and UFH225 animals. *p < 0.05, **p < 0.01 versus CCI, #p < 0.05, ##p < 0.01 versus UFH225, †p < 0.05, ††p < 0.01 versus UFH75, Φp < 0.05 versus NoCCI.
Hemodynamics, Hemoglobin
No differences were found among groups in mean arterial blood pressure at 96 hours (NoCCI: 75.5 ± 18.0 mm Hg, CCI: 71.2 ± 19.4 mm Hg, UFH75: 72.1 ± 11.1 mm Hg, UFH225: 58.5 ± 13.0 mm Hg; p > 0.05 for all comparisons), although the lowest blood pressure observed was in the injured animals treated with high-dose UFH (225 U/kg). As compared with Hb levels in uninjured animals (13.0 ± 1.2 mg/dL), levels were significantly lower only in UFH225 (10.6 ± 1.2 mg/dL, p = 0.049) 96 hours after injury/sham craniotomy. Hemoglobin in UFH75 animals (12.4 ± 2.2 mg/dL) and in CCI animals (12.1 ± 1.5 mg/dL) was similar to that of uninjured animals.
Immunohistochemical Analysis of Brain PMN Sequestration
Ninety-six hours after TBI, brain tissue at the level of the contusion was assessed using immunohistochemistry specific for a marker of PMNs (Ly6g). Figure 4 displays a typical stained section with particular concentration of PMNs in the penumbral portion of the injured cerebral hemisphere. Compared with untreated injured animals (124.2 ± 44.1 PMNs/HPF), both UFH75 (p = 0.045) and UFH225 (p = 0.004) groups demonstrated significantly decreased cerebral pericontusional PMN sequestration (Fig. 4, B and C).
Figure 4.
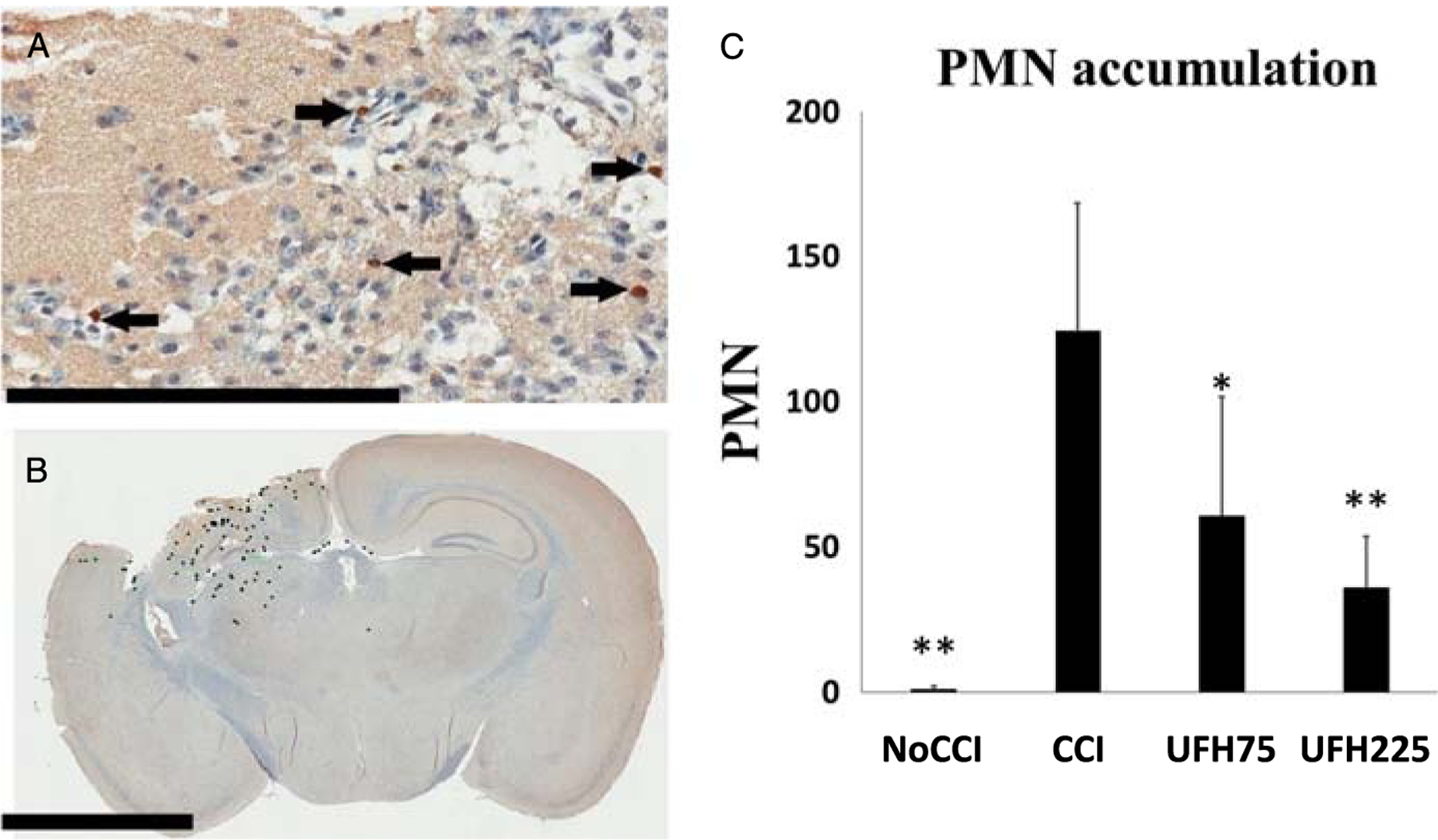
(A) Representative immunohistochemistry images of coronal brain sections demonstrating PMNs in the contusional/pericontusional regions as 1A8-positive cells indicated by black arrows (scale bar 200 μm). (B) Mean PMN counts were quantified by a blinded observer using annotated whole-brain sections in the coronal plane at the level of the contusion (scale bar 3 mm). (C) Compared with untreated injured animals, both UFH treatment groups demonstrated significantly less accumulation of PMNs in penumbral brain tissue. *p < 0.05, **p < 0.01 versus CCI.
DISCUSSION
In the present study, we report how a prophylactic regimen of repeated heparin administration after TBI resulted in reduced cerebral edema and PMN sequestration and was associated with improved cognitive recovery persisting 4 days after injury. While higher-dose heparin resulted in similarly reduced penumbral PMN accumulation, it did not result in diminished brain edema or in late cognitive recovery and instead was associated with lower Hb levels and a tendency to lower systemic blood pressures. Taken together, these findings suggest that prophylactic UFH administered early after TBI may improve neurologic recovery through reducing brain edema and brain leukocyte accumulation, whereas higher doses may result in bleeding, ultimately negating these beneficial effects.
Following the deformation of brain tissue that occurs with severe TBI, progressive brain swelling may occur leading to intracranial hypertension, cerebral herniation, and ultimately brain death.4,19 Loss of integrity of the BBB is at the core of this progression to secondary brain injury, resulting in pervasively increased neurovascular permeability. In physiologic conditions, an intact BBB restricts extravascular passage of circulating leukocytes, restricting their entry and accumulation into the central nervous system. However, after BBB breakdown, leukocyte recruitment may self-perpetuate the release of inflammatory chemokines and cytokines, promoting further tissue leukocyte recruitment and sequestration and further BBB injury.5 We have previously shown how leukocyte-endothelial interactions are observed in penumbral regions of the brain after injury and how this is concurrently associated with greater macromolecular leakage in vivo.20 In particular, significant PMNs rolling on endothelium can be observed adjacent to the injured tissue, which is associated with significant local vasogenic edema in the injured cerebral hemisphere.15,21,22 In addition, leukocyte penetration into the injured parenchymal tissue may activate resident astrocytes and microglia, leading to further local tissue injury and disruption of BBB integrity.7 Different rodent models have demonstrated PMN accumulation in the traumatized hemisphere as early as 4 hours to 8 hours after TBI, peaking at 24 hours to 48 hours and declining at 7 days.23,24 Thus, controlling tissue accumulation of activated circulating leukocytes to arrest ongoing microvascular inflammation and BBB break-down presents an ideal opportunity for therapeutic intervention.
Patients with TBI have a significant risk of VTE,9,25 and pharmacological thromboprophylaxis with unfractionated heparin or low-molecular-weight heparins has been shown to reduce this risk.26,27 However, early heparinoid administration after brain injury is often withheld, fearing worsening of intracranial bleeding. Thus, dose, timing, and nature of heparinoid administration following brain injury are not clearly established and remain guided by local provider preference.
Unfractionated heparins have not only anticoagulant activities, but also immunomodulatory activity. Early studies investigating hemorrhagic shock found heparin administration before induction of shock to prevent endothelial dysfunction, facilitating microvascular rheology and flow and improving end-organ function.10–12 In subsequent reports, several animal studies demonstrated how heparin blunts leukocyte-endothelial interactions in the microcirculation. In particular, heparin was found to inhibit complement activation, disrupting CD11b/CD18–mediated leukocyte adhesion.28 Heparin may also inhibit surface expression of the leukocyte integrins, CD18/CD11b,29 and their endothelial counterligands, ICAM-1 and VCAM-1,30,31 thereby blocking EC-PMN interactions in the microcirculation. Using in vivo microscopy to observe the pial surface of the CCI-injured rodents, we previously demonstrated how UFH administered for 2 days after injury reduced live PMNs circulating in penumbral brain tissue—specifically PMNs rolling on endothelium.15 In that study, the same neurovasculature that demonstrated UFH-related reductions in EC-PMN interactions was also found to simultaneously display less macromolecular leakage when compared with that of untreated animals. In the present study, we sought to evaluate a greater period of 4 days after injury and to determine if the mobilization of circulating leukocytes to the injured brain was also seen as blunted cerebral tissue sequestration of neutrophils at this greater time interval. We were able to demonstrate that PMN sequestration was decreased by UFH up to 4 days after injury on histological analysis of the peri-injury brain tissue. We also demonstrated persistence of diminished brain edema in the injured hemisphere at this extended time period. These findings mirror another study demonstrating reduced ipsilateral cerebral edema with heparin administration in subarachnoid hemorrhage.32 Similarly, heparinoids administered in a rodent cerebral ischemia model resulted in smaller cortical lesion size and diminished surrounding brain edema.33 Interestingly, in our study, postmortem reductions in brain edema were correlated with prior improved cognitive recovery (GNT scores) but only with standard prophylactic UFH doses. Higher UFH doses, on the contrary, did not result in similar reductions in ipsilateral hemisphere wet-to-dry ratios or in cognitive recovery at 4 days after brain injury. While we did not perform histological analysis to evaluate the extent of cerebral bleeding with the higher UFH dose, mice in this group demonstrated lower Hb levels and a trend of reduced systemic blood pressures at 4 days after injury, suggesting that this was from greater bleeding in these animals. Further suggestive of the ill effects of higher-dose UFH was the significant lack of weight loss recovery in these animals 48 hours after injury. In these animals, hypoactivity or poor feeding due to anemia or obtundation from increased brain hemorrhage secondary to greater anticoagulation may have led to the inability to regain lost body weight.
As a small animal protocol, this study has important limitations. First, it was conducted in mice, and there is no definitive knowledge to ensure that both TBI and brain heparin effects occur in mice identically as in human brain injury. Second, repeated UFH administration lasted from 2 hours to 36 hours after CCI, while animals were killed 4 days after injury. In addition, time intervals between UFH heparin doses were not exactly 8 hours, which would have otherwise been a typical dosing frequency. In clinical settings, patients initiated on prophylactic heparinoids would continue receiving these agents every 8 hours, at least until hospital discharge. We chose to use the same 2-day regimen of UFH as in previous analyses15 to determine if that regimen also resulted in improved cognitive recovery, lower brain edema, and reduced leukocyte histological accumulation at the longer time frame. Nonetheless, administering UFH for an additional 2 days until termination of the observation period (4 days) may have increased the risk of brain hemorrhage and cancelation of the beneficial effects related to reduced tissue water. Third, we did not evaluate or quantify the amount of intracranial hemorrhage following injury with or without UFH. Using a histological grading of hemorrhagic contusion or imaging with computed tomography or magnetic resonance imaging would have been a better way to assess bleeding effects of variable UFH doses on injured brain. Nonetheless, we measured Hb levels, not the best surrogate of bleeding, albeit adequate because it was measured several days after injury, allowing for fluid redistribution and equilibration. Fourth, we did not measure systemic white blood cell counts. Previous studies have reported a significant leukocytosis after TBI34,35 and differential circulating LEU levels may have influenced the unambiguous differences in sequestered PMNs demonstrated via immunohistochemistry. Finally, we did not evaluate whether our standardized CCI model16 resulted in variable subarachnoid or intra-axial cerebral bleeding in different animals. We have used the current model in our laboratory for several years and have not had the ability to observe such differences among animals injured with the same CCI parameters.
In conclusion, we have demonstrated that standard clinical prophylactic doses of UFH repeatedly administered after TBI prevented accumulation of neutrophils and edema in injured brain tissue as late as 4 days after injury. This was directly correlated to persistently improved cognitive recovery, during the same time frame. Higher UFH doses resulted in a drop in Hb 4 days after injury, suggesting increased bleeding complications perhaps negating any brain water-reducing effects of UFH. Early administration of low-dose prophylactic UFH following TBI may blunt leukocyte-related cerebral swelling and slow progression of secondary brain injury. Further studies are needed to confirm if the antineuroinflammatory properties of heparin can be leveraged in brain injury without greater risk of bleeding or hematoma expansion.
ACKNOWLEDGMENTS
The authors thank Ms. Robin Armstrong for her technical and organizational assistance.
EDITORIAL CRITIQUE
Dr. Nagata and colleagues have suggested that the loss of integrity of the blood brain barrier through the activation of leukocytes, specifically PMNs, is one key manifestation in the progression of secondary brain injury as this activation promotes microvascular injury through the release of metalloproteinases, proteases, inflammatory cytokines, and reactive oxygen species. Thus preventing PMN migration into the severely injured brain could impede the progression of secondary brain injury and improve outcomes.
By using a controlled cortical impact device in a mouse model, the authors have found that repeated injections of low dose unfractionated heparin (UFH) after severe TBI reduces tissue leukocyte sequestration and cerebral edema in the injured mouse brain resulting in improved cognitive recovery.
While the authors are to be commended for providing new data on the prevention of secondary brain injury in a mouse model, it is important to note that the authors did not describe the type of brain injury that occurred within their model. There are several types of brain injury including subarachnoid hemorrhage, epidural hematomas, subdural hematomas, diffuse axonal injury, and intraparenchymal hemorrhage and they all behave differently with respect to their pathophysiology. Thus, because the type of brain injury was not described, one cannot extrapolate the results of this study to apply to all TBI.
Second, hemoglobin is not the best surrogate of bleeding, as acknowledged by the authors; thus, this study did not demonstrate whether multiple administrations of UFH within such a short period of time from injury would lead to progression of the TBI.
While I congratulate the authors on describing the beneficial effect of UFH on PMN sequestration in a mouse model, we must be cautiously optimistic and address the concerns outlined above before the implementation of a clinical trial.
Footnotes
DISCLOSURE
The authors declare no conflicts of interest.
REFERENCES
- 1.Rosenfeld JV, Maas AI, Bragge P, Morganti-Kossmann MC, Manley GT, Gruen RL. Early management of severe traumatic brain injury. Lancet. 2012;380(9847):1088–1098. [DOI] [PubMed] [Google Scholar]
- 2.Logsdon AF, Lucke-Wold BP, Turner RC, Huber JD, Rosen CL, Simpkins JW. Role of microvascular disruption in brain damage from traumatic brain injury. Compr Physiol. 2015;5(3):1147–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang ZG, Sun X, Zhang QZ, Yang H. Neuroprotective effects of ultra-low-molecular-weight heparin on cerebral ischemia/reperfusion injury in rats: involvement of apoptosis, inflammatory reaction and energy metabolism. Int J Mol Sci. 2013;14(1):1932–1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shlosberg D, Benifla M, Kaufer D, Friedman A. Blood-brain barrier break-down as a therapeutic target in traumatic brain injury. Nat Rev Neurol. 2010;6(7):393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scholz M, Cinatl J, Schadel-Hopfner M, Windolf J. Neutrophils and the blood-brain barrier dysfunction after trauma. Med Res Rev. 2007;27(3): 401–416. [DOI] [PubMed] [Google Scholar]
- 6.Pascual JL, Murcy MA, Li S, Gong W, Eisenstadt R, Kumasaka K, Sims C, Smith DH, Browne K, Allen S, et al. Neuroprotective effects of progesterone in traumatic brain injury: blunted in vivo neutrophil activation at the blood-brain barrier. Am J Surg. 2013;206(6):840–845; discussion 5–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Corps KN, Roth TL, McGavern DB. Inflammation and neuroprotection in traumatic brain injury. JAMA Neurol. 2015;72(3):355–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pun PB, Lu J, Moochhala S. Involvement of ROS in BBB dysfunction. Free Radic Res. 2009;43(4):348–364. [DOI] [PubMed] [Google Scholar]
- 9.Reiff DA, Haricharan RN, Bullington NM, Griffin RL, McGwin G Jr, Rue LW 3rd. Traumatic brain injury is associated with the development of deep vein thrombosis independent of pharmacological prophylaxis. J Trauma. 2009;66(5):1436–1440. [DOI] [PubMed] [Google Scholar]
- 10.Rana MW, Singh G, Wang P, Ayala A, Zhou M, Chaudry IH. Protective effects of preheparinization on the microvasculature during and after hemorrhagic shock. J Trauma. 1992;32(4):420–426. [DOI] [PubMed] [Google Scholar]
- 11.Wang P, Ba ZF, Chaudry IH. Endothelial cell dysfunction occurs after hemorrhage in nonheparinized but not in preheparinized models. J Surg Res. 1993;54(5):499–506. [DOI] [PubMed] [Google Scholar]
- 12.Wang P, Singh G, Rana MW, Ba ZF, Chaudry IH. Preheparinization improves organ function after hemorrhage and resuscitation. Am J Physiol. 1990;259(3 Pt 2):R645–R650. [DOI] [PubMed] [Google Scholar]
- 13.Weber JR, Angstwurm K, Rosenkranz T, Lindauer U, Freyer D, Burger W, Busch C, Einhaupl KM, Dirnagl U. Heparin inhibits leukocyte rolling in pial vessels and attenuates inflammatory changes in a rat model of experimental bacterial meningitis. J Cereb Blood Flow Metab. 1997;17(11):1221–1229. [DOI] [PubMed] [Google Scholar]
- 14.Yanaka K, Spellman SR, McCarthy JB, Oegema TR Jr, Low WC, Camarata PJ. Reduction of brain injury using heparin to inhibit leukocyte accumulation in a rat model of transient focal cerebral ischemia. I. Protective mechanism. J Neurosurg. 1996;85(6):1102–1107. [DOI] [PubMed] [Google Scholar]
- 15.Nagata K, Kumasaka K, Browne KD, Li S, St-Pierre J, Cognetti J, Marks J, Johnson VE, Smith DH, Pascual JL. Unfractionated heparin after TBI reduces in vivo cerebrovascular inflammation, brain edema and accelerates cognitive recovery. J Trauma Acute Care Surg. 2016;81(6):1088–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith DH, Soares HD, Pierce JS, Perlman KG, Saatman KE, Meaney DF, Dixon CE, McIntosh TK. A model of parasagittal controlled cortical impact in the mouse: cognitive and histopathologic effects. J Neurotrauma. 1995;12(2):169–178. [DOI] [PubMed] [Google Scholar]
- 17.Garcia JH, Wagner S, Liu KF, Hu XJ. Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Statistical validation. Stroke. 1995;26(4):627–634; discussion 35. [DOI] [PubMed] [Google Scholar]
- 18.Manaenko A, Lekic T, Ma Q, Ostrowski RP, Zhang JH, Tang J. Hydrogen inhalation is neuroprotective and improves functional outcomes in mice after intracerebral hemorrhage. Acta Neurochir Suppl. 2011;111:179–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pop V, Badaut J. A neurovascular perspective for long-term changes after brain trauma. Transl Stroke Res. 2011;2(4):533–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kumasaka K, Marks JA, Eisenstadt R, Murcy MA, Samadi D, Li S, Johnson V, Browne KD, Smith DH, Schwab CW, et al. In vivo leukocyte-mediated brain microcirculatory inflammation: a comparison of osmotherapies and progesterone in severe traumatic brain injury. Am J Surg. 2014;208(6): 961–968; discussion 7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li S, Marks JA, Eisenstadt R, Kumasaka K, Samadi D, Johnson VE, Holena DN, Allen SR, Browne KD, Smith DH, Pascual JL. Enoxaparin ameliorates post-traumatic brain injury edema and neurologic recovery, reducing cerebral leukocyte endothelial interactions and vessel permeability in vivo. J Trauma Acute Care Surg. 2015;79(1):78–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li S, Eisenstadt R, Kumasaka K, Johnson VE, Marks J, Nagata K, Browne KD, Smith DH, Pascual JL. Does enoxaparin interfere with HMGB1 signaling after TBI? A potential mechanism for reduced cerebral edema and neurologic recovery. J Trauma Acute Care Surg. 2016; 80(3):381–387; discussion 7–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carlos TM, Clark RS, Franicola-Higgins D, Schiding JK, Kochanek PM. Expression of endothelial adhesion molecules and recruitment of neutrophils after traumatic brain injury in rats. J Leukoc Biol. 1997;61(3): 279–285. [DOI] [PubMed] [Google Scholar]
- 24.Chatzipanteli K, Alonso OF, Kraydieh S, Dietrich WD. Importance of post-traumatic hypothermia and hyperthermia on the inflammatory response after fluid percussion brain injury: biochemical and immunocytochemical studies. J Cereb Blood Flow Metab. 2000;20(3):531–542. [DOI] [PubMed] [Google Scholar]
- 25.Knudson MM, Ikossi DG, Khaw L, Morabito D, Speetzen LS. Thromboembolism after trauma: an analysis of 1602 episodes from the American College of Surgeons National Trauma Data Bank. Ann Surg. 2004;240(3):490–496; discussion 6–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Farooqui A, Hiser B, Barnes SL, Litofsky NS. Safety and efficacy of early thromboembolism chemoprophylaxis after intracranial hemorrhage from traumatic brain injury. J Neurosurg. 2013;119(6):1576–1582. [DOI] [PubMed] [Google Scholar]
- 27.Kim L, Schuster J, Holena DN, Sims CA, Levine J, Pascual JL. Early initiation of prophylactic heparin in severe traumatic brain injury is associated with accelerated improvement on brain imaging. J Emerg Trauma Shock. 2014;7(3):141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rao NV, Argyle B, Xu X, Reynolds PR, Walenga JM, Prechel M, Prestwich GD, MacArthur RB, Walters BB, Hoidal JR, et al. Low anticoagulant heparin targets multiple sites of inflammation, suppresses heparin-induced thrombo-cytopenia, and inhibits interaction of RAGE with its ligands. Am J Physiol Cell Physiol. 2010;299(1):C97–C110. [DOI] [PubMed] [Google Scholar]
- 29.Diamond MS, Alon R, Parkos CA, Quinn MT, Springer TA. Heparin is an adhesive ligand for the leukocyte integrin Mac-1 (CD11b/CD1). J Cell Biol. 1995;130(6):1473–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miller SJ, Hoggat AM, Faulk WP. Heparin regulates ICAM-1 expression in human endothelial cells: an example of non–cytokine-mediated endothelial activation. Thromb Haemost. 1998;80(3):481–487. [PubMed] [Google Scholar]
- 31.Lee JH, Kim CH, Seo GH, Lee J, Kim JH, Kim DG, Ahn YS. Heparin attenuates the expression of TNFalpha-induced cerebral endothelial cell adhesion molecule. Korean J Physiol Pharmacol. 2008;12(5): 231–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Altay O, Suzuki H, Hasegawa Y, Sorar M, Chen H, Tang J, Zhang JH. Effects of low-dose unfractionated heparin pretreatment on early brain injury after subarachnoid hemorrhage in mice. Acta Neurochir Suppl. 2016;121:127–130. [DOI] [PubMed] [Google Scholar]
- 33.Stutzmann JM, Mary V, Wahl F, Grosjean-Piot O, Uzan A, Pratt J. Neuroprotective profile of enoxaparin, a low molecular weight heparin, in in vivo models of cerebral ischemia or traumatic brain injury in rats: a review. CNS Drug Rev. 2002;8(1):1–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rhind SG, Crnko NT, Baker AJ, Morrison LJ, Shek PN, Scarpelini S, Rizoli SB. Prehospital resuscitation with hypertonic saline-dextran modulates inflammatory, coagulation and endothelial activation marker profiles in severe traumatic brain injured patients. J Neuroinflammation. 2010;7:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liao Y, Liu P, Guo F, Zhang ZY, Zhang Z. Oxidative burst of circulating neutrophils following traumatic brain injury in human. PLoS One. 2013; 8(7):e68963. [DOI] [PMC free article] [PubMed] [Google Scholar]