

Abstract

A set of unsymmetrical heteroaryl 1,2-diketones were synthesized by a heteroarylation/oxidation sequence with up to 65% isolated yields. Palladium catalyst XPhos Pd G4 and SeO2 were the key reagents used in this methodology, and microwave irradiation was utilized to facilitate an efficient and ecofriendly process. The application of heteroaryl 1,2-diketones is demonstrated through the synthesis of an unsymmetrical 2-phenyl-3-(pyridin-3-yl)quinoxaline (5a) from 1-phenyl-2-(pyridin-3-yl)ethane-1,2-dione (4a). The lowest energy conformations of 4a and 5a were located using Density Functional Theory (DFT) at the M06-2X/def2-TZVP level of theory. Two lowest energy conformations of 4a differ with respect to the position of the N atom in the pyridyl ring and 0.27 kcal/mol energy difference between them corresponds to 60.4 and 39.6% at 50 °C in toluene. Four lowest energy conformations for 5a have the energy differences of 0.01, 0.03 and 0.07 kcal/mol that corresponds to 26.0, 25.7, 24.9 and 23.4%, respectively. A comparison of 4a and 5a to the less hindered analogs (oxalyl chloride and oxalic acid) is used to investigate the structural features and bonding using Natural Bond Orbital (NBO) analysis.
Introduction
The 1,2-diketones are versatile building blocks and are used as organic intermediates in the total synthesis of natural products, preparation of bioactive molecules, etc.1−4 A well-known 1,2-diketone is benzil or 1,2-diphenylethane-1,2-dione (PhCO)2, which can be synthesized from deoxybenzoins, alkynes, aldehydes, or α-oxo acid chloride.5−7 Representative reactions for 1,2-diketones are their condensation reactions with bifunctional nucleophiles such as diamines and thioureas to form heterocyclic rings, diamines and diimines (Scheme 1).8 Additionally, 1,2-diketones have demonstrated some uncommon yet interesting reactions including addition/oxy Cope rearrangement to make 1,6-diketones, and condensation with 1,3-diphenylacetone to make tetraphenylcyclopentadienone (Scheme 1).9−11 By using different heteroaryl substrates, appealing molecular entities can be readily prepared and utilized in many applications, for example pharmaceutical reagents, catalytic ligands and chemical sensors.12
Scheme 1. Synthetic Application of 1,2-Diketone Compounds.

During our previous research using acetophenone 1a and 3-iodopyridine 2a to form heteroarylation product 3a,13,14 an unexpected heteroaryl 1,2-diketone structure 4a was observed (Scheme 2). The structure of this diketone 4a was confirmed by 13C NMR (193.0 and 192.8 ppm for carbonyl carbons) and High Resolution Mass Spectroscopy. This interesting observation inspired us to develop a route for the synthesis of unsymmetrical, heteroaryl 1,2-diketone compounds. Though a wide variety of methods have been developed to synthesize 1, 2-diketones,15−20 the literature on synthetic routes to heteroaryl 1,2-diketones are sparse.21,22 Katritzky reported the synthesis of heteroaryl 1,2-diketones from heteroaryl(aryl)methyl benzotriazoles and esters using BuLi as a key agent in 2005.23 Kumar investigated a one-pot, two-step procedure to synthesize heteroaryl 1,2-diketones from nitriles and organoboron reagents.24 To the best of the author’s knowledge, these are the closest examples on the synthesis of a set of heteroaryl 1,2-diketones from simple, commercially available starting materials.
Scheme 2. Direct α-heteroarylation of Acetophenone (1a) With 3-Iodopyridine (2a) via Palladium-Catalysis.

Herein we report our recent research findings on the construction of unsymmetrical heteroaryl 1,2-diketones from commercially available ketones and heteroaryl halides. The optimization, synthesis and potential application of heteroaryl 1,2-diketone compounds were investigated in this study. A special feature about this reported methodology is that it allows the synthesis of rarely reported alkyl heteroaryl 1,2-diketones with minimal effort. The lowest energy conformations of the heteroaryl 1,2-diketone and its quinoxaline derivative are located using density functional theory (DFT) calculations. Natural bond orbital (NBO) analysis was applied in order to gain insight into electron delocalizations.
Results and Discussion
Reaction Condition Optimization for the Oxidation Step
The oxidation reaction of a heteroaryl ketone to a diketone might be induced by accidental exposure to air. Aerobic oxidations have been reported to convert vinyl or alkyl groups, especially benzylic alkyl groups to alcohols, aldehydes, ketones, carboxylic acids, etc.25,26 The oxidation of ketones to diketones using catalytic 1,4-diazabicyclo[2.2.2]octane (DABCO) in the presence of air in a recent report showed similarities with our reaction system.27 The formation of diketone could also be facilitated by oxidizing agents such as I2.28,29 Some mild oxidizing agents have been reported to successfully facilitate selective oxidation for palladium-catalyzed reactions.30,31 Practically, iodine I2 is often used in combination with other oxidants and I2 functions as a promoter.32,33 Thus, various chemicals were tested in combination with I2 in our study to further promote the oxidation step shown in Scheme 2. The addition of I2, DMSO and CuO directly into the heteroarylation reaction mixture produced the desired products, though the yields were very low (Table 1, Entry 1).28,34 In order to obtain better yields, the heteroarylation product was briefly worked up with a simple extraction and then the crude product was carried forward to the oxidation step. This change has dramatically improved the overall yield (Table 1, Entry 2) for this heteroarylation/oxidation process. A survey of the literature brought to our attention selenium oxide (SeO2) which was used to obtain 1,2-diketones from natural product scaffolds such as camphor.35−37 We tested SeO2 and found the combination of SeO2 and 1,4-dioxane provided the best yields and purity. While the overall yield for the two-step heteroarylation/oxidation sequence was reasonable (Table 1, Entry 3), carrying out the SeO2 oxidation step only when purified 3a was utilized led to a better yield (Table 1, Entry 4), indicating the high efficiency of this step. Several other external oxidants were also tested including 1,4-benzoquinone, PhI(OAc)2, mCPBA and H2O2, but these options did not give promising results. Considering the ease of operation, SeO2 and dioxane was established as the optimal reaction conditions for the preparation of heteroaryl 1,2-diketones.
Table 1. Reaction Condition Optimization for the Oxidation Stepa.

| entry | oxidant/additive/solvent | T (oC) | yield (%)b |
|---|---|---|---|
| 1c | 1.1 equiv I2, 1.1 equiv CuO, Toluene/DMSO | 50 | 8% |
| 2 | 1.1 equiv I2, 1.1 equiv CuO, DMSO | 50 | 46% |
| 3 | 1.5 equiv SeO2, Dioxane | 50 | 59% |
| 4d | 1.5 equiv SeO2, Dioxane | 50 | 89%e |
Unless otherwise noted, all reactions were conducted with 1.1 equiv oxidant, 1.1 equiv additive, 1 mL solvent, stir, overnight. The oxidation step was performed on crude 3a after work-up.
Yield reported represents overall transformation of 1a and 2a to 4a.
Reagents were added directly to the step 1 reaction mixture without work-up.
The oxidation step was performed on purified 3a.
Yield reported is specifically the transformation of 3a to 4a.
Ketone and Heteroaryl Halide Substrate Scope Investigation
After optimizing the reaction conditions for 1,2-diketone 4a using the heteroarylation/oxidation sequence, we proceeded to investigate the substrate scope using various ketones and heteroaryl halides. The first step, heteroarylation, was conducted in a sealed reaction vessel under microwave irradiation using a microwave reactor. The reaction temperature was monitored by an external surface IR sensor, and the temperature was maintained at the expected reaction temperature in each experiment. The crude product from the heteroarylation step was subsequently utilized in the second step in the presence of SeO2 in dioxane to form the 1,2-diketone product, which was purified by automated flash chromatography. About twenty 1,2-heteroaryl diketones were successfully synthesized and purified in up to 65% yields over the course of the two reactions (Scheme 3). Most of these molecules have not been reported previously, therefore they have the potential to lead to the discovery of interesting molecular entities upon further structural modifications.
Scheme 3. Substrate Scope Study for the Synthesis of Heteroaryl 1,2-Diketones via Palladium-Catalysis.

Unless otherwise noted, all reactions were conducted under the following conditions: (1) 1 mol % XPhos Pd G4 catalyst, 2.4 equivalent NaOtBu, 1.0 equivalent heteroaryl halide, 1.1 equivalent ketone, toluene, microwave irradiation, 150 °C for 10 min; (2) SeO2, dioxane, 50 °C, overnight.
1.5 equivalent of ketones and 2.9 equivalent of NaOtBu were used.
It is noteworthy to point out the following observations from our study. A direct comparison of the reactivity for 3-iodopyridine, 3-bromopyridine and 3-chloropyridine showed that heteroaryl iodide was more reactive than heteroaryl bromide, which was more reactive than heteroaryl chloride (Scheme 3, compound 4a). This aryl halide reactivity trend is consistent with most other palladium-catalyzed cross-coupling processes. Most ketones investigated in this study are good substrates. Acetophenone with methyl, methoxy, bromine, chlorine and fluorine substitutions produced the desired products (compounds 4b–4h) with up to 48% overall yield. Comparing compounds 4b, 4d, 4e with 4c, 4f, 4g, 4h, it appears that electron withdrawing groups or ortho-substituents lower the yields. The developed mild reaction conditions can tolerate sensitive functional groups, as indicated by the synthesis of the TBSO-protected heteroaryl 1,2-diketone 4i. This will extend the substrate scope to heteroaryl diketones with two or more hydroxyl groups which are of potential biological interest.38,39 The reactions using volatile ketones such as methyl isopropyl ketone, pinacolone, or methyl isobutyl ketone proved to be the most convenient considering the ease of operation: the excess ketones and other reagents can be removed by simple extraction and evaporation, and the pure products 4j–4l are obtained without the need to use flash chromatography. This unique advantage presents an opportunity to quickly construct a small pool of alkyl heteroaryl 1,2-diketones from commercially available reagents. Generally speaking, aliphatic substrates 4j and 4k provided lower yields than the aryl substrates; yet, 4l gave the highest yield. Reactions with heteroaryl ketone substrates such as 3-acetyl-2,5-dimethylfuran and 3-acetyl-2,5-dimethylthiophene proceeded smoothly and yielded the expected products 4m and 4n. However, some heteroaryl ketones are challenging and the reactions with 2, 3 or 4-acetylpyridines have not been successful yet. In addition to 3-iodopyridine, we also tested the optimized reaction conditions on several other heteroaryl substrates. The six-membered heteroaryl halide 2-iodopyrazine produced the expected product 4o. The bicyclic heteroaryl halides were also investigated and these reactions showed reasonable yields: 6-bromoquinoline produced 4p, 6-bromoquinoxaline produced 4q, 4-bromoisoquinoline produced 4r and 3-bromoquinoline produced 4s. Compared with compounds 4p, 4r and 4s, the yields for compounds 4o and 4q were significantly lower, possibly due to the increased chance of catalyst poisoning. The two carbonyl groups in these 1,2-diketone compounds showed characteristic IR absorption signals. For example, aryl heteroaryl 1,2-diketones such as 4c, 4e, 4f, 4n and 4o showed two signals around 1680 and 1660 cm–1. Alkyl heteroaryl 1,2-diketones such as 4j, 4k and 4l showed two signals around 1700 and 1680 cm–1. The two distinctive carbonyl signals on the IR spectra clearly support the unsymmetrical structures of these heteroaryl 1,2-diketones.
Applications and Structural Features of 4a and 5a
To demonstrate the possible application of these diketone compounds, a condensation reaction was performed by refluxing 4a with an equimolar amount of o-phenylenediamine and catalytic TsOH (0.1 eq.) in toluene overnight. After a simple aqueous wash and rotatory evaporation, the desired quinoxaline derivative 5a was obtained in high yields and good purity (Scheme 4). Quinoxaline, or benzopyrazine, is a useful scaffold for drug candidates or catalytic ligand design.40,41 For example, quinoxaline can be oxidized by KMnO4 into a pyrazine scaffold which can be further modified into drug molecules such as pyrazinamide and morinamide for the treatment of tuberculosis. Quinoxaline and its derivatives have been recently reviewed for their pharmaceutical importance as an emerging class of antimycobacterials.42
Scheme 4. Demonstration of a Possible Application of Heteroaryl Diketone in the Synthesis of Heterocycles.

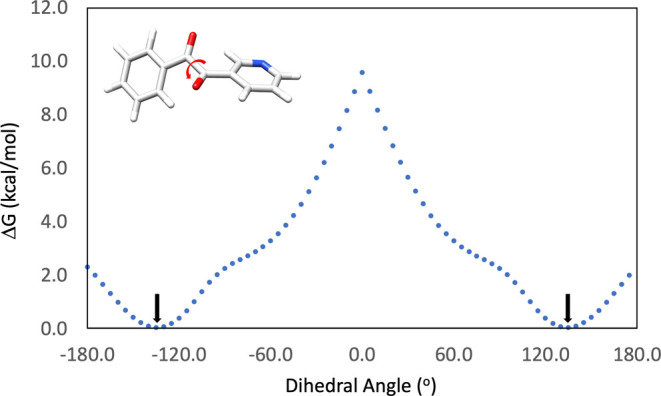
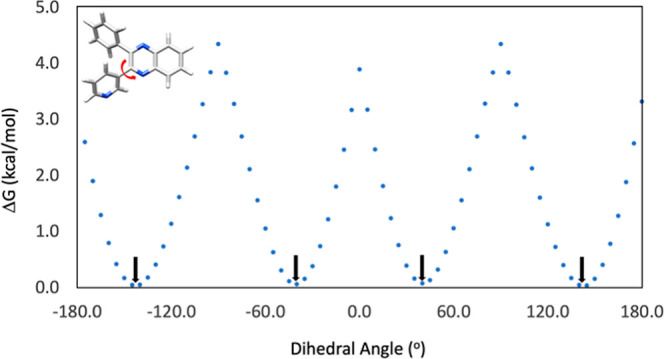
To gain insight on the structural features of the diketones and their products, geometry optimizations and frequency calculations for the heteroaryl 1,2-diketone 4a and its quinoxaline derivative 5a were performed at the M06-2X/def2-TZVP level of theory, which was previously reported to be both accurate and efficient for similar organic molecules.43 The lowest energy conformations of 4a and 5a were located with a dihedral scan around the central carbon–carbon bond in 4a (Scheme 5) and around the pyridyl ring in 5a (Scheme 6), followed by fully optimizing the structures around the energy minima.
Scheme 5. Dihedral Scan Around the Central Carbon–Carbon Bond in 4a.
Scheme 6. Dihedral Scan Around the Pyridyl Ring in 5a.
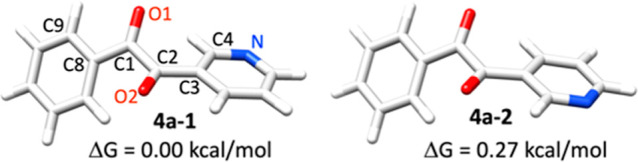
There are two lowest energy conformations of 4a (Table 2) that differ with respect to the position of the N atom in the pyridyl ring. The energy difference between these conformations is only 0.27 kcal/mol which corresponds to 60.4 and 39.6% of the conformers populated at 50 °C in toluene based on the Boltzmann distribution.
Table 2. Selected Interatomic Distances (Å) for the Lowest Energy Conformations of 4a.
| C1–C2 | C1–O1 | C1–C8 | C2–O2 | C2–C3 | |
|---|---|---|---|---|---|
| 4a-1 | 1.536 | 1.207 | 1.481 | 1.207 | 1.483 |
| 4a-2 | 1.537 | 1.208 | 1.482 | 1.207 | 1.483 |
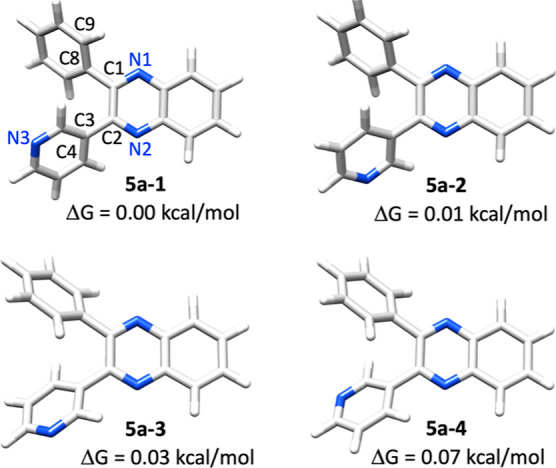
For the condensation product 5a, there are four lowest energy conformations that differ not only with respect to the position of the N atom in the pyridyl ring (Table 3), but also with respect to the twist angle of the phenyl and the pyridyl ring. The energy differences between these conformations are very small with only 0.01, 0.03 and 0.07 kcal/mol higher than the lowest energy conformer. The corresponding percentages of these four conformers are 26.0, 25.7, 24.9 and 23.4% at 50 °C in toluene, respectively.
Table 3. Selected Interatomic Distances (Å) for the Lowest Energy Conformations of 5a.
| C1–C2 | C1–N1 | C1–C8 | C2–N2 | C2–C3 | |
|---|---|---|---|---|---|
| 5a-1 | 1.445 | 1.307 | 1.486 | 1.307 | 1.484 |
| 5a-2 | 1.445 | 1.306 | 1.486 | 1.306 | 1.484 |
| 5a-3 | 1.445 | 1.306 | 1.486 | 1.306 | 1.484 |
| 5a-4 | 1.445 | 1.307 | 1.486 | 1.307 | 1.484 |
Different from their less hindered analogs such as oxalyl chloride or oxalic acid (see Tables S5 for the optimized structures), the lowest energy conformers of 4a did not show a planar structure. The central carbon–carbon bond length is 1.54 Å. The two (hetero)aryl rings are twisted with respect to one another with a dihedral angle (C8–C1–C2–C3) of 122.1 and 124.2° in 4a-1 and 4a-2, respectively, due to steric hindrance. Its condensation product 5a showed similar structural feature. The dihedral angle between two (hetero)aryl rings (C8–C1–C2–C3) are −8.7, −8.7, 8.4 and 8.9° in the lowest energy conformers of 5a. The unsymmetrical, non-planar geometry of compounds 4a and 5a provide opportunities to further modify these molecules into interesting chiral ligands or auxiliaries in enantioselective catalytic system design.
To investigate the conjugation throughout the molecules, Natural bond orbital (NBO) analysis was conducted on the lowest energy conformers of 4a and 5a. As a reference, this analysis has been extended to the less hindered analogs, oxalyl chloride and oxalic acid. A summary of the Natural population analysis (NPA) is shown in Table 4, occupancies of NBOs are summarized in Table 5 and second-order perturbation theory (PT2F) analysis of the Fock matrix in the NBO basis (E(2) kcal/mol) from the donor NBO(i) to the acceptor NBO(j) is summarized in Table 6.
Table 4. Summary of Natural Population Analysis (NPA).
| C1 | C2 | O1 | O2 | |
|---|---|---|---|---|
| (COCl)2 | 0.41692 | 0.41692 | –0.42762 | –0.42762 |
| (CO2H)2 | 0.67842 | 0.67842 | –0.53327 | –0.53327 |
| 4a-1 | 0.48922 | 0.49227 | –0.51143 | –0.51193 |
| 4a-2 | 0.49035 | 0.49109 | –0.51701 | –0.50676 |
| C1 | C2 | N1 | N2 | |
| 5a-1 | 0.18610 | 0.17937 | –0.36492 | –0.36699 |
| 5a-2 | 0.18381 | 0.17897 | –0.36498 | –0.36321 |
| 5a-3 | 0.18382 | 0.17895 | –0.36489 | –0.36698 |
| 5a-4 | 0.18609 | 0.17934 | –0.36489 | –0.36698 |
Table 5. Occupancy of Natural Bond Orbitals (NBO).
| (COCl)2 | (CO2H)2 | 4a-1 | 4a-2 | |
|---|---|---|---|---|
| σC1-C2 | 1.97662 | 1.97700 | 1.98049 | 1.98101 |
| πC1-O1 | 1.98351 | 1.98654 | 1.97346 | 1.97355 |
| πC2-O2 | 1.98351 | 1.98654 | 1.97420 | 1.97334 |
| nO1 | 1.80284 | 1.84460 | 1.87959 | 1.88102 |
| nO2 | 1.80284 | 1.84460 | 1.88060 | 1.88017 |
| σ*C1–C2 | 0.17564 | 0.15133 | 0.11595 | 0.11654 |
| π*C1–O1 | 0.13205 | 0.18802 | 0.11226 | 0.11300 |
| π*C2–O2 | 0.13205 | 0.18802 | 0.10870 | 0.10772 |
| 5a-1 | 5a-2 | 5a-3 | 5a-4 | |
|---|---|---|---|---|
| σC1-C2 | 1.98076 | 1.98085 | 1.98084 | 1.98076 |
| πC1-N1 | 1.78124 | 1.78298 | 1.78299 | 1.78123 |
| πC2-N2 | 1.78470 | 1.78437 | 1.78432 | 1.78467 |
| nN1 | 1.91369 | 1.91395 | 1.91398 | 1.91370 |
| nN2 | 1.91411 | 1.91415 | 1.91419 | 1.91410 |
| σ*C1–C2 | 0.06392 | 0.06441 | 0.06443 | 0.06392 |
| π*C1–N1 | 0.27313 | 0.27315 | 0.27309 | 0.27311 |
| π*C2–N2 | 0.27890 | 0.27775 | 0.27780 | 0.27893 |
Table 6. Second Order Perturbation Theory Analysis of Fock Matrix in NBO Basis (E(2) Kcal/Mol) From the Donor NBO(i) to the Acceptor NBO(j).
| donor/acceptor | (COCl)2 | (CO2H)2 | 4a-1 | 4a-2 |
|---|---|---|---|---|
| nO1/σ*C1–C2 | 35.00 | 33.98 | 28.92 | 28.73 |
| nO2/σ*C1–C2 | 35.00 | 33.98 | 28.60 | 28.84 |
| πC1-O1/π*C2–O2 | 5.50 | 4.80 | 3.50 | 3.58 |
| πC2-O2/π*C1–O1 | 5.50 | 4.80 | 3.32 | 3.52 |
| 5a-1 | 5a-2 | 5a-3 | 5a-4 | |
| nN1/σ*C1–C2 | 14.92 | 14.92 | 14.93 | 14.92 |
| nN2/σ*C1–C2 | 14.85 | 14.90 | 14.90 | 14.85 |
| πC1-N1/π*C2–N2 | 17.38 | 17.49 | 17.50 | 17.38 |
| πC2-N2/π*C1–N1 | 17.65 | 17.56 | 17.56 | 17.65 |
The atomic charges calculated with NPA indicate a partial positive charge on C1 and C2, and a partial negative charge on O1/N1 and O2/N2. When 4a-1 and 4a-2 are compared with oxalyl chloride and oxalic acid, the charges on C and O are found to be in between those for oxalyl chloride and oxalic acid. On the other hand, the charges for C and N in the lowest energy conformers of 5a is less positive and less negative, respectively than oxalyl chloride, oxalic acid and the lowest energy conformers of 4a. This is consistent with the occupancy of the NBOs, particularly the oxygen and nitrogen lone pair electrons (nO/N) and their participation in the conjugation.
The electron delocalization in these molecules results in a noticeable change in the occupancy of the NBO. The occupancy of the oxygen lone pair electrons (1.80–1.88) is slightly less than the occupancy of the nitrogen lone pair electrons (1.91), which is consistent with the greater partial negative charge on the oxygen compared to nitrogen.
The transfer of electron density from the oxygen/nitrogen lone pair (nO/N) is accompanied by a change in the occupancy of the σ* antibonding orbital of C1–C2 (0.12–0.18), the π* antibonding orbitals of C1–O1 (0.11–0.19) and C2–O2 (0.11–0.19) in oxalyl chloride, oxalic acid and the lowest energy conformers of 4a and in both the σ* antibonding orbital of C1–C2 (0.06), and the π* antibonding orbitals of C1–N1(0.27) and C2–N2 (0.28) in the lowest energy conformers of 5a. There is also considerable electron delocalization from the πC1-N1 (1.78) and πC2-N2 (1.78) orbitals to the π*C2–N2 (0.28) and π*C1–N1 (0.27) orbitals in the lowest energy conformers of 5a. To evaluate the electron delocalization, the individual donor–acceptor interactions were quantified by using second-order perturbation theory analysis.
The occupancy of σ*C1–C2 orbitals is mainly due to the electron donation from the oxygen lone pair electrons, nO1 → σ*C1–C2 and nO2 → σ*C1–C2 and nitrogen lone pair electrons, nN1 → σ*C1–C2 and nN2 → σ*C1–C2. There is also electron donation from the πC1-O1 orbital to the π*C2–O2 orbital and the πC2-O2 orbital to the π*C1–O1 orbital in oxalyl chloride, oxalic acid and the lowest energy conformers of 4a. Similarly, in the lowest energy conformers of 5a, there is electron donation from the πC1-N1 orbital to the π*C2–N2 orbital and the πC2-N2 orbital to the π*C1–N1 orbital. The magnitude of the delocalization energies is an indication of the importance of conjugation on the electronic structure of these molecules.
The slightly higher delocalization energy for nO1 → σ*C1–C2 and nO2 → σ*C1–C2 (35.00 kcal/mol) in oxalyl chloride and oxalic acid as compared to the delocalization energies of 28.60–28.92 kcal/mol in the lowest energy conformers of 4a is consistent with the planar geometries of the oxalyl chloride and oxalic acid and the greater extent of orbital overlap. In the lowest energy conformers of 5a, although the lone pair electrons of N1 and N2 are in the same plane as σ*C1–C2, the lowest delocalization energy for nN1 → σ*C1–C2 and nN2 → σ*C1–C2 (14.85–14.93 kcal/mol) is due to these atoms being part of a ring structure and interacting with other atoms and bonds. In the lowest energy conformers of 5a, the electron donation between π bonding orbitals and π* antibonding orbitals of C1–N1 and C2–N2 contributes more to the conjugation in this molecule. The magnitude of the delocalization energies in these interactions, 17.38–17.65 kcal/mol, is consistent with the greater electron delocalization from the πC1-N1 and πC2-N2 orbitals compared to the nN1 and nN1 lone pair electrons.
The HOMO and LUMO of oxalyl chloride and the lowest energy conformers of 4a and 5a are shown in Figure 1. The HOMO-1, HOMO, LUMO and LUMO+1 of oxalyl chloride, oxalic acid, the lowest energy conformers of 4a and 5a (Scheme S1–Scheme S8) and their energies (Table S6) are reported in the Supporting Information Although the lowest energy conformer of 4a does not have a planar geometry like oxalyl chloride, the HOMO and LUMO are very similar and the twist in the lowest energy conformer of 4a does not prevent the conjugation, as can easily be seen from the overlap of the orbitals. The visual differences in the HOMO and LUMO of the lowest energy conformer of 5a can be attributed to the nitrogen atoms being part of a ring structure and interacting with other atoms and bonds in the quinoxaline derivative. Compared to the less hindered analogs (oxalyl chloride and oxalic acid), 4a is surprisingly not planar. The conformational landscape of 4a and 5a provided insight regarding the most stable conformers and their rotational barriers. The conjugation in 4a and 5a were investigated in comparison to oxalyl chloride and oxalic acid using NBO analysis. Although these molecules deviate from planarity in oxalyl chloride or oxalic acid, there is still considerable delocalization among the carbonyl groups which suggests similar affinity toward nucleophiles.
Figure 1.
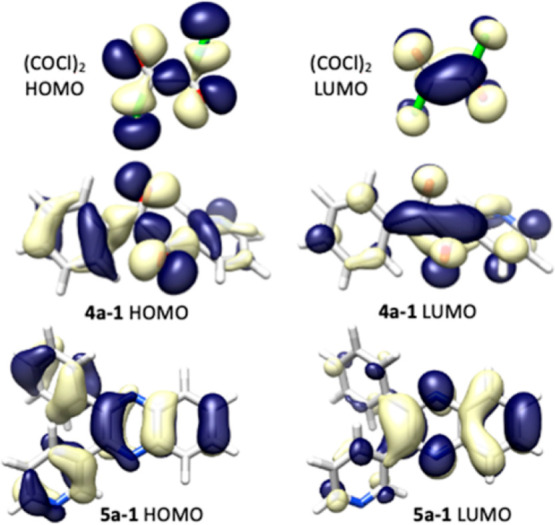
A comparison of HOMO and LUMO of oxalyl chloride and the lowest energy conformers of 4a and 5a.
In summary, a heteroarylation/oxidation reaction sequence has been developed to synthesize 1, 2-diketones with heteroaryl groups. The features of this method include mild neutral conditions, ease of practical operation, and access to unsymmetrical heteroaryl diketones. This method has been applied in a condensation reaction with diamines to make a biologically interesting quinoxaline derivative. Using DFT calculations, two lowest energy conformers for the 1,2-diketone 4a and four for the quinoxaline derivative 5a were located. The structural features and bonding in these lowest energy conformers demonstrate the impacts of their non-planar geometries on the electronic structure of the molecules and should help to inform future derivation reactions.
Methods Experimental Section
General Information
Unless otherwise noted, the chemical reagents were obtained from commercial vendors and used without further purification. Pd catalysts and NaOtBu were kept in a glovebox under N2. Toluene solvent was vigorously purged with argon for 2 h before use. The MultiwavePro microwave reaction system from Anton Paar Instruments was utilized in this study to conduct the microwave-assisted reactions. The microwave reaction vessels consist of a disposable Wheaton glass vial (Item# 224882), a special PEEK screw cap, and a PTFE seal (reaction volume 0.3–3 mL, operation pressure 20 bar). Four silicon carbide (SiC) 96-well plates on a rotor (4 × 24MG5) were used for homogeneous heating. Column chromatography was performed using pre-packed RediSep Rf Silica columns on a CombiFlash Rf Flash Chromatography system (Teledyne Isco). A Joel 500 MHz spectrometer was used to obtain NMR spectra. Chemical shifts were reported in parts per million (ppm) relative to the tetramethylsilane (TMS) signal at 0.00 ppm. Coupling constants, J, were reported in hertz (Hz). A Micromass Q-TOF 2 or a Thermo Scientific LTQ-FT mass spectrometer was employed to obtain high resolution mass spectra and electrospray (ES) mode was used. Infrared spectra were recorded on a Thermo Scientific Nicolet iS 10 FTIR Spectrometer.
General procedure for synthesis of 1,2-diketones via palladium-catalyzed heteroarylation followed by a Riley Oxidation.
Microwave reaction vials (standard Wheaton glass vials, Item# 224882) containing stirring bars were dried in an oven overnight before use. In the following sequence and inside a glove box, 2.4 equiv NaOtBu, 1.1 equiv Ketone, 3.0 mL toluene and 1 mol % XPhos Pd G4 catalyst were added to a dried microwave reaction vial. The reaction mixture was stirred at room temperature for 10 min before the addition of 1.1 equiv Heteroaryl halide. After the reaction vial was secured with a Teflon seal (Anton Paar Cat No 41186) and closed finger-tight with a PEEK cap (Anton Paar Cat No 41188), it was transferred from the glovebox to the MultiwavePro microwave reaction system (Anton Paar USA, Inc). The reaction mixture was subject to microwave irradiation at 150 °C for 10 min. After cooling to ambient temperature, the reaction mixture was transferred to a separatory funnel. The crude product was extracted with diethyl ether (25 mL) and washed three times with saturated NH4Cl (3 × 25 mL). The ether layer was dried over anhydrous MgSO4, followed by rotatory evaporation to obtain the crude product. To a 20 mL scintillation vial, the crude product, 1,4-dioxane (3 mL), and 1.5 equiv SeO2 was added and then stirred at 50 °C overnight. After filtering through Celite, the filtrate was transferred to a separatory funnel followed by the addition of diethyl ether (25 mL). The crude product was washed three times with water (3 × 25 mL). The ether layer was collected and dried over anhydrous MgSO4 before removing the solvent by rotatory evaporation. Automated flash column chromatography was utilized to obtain the purified product (0–100% ethyl acetate: hexanes or 0–20% MeOH/CH2Cl2).
1-Phenyl-2-(pyridin-3-yl)ethane-1,2-dione (4a).19,21
Synthesized according to the general procedure described above. Yield, 59%. 1H NMR (500 MHz, CDCl3, ppm): δ 9.05 (s, 1H), 8.73 (dd, J = 4.8, 1.6 Hz, 1H), 8.17 (dt, J = 8.0 Hz, 2.0 Hz, 1H), 7.88 (d, J = 7.2 Hz, 2H), 7.56 (t, J = 7.4 Hz, 1H), 7.41 (t, J = 7.8 Hz, 2H), 7.35 (dd, J = 7.9 Hz, 4.9 Hz, 1H). 13C{1H} NMR (125 MHz, CDCl3, ppm): δ 193.0, 192.8, 154.9, 151.4, 136.9, 135.3, 132.5, 130.1, 129.2, 128.7, 123.9. HRMS calcd for C13H10NO2 [M + H] 212.0712; found, 212.0718.
1-(Pyridin-3-yl)-2-(m-tolyl)ethane-1,2-dione (4b)
Microwave reaction vials (standard Wheaton glass vials, Item# 224882) containing stirring bars were dried in an oven overnight before use. In the following sequence and inside a glove box, 2.4 equiv NaOtBu, 1.1 equiv ketone, 3.0 mL toluene and 1 mol % XPhos Pd G4 catalyst were added to a dried microwave reaction vial. The reaction mixture was stirred at room temperature for 10 min before the addition of 1.1 equiv heteroaryl halide. After the reaction vial was secured with a Teflon seal (Anton Paar Cat No 41186) and closed finger-tight with a PEEK cap (Anton Paar Cat No 41188), it was transferred from the glovebox to the MultiwavePro microwave reaction system (Anton Paar USA, Inc). The reaction mixture was subject to microwave irradiation at 150 °C for 10 min. After cooling to ambient temperature, the reaction mixture was transferred to a separatory funnel. The crude product was extracted with diethyl ether (25 mL) and washed three times with saturated NH4Cl (3 × 25 mL). The ether layer was dried over anhydrous MgSO4, followed by rotatory evaporation to obtain the crude product. A microwave reaction vial was charged with the crude product, DMSO (1 mL), 1.1 equiv I2 and 1.1 equiv CuO. After microwave irradiation at 150 °C for 10 min, the crude product was transferred to a separatory funnel and extracted with EtOAc (3 × 15 mL)/NH4Cl (5 mL). The final product was purified using flash column chromatography (0–100% ethyl acetate: hexanes). Yield, 50%. 1H NMR (500 MHz, CDCl3, ppm): δ 9.17 (s,1H), 8.88 (dd, J = 4.8 Hz, 1.7 Hz, 1H), 8.31 (dt, J = 8.0 Hz, 1.9 Hz, 1H), 7.82–7.76 (m, 2H), 7.50 (dd, J = 7.2 Hz, 5.8 Hz, 2H), 7.42 (t, J = 7.6 Hz, 1H), 2.42 (s, 3H). 13C{1H} NMR (125 MHz, CDCl3, ppm): δ 193.3, 193.0, 154.8, 151.4, 139.3, 137.0, 136.2, 132.6, 130.4, 129.1, 128.9, 127.5, 124.0, 21.4. HRMS calcd for C14H12NO2 [M + H] 226.0868; found, 226.0860.
1-(2-methoxyphenyl)-2-(pyridin-3-yl)ethane-1,2-dione (4c)
Synthesized according to the general procedure described above. Yield, 37%. 1H NMR (500 MHz, CDCl3, ppm): δ 9.09 (s, 1H), 8.82 (dd, J = 4.8 Hz, 1.6 Hz, 1H), 8.22 (dt, J = 7.9 Hz, 1.9 Hz, 1H), 7.99 (dd, J = 7.8 Hz, 1.6 Hz, 1H), 7.61 (t, J = 7.0 Hz, 1H), 7.45 (dd, J = 8.0 Hz, 4.9 Hz, 1H), 7.13 (t, J = 7.6 Hz, 1H), 6.94 (d, J = 8.6 Hz, 1H), 3.58 (s, 3H). 13C{1H} NMR (125 MHz, CDCl3, ppm): δ 193.8, 192.1, 160.4, 154.1, 151.0, 136.9, 136.5, 130.7, 128.7, 123.8, 123.6, 121.9, 112.4, 55.8. HRMS calcd for C14H12NO3 [M + H] 242.0817; found, 242.0817.
1-(3-methoxyphenyl)-2-(pyridin-3-yl)ethane-1,2-dione (4d)
Synthesized according to the general procedure described above. Yield, 40%. 1H NMR (500 MHz, CDCl3, ppm): δ 9.15 (s, 1H), 8.87 (dd, J = 4.8 Hz, 1.4 Hz, 1H), 8.30 (dt, J = 8.0 Hz, 1.9 Hz, 1H), 7.55 (s, 1H), 7.50 (t, J = 6.5 Hz, 2H), 7.42 (t, J = 7.9 Hz, 1H), 7.24 (dd, J = 8.2 Hz, 2.6 Hz, 1H), 3.88 (s, 3H). 13C{1H} NMR (125 MHz, CDCl3, ppm): δ 192.9, 192.7, 160.3, 154.7, 151.3, 137.1, 133.8, 130.3, 128.9, 124.1, 123.4, 122.4, 113.1, 55.7. HRMS calcd for C14H12NO3 [M + H] 242.0817; found, 242.0811.
1-(4-methoxyphenyl)-2-(pyridin-3-yl)ethane-1,2-dione (4e)
Synthesized according to the general procedure described above. Yield, 48%. 1H NMR (500 MHz, CDCl3, ppm): δ 9.16 (s, 1H), 8.85 (dd, J = 4.8 Hz, 1.7 Hz, 1H), 8.29 (dt, J = 8.0 Hz, 2.0 Hz, 1H), 7.97 (d, J = 8.9 Hz, 2H), 7.47 (ddd, J = 8.9 Hz, 4.9 Hz, 0.8 Hz, 1H), 6.99 (d, J = 9.0 Hz, 2H), 3.90 (s, 3H). 13C{1H} NMR (125 MHz, CDCl3, ppm): δ 193.1, 191.5, 165.4, 154.7, 151.4, 137.0, 132.7, 129.0, 125.7, 123.9, 114.6, 55.8. HRMS calcd for C14H12NO3 [M + H] 242.0817; found, 242.0809.
1-(4-bromophenyl)-2-(pyridin-3-yl)ethane-1,2-dione (4f)
Synthesized according to the general procedure described above. Yield, 25%. 1H NMR (500 MHz, CDCl3, ppm): δ 9.13 (s, 1H), 8.84 (dd, J = 4.8 Hz, 1.7 Hz, 1H), 8.26 (dt, J = 8.0 Hz, 2.0 Hz, 1H), 7.84 (d, J = 8.6 Hz, 2H), 7.66 (d, J = 8.6 Hz, 2H), 7.46 (ddd, J = 8.0 Hz, 4.9 Hz, 0.7 Hz, 1H). 13C{1H} NMR (125 MHz, CDCl3, ppm): δ 192.0, 191.6, 154.9, 151.3, 137.2, 132.7, 131.5, 131.3, 131.1, 128.7, 124.1. HRMS calcd for C13H9NO2Br [M + H] 289.9817; found, 289.9804.
1-(4-chlorophenyl)-2-(pyridin-3-yl)ethane-1,2-dione (4g)
Synthesized according to the general procedure described above. Yield, 34%. 1H NMR (500 MHz, CDCl3, ppm): δ 9.20 (s, 1H), 8.90 (d, J = 4.4 Hz, 1H), 8.38 (d, J = 8.0 Hz, 1H), 7.97 (d, J = 8.5 Hz, 2H), 7.56 (dd, J = 8.0 Hz, 5.0 Hz, 1H), 7.53 (d, J = 8.6 Hz, 2H). 13C{1H} NMR (125 MHz, CDCl3, ppm): δ 192.0, 191.4, 154.8, 151.3, 142.2, 137.2, 131.5, 130.9, 129.7, 128.7, 124.1. HRMS calcd for C13H9NO2Cl [M + H] 246.0322; found, 246.0318.
1-(4-fluorophenyl)-2-(pyridin-3-yl)ethane-1,2-dione (4h)
Microwave reaction vials (standard Wheaton glass vials, Item# 224882) containing stirring bars were dried in an oven overnight before use. In the following sequence and inside a glove box, 2.4 equiv NaOtBu, 1.1 equiv Ketone, 3.0 mL toluene and 1 mol % XPhos Pd G4 catalyst were added to a dried microwave reaction vial. The reaction mixture was stirred at room temperature for 10 min before the addition of 1.1 equiv heteroaryl halide. After the reaction vial was secured with a Teflon seal (Anton Paar Cat No 41186) and closed finger-tight with a PEEK cap (Anton Paar Cat No 41188), it was transferred from the glovebox to the MultiwavePro microwave reaction system (Anton Paar USA, Inc). The reaction mixture was subject to microwave irradiation at 150 °C for 10 min. After cooling to ambient temperature, the reaction mixture was transferred to a separatory funnel. The crude product was extracted with diethyl ether (25 mL) and washed three times with saturated NH4Cl (3 × 25 mL). The ether layer was dried over anhydrous MgSO4, followed by rotatory evaporation to obtain the crude product. A microwave reaction vial was charged with the crude product, DMSO (1 mL), 1.1 equiv I2 and 1.1 equiv CuO. After microwave irradiation at 150 °C for 10 min, the crude product was transferred to a separatory funnel and extracted with EtOAc (3 × 15 mL)/NH4Cl (5 mL). The final product was purified using flash column chromatography (0–100% ethyl acetate: hexanes). Yield, 43%. 1H NMR (500 MHz, CDCl3, ppm): δ 9.18 (s, 1H), 8.89 (d, J = 3.9 Hz, 1H), 8.32 (dt, J = 8.0 Hz, 1.9 Hz, 1H), 8.08–8.02 (m, 2H), 7.50 (dd, J = 8.0 Hz, 4.8 Hz, 1H), 7.22 (t, J = 8.6 Hz, 2H). 13C{1H} NMR (125 MHz, CDCl3, ppm): δ 192.1, 191.4, 155.0, 151.4, 142.2, 137.1, 131.5, 131.0, 129.7, 128.7, 124.0. HRMS calcd for C13H9NO2F [M + H] 230.0617; found, 230.0609.
1-(3,4-bis((tert-butyldimethylsilyl)oxy)phenyl)-2-(pyridin-3-yl)ethane-1,2-dione (4i)
Synthesized according to the general procedure described above. Yield, 57%. 1H NMR (500 MHz, CDCl3, ppm): δ 9.16 (s, 1H), 8.85 (dd, J = 4.9 Hz, 1.6 Hz, 1H), 8.32 (dt, J = 8.0 Hz, 1.9 Hz, 1H), 7.53 (d, J = 2.2 Hz, 1H), 7.50 (dd, J = 7.8 Hz, 5.0 Hz, 1H), 7.44 (dd, J = 8.4 Hz, 2.2 Hz, 1H), 6.89 (d, J = 8.4 Hz, 1H), 0.98 (s, 18H), 0.24 (s, 6H), 0.22 (s, 6H). 13C{1H} NMR (125 MHz, CDCl3, ppm): δ 192.9, 191.4, 154.5, 154.2, 151.0, 147.8, 137.4, 129.2, 126.2, 125.6, 124.1, 121.6, 120.9, 25.9, 25.8, 18.6, 18.5, −4.0, −4.1. HRMS calcd for C25H36NO4Si2 [M + H] 472.2339; found, 472.2336.
3-Methyl-1-(pyridin-3-yl)butane-1,2-dione (4j)
Synthesized according to the general procedure described above with the exception of the amount of ketone (1.5 equiv) and base (2.9 equiv) used. Yield, 29%. 1H NMR (500 MHz, CDCl3, ppm): δ 9.12 (s, 1H), 8.81 (dd, J = 4.7 Hz, 1.4 Hz, 1H), 8.24 (dt, J = 8.0 Hz, 1.8 Hz, 1H), 7.43 (dd, J = 8.0 Hz, 4.9 Hz, 1H), 3.46–3.35 (m, J = 7.0 Hz, 1H), 1.18 (d, J = 7.0 Hz, 6H). 13C{1H} NMR (125 MHz, CDCl3, ppm): δ 204.9, 191.7, 154.6, 151.5, 137.2, 128.5, 123.8, 36.1, 17.0. HRMS calcd for C10H12NO2 [M + H] 178.0868; found, 178.0861.
4-Methyl-1-(pyridin-3-yl)pentane-1,2-dione (4k)
Synthesized according to the general procedure described above with the exception of the amount of ketone (1.5 equiv) and base (2.9 equiv) used. Yield, 24%. 1H NMR (500 MHz, CDCl3, ppm): δ 9.20 (s, 1H), 8.81 (dd, J = 4.8 Hz, 1.6 Hz, 1H), 8.30 (dt, J = 8.0 Hz, 1.8 Hz, 1H), 7.43 (dd, J = 8.0 Hz, 4.8 Hz, 1H), 2.78 (d, J = 6.9 Hz, 2H), 2.31–2.20 (m, J = 6.7 Hz, 1H), 0.99 (d, J = 6.6 Hz, 6H). 13C{1H} NMR (125 MHz, CDCl3, ppm): δ 201.4, 195.3, 154.0, 151.4, 137.9, 128.2, 123.9, 46.9, 24.3, 22.7. HRMS calcd for C11H14NO2 [M + H] 192.1025; found, 192.1018.
3,3-Dimethyl-1-(pyridin-3-yl)butane-1,2-dione (4l)
Synthesized according to the general procedure described above with the exception of the amount of ketone (1.5 equiv) and base (2.9 equiv) used. Yield, 65%. 1H NMR (500 MHz, CDCl3, ppm): δ 8.97 (s, 1H), 8.81 (dd, J = 4.8 Hz, 1.7 Hz, 1H), 8.13 (dt, J = 8.0 Hz, 2.0 Hz, 1H), 7.43 (dd, J = 7.9 Hz, 4.9 Hz, 1H), 1.29 (s, 9H). 13C{1H} NMR (125 MHz, CDCl3, ppm): δ 209.3, 193.7, 154.7, 151.1, 136.5, 128.7, 123.9, 42.9, 26.1. HRMS calcd for C11H14NO2 [M + H] 192.1025; found, 192.1020.
1-(2,5-Dimethylfuran-3-yl)-2-(pyridin-3-yl)ethane-1,2-dione (4m)
Synthesized according to the general procedure described above. Yield, 21%. 1H NMR (500 MHz, CDCl3, ppm): δ 9.19 (s, 1H), 8.85 (dd, J = 4.8 Hz, 1.7 Hz, 1H), 8.31 (dt, J = 8.0 Hz, 2.0 Hz, 1H), 7.47 (dd, J = 8.0 Hz, 4.9 Hz, 1H), 6.28 (s, 1H), 2.58 (s, 3H), 2.27 (s, 3H). 13C{1H} NMR (125 MHz, CDCl3, ppm): δ 191.8, 187.5, 161.4, 154.6, 151.6, 151.5, 137.2, 128.5, 123.8, 118.7, 105.9, 14.7, 13.1. HRMS calcd for C13H12NO3 [M + H] 230.0817; found, 230.0807.
1-(2,5-Dimethylthiophen-3-yl)-2-(pyridin-3-yl)ethane-1,2-dione (4n)
Synthesized according to the general procedure described above. Yield, 42%. 1H NMR (500 MHz, CDCl3, ppm): δ 9.16 (s, 1H), 8.85 (dd, J = 4.8 Hz, 1.7 Hz, 1H), 8.30 (dt, J = 8.0 Hz, 2.0 Hz, 1H), 7.48 (ddt, J = 7.8 Hz, 4.9 Hz, 0.8 Hz, 1H), 6.91 (s, 1H), 2.76 (s, 3H), 2.38 (s, 3H). 13C{1H} NMR (125 MHz, CDCl3, ppm): δ 192.5, 187.6, 154.6, 152.9, 151.5, 137.1, 136.7, 131.5, 128.6, 126.7, 123.9, 16.2, 14.9. HRMS calcd for C13H12NO2S [M + H] 246.0589; found, 246.0585.
1-Phenyl-2-(pyrazin-2-yl)ethane-1,2-dione (4o)
Synthesized according to the general procedure described above. Yield, 23%. 1H NMR (500 MHz, CDCl3, ppm): δ 9.40 (s, 1H), 8.82 (d, J = 2.3 Hz, 1H), 8.64 (dd, J = 2.4 Hz, 1.4 Hz, 1H), 7.96 (dd, J = 8.4 Hz, 1.2 Hz, 2H), 7.68 (t, J = 7.5 Hz, 1H), 7.54 (t, J = 7.8 Hz, 2H). 13C{1H} NMR (125 MHz, CDCl3, ppm): δ 194.5, 194.0, 148.8, 146.4, 144.8, 144.3, 135.2, 132.8, 129.8, 129.2. HRMS calcd for C12H9N2O2 [M + H] 213.0664; found, 213.0657.
1-Phenyl-2-(quinolin-6-yl)ethane-1,2-dione (4p)
Synthesized according to the general procedure described above. Yield, 52%. 1H NMR (500 MHz, CDCl3, ppm): δ 9.05 (dd, J = 4.2 Hz, 1.7 Hz, 1H), 8.43 (s, 1H), 8.35 (dd, J = 8.9 Hz, 1.9 Hz, 1H), 8.25 (t, J = 7.3 Hz, 2H), 8.04 (d, J = 7.2 Hz, 2H), 7.69 (t, J = 7.4 Hz, 1H), 7.54 (t, J = 4.2 Hz, 2H), 7.50 (q, J = 4.2 Hz, 1H). 13C{1H} NMR (125 MHz, CDCl3, ppm): δ 194.2, 193.8, 153.6, 150.9, 137.9, 135.2, 133.2, 133.0, 131.0, 130.9, 130.1, 129.2, 127.7, 127.6, 122.4. HRMS calcd for C17H12NO2 [M + H] 262.0868; found, 262.0866.
1-Phenyl-2-(quinoxalin-6-yl)ethane-1,2-dione (4q)
Synthesized according to the general procedure described above. Yield, 24%. 1H NMR (500 MHz, CDCl3, ppm): δ 8.96 (d, J = 1.6 Hz, 1H), 8.94 (d, J = 1.6 Hz, 1H), 8.65 (d, J = 1.8 Hz, 1H), 8.42 (dd, J = 8.9 Hz, 2.0 Hz, 1H), 8.27 (d, J = 8.8 Hz, 1H), 8.04 (d, J = 8.0 Hz, 2H), 7.70 (t, J = 7.4 Hz, 1H), 7.55 Hz (t, J = 7.8 Hz, 2H). 13C{1H} NMR (125 MHz, CDCl3, ppm): δ 193.8, 193.7, 147.4, 146.4, 146.0, 142.5, 135.3, 134.3, 133.8, 132.8, 131.0, 130.1, 129.3, 128.2. HRMS calcd for C16H11N2O2 [M + H] 263.0821; found, 263.0821.
1-(Isoquinolin-4-yl)-2-phenylethane-1,2-dione (4r)
Synthesized according to the general procedure described above. Yellow solid. Yield, 58%. 1H NMR (500 MHz, CDCl3, ppm): δ 9.42 (s, 1H), 9.22 (d, J = 8.6 Hz, 1H), 8.88 (s, 1H), 8.09 (d, J = 8.2 Hz, 1H), 8.03 (d, J = 7.4 Hz, 2H), 7.96 (t, J = 7.4 Hz, 1H), 7.77 (t, J = 7.6 Hz, 1H), 7.68 (t, J = 7.4 Hz, 1H), 7.53 (t, J = 7.9 Hz, 2H). 13C{1H} NMR (125 MHz, CDCl3, ppm): δ 196.6, 193.6, 158.8, 150.6, 135.2, 133.9, 133.2, 133.1, 130.2, 129.2, 128.9, 128.8, 128.7, 125.2, 123.0. HRMS calcd for C17H12NO2 [M + H] 262.0868; found, 262.0864.
1-Phenyl-2-(quinolin-3-yl)ethane-1,2-dione (4s)
Microwave reaction vials (standard Wheaton glass vials, Item# 224882) containing stirring bars were dried in an oven overnight before use. In the following sequence and inside a glove box, 2.4 equiv NaOtBu, 1.1 equiv Ketone, 3.0 mL toluene and 1 mol % XPhos Pd G4 catalyst were added to a dried microwave reaction vial. The reaction mixture was stirred at room temperature for 10 min before the addition of 1.1 equiv Heteroaryl halide. After the reaction vial was secured with a Teflon seal (Anton Paar Cat No 41186) and closed finger-tight with a PEEK cap (Anton Paar Cat No 41188), it was transferred from the glovebox to the MultiwavePro microwave reaction system (Anton Paar USA, Inc). The reaction mixture was subject to microwave irradiation at 150 °C for 10 min. After cooling to ambient temperature, the reaction mixture was transferred to a separatory funnel. The crude product was extracted with diethyl ether (25 mL) and washed three times with saturated NH4Cl (3 × 25 mL). The ether layer was dried over anhydrous MgSO4, followed by rotatory evaporation to obtain the crude product. A microwave reaction vial was charged with the crude product, DMSO (1 mL), 1.1 equiv I2 and 1.1 equiv CuO. After microwave irradiation at 150 °C for 10 min, the crude product was transferred to a separatory funnel and extracted with EtOAc (3 × 15 mL)/NH4Cl (5 mL). The final product was purified using flash column chromatography. The final product was purified using column chromatography (0–100% ethyl acetate: hexanes). Yield, 36%. 1H NMR (500 MHz, CDCl3, ppm): δ 9.50 (s, 1H), 8.74 (s, 1H), 8.19 (d, J = 8.7 Hz, 1H), 8.06 (d, J = 7.1 Hz, 2H), 7.93 (d, J = 8.1 Hz, 1H), 7.90 (t, J = 7.0 Hz, 1H), 7.71 (t, J = 7.4 Hz, 1H), 7.65 (t, J = 7.5 Hz, 1H), 7.56 (t, J = 7.8 Hz, 2H). 13C{1H} NMR (125 MHz, CDCl3, ppm): δ 193.2, 192.9, 150.5, 149.3, 140.3, 135.4, 133.1, 132.8, 130.2, 129.8, 129.8, 129.3, 128.0, 126.7, 125.6. HRMS calcd for C17H12NO2 [M + H] 262.0868; found, 262.0864.
2-Phenyl-3-(pyridin-3-yl)quinoxaline (5a)
To a 20 mL vial containing 1-phenyl-2-(pyridin-3-yl)ethane-1,2-dione (67.3 mg, 0.32 mmol), was added benzene-1,2-diamine (34.9 mg, 0.32 mmol) and toluene (5 mL). With stirring, tosic acid monohydrate (5.7 mg, 0.03 mmol) and 3–4 crushed 3 Å sieves were added. After the solution was stirred at reflux overnight, it was cooled to room temperature and filtered. The filtrate was diluted with ether (20 mL) and washed with saturated sodium bicarbonate (20 mL) and water (2 × 20 mL). The organic phase was dried over magnesium sulfate, filtered, and concentrated in vacuo to afford the product (88% yield) as an orange solid. 1H NMR (500 MHz, CDCl3, ppm): δ 8.80 (s, 1H), 8.61 (dd, J = 4.8 Hz, 1.4 Hz, 1H), 8.23–8.16 (m, 2H), 7.85 (dt, J = 8.0 Hz, 2.0 Hz, 1H), 7.83–7.79 (m, 2H), 7.51 (d, J = 6.2 Hz, 2H), 7.43–7.35 (m, 3H), 7.29 (dd, J = 7.8 Hz, 4.9 Hz, 1H). 13C{1H} NMR (125 MHz, CDCl3, ppm): δ 153.4, 150.4, 150.3, 149.4, 141.6, 141.4, 138.5, 137.5, 135.2, 130.7, 130.4, 129.9, 129.4, 129.4, 129.4, 128.8, 123.1. HRMS calcd for C19H14N3 [M + H] 284.1188; found, 284.1183.
Computational Details
Gaussian 16 suite of programs was used for all density functional theory calculations.44 Gaussian input files were generated using GaussView6.45 Structures 4a and 5a were optimized in gas phase with no symmetry constraints using redundant internal coordinates 44 and M06-2X hybrid functional.46 in gas phase with DFT and a wave function incorporating the hybrid functional of Truhlar and Zhao, M06-2X.47 Def2-TZVP basis set was used to represent all atoms.48,49 Structures were reoptimized with solvation model based on density (SMD)50 to evaluate the solvent effects on the geometries and energies in toluene. Optimized geometries were confirmed with zero imaginary frequencies and zero-point energies, thermal and entropic corrections were calculated by frequency calculations with the same basis set. Gibbs free energies at 298.15 K and 1 atm and with Truhlar’s quasi-harmonic corrections are used throughout the text.51,52 Natural bond orbital analysis was calculated using the natural population analysis method53 as implemented within G16. Optimized structures and molecular orbitals were illustrated using UCSF Chimera.54
Acknowledgments
The research presented in this work was supported by the National Science Foundation (CHE-1760393). The authors are grateful to the NKU Center for the Integration of Science and Mathematics, NKU UR-STEM Program and the Department of Chemistry and Biochemistry for financial and logistical support. The authors also want to thank the School of Chemical Sciences Mass Spectrometry Laboratory at the University of Illinois at Urbana-Champaign for obtaining HRMS data. T.A.A. acknowledges the Texas Advanced Computing Center (TACC) at The University of Texas at Austin for providing HPC resources that have contributed to the research results reported within this paper (URL: http://www.tacc.utexas.edu) and support through a Departmental Grant from the Robert A. Welch Foundation (Grant no. BX-0048) at the Department of Chemistry at the University of Texas Rio Grande Valley.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c02914.
1H and 13C {1H} NMR spectra of all of the products. Electronic energies, thermochemical corrections and solvation energies for the optimized stationary points, selected dihedral angles (°) for the lowest energy conformers of 4a and 5a, selected interatomic distances (Å) and dihedral angles (°) for the lowest energy conformers of oxalyl chloride or oxalic acid, complete set of HOMO-1, HOMO, LUMO and LUMO+1 and their energies, summaries of NPA, NBO and PT2F analysis (PDF).
Mol2 file names for the optimized structures (mol2) (ZIP).
The authors declare no competing financial interest.
Supplementary Material
References
- Trost B. M.; Dong G.; Vance J. A. Cyclic 1,2-Diketones as Core Building Blocks: A Strategy for the Total Synthesis of (−)-Terpestacin. Chem.–Eur. J. 2010, 16, 6265–6277. 10.1002/chem.200903356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulman Page P. C.; Graham A. E.; Park B. K. A convenient preparation of symmetrical and unsymmetrical 1,2-diketones: application to fluorinated phenytoin synthesis. Tetrahedron 1992, 48, 7265–7274. 10.1016/s0040-4020(01)88265-x. [DOI] [Google Scholar]
- Hyatt J. L.; Stacy V.; Wadkins R. M.; Yoon K. J. P.; Wierdl M.; Edwards C. C.; Zeller M.; Hunter A. D.; Danks M. K.; Crundwell G.; Potter P. M. Inhibition of Carboxylesterases by Benzil (Diphenylethane-1,2-dione) and Heterocyclic Analogues Is Dependent upon the Aromaticity of the Ring and the Flexibility of the Dione Moiety. J. Med. Chem. 2005, 48, 5543–5550. 10.1021/jm0504196. [DOI] [PubMed] [Google Scholar]
- Yang W.; Xia J.; Zhou G.; Jiang D.; Li Q.; Wang S.; Zheng X.; Li X.; Li X.; Shen Y. Luminescent oxygen-sensing film based on β-diketone-modified Eu(III)-doped yttrium oxide nanosheets. Sens. Actuators, B 2018, 257, 340–346. 10.1016/j.snb.2017.10.150. [DOI] [Google Scholar]
- Min H.; Palani T.; Park K.; Hwang J.; Lee S. Copper-catalyzed direct synthesis of diaryl 1,2-diketones from aryl iodides and propiolic acids. J. Org. Chem. 2014, 79, 6279–6285. 10.1021/jo501089k. [DOI] [PubMed] [Google Scholar]
- Kashiwabara T.; Tanaka M. Synthesis of 1,2-Diketones by the Transition Metal-Catalyst-Free Reaction of α-Oxo Acid Chlorides or Oxalyl Chloride with Organostannanes. J. Org. Chem. 2009, 74, 3958–3961. 10.1021/jo802814r. [DOI] [PubMed] [Google Scholar]
- Jing X.; Pan X.; Li Z.; Shi Y.; Yan C. Novel One-Pot Procedure for the Synthesis of 1,2-Diketones. Synth. Commun. 2009, 39, 492–496. 10.1080/00397910802398264. [DOI] [Google Scholar]
- Mobinikhaledi A.; Amiri A. K. TMSCl-catalysed condensation of α-diketone compounds with urea/thiourea derivatives under solvent-free conditions. J. Chem. Sci. 2013, 125, 1055–1062. 10.1007/s12039-013-0471-1. [DOI] [Google Scholar]
- Clausen R. W.; Wartchow R.; Butenschön H. Holger Butenschön, Reactions of 1,2-Diketones with Vinyllithium: Addition Reactions and Dianionic Oxy Cope Rearrangements of Cyclic and Acyclic Substrates. Eur. J. Org. Chem. 2001, 2001, 93–113. . [DOI] [Google Scholar]
- Blackburn C.; Childs R. F.; Kennedy R. A. Lewis acid complexes of 1,2-diketones and their derivatives. The synthesis of seven-membered rings. Can. J. Chem. 1983, 61, 1981–1986. 10.1139/v83-342. [DOI] [Google Scholar]
- Walsh C. J.; Mandal B. K. Improved Synthesis of Unsymmetrical, Heteroaromatic 1,2-Diketones and the Synthesis of Carbazole Ring Substituted Tetraaryl Cyclopentadieneones. J. Org. Chem. 1999, 64, 6102–6105. 10.1021/jo9904213. [DOI] [Google Scholar]
- da Silva L. C.; Machado V. G.; Menezes F. G. Quinoxaline-based chromogenic and fluorogenic chemosensors for the detection of metal cations. Chem. Pap. 2021, 75, 1775–1793. 10.1007/s11696-020-01484-9. [DOI] [Google Scholar]
- Quillen A.; Nguyen Q.; Neiser M.; Lindsay K.; Rosen A.; Ramirez S.; Costan S.; Johnson N.; Do T. D.; Rodriguez O.; Rivera D.; Atesin A.; Ateşin T. A.; Ma L. Palladium-Catalyzed Direct α-C(sp3) Heteroarylation of Ketones under Microwave Irradiation. J. Org. Chem. 2019, 84, 7652–7663. 10.1021/acs.joc.9b00446. [DOI] [PubMed] [Google Scholar]
- Rosen A.; Lindsay K.; Quillen A.; Nguyen Q.; Neiser M.; Ramirez S.; Costan S.; Johnson N.; Do T. D.; Ma L. A Microwave-Assisted Direct Heteroarylation of Ketones Using Transition Metal Catalysis. J. Visualized Exp. 2020, 156, e60441 10.3791/60441. [DOI] [PubMed] [Google Scholar]
- Clarke H. T.; Dreger E. E.. Benzil. Organic Syntheses, 1926; Vol. 6, pp 6–7. [Google Scholar]
- Khurana J. M.; Kandpal B. M. A novel method of synthesis of 1,2-diketones from 1,2-diols using N-bromosuccinimide. Tetrahedron Lett. 2003, 44, 4909–4912. 10.1016/s0040-4039(03)01075-x. [DOI] [Google Scholar]
- Habel L. W.; De Keersmaecker S.; Wahlen J.; Jacobs P. A.; De Vos D. E. Synthesis of vinyl 1,2-diketones. Tetrahedron Lett. 2004, 45, 4057–4059. 10.1016/j.tetlet.2004.03.157. [DOI] [Google Scholar]
- Matsuda T.; Oyama S. Synthesis of unsymmetrical benzils via palladium-catalysed alpha-arylation-oxidation of 2-hydroxyacetophenones with aryl bromides. Org. Biomol. Chem. 2020, 18, 3679–3683. 10.1039/d0ob00575d. [DOI] [PubMed] [Google Scholar]
- Zhai Y.; Su Z.; Jiang H.; Rong J.; Qiu X.; Tao C. B2pin2-mediated copper-catalyzed oxidation of alkynes into 1,2-diketones using molecular oxygen. Tetrahedron Lett. 2019, 60, 843–846. 10.1016/j.tetlet.2019.02.023. [DOI] [Google Scholar]
- Lee J. C.; Park H.-J.; Park J. Y. Rapid microwave-promoted solvent-free oxidation of α-methylene ketones to α-diketones. Tetrahedron Lett. 2002, 43, 5661–5663. 10.1016/s0040-4039(02)01130-9. [DOI] [Google Scholar]
- Zhou J.; Tao X.-Z.; Dai J.-J.; Li C.-G.; Xu J.; Xu H.-M.; Xu H.-J. Electrochemical synthesis of 1,2-diketones from alkynes under transition-metal-catalyst-free conditions. Chem. Commun. 2019, 55, 9208–9211. 10.1039/c9cc03996a. [DOI] [PubMed] [Google Scholar]
- Dubovtsev A. Y.; Dar’in D. V.; Krasavin M.; Kukushkin V. Y. Gold-Catalyzed Oxidation of Internal Alkynes into Benzils and its Application for One-Pot Synthesis of Five-, Six-, and Seven-Membered Azaheterocycles. Eur. J. Org. Chem. 2019, 2019, 1856–1864. 10.1002/ejoc.201900108. [DOI] [Google Scholar]
- Katritzky A. R.; Zhang D.; Kirichenko K. Synthesis of Heteroaryl 1,2-Diketones. J. Org. Chem. 2005, 70, 3271–3274. 10.1021/jo0477998. [DOI] [PubMed] [Google Scholar]
- Kumar Y.; Jaiswal Y.; Kumar A. Two-Step One-Pot Synthesis of Unsymmetrical (Hetero)Aryl 1,2-Diketones by Addition-Oxygenation of Potassium Aryltrifluoroborates to (Hetero)Arylacetonitriles. Eur. J. Org. Chem. 2018, 2018, 494–505. 10.1002/ejoc.201701625. [DOI] [Google Scholar]
- Ishii Y.; Sakaguchi S.. Aerobic Oxidations and Related Reactions Catalyzed by N-Hydroxyphthalimide.Modern Oxidation Methods, 2004; pp 119–163. [Google Scholar]
- Li S.; Zhu B.; Lee R.; Qiao B.; Jiang Z. Visible light-induced selective aerobic oxidative transposition of vinyl halides using a tetrahalogenoferrate(iii) complex catalyst. Org. Chem. Front. 2018, 5, 380–385. 10.1039/c7qo00798a. [DOI] [Google Scholar]
- Jiang H.; Qi C.; Huang L.; Chen Z.; Chen H. DABCO-Catalyzed Oxidation of Deoxybenzoins to Benzils with Air and One-Pot Synthesis of Quinoxalines. Synthesis 2011, 2011, 387–396. 10.1055/s-0030-1258375. [DOI] [Google Scholar]
- Ren X.; Du H. Chiral Frustrated Lewis Pairs Catalyzed Highly Enantioselective Hydrosilylations of 1,2-Dicarbonyl Compounds. J. Am. Chem. Soc. 2016, 138, 810–813. 10.1021/jacs.5b13104. [DOI] [PubMed] [Google Scholar]
- Yin G.; Zhou B.; Meng X.; Wu A.; Pan Y. Efficient C–C Double-Bond Formation Reaction via a New Synthetic Strategy: A Self-Sorting Tandem Reaction. Org. Lett. 2006, 8, 2245–2248. 10.1021/ol060541e. [DOI] [PubMed] [Google Scholar]
- Newhouse T.; Baran P. S. If C-H bonds could talk: selective C-H bond oxidation. Angew. Chem. 2011, 50, 3362–3374. 10.1002/anie.201006368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito Y.; Hirao T.; Saegusa T. Synthesis of .alpha.,.beta.-unsaturated carbonyl compounds by palladium(II)-catalyzed dehydrosilylation of silyl enol ethers. J. Org. Chem. 1978, 43, 1011–1013. 10.1021/jo00399a052. [DOI] [Google Scholar]
- Li W.; Zhang J.; He J.; Xu L.; Vaccaro L.; Liu P.; Gu Y. I2/DMSO-Catalyzed Transformation of N-tosylhydrazones to 1,2,3-thiadiazoles. Front. Chem. 2020, 8, 466. 10.3389/fchem.2020.00466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W.; Chen C.; Zhou P. NN-Dimethylformamide (DMF as a Source of Oxygen To Access alpha-Hydroxy Arones via the alpha-Hydroxylation of Arones. J. Org. Chem. 2017, 82, 2219–2222. 10.1021/acs.joc.6b02751. [DOI] [PubMed] [Google Scholar]
- Chebolu R.; Bahuguna A.; Sharma R.; Mishra V. K.; Ravikumar P. C. An unusual chemoselective oxidation strategy by an unprecedented exploration of an electrophilic center of DMSO: a new facet to classical DMSO oxidation. Chem. Commun. 2015, 51, 15438–15441. 10.1039/c5cc05713b. [DOI] [PubMed] [Google Scholar]
- Riley H. L.; Morley J. F.; Friend N. A. C. Selenium dioxide, a new oxidising agent. Part I. Its reaction with aldehydes and ketones. J. Chem. Soc. 1932, 1875–1883. 10.1039/jr9320001875. [DOI] [Google Scholar]
- Ramajayam R.; Giridhar R.; Yadav M. R.; Balaraman R.; Djaballah H.; Shum D.; Radu C. Synthesis, antileukemic and antiplatelet activities of 2,3-diaryl-6,7-dihydro-5H-1,4-diazepines. Eur. J. Med. Chem. 2008, 43, 2004–2010. 10.1016/j.ejmech.2007.11.023. [DOI] [PubMed] [Google Scholar]
- Zarghi A.; Arfaei S.; Shirazi F. H. Design, synthesis, and cytotoxic activities of new 2,4,5-triarylimidazoles. Med. Chem. Res. 2013, 22, 3897–3904. 10.1007/s00044-012-0391-5. [DOI] [Google Scholar]
- Staedler D.; Chapuis-Bernasconi C.; Dehmlow H.; Fischer H.; Juillerat-Jeanneret L.; Aebi J. D. Cytotoxic Effects of Combination of Oxidosqualene Cyclase Inhibitors with Atorvastatin in Human Cancer Cells. J. Med. Chem. 2012, 55, 4990–5002. 10.1021/jm300256z. [DOI] [PubMed] [Google Scholar]
- Hofmann E.; Webster J.; Do T.; Kline R.; Snider L.; Hauser Q.; Higginbottom G.; Campbell A.; Ma L.; Paula S. Hydroxylated chalcones with dual properties: Xanthine oxidase inhibitors and radical scavengers. Bioorg. Med. Chem. 2016, 24, 578–587. 10.1016/j.bmc.2015.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian M.; Li Q.; Zhu Y.; Yin G.; Wu A. Logic design and synthesis of quinoxalines via the integration of iodination/oxidation/cyclization sequences from ketones and 1,2-diamines. Tetrahedron 2012, 68, 9598–9605. 10.1016/j.tet.2012.09.056. [DOI] [Google Scholar]
- Montana M.; Montero V.; Khoumeri O.; Vanelle P. Quinoxaline Derivatives as Antiviral Agents: A Systematic Review. Molecules 2020, 25, 2784. 10.3390/molecules25122784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keri R. S.; Pandule S. S.; Budagumpi S.; Nagaraja B. M. Quinoxaline and quinoxaline-1,4-di-N-oxides: An emerging class of antimycobacterials. Arch. Pharm. 2018, 351, 1700325. 10.1002/ardp.201700325. [DOI] [PubMed] [Google Scholar]
- St John S. t.; Kim Y.; Etz Y.; Kim B. D.; Paton S.; Paton R. S. Quantum chemical calculations for over 200,000 organic radical species and 40,000 associated closed-shell molecules. Sci. Data 2020, 7, 244. 10.1038/s41597-020-00588-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson G. A.; Nakatsuji H.; Li X.; Caricato M.; Marenich A. V.; Bloino J.; Janesko B. G.; Gomperts R.; Mennucci B.; Hratchian H. P.; Ortiz V.; Izmaylov A. F.; Sonnenberg J.; Williams L.; Ding F.; Lipparini F.; Egidi F.; Goings J.; Peng B.; Petrone A.; Henderson T.; Ranasinghe D.; Zakrzewski V. G.; Gao J.; Rega N.; Zheng G.; Liang W.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Throssell K.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M. J.; Heyd J.; Brothers E. N.; Kudin K. N.; Staroverov V. N.; Keith T. A.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A. P.; Burant C.; Iyengar S. S.; Tomasi J.; Cossi M.; Millam M.; Klene M.; Adamo C.; Cammi R.; Ochterski W.; Martin R. L.; Morokuma K.; Farkas O.; Foresman B.; Fox D. J.. Gaussian 16; Rev. C.01: Wallingford, CT, 2016.
- GaussView Version 6 ; Dennington R.; Keith T. A.; Millam J. M.; Semichem Inc.: Shawnee Mission, KS, 2016. [Google Scholar]
- Peng C.; Ayala P. Y.; Schlegel H. B.; Frisch M. J. Using redundant internal coordinates to optimize equilibrium geometries and transition states. J. Comput. Chem. 1996, 17, 49–56. . [DOI] [Google Scholar]
- Zhao Y.; Truhlar D. G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. 10.1007/s00214-007-0310-x. [DOI] [Google Scholar]
- Weigend F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. 10.1039/b515623h. [DOI] [PubMed] [Google Scholar]
- Weigend F.; Ahlrichs R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- Marenich A. V.; Cramer C. J.; Truhlar D. G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
- Ribeiro R. F.; Marenich A. V.; Cramer C. J.; Truhlar D. G. Use of solution-phase vibrational frequencies in continuum models for the free energy of solvation. J. Phys. Chem. B 2011, 115, 14556–14562. 10.1021/jp205508z. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Truhlar D. G. Computational characterization and modeling of buckyball tweezers: density functional study of concave–convex π···π interactions. Phys. Chem. Chem. Phys. 2008, 10, 2813–2818. 10.1039/b717744e. [DOI] [PubMed] [Google Scholar]
- Glendening E. D.; Reed A. E.; Carpenter J. E.; Weinhold F.. NBO version 3.1; Gaussian Inc.: Pittsburgh, 2003.
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.