

Abstract
Glutamine phosphoribosylpyrophosphate amidotransferase from Bacillus subtilis is a member of an N-terminal nucleophile hydrolase enzyme superfamily, several of which undergo autocatalytic propeptide processing to generate the mature active enzyme. A series of mutations was analyzed to determine whether amino acid residues required for catalysis are also used for propeptide processing. Propeptide cleavage was strongly inhibited by replacement of the cysteine nucleophile and two residues of an oxyanion hole that are required for glutaminase function. However, significant propeptide processing was retained in a deletion mutant with multiple defects in catalysis that was devoid of enzyme activity. Intermolecular processing of noncleaved mutant enzyme subunits by active wild-type enzyme subunits was not detected in hetero-oligomers obtained from a coexpression experiment. While direct in vitro evidence for autocatalytic propeptide cleavage was not obtained, the results indicate that some but not all of the amino acid residues that have a role in catalysis are also needed for propeptide processing.
Glutamine phosphoribosylpyrophosphate (PRPP) amidotransferase is the key regulatory enzyme of de novo purine nucleotide synthesis. Genes encoding the enzyme have been cloned from more than 20 organisms, including bacteria, archaea, and eucarya (30). However, only the enzymes from Bacillus subtilis and Escherichia coli have been purified to homogeneity and characterized biochemically. X-ray structures have been determined for the B. subtilis (27) and E. coli (22) enzymes. The B. subtilis and E. coli enzymes are representative of two distinctive but homologous glutamine PRPP amidotransferase classes. Enzymes of the B. subtilis class are synthesized with an N-terminal propeptide and, in addition, acquire an Fe-S cluster. Enzymes of the E. coli class have neither a propeptide nor an Fe-S cluster. Cleavage of the propeptide is essential for generating the active enzyme containing an N-terminal cysteine that functions as a nucleophile in catalysis. Brannigan et al. (2) initially recognized that three structurally defined amidohydrolases—penicillin acylase, the 20S proteasome, and B. subtilis glutamine PRPP amidotransferase—share a common fold in which the N-terminal nucleophile and other catalytic groups occupy equivalent sites. These enzymes, as well as glycosylasparaginase (23), comprise an N-terminal nucleophile (Ntn) hydrolase structural superfamily. Several of the Ntn hydrolases are synthesized as inactive proenzymes that are posttranslationally processed to generate the mature active enzyme with an N-terminal nucleophile. Several reports have provided strong evidence that 20S proteasome β subunits (3, 12, 21, 26) and glycosylasparaginase (9–11, 29) undergo autocatalytic proenzyme processing. For B. subtilis glutamine PRPP amidotransferase (18, 28) and penicillin acylase (6), the possibility of autocatalytic propeptide cleavage has been suggested from observations that expression of cloned genes in a different host results in active enzyme and that mutations distant from the cleavage site can prevent processing.
Thus far, it has not been possible to detect processing of B. subtilis glutamine PRPP amidotransferase proenzyme in vitro (16). In this study we have sought to determine whether amino acids that are required for glutamine PRPP amidotransferase catalysis also have a role in propeptide processing. Glutamine PRPP amidotransferase catalyzes the synthesis of phosphoribosylamine (PRA) from PRPP and glutamine or NH3 as shown by the following equations.
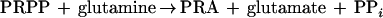 |
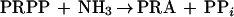 |
The mature enzyme contains two domains (30). An N-terminal glutamine domain converts glutamine to glutamate plus NH3. The C-terminal phosphoribosyltransferase (PRTase) domain binds PRPP and utilizes NH3 derived from glutamine or supplied externally for the synthesis of PRA. Binding of PRPP is required for glutamine hydrolysis. PRPP binding results in conformational changes that shield the PRPP site from solvent, restructure the glutamine site, and form a 20-Å channel which connects the glutamine and PRTase sites and serves as a route for NH3 transfer (14). The mutational analysis in this study indicates that some but not all elements of catalysis are required for propeptide processing.
MATERIALS AND METHODS
Strains and plasmids.
E. coli BL834(DE3) (7) was used for the expression of wild-type and mutant purF genes. For site-directed mutagenesis, genes were cloned into plasmids pUC118 or pET14b. Plasmids pTrc99A and pT7-7 were used as vectors for enzyme production. Other specialized plasmids that were constructed for this work are described in the following sections.
Construction of plasmids to analyze propeptide processing.
To increase the length of the glutamine PRPP amidotransferase propeptide, the B. subtilis purF gene was first cloned into pET14b (Novagen). Plasmid pGZ1, which contains B. subtilis purF in a 1.65-kb EcoRI-HindIII fragment in pUC18 (19) was the template for a PCR reaction in which an NdeI site was incorporated into the ATG translation start site and a BamHI site downstream of the gene for transcription termination. The NdeI-BamHI PCR product was ligated into the corresponding sites of pET14b. Digestion with NcoI-BamHI released a 1.5-kb purF gene containing a 20-codon 5′ extension. The fragment was ligated into pTrc99A to yield plasmid pTrcBF. To add a C-terminal His tag, an NcoI-HindIII purF fragment was transferred from pTrcBF to a pUC118 derivative containing an NcoI site (pUCNco). The His tag was added by site-directed mutagenesis (15) with the oligonucleotide, 5′-CTTGCATGCCTGCAGTTAGTGGTGGTGGTGGTGGTGGGATCCTTCTTTGGTTAATACTGC (histidine codons are underlined) to yield plasmid pUCBFc. Further purF site-directed mutagenesis to examine the roles of residues in propeptide processing was carried out with plasmid pUCBFc.
A PCR reaction was used to delete the PRTase flexible loop: residues Leu303 to Leu329 (27). Plasmid pUCBFc was the DNA template. The primers were 5′-GGTGGTGCTAGCCCCGCCAAGCTCATACGGAATGCC and 5′-GGTGGTGCTAGCGGCTCTGCGGTGGGCGGGGTTGTA, each containing an NheI site (underlined). The 4.5-kb PCR product was isolated by using a Qiagen PCR purification kit and then digested with NheI and self-ligated. The sequence change is shown: wild type, (302) GLIKNRYVGRTFIQPSQALREQGVRMKLSA (331); deletion, (302) GGAS SA (307)
The resulting plasmid was named pUC(Δloop)c. All mutations were confirmed by sequencing (24) the 100 to 200 bp overlapping the position of the mutation. Confirmed plasmids were digested with NcoI-HindIII, and the 1.5-kb purF fragment was returned to pTrc99A for enzyme production. The plasmid designations are pTrcBFc for purF+, pTrc(Δloop)c, pTrc(N102D)c, pTrc(H25Q)c, pTrc(H70Y)c, pTrc(H70N)c, and pTrc(H101Q)c.
Deletion of the propeptide for measurement of enzyme activity.
For measurement of enzyme activity, codons for the propeptide were deleted from the purF gene in plasmids pUCBFc, pUC(Δloop)c, pUC(N102D)c, pUC(H25Q)c, pUC(H70Y)c, pUC(H70N)c, and pUC(H101Q)c. First, an NdeI site was introduced at the position of the translation initiation codon of the mature enzyme by site-directed mutagenesis (15). Next, each plasmid was digested with NdeI-HindIII, and the inserts were transferred to pT7-7. The resulting plasmids were designated ΔP/H6 (wild type with His tag), ΔP(Δloop)H6, ΔP(N102D)H6, ΔP(H25Q)H6, ΔP(H70N)H6, ΔP(H70Y)H6, and ΔP(H101Q)H6.
Construction of a coexpression system.
Plasmids containing a bicistronic operon containing mutant purF followed by active purF were constructed by the following steps. First, an XbaI site was introduced at the 3′ end of pUC(N102D)c by site-directed mutagenesis (15). The NcoI-XbaI purF fragment from pUC(N102D)c and XbaI-HindIII-cut wild-type purF DNA from pT7BF were mixed and ligated into pTrc99A that had been digested with NcoI-HindIII. The resulting coexpression plasmid pTrc(N102D)c-wt contained purF with a N102D mutation, a 31-codon propeptide sequence, and a 3′ six-histidine codon sequence followed by a wild-type purF gene. A second coexpression plasmid was constructed in which the downstream wild-type purF was replaced with purF+ containing a propeptide deletion. Wild-type purF was excised from pTrc(N102D)c-wt by digestion with XbaI-HindIII and was replaced with the corresponding purF+ fragment containing a propeptide deletion from pT7BFΔ. The bicistronic plasmid was named pTrc(N102D)c-Δwt.
Enzyme production and purification.
Wild-type and mutant purF genes were expressed in E. coli BL834(DE3). Plasmid-bearing cells were initially grown in 5 ml of Luria-Bertani medium containing 100 μg of ampicillin per ml at 37°C. A portion (1 ml) of overnight culture was used to inoculate 250 ml of the same medium. The cells were grown at 30°C for 15 h, harvested by centrifugation, and suspended in 10 ml of buffer A (20 mM Tris-HCl, pH 8.0; 100 mM NaCl; 2 mM AMP). All buffers used for enzyme purification were sparged with nitrogen gas prior to use to minimize oxidation of the enzyme’s Fe-S cluster. The cells were broken with a French press at 1,000 lb/in2, and cell debris was removed by centrifugation at 27,000 × g for 30 min. Glutamine PRPP amidotransferase was purified by metal chelate affinity chromatography with Talon Co2+ affinity resin (Clontech) according to the protocol recommended by the supplier. Briefly, 8 ml of extract was incubated with 2 ml of resin for 20 min at 4°C. After being washed with 20 ml of buffer A two times, the resin was packed into two small columns. Enzyme from each column was eluted with 3 ml of buffer A containing 100 mM imidazole. The last 2 ml of eluant was collected and concentrated with a Centricon. After addition of 2 ml of buffer (50 mM Tris-HCl, pH 7.5; 2 mM AMP), the enzyme was concentrated again. First, 1 ml of 50 mM Tris-HCl (pH 7.5) was added, and then the sample was centrifuged to a final volume of 150 to 200 μl. Protein concentration was determined (1) by using bovine serum albumin as a standard. The enzyme was purified to approximately 90% homogeneity as judged by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The yield was 5 to 6 mg of protein per 250 ml of culture.
Enzyme assay.
Activity with glutamine as a substrate was determined by measuring the production of glutamate as described earlier (4). Activity with NH3 as a substrate was determined by coupling PRA production to the synthesis of glycinamide ribonucleotide with glycinamide ribonucleotide synthetase (25). A unit of activity is defined as the amount of enzyme forming 1 μmol of product/min at 37°C. Specific activity is given as units per milligram of protein.
Assays for propeptide processing.
In preliminary experiments, propeptide processing was assayed by using enzymes with the normal 11-amino-acid propeptide and no His tag. Wild-type B. subtilis enzyme was produced in E. coli B834(DE3) from pET-BsF (5) or the corresponding plasmids with R72A or D127A amino acid replacements. Cells were grown as previously described, and proteins in cell lysates were resolved by SDS-PAGE (5). Proteins were transferred to a polyvinylidine membrane, and 5 to 10 cycles of Edman degradation were determined from the protein band containing incompletely resolved proenzyme and mature enzyme. The relative amounts of proenzyme and mature enzyme were calculated from the amino acid quantitation averaged over at least five cycles of degradation.
Except for the preliminary experiments just described, propeptide processing was evaluated by using wild-type and mutant enzymes modified with a 31-amino-acid propeptide and a C-terminal His tag. Cells were grown and enzyme was isolated as described above. Proenzyme and mature enzyme were resolved by SDS–8% PAGE (5).
RESULTS
Analysis of glutamine PRPP amidotransferase propeptide processing.
Two modifications were incorporated into glutamine PRPP amidotransferase in order to facilitate the analysis of propeptide processing by SDS-PAGE. First, the length of the propeptide was extended to improve the electrophoretic separation of the precursor from the mature enzyme. The B. subtilis glutamine PRPP amidotransferase propeptide was increased from 11 to 31 amino acids by fusing a 20-amino-acid peptide to the N terminus of the proenzyme. With this increased mass of 2.18 kDa there was improved electrophoretic resolution of the 53.8-kDa proenzyme and the 50.4-kDa mature species. Second, a C-terminal His tag was added to permit a rapid one-step enzyme purification. The main problem in assessing propeptide processing from crude cell extracts by SDS-PAGE was the background of E. coli proteins of 50 to 60 kDa, which interfered with the identification of the precursor and mature forms of the amidotransferase. By using the C-terminal His tag, the precursor and mature glutamine PRPP amidotransferase species could be rapidly separated from E. coli proteins by a one-step affinity purification step.
The SDS-gel electrophoresis profiles in Fig. 1 show the resolution of the modified wild-type enzyme (lane 1) from three largely unprocessed mutants in lanes 2 to 4. As shown in Fig. 1, lane 1, there was essentially complete propeptide processing of the modified B. subtilis enzyme overproduced in E. coli. Therefore, neither the propeptide extension nor the C-terminal His tag adversely affected propeptide processing.
FIG. 1.

Enzymes with mutations in the N-terminal nucleophile and residues for the oxyanion hole are not processed. The wild-type enzyme from plasmid pTrcBFc (lane 1) and enzymes with mutations C1T (lane 2), G103A (lane 3), and N102D (lane 4) were resolved by electrophoresis on an SDS–8% polyacrylamide gel. Arrows identify the proenzyme and mature enzyme species.
Relationship between enzyme catalysis and propeptide processing.
We have mutated amino acid residues that participate in catalysis in order to determine the relationship between the amino acids required for enzyme catalysis and those needed for propeptide processing. There are four initial steps in catalysis that are necessary for the hydrolysis of glutamine and amide transfer to form PRA (30). These steps are (i) enzyme activation by PRPP, (ii) covalent catalysis by Cys1, (iii) stabilization of a tetrahedral glutaminyl oxyanion, and (iv) general acid-base catalysis. Mutations were constructed to disable each of these steps, and their effect on propeptide processing was determined.
(i) Enzyme activation by PRPP.
The key structural change in the transition from inactive to active enzyme is an ordering of a PRTase domain flexible loop (14). PRPP binding triggers the flexible loop to close the site and sequester the bound PRPP from the solvent. The rearranged PRTase loop also contributes to the formation of a 20-Å channel that connects the glutamine and PRPP sites. To examine the relationship between enzyme activation and propeptide processing, a deletion of residues 303 to 329 that removes most of the flexible loop was constructed. In order to assess the effects of mutations on catalysis, independent of the requirement for propeptide cleavage, it was necessary to delete the sequence encoding the propeptide from the cloned purF gene. Table 1 shows the results of a wild-type control to compare the enzyme activity of the native wild-type amidotransferase and the enzyme synthesized without a propeptide and containing a C-terminal His tag (designated ΔP/H6) for rapid purification. The two enzymes had identical activities with glutamine, although the modifications reduced the activity with NH3. Since the assay of NH3-dependent activity involves a coupling reaction with GAR synthetase, one possibility is that the C-terminal His tag interferes with coupling, thus accounting for the reduced NH3-dependent activity of the modified enzyme. Table 1 also shows that deletion of the PRTase domain flexible loop abolished all activity in the ΔP(Δloop)/H6 enzyme, as expected for a mutation that should disrupt the PRPP site and prevent the transition of the inactive to active conformer.
TABLE 1.
Activity of wild-type and mutant enzymes
| Enzymea | Activity (U/mg) with:
|
|
|---|---|---|
| Glutamine | NH3 | |
| Wild type | 22.2 | 27.1 |
| ΔP/H6 | 22.2 | 10.5 |
| ΔP(Δloop)H6 | 0 | 0 |
| ΔP(N102D)H6 | 0.02 | 10.9 |
| ΔP(H25Q)H6 | 7.80 | 12.8 |
| ΔP(H70N)H6 | 1.29 | 7.43 |
| ΔP(H70Y)H6 | 0.18 | 7.96 |
| ΔP(H101Q)H6 | 0.38 | 10.1 |
Enzymes with a ΔP prefix were produced as propeptide deletions and contained a C-terminal His tag, H6. Mutations are given in parentheses.
As shown in Fig. 2, the Δloop/H6 enzyme with the flexible loop deletion was approximately 50% processed. The upper band in Fig. 2, lane 1, corresponds to unprocessed proenzyme. The electrophoretic migration of this proenzyme was similar to that of a processed H70N/H6 amidotransferase mutant (lane 2), a finding which is consistent with their similar masses. These results indicate that the inactive conformer, lacking an intact PRPP site and residues that are needed for the NH3 channel that connects the glutamine and PRPP sites, can undergo propeptide processing. The glutamine- and NH3-dependent catalytic activities therefore do not have an obligatory role in propeptide processing.
FIG. 2.
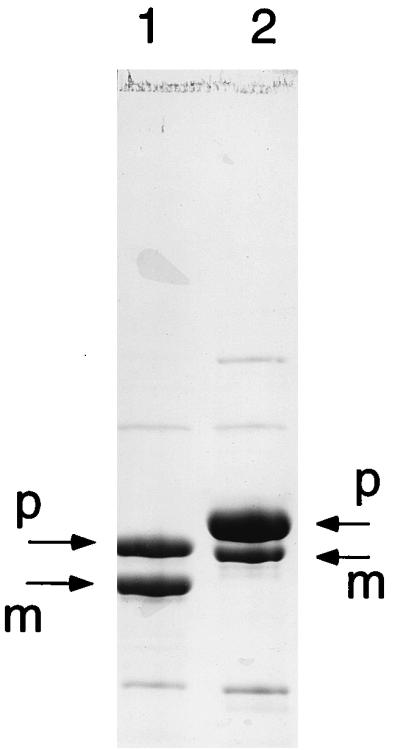
Processing of PRTase flexible-loop-deleted glutamine PRPP amidotransferase. The PRTase flexible-loop-deleted enzyme (lane 1) and an H70N enzyme (lane 2) as a size marker were resolved by electrophoresis on an SDS–8% polyacrylamide gel. Arrows identify the proenzyme and mature enzyme species.
(ii) Covalent catalysis by Cys1.
The role of Cys1 in catalysis is nucleophilic attack on the carboxamide of glutamine to form a tetrahedral oxyanion intermediate (30). In light of the capacity of an N-terminal threonine to function as a nucleophile in other Ntn hydrolases (8–11, 17), a C1T mutant amidotransferase was constructed. The C1T mutant enzyme had no detectable activity with glutamine or with NH3 and, as shown in Fig. 1, lane 2, the proenzyme was completely unprocessed. Previous work has shown that C1F (20) and C1S (28) mutant enzymes are not processed and have no activity with glutamine. The C1F mutant retained about 40% of NH3 activity relative to the wild type, whereas the C1S enzyme had about 1% of the wild-type activity.
(iii) Stabilization of the tetrahedral glutaminyl oxyanion.
According to the current mechanism (30), attack of Cys1 thiolate on the Cδ of glutamine results in formation of a covalent enzyme-glutaminyl intermediate. In the E. coli glutamine PRPP amidotransferase, the resulting tetrahedral oxyanion is stabilized by hydrogen bonds to Nδ of Asn101 and the backbone NH of Gly102 (13). The corresponding residues in the B. subtilis amidotransferase are Asn102 and Gly103. Data in Table 1 show nearly complete loss of activity with glutamine in the N102D mutant B. subtilis enzyme, whereas activity with NH3 is comparable to that for the ΔP/H6 parent enzyme. This result points to similar oxyanion stabilization in the B. subtilis and E. coli enzymes. The activity of a G103A mutant enzyme was not determined but is expected to have a similar defect in glutamine-dependent activity, as seen for the E. coli glutamine PRPP amidotransferase (13). This conclusion is based on the fact that the atomic positions of all catalytic residues are identical within experimental error in the glutamine domain of the two enzymes (22). The data in Fig. 1 (lanes 3 and 4) indicate a strong inhibition of propeptide processing in N102D and G103A mutants. These results suggest that the oxyanion hole that participates in catalysis also has a role in propeptide processing.
(iv) General acid or base catalysis.
A general acid or base group is required for two steps in catalysis at the glutamine site. First, a proton is donated to the leaving amide to yield NH3 upon collapse of the tetrahedral oxyanion intermediate and, second, a proton is accepted from water to generate hydroxyl ion for hydrolysis of the resultant thioester and the release of glutamate. Because no appropriate amino acid side chains are in close enough proximity to Cys1 in the X-ray structures of the inactive conformer of the B. subtilis enzyme nor in the active state of the E. coli enzyme, the proposal was made that the α-amino group of Cys1 functions as a proton donor or acceptor (30). The α-amino group of Cys1 is suitably positioned to mediate proton transfers directly or via a bridging H2O molecule. A general acid or base is also needed for the steps of propeptide cleavage, yet the α-amino group of Cys1 is in peptide linkage and is therefore not available to assume this function. To search for a potential amino acid side chain that might function as a proton acceptor and donor in propeptide cleavage, we examined propeptide processing in a series of mutants. Each of the three conserved histidine residues in the glutamine domain and two conserved glutamate residues in the propeptide were targeted for mutagenesis. The gel profile in Fig. 3 shows the results for propeptide processing in these mutants. Of the three conserved histidines, only His70 was necessary to maintain propeptide processing. H70Y and H70N mutant enzymes were largely unprocessed. On the other hand, H25Q and H101Q mutations had little if any effect on propeptide processing. Of several replacements for Glu−2, an E−2G amidotransferase was about 50% processed. Glu−2 refers to amino acid position minus two in the propeptide, numbered relative to Cys1 in the mature enzyme. Intermediate levels of propeptide processing were also seen for enzymes with E−2D, E−2H, and E−2Q replacements (data not shown). An E−2G E−1A double mutant was also about 50% processed. These results indicate that Glu−2 and Glu−1 have no obligatory role in propeptide processing and are thus not likely to function as general acid/base groups in the reaction. The role of His70 is uncertain.
FIG. 3.

Effect on processing of mutations in conserved histidine residues in the glutaminase domain and in conserved glutamates in the propeptide. Enzymes with mutations in H25Q (lane 1), H70Y (lane 2), H70N (lane 3), H101Q (lane 4), E2G (lane 5), E-2G/E-1A (lane 6), and the wild type (lane 7) were resolved by electrophoresis on an SDS–8% polyacrylamide gel. Arrows identify the proenzyme and mature enzyme species.
Data in Table 1 summarize the effect of the His70 and His101 mutations on glutamine PRPP amidotransferase catalysis. The enzyme with a H25Q replacement retained 35% of its activity with glutamine, whereas H70N, H70Y, and H101Q mutations lost between 90 and 99% of the glutamine-dependent activity. Activity with NH3 was retained at or near the level of the ΔP/H6 parent in each of these mutant enzymes.
Substrate binding.
Arg73 and Asp127 are key residues required for glutamine binding to E. coli glutamine PRPP amidotransferase (13). The corresponding B. subtilis amidotransferase residues (Arg72 and Asp127) were replaced to determine if the site for glutamine binding was required for propeptide processing. These experiments were carried out with enzymes having the wild-type 11-amino-acid propeptide and no C-terminal His tag. After SDS-PAGE, proteins were transferred to a polyvinylidine difluoride membrane, and N-terminal sequences were determined for incompletely resolved proenzyme and mature enzyme. By this method there was 91% processing for wild-type, 77% for R72A, and 67% for D127A enzymes. An intact glutamine binding site is thus not required for propeptide processing.
A test of intermolecular processing.
The requirement for several catalytic residues to convert proenzyme into mature enzyme points to an autocatalytic propeptide processing mechanism. Autocatalysis could be intra- or intermolecular. A test of intermolecular autocatalysis was carried out in vivo by determining if wild-type subunits were able to cleave propeptide from a mutant subunit during or after assembly of hetero-oligomeric enzyme. Two plasmids were constructed for this experiment. The first, pTrc(N102D)c-wt, encodes in a bicistronic operon the N102D mutant enzyme with a 31-amino-acid propeptide plus a C-terminal His tag followed by the native wild-type amidotransferase containing the natural 11-amino-acid propeptide. The second plasmid is similar except the gene encoding the native wild type was replaced with one encoding wild-type enzyme without a propeptide. Coexpression of the two genes was designed to produce hetero-oligomers that could be purified by using the His tag on the N102D subunit. Lanes 1 to 3 in Fig. 4 show the results of N102D coexpression with wild-type purF, and lanes 4 to 6 show coexpression in which wild-type purF lacked codons for the propeptide. Two protein bands from the cell extract were identified in lane 1. The top band corresponds to unprocessed N102D subunit with the His tag, and the lower band corresponds to the mature enzyme subunit, either wild type or N102D. It is not possible to distinguish whether the mature subunit in lane 1 is wild type or N102D without the further analysis shown in lanes 2 and 3. Lane 2 shows the composition of the enzyme when affinity purified under native conditions. The upper band is the N102D proenzyme subunit, and the lower band is the mature subunit, either wild type (no His tag) or N102D with the His tag. To determine whether there was any mature N102D subunit in the lower band, the purified enzyme was denatured in 8 M urea, isolated by affinity chromatography, and electrophoresed in lane 3. The result in lane 3 shows that after denaturation, only proenzyme subunits were purified. Thus, there were no detectable mature N102D subunits in the lower band in lane 2. Very similar results, shown in lanes 4 to 6, were obtained for the coexpression of genes encoding N102D proenzyme and wild-type amidotransferase synthesized without a propeptide. Based on the relative staining of the bands in lanes 2 and 5 corresponding to the proenzyme subunit and the mature enzyme subunit, it would appear that the hetero-oligomer stoichiometry was likely one N102D proenzyme and three mature wild-type subunits. This stoichiometry is in accord with the construction of the bicistronic operon that was designed to yield a higher synthesis rate for wild-type subunits resulting from a stronger ribosome binding site as outlined in Materials and Methods. This experiment demonstrates that the precursor N102D mutant subunit has sufficient native structure to form hetero-oligomers with wild-type subunits but that active wild-type subunits could not catalyze trans propeptide cleavage of the N102D propeptide under these conditions.
FIG. 4.

No processing of mutant N102D subunits by wild-type subunits in a hetero-oligomer. In experiment 1 (lanes 1 to 3) hetero-oligomers were made in vivo from plasmid pTrc(N102D)c-wt encoding N102D proenzyme with a His tag and proenzyme wild type (no His tag). Lane 1, extract; lane 2, proteins purified by using the His tag under native conditions; lane 3, proteins purified by using the His tag under denaturing conditions. In experiment 2 (lanes 4 to 6) heterooligomers were made in vivo from plasmid pTrc(N102D)c-Δwt encoding N102D proenzyme with a His tag and mature wild-type enzyme. Samples in lanes 4 to 6 correspond to the samples in lanes 1 to 3. Proteins were resolved by electrophoresis on an SDS–8% polyacrylamide gel. Arrows mark the positions of the proenzyme and mature enzyme species.
DISCUSSION
Since we have not detected glutamine PRPP amidotransferase propeptide processing in vitro (16), further characterization of enzyme maturation in cells should prove useful. An objective of this work was to determine if cleavage of the propeptide amide bond requires the same catalytic groups as glutamine amide hydrolysis, an initial step in the reaction to synthesize PRA. The interpretation of these results leads to the conclusion that some, although not all, of the residues needed for glutamine hydrolysis are involved in propeptide cleavage, as explained in the discussion that follows. Binding of PRPP to its catalytic site activates glutamine hydrolysis by structural changes that lower the Km for glutamine by 100-fold and increase the Vm by 3-fold (13). A deletion of residues which in the nonactive conformer comprises a PRTase flexible loop and are needed to form a functional PRPP site, a functional glutaminase site, and the channel to connect the two sites abolished glutaminase activity and NH3-dependent synthesis of PRA, as shown in Table 1. Yet, as seen in Fig. 1, approximately one-half of this mutant enzyme was processed. Therefore, a functional glutaminase site is not obligatory for propeptide cleavage. On the other hand, the perturbations in this mutant of the glutamine site could explain the approximately 50% inhibition of propeptide processing. Cys1, the Ntn, and Asn102 plus Gly103, residues that provide oxyanion stabilization, are two components of enzyme catalysis that are clearly necessary for propeptide processing (Fig. 1). It would therefore appear that these residues retain some capacity to function in propeptide cleavage in the PRTase flexible loop deletion. The role of Cys1 in catalysis is nucleophilic attack on the carboxamide of glutamine to yield a tetrahedral oxyanion intermediate. This intermediate is stabilized by interactions with the amide side chain of Asn102 and the backbone NH of Gly103. It is therefore reasonable to conclude that the Cys1 thiolate performs a similar nucleophilic attack on the peptide bond between Glu−1 and Cys1. The resulting tetrahedral oxyanion might be stabilized by interactions with Asn102 and Gly103. The partial inhibition of propeptide processing by the PRTase flexible loop deletion suggests that the repositioning of active-site residues by PRPP binding that is essential for glutamine hydrolysis increases the efficiency of propeptide cleavage but is not obligatory. Glutamine binding is unlikely to have a role in propeptide processing, since mutations that increase the Km for glutamine by 60- to 130-fold had only small effects on propeptide cleavage.
To complete the peptide bond cleavage by this proposed mechanism general acid and general base groups are required to protonate the NH of Cys1 upon cleavage of the backbone and to accept a proton from H2O to generate the hydroxyl ion needed for thioester hydrolysis, respectively. In the structure-based mechanism for catalysis the free α-amino group of the N-terminal cysteine is proposed to carry out the comparable proton transfer steps for glutamine hydrolysis (30). However, since the α-amino of Cys1 is in peptide linkage in the proenzyme it is unavailable to function as a general acid or base group. We investigated whether conserved histidine or glutamate side chains might carry out this function. Of three conserved histidines in the glutamine domain, only replacement of His70 had a major effect on propeptide cleavage. It is uncertain, however, whether His70 could have a direct role as a general base and acid group in propeptide cleavage. This is because glutaminase function was inhibited in the two His70 mutants in parallel with the inhibition of propeptide cleavage. Since there is no presently known direct role for His70 in the structure-based catalytic mechanism (30), its replacement may indirectly affect catalysis and by analogy indirectly affect propeptide cleavage. Essential roles of the two highly conserved glutamate residues at positions −1 and −2 in the propeptide are unlikely, as shown by the significant processing that was retained in the Glu−1 Glu−2 mutant enzyme. The question of the general acid or base group needed for propeptide cleavage is therefore unresolved.
Direct evidence for autocatalytic processing has been obtained for two Ntn hydrolases, glycosylasparagenase (9, 10, 29) and the 20S proteasome (3, 21, 26). The amino acid residue in the −2 position of Flavobacterium meningosepticum glycosylasparaginase (His150) was shown to be required for intermolecular autoproteolysis and is assumed to function as the base that activates Thr152 for nucleophilic attack on the Asp151 peptide backbone (9, 10). The resulting cleavage of the proenzyme generates an active β subunit with an N-terminal threonine. The acid or base group required for this step has not been identified. Thr152 Oγ functions as the nucleophile and the α-amino group as the base for amidohydrolase catalysis. The backbone NH of Gly204 and perhaps Thr203 Oγ make up the oxyanion hole (11, 23, 29).
A number of in vitro and in vivo experiments have provided strong evidence for the autocatalytic processing of proteosome β subunits (21, 26), which are assembled into a hetero-oligomeric (α7)2(β7)2 barrel-shaped structure (8, 17). Each of the seven precursor β subunits from these enzymes are identical, and they are processed intermolecularly during folding and assembly of the (α7)2(β7)2 hetero-oligomer. Thr1 is the N-terminal nucleophile for processing and for catalysis. The α-amino group of Thr1 is implicated as the base for catalysis and is possibly assisted by Lys33. However, the base used for propeptide cleavage has not been defined for the archaea enzymes. In the more complex yeast proteasome, with seven different β subunits, propeptide cleavages can occur by intra- and intermolecular mechanisms (8, 12). A structure-based mechanism for intramolecular propeptide cleavage invokes Thr1 as a nucleophile and a water molecule as a general base for activation of Thr1 and for cleavage of the peptide bond (8). Lys33 Nζ and Gly47N are correctly positioned to function as an oxyanion hole.
Our experiments did not detect processing of an inactive N102D proenzyme by an active wild-type glutamine PRPP amidotransferase when genes for the two enzymes were coexpressed from a bicistronic operon. Inactive Thermoplasma proteasome β subunits were processed by active subunits in an experiment of similar design (26). Once they were assembled into α7 and β7 ring structures, no further processing took place. It was concluded that processing probably occurs before β subunits have reached their final folded state and are assembled into proteosomes. Glutamine PRPP amidotransferase is also processed prior to the formation of stable dimers or tetramers. Highly expressed wild-type enzyme is a mixture of processed and unprocessed subunits (5). Further processing was not observed in cells incubated for extended periods of time or in cell extracts incubated in the presence or absence of oxygen.
ACKNOWLEDGMENTS
This work was supported by Public Health Service grants GM24658 (H.Z.) and DK42303 (J.L.S.). Protein sequencing was carried out by the Purdue Laboratory for Macromolecular Structure, which is supported by the Diabetes Research and Training Center (Public Health Service grant DK20542).
We thank Sihong Chen for help with enzyme assays and for sharing her expertise with the enzyme, and we thank Jay Bertrand for structural insights.
Footnotes
Journal paper 15893 from the Purdue University Agricultural Experiment Station.
REFERENCES
- 1.Bradford M M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 2.Brannigan J A, Dodson G, Duggleby H J, Moody P C E, Smith J L, Tomchick D R, Murzin A G. A protein catalytic framework with an N-terminal nucleophile is capable of self-activation. Nature. 1995;378:416–419. doi: 10.1038/378416a0. [DOI] [PubMed] [Google Scholar]
- 3.Chen P, Hochstrasser M. Autocatalytic subunit processing couples active site formation in the 20S proteasome to completion of assembly. Cell. 1996;86:961–972. doi: 10.1016/s0092-8674(00)80171-3. [DOI] [PubMed] [Google Scholar]
- 4.Chen S, Tomchick D R, Wolle D, Hua P, Smith J L, Switzer R L, Zalkin H. Mechanism of the synergistic end-product regulation of Bacillus subtilis glutamine phosphoribosylpyrophosphate amidotransferase by nucleotides. Biochemistry. 1997;36:10718–10726. doi: 10.1021/bi9711893. [DOI] [PubMed] [Google Scholar]
- 5.Chen S, Zheng L, Dean D R, Zalkin H. Role of Nifs in maturation of glutamine phosphoribosyltransferase. J Bacteriol. 1997;179:7587–7590. doi: 10.1128/jb.179.23.7587-7590.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi K S, Kim J A, Kang H S. Effects of site-directed mutations on processing and activities of penicillin G acylase from Escherichia coli ATCC 11105. J Bacteriol. 1972;174:6270–6276. doi: 10.1128/jb.174.19.6270-6276.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doherty A J, Ashford S R, Brannigan J A, Wigley D B. A superior host strain for the overexpression of cloned genes using the T7 promoter based vectors. Nucleic Acids Res. 1995;23:2074–2075. doi: 10.1093/nar/23.11.2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Groll M, Ditzel L, Löwe J, Stock D, Bochtler M, Bartunik H D, Huber R. Structure of the 20S proteasome from yeast at 2.4 Å resolution. Nature. 1997;386:463–471. doi: 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- 9.Guan C, Cui T, Rao V, Liao W, Benner J, Lin C-L, Comb D. Activation of glycosylasparaginase. Formation of active N-terminal threonine by intramolecular autoproteolysis. J Biol Chem. 1996;271:1731–1737. doi: 10.1074/jbc.271.3.1732. [DOI] [PubMed] [Google Scholar]
- 10.Guan C, Liu Y, Shao Y, Cui T, Liao W, Ewel A, Whitaker R, Paulus H. Characterization and functional analysis of the cis-autoproteolysis active center of glycosylasparaginase. J Biol Chem. 1998;273:9695–9702. doi: 10.1074/jbc.273.16.9695. [DOI] [PubMed] [Google Scholar]
- 11.Guo H-C, Xu Q, Buckley D, Guan C. Crystal structures of Flavobacterium glycosylasparaginase. An N-terminal nucleophile hydrolase activated by intramolecular proteolysis. J Biol Chem. 1998;273:20205–20212. doi: 10.1074/jbc.273.32.20205. [DOI] [PubMed] [Google Scholar]
- 12.Heinemeyer W, Fischer M, Krimmer T, Stachon U, Wolf D H. The active sites of the eukaryotic 20S proteasome and their involvement in subunit precursor processing. J Biol Chem. 1997;272:25200–25209. doi: 10.1074/jbc.272.40.25200. [DOI] [PubMed] [Google Scholar]
- 13.Kim J H, Krahn J M, Tomchick D R, Smith J L, Zalkin H. Structure and function of the glutamine phosphoribosylpyrophosphate amidotransferase glutamine site and communication with the phosphoribosylpyrophosphate site. J Biol Chem. 1996;271:15549–15557. doi: 10.1074/jbc.271.26.15549. [DOI] [PubMed] [Google Scholar]
- 14.Krahn J M, Kim J H, Burns M R, Parry R J, Zalkin H, Smith J L. Coupled formation of an amidotransferase interdomain ammonia channel and a phosphoribosyltransferase active site. Biochemistry. 1997;36:11061–11068. doi: 10.1021/bi9714114. [DOI] [PubMed] [Google Scholar]
- 15.Kunkel T A, Roberts J D, Koukour R A. Rapid and efficient site-specific mutagenesis without phenotypic selection. Methods Enzymol. 1987;154:367–382. doi: 10.1016/0076-6879(87)54085-x. [DOI] [PubMed] [Google Scholar]
- 16.Li, S., and H. Zalkin. 1998. Unpublished results.
- 17.Löwe J, Stock D, Jap B, Zwicki P, Baumeister W, Huber R. Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4Å resolution. Science. 1995;268:533–539. doi: 10.1126/science.7725097. [DOI] [PubMed] [Google Scholar]
- 18.Makaroff C A, Paluh J L, Zalkin H. Mutagenesis of ligands to the [4Fe-4S] center of Bacillus subtilis glutamine phosphoribosylpyrophosphate amidotransferase. J Biol Chem. 1986;261:11416–11423. [PubMed] [Google Scholar]
- 19.Makaroff C A, Zalkin H, Switzer R L, Vollmer S J. Cloning of Bacillus subtilis glutamine phosphoribosylpyrophosphate amidotransferase gene in Escherichia coli. Nucleotide sequence determination and properties of the plasmid-encoded enzyme. J Biol Chem. 1983;258:10586–10593. [PubMed] [Google Scholar]
- 20.Mäntsälä P, Zalkin H. Glutamine amidotransferase function. Replacement of the active site cysteine in glutamine phosphoribosylpyrophosphate amidotransferase by site-directed mutagenesis. J Biol Chem. 1984;259:14230–14236. [PubMed] [Google Scholar]
- 21.Maupin-Furlow J A, Aldrich H C, Ferry J G. Biochemical characterization of the 20S proteasome from the methanoarchaeon Methanosarcina thermophila. J Bacteriol. 1998;180:1480–1487. doi: 10.1128/jb.180.6.1480-1487.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muchmore C R A, Krahn J M, Kim J H, Zalkin H, Smith J L. Crystal structure of glutamine phosphoribosylpyrophosphate amidotransferase from Escherichia coli. Protein Sci. 1998;7:39–51. doi: 10.1002/pro.5560070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oinonen C, Tikkanen R, Rouvinen J, Peltonen L. Three-dimensional structure of human lysosomal aspartylglucosaminidase. Nat Struct Biol. 1995;2:1102–1108. doi: 10.1038/nsb1295-1102. [DOI] [PubMed] [Google Scholar]
- 24.Sanger F, Nicklen S, Coulson A R. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schendel F J, Cheng Y S, Otvos J D, Wehrli S, Stubbe J. Characterization and chemical properties of phosphoribosylamine, an unstable intermediate in the de novo purine biosynthetic pathway. Biochemistry. 1988;27:2614–2623. doi: 10.1021/bi00407a052. [DOI] [PubMed] [Google Scholar]
- 26.Seemüller E, Lupas A, Baumeister W. Autocatalytic processing of the 20S proteasome. Nature. 1996;382:468–470. doi: 10.1038/382468a0. [DOI] [PubMed] [Google Scholar]
- 27.Smith J L, Zaluzec E J, Wery J-P, Niu L, Switzer R L, Zalkin H, Satow Y. Structure of the allosteric regulatory enzyme of purine biosynthesis. Science. 1994;264:1427–1433. doi: 10.1126/science.8197456. [DOI] [PubMed] [Google Scholar]
- 28.Souciet J-L, Hermodson M A, Zalkin H. Mutational analysis of the glutamine phosphoribosylpyrophosphate amidotransferase pro-peptide. J Biol Chem. 1988;263:3323–3327. [PubMed] [Google Scholar]
- 29.Tikkanen, R., A. Riikonen, C. Oinonen, J. Rouvinen, and L. Peltonen. Functional analyses of active site residues of human lysosomal aspartylglucosaminidase: implications for catalytic mechanism and autocatalytic activation. EMBO J. 15:2954–2960. [PMC free article] [PubMed]
- 30.Zalkin H, Smith J L. Enzymes using glutamine as an amide donor. Adv Enzymol Relat Areas Mol Biol. 1998;72:87–144. doi: 10.1002/9780470123188.ch4. [DOI] [PubMed] [Google Scholar]