

Abstract
Myocardial infarction (MI) is a cardiovascular disease with high morbidity and mortality. Clinically, rehabilitation after massive MI often has a poor prognosis. Therefore, it is necessary to explore the therapeutic methods of myocardial protection after MI. As a first-line treatment for type 2 diabetes, metformin has been found to have a certain protective effect on myocardial tissue. However, its pharmacological mechanism remains unclear. In this study, we investigated key factors that reduced MI with metformin. Through in vivo, in vitro, and in silico analyses, we identified HSF1 as a key target for metformin. HSF1 could up-regulate the transcriptional level of AMPKα2 through transcriptional activation and stimulate the activity of the downstream AMPK/mTOR signaling pathway. Metformin stimulated cardiomyocytes to form stress granules (SGs), and knockdown of HSF1 reversed this process. Furthermore, HSF1 exhibited better in vitro affinity for metformin than AMPK, suggesting that HSF1 may be a more sensitive target for metformin.
Keywords: myocardial infarction (MI), metformin, HSF1, AMPK, stress granules
Graphical abstract
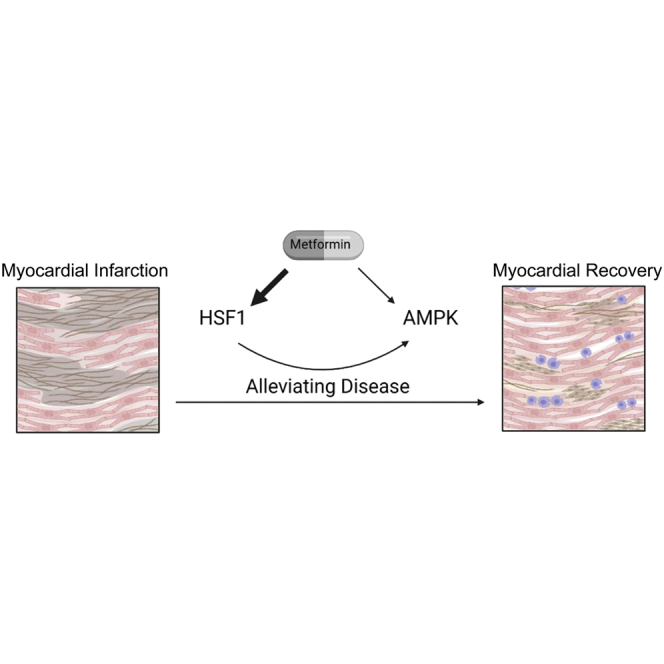
Metformin can effectively inhibit the progression of myocardial infarction and has certain benefits for the recovery of cardiac function. We identified an important target of metformin in exerting cardioprotective effects—HSF1. In mouse cardiomyocytes, HSF1 is a more selective metformin target than AMPK.
Introduction
Myocardial infarction (MI) is a cardiovascular disease with high morbidity and mortality.1 Most MIs are based on coronary artery disease, complicated by a sharp reduction or interruption of blood supply, which makes the corresponding myocardium serious and continuous ischemia leading to myocardial necrosis.2 The necrosis of adult myocardial tissue is often irreversible, and clinically, the prognosis of myocardial recovery treatment after a large area of MI is often poor.3 Therefore, understanding of the mechanisms of myocardial protection is essential for the development of reasonable drug therapy and improving the patient’s prognosis.
Recent studies have shown that there are varying degrees of association between hypoglycemic drugs and cardiovascular events, and some drugs show clear cardiovascular benefits.4 Among them, metformin has a clear effect on the prevention of atherosclerotic plaque and the protection of myocardium.5 A retrospective cohort study showed that among diabetic patients discharged from hospital after acute MI, the non-mortality rate of discharged patients with metformin adherence at 30 days, 6 months, and 1 year was significantly lower than that of patients with discontinuation after discharge, and the rate of re-admissions due to cardiovascular events had decreased.6 In diabetic mice models, treatment with metformin for 4 weeks normalized AMPK phosphorylation levels, and in AMPKα2 knockout mice, 26S proteosome levels increased.7 This indicates that the beneficial effect of metformin on vascular endothelial function is related to the activation of AMPK. However, the role of metformin is currently not fully understood. The study of the drug in myocardial protection will help reveal the pathophysiological mechanism of myocardial injury and repair, and further expand its application fields.
In many pathological conditions (such as ischemic injury, oxidative stress, pressure overload, etc.), heat shock factor (HSF) can protect myocardium injury.8,9 The mammalian genome encodes HSF1 and HSF2 as well as other tissue-specific paralogs. Among them, HSF2 can regulate HSF1 and form heteromers, as well as alter downstream target genes induced by heat shock response (HSR). Functionally, HSF1 has been shown to promote the progression of malignant tumors, while HSF2 inhibits tumor progression.10 HSF1 is an important transcription factor that regulates the expression of heat shock proteins under various stress factors. HSR mediated by HSF1 is a molecular stress defense response formed in the long-term evolution of organisms.11 In cardiovascular aspect, HSF1 can alleviate heart failure by actively regulating the AC6/cAMP/PKA pathway in a pressure overload heart failure model.12 In addition, HSF1 can reduce myocardial cell damage and ultimately decrease the risk of heart failure.13 However, more details about the mechanism of HSF1 in cardioprotection are still lacking.
In the present study, we identify that HSF1 is a crucial target for metformin in alleviating MI in mice. We found that the myocardial injury in HSF1 knockout mice treated with metformin progressed faster. Moreover, HSF1 could activate AMPKα2 transcription and its downstream signaling pathways. In addition, knocking down of HSF1 could block metformin’s stimulation of cardiomyocytes to form stress granules (SGs). In the comparison between HSF1 and AMPK, metformin had better affinity with HSF1 than AMPK. Therefore, HSF1 played a more important role in the treatment of MI in mice with metformin. This study provides new ideas for probing the pharmacological mechanism of metformin and developing new drug targets.
Results
Metformin could reduce myocardial injury after MI in mice
First, we tried to verify whether metformin could have a therapeutic effect on the MI mice. It was shown that metformin could significantly reduce the scope of MI. The MI area of the left ventricle in the longitudinal section of the untreated mice accounted for 33.90% ± 2.22% of the left ventricular area. In the metformin-treated group, this index decreased to 11.23% ± 2.46%. (Figure 1B, ∗∗∗p < 0.001).
Figure 1.

Metformin could reduce myocardial injury after myocardial infarction in mice
(A) Simulation process diagram. (B) Masson staining of representative left ventricular section. The right panel shows infarct sizes. LV, left ventricle; INF, infarct area. Scale bar, 2,000 μm; n = 3; ∗∗∗p < 0.001. (C) Transmission electron microscopy photo of infarcted myocardial edge tissue. Scale bar, 1 μm.
The effect of metformin on myocardial tissue adjacent to infarction was observed by electron microscope. The results showed that the marginal tissue of the infarcted myocardium without drug treatment was dissolved and destroyed, a large number of vacuoles appeared, the texture was unclear, and the mitochondria were obviously swollen. The myocardium of the infarcted myocardium in the metformin treatment group was relatively regular, the degree of myofibrolysis was significantly reduced, the texture was relatively clear, and the mitochondrial structure was normal (Figure 1C). In addition, the systolic blood pressure of MI mice was significantly higher than that of the sham operation group. However, metformin treatment could alleviate this effect (Figure S1A). In isolated and cultured mouse primary cardiomyocytes, we found that metformin reduced the apoptosis rate of cardiomyocytes (early and late apoptosis, Q2+Q3, Figure S1B). These data suggest that metformin has a protective effect on the myocardial tissue at the edge of MI and effectively slows down the expansion of the infarct area.
HSF1 had a cardioprotective effect on MI mice
Considering the protective effect of HSF1 on biological organisms, we tried to explore the changes of HSF1 during the treatment of MI in mice. Proteins from the tissue surrounding infarcted myocardium was extracted for western blot analysis. We found that p-HSF1Ser326/HSF1 levels were significantly reduced in MI model mice compared with the sham-operated group. After metformin treatment, the level of p-HSF1Ser326/HSF1 was significantly increased (Figures 2A and S2A). Since HSF1 could be activated after phosphorylation, it was speculated that metformin could promote the phosphorylation of HSF1 at Ser326 and activate it to protect the myocardium.
Figure 2.

HSF1 had protective effect on mouse myocardium
(A) Expression levels of p-HSF1Ser326/HSF1 in tissues adjacent to infarcted myocardium in different treatment groups. (B) mRNA levels of HSF1 in tissues near infarcted myocardium in different treatment groups. (n = 3; SD) (C) Subcellular distribution of HSF1 in mouse cardiomyocytes. Red represents HSF1 fluorescence and blue represents DAPI. Scale bar, 50 μm. (D) Expression levels of HSF1 in the cytoplasm and nucleus, respectively. Cytoplasmic proteins were homogenized with β-actin as an internal reference, and nuclear proteins were homogenized with histone H3 as an internal reference. (E) Masson staining of representative left ventricular section. The right panel shows infarct sizes. LV, left ventricle; INF, infarct area. Scale bar, 2,000 μm; n = 3; ∗∗∗p < 0.001. (F) Representative pictures of transmission electron microscopy of tissue adjacent to infarcted myocardium in wild-type mice (WT) and HSF1-knockout homozygous mice (HSF1−/−). Scale bar, 1 μm.
RNA was extracted from the above-mentioned experimental specimens and tested by qPCR. In different treatment groups, there was no significant difference in the expression of HSF1 mRNA between any two groups (Figure 2B). This indicated that metformin failed to affect HSF1 at the RNA level.
Considering that the activation of HSF1 leads to an increase in its nuclear entry and activates its transcriptional regulatory activity,14 we further tried to explore whether metformin had an effect on the spatial distribution of HSF1 in subcellular locations.
We found that under serum-free culture conditions (nutrient deprivation), the red fluorescence of HSF1 was mainly distributed in the cytoplasm, with a small amount in the nuclear region. After metformin treatment, the red fluorescence of HSF1 mostly overlapped with the blue fluorescent DAPI staining of the nucleus. Merging the fluorescence of the two colors, it could be seen that the metformin-treated nuclei were red and blue overlapping cross colors (Figure 2C).
By extracting the nuclear and cytoplasmic components, western blot was used to detect the protein expression of HSF1 in the nucleus and cytoplasm, respectively. We found that metformin significantly reduced the expression of HSF1 in the cytoplasm. Among nuclear proteins, metformin promoted HSF1 expression (Figure 2D).
To illustrate the importance of HSF1 for myocardial protection in mice during metformin treatment, we selected 8-week-old C57BL/6 male wild-type (WT) and HSF1−/− mice, and established surgical models of MI, respectively. In addition, we performed compensation experiments for the overexpression of HSF1 by lentivirus on the two groups of mice. The above groups were given metformin treatment together. Finally, the mice were killed on the seventh day after the administration. The hearts were taken out, and Masson staining was performed after fixation. In metformin-treated WT mice, the infarct area of myocardium accounted for 7.28% ± 0.89% of the left ventricle; in the metformin-treated HSF1−/− mice, the infarct area accounted for 16.76% ± 1.05% (∗∗∗p < 0.001, Figure 2E). In the compensation experiment of overexpression of HSF1, we found that in HSF1−/− mice, overexpression of HSF1 could significantly reduce the infarct area of myocardium (13.93% ± 0.43% versus 16.76% ± 1.05%, ∗∗∗p < 0.001, Figure 2E). In the WT mice, overexpression of HSF1 also showed a similar trend (2.43% ± 0.35% versus 7.28% ± 0.89%, ∗∗∗p < 0.001, Figure 2E). Transmission electron microscopy (Figure 2F) assay showed that compared with the WT group, the sarcomere destruction of the infarcted myocardial margin tissue in the HSF1−/− group was more obvious, and the mitochondria were significantly enlarged and deformed. These results demonstrated that HSF1 deficiency could dampen the protective effect of metformin against MI.
Metformin stimulated cardiomyocytes to produce SGs through HSF1
The production process of SGs is considered as a self-protection mechanism of eukaryotic cells in response to stimuli.15 Here, we initially explored whether metformin induced SGs and the role of HSF1. Cardiomyocytes were treated with metformin (100 μM) for 24 h. Cells were fixed, permeabilized, and processed for immunofluorescence using antibodies against different SG markers (FMRP, G3BP, FXR1, and DDX3). We observed that metformin did stimulate the formation of SGs in mice cardiomyocytes, and when HSF1 was knocked down, the formation of SGs was blocked (Figure 3).
Figure 3.

Cardiomyocytes were treated with MET (100 μM) for 24 h
Cells were fixed, permeabilized, and processed for immunofluorescence using antibodies against different SG markers (FMRP, G3BP, FXR1, and DDX3). DAPI is used as a marker for nuclei. The indicated percentage of SG-positive cells (>3 granules/cell) representing more than 1,000 cells of three independent experiments. UNT, untreated; MET, metformin. Scale bar, 20 μm.
HSF1 was the upstream regulator of AMPK/mTOR pathway in the myocardium of MI mice
In the primary cardiomyocytes of newborn mice cultured in vitro, the expression level of AMPK-related mRNA was detected by means of overexpression and knockdown of HSF1. We found that compared with the normal cultured group, when HSF1 was knocked down, the mRNA level of AMPKα2 was significantly reduced, while the mRNA level of mTOR was significantly increased. On the contrary, when HSF1 was overexpressed, the mRNA level of AMPKα2 was significantly increased, while the mRNA level of mTOR was significantly decreased (Figure 4A, ∗∗∗p < 0.001). A separate comparison of the RNA expression levels of HSF1 and AMPKα2 showed that the RNA expression levels of HSF1 and AMPKα2 were positively correlated (Figure 4B, R2 = 0.7343). Overexpression of HSF1 (Ad-HSF1) could promote the formation of LC3 fluorescent puncta in cardiomyocytes (GFP-LC3 adenovirus transfection). When HSF1 (si-HSF1) was inhibited, the green fluorescent puncta representing LC3 were significantly reduced (Figure 4C).
Figure 4.

HSF1 was the upstream regulator of AMPK/mTOR pathway
(A) The mRNA expression levels of HSF1, AMPK, and mTOR in the cardiomyocytes of HSF1 overexpression group and its control group (Ad-HSF1 and Ad-NC), HSF1 knockdown group and its control group (si-HSF1 and si-NC) mice. (n = 3; SD; ∗∗∗p < 0.001) (B) Scatterplot of the correlation analysis between HSF1 and AMPK mRNA expression in mouse cardiomyocytes, measured by qPCR. (C) Punctate accumulation of GFP-LC3 in mouse cardiomyocytes at 48 h in different treatment groups (Ad-NC, Ad-HSF1, si-NC, and si-HSF1). (D) Expression levels of HSF1, AMPK/mTOR pathway, autophagy-related protein p62, and LC3II/I in mouse cardiomyocytes. (E) Expression levels of HSF1 and AMPK proteins in mouse cardiomyocytes after overexpression and knockdown of AMPK (Ad-NC, Ad-AMPK, si-NC, and si-AMPK). (F) Expression levels of mTOR and autophagy-related proteins p62, LC3II/I in mouse cardiomyocytes after overexpression and knockdown of AMPK or HSF1 (Ad-HSF1, si-HSF1, Ad-AMPK, and si-AMPK).
In order to clarify whether HSF1 was the upstream regulator of AMPKα2, we interfered with HSF1 and AMPKα2 at the same time, and added a rescue verification experiment to determine the up- or downstream relationship between HSF1 and AMPKα2. When HSF1 was overexpressed, the level of AMPK protein increased, the level of mTOR protein decreased, the expression of p62 decreased, and the ratio of LC3II/I increased. Conversely, when HSF1 was knocked down, AMPK protein level decreased, mTOR protein level increased, p62 expression increased, and the ratio of LC3II/I decreased (Figures 4D and S2B–S2E). Overexpression or knockdown of AMPKα2 failed to change the protein expression of HSF1 (Figure 4E).
However, when HSF1 was overexpressed and AMPKα2 was knocked down at the same time, the expression of mTOR and p62 increased, while the LC3II/I ratio decreased. On the contrary, when HSF1 was knocked down and AMPKα2 was overexpressed, the expression of mTOR, p62, and LC3II/I was the same as overexpression of AMPKα2 (Figures 4F and S2F–S2H). Therefore, HSF1 could act as an upstream regulator of AMPKα2 to regulate the activity of its downstream signal pathways.
HSF1 regulated the transcription of AMPKα2 through binding to its promoter
After screening by bioinformatics, we found HSF1 as a transcription factor-specific motif in the promoter region of AMPKα2, and designed a reporter gene experiment to verify it. We found that HSF1 could specifically bind to the "GAACGTTC" fragment at position −340 in the promoter region of AMPKα2, and up-regulate the transcription of AMPKα2 (Figures 5A–5D). The results of chromatin immunosuppression (ChIP)-qPCR reverse verification of HSF1 binding sites suggested that HSF1 could significantly enrich the DNA fragments between −446 and −264 in the AMPKα2 promoter region compared with the immunoglobulin (Ig)G group (Figures 5E and 5F).
Figure 5.

AMPKα2 promoter reporters were transactivated by HSF1
(A) Diagram shows the relative positions of full-length (FL) and fragments of AMPKα2 promoter reporters. (B and C) Reporter assay response of AMPKα2 promoter FL and the individual fragments was investigated. (D) Reporter assays of the P2 fragment of the AMPKα2 promoter containing a mutated TFBM as indicated. (E) A schematic illustrates the relative positions of qPCR probes to putative motif (pm) for ChIP-QPCR experiments. (F) Antibody-pulled down chromatins were analyzed by qPCR. RLU, relative light unit. Data were mean ± SD of three independent experiments. (n = 3; SD; ∗∗∗p < 0.001)
Metformin promoted autophagy in mouse cardiomyocytes through AMPK/mTOR pathway
In order to confirm whether AMPK/mTOR was involved in the protective effect of metformin, we detected the expression levels of AMPK, mTOR, and phosphorylated proteins p-AMPKThr172 and p-mTORSer2448 in the sham operation group, the untreated group of surgical modeling, and the metformin treatment group after surgical modeling. Compared with the sham operation group, the p-AMPKThr172/AMPK ratio of the marginal tissue of the infarcted myocardium in the MI model group was significantly reduced, and the p-mTORSer2448/mTOR ratio was increased. However, in the metformin treatment group, the p-AMPKThr172/AMPK ratio was significantly higher than those of the first two groups, and the p-mTORSer2448/mTOR ratio was significantly lower than those of the first two groups (Figures 6A, S3A, and S3B).
Figure 6.

Metformin promoted autophagy in mouse cardiomyocytes through the AMPK/mTOR pathway
(A) AMPK/mTOR pathway and its phosphorylated protein expression levels in tissues adjacent to infarcted myocardium in different treatment groups. (B) qPCR was used to detect the knockdown effect of si-AMPK in primary mouse cardiomyocytes. (n = 3; SD; ∗∗∗p < 0.001) (C) Comparison of different treatments on GFP-LC3 punctate aggregation of mouse cardiomyocytes in vitro (negative control, 1 μM met, 100 μM met and si-AMPK + 100 μM met). The left side shows representative photos of GFP-LC3 mouse cardiomyocytes. Scale bar, 20 μm. The percentage of cells with GFP-LC3 punctate aggregation is shown on the right. (n = 3) (D) Autophagy-related proteins LC3II/I and p62, AMPK/mTOR pathway, and their phosphorylated protein levels in mouse cardiomyocytes cultured in vitro. (E) Masson staining of representative left ventricular section. The right panel shows infarct sizes. LV, left ventricle; INF, infarct area. Scale bar, 2,000 μm; n = 3; ∗∗∗p < 0.001. (F) Transmission electron microscope representative pictures of the tissues adjacent to the infarcted myocardium in met + lv/sh-scramble group and met + lv/sh-AMPKα2 group. Scale bar, 1 μm.
Autophagy-related markers were detected in the myocardium adjacent to infarcted myocardium. Transmission electron microscopy observed myocardial tissue adjacent to MI. We found that metformin and the rapamycin (autophagy activator) had similar effects (Figure S1C). Compared with the sham-operated untreated group, the mRNA expression level of Becn1 and Atg5 in the myocardium adjacent to infarcted myocardium decreased, while the mRNA expression level of mTOR increased. In the metformin treatment group, the mRNA expression levels of Becn1 and Atg5 increased, while the mRNA expression levels of mTOR decreased (Figure S1D) compared with the sham-operated untreated group. Compared with the sham-operated untreated group, the level of LC3II/I decreased in the non-metformin treatment group, and increased in the metformin treatment group. (Figure S1E). It showed that metformin could enhance the autophagy level of the adjacent myocardium of infarcted myocardium, which may contribute to the protection of myocardial tissue.
In primary mouse cardiomyocytes cultured with serum, a high dose of metformin (100 μM) could more effectively stimulate the formation of autophagosomes than a low dose of metformin (1 μM). However, when AMPKα2 was knocked down (verified by qPCR, Figure 6B), the autophagy activation effect of metformin was blocked (Figure 6C).
In addition, knocking down of AMPKα2 (si-AMPKα2) could inhibit the phosphorylation ratio of AMPK at Thr172 in the metformin low-dose (1 μM) group, thereby inhibiting the activation of the AMPK/mTOR signal pathway, blocking the reduction of p62 and LC3II/I ratio. However, a high dose of metformin (100 μM) increased the overall protein expression of LC3, but failed to activate the AMPK/mTOR signal pathway, as the LC3II/I ratio and p62 showed no significant increase or decrease (Figure 6D). Therefore, we speculated that AMPK/mTOR was indispensable for metformin to activate autophagy in mice cardiomyocytes. Knockdown of AMPKα2 could effectively block this process. Although the overall expression of LC3 protein increased, the p62 protein did not decrease, indicating that the autophagy was blocked after it was initiated.
In order to confirm the above hypothesis, we used a mouse MI model combined with lentiviral vectors to knock down AMPKα2 in vivo. Seven days after metformin treatment of MI, lentivirus was injected via the tail vein, and the dose of lentivirus injection was 2 × 108 U/mouse. Two weeks later, samples were taken to assess the condition of MI in mice. The proportion of the infarct area in the AMPKα2 knockdown group was significantly higher than that of the control group (31.44% ± 2.64% versus 12.31% ± 1.83%, p < 0.001, Figure 6E). The sarcomere of the infarcted myocardium marginal tissue in the lentiviral knockout AMPKα2 group was more damaged, and the mitochondria were significantly enlarged and deformed. This showed that knocking out AMPKα2 could weaken the therapeutic effect of metformin (Figure 6F).
The combination of metformin-HSF1 showed a stronger binding effect than the combination of metformin-AMPK
The three-dimensional structure of AMPK crystal mainly consists of three subunits: α, β, and γ. The active site is mainly composed of α subunits. The hydrophobic groove of the protein is located between the subunits of the protein, and metformin acts on this active site (Figure 7A). The three-dimensional structure of the HSF1 crystal is mainly composed of four alpha helices, 20 beta sheets, and random coils, and its active site is mainly composed of beta sheets and random coils. The hydrophobic groove of the protein is located deep into the surface of the protein, and the small molecule ligand acts on this active site to antagonize or activate this target to act on the signal pathway (Figure 7B).
Figure 7.

The combination of metformin-HSF1 showed a stronger binding effect than the combination of metformin-AMPK
(A and B) Schematic diagram of metformin ligand binding AMPK (A) and HSF1 (B) protein (pymol mapping); the protein is displayed in cartoon form. (C and D) Schematic diagram of the amino acid sites of metformin ligand binding to AMPK (C) and HSF1 (D) protein (pymol mapping). (E and F) Concentration gradient curve of compound metformin and protein AMPK (E) and HSF1 (F). (G) Binding curves of two proteins (AMPK and HSF1) at 3,200 nM and metformin, respectively. (H) Histogram of the maximum binding signal between the two proteins at 3,200 nM and metformin on the chip.
According to the flexible docking method, through 100 calculations, the form with the lowest binding free energy was selected. The docking free energy of metformin and HSF1 was −6.88 kal/M, which was lower than the free energy of docking metformin and AMPK −3.2 kal/M. This showed that metformin and HSF1 may have a higher affinity than AMPK (Table 1).
Table 1.
Comparison of the docking energy of metformin with AMPK and HSF1
| Protein symbol | Ligand structure | Docking energy (kal/M) | Interaction force | Combination type |
|---|---|---|---|---|
| AMPK | 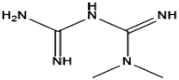 |
−3.2 | Hydrophobic interaction, van der force, hydrogen bond | Non-covalent bonding |
| HSF1 | −6.88 |
By analyzing the results of 100 docking times, the energy-optimal conformation was selected. Metformin entered the active site of HSF1 target protein mainly through hydrophobic and van der chemistries. It mainly interacted with VAL24, ASP43, PHE45, PHE53, HIS56, GLU103, GLN115, and other amino acid residues at the active site of the protein. Through molecular docking, the binding conformation of metformin and HSF1 target protein was simulated, and it acted on the active site of the protein in a non-covalent manner. The hydrogen atom on the ligand could form a hydrogen bond with the oxygen on the ASP43 amino acid, the hydrogen bond length was 2.0 A; it formed a hydrogen bond with GLU103, the hydrogen bond length was 1.8 A and 1.9 A, respectively. The formation of hydrogen bonds enhanced the ability of metformin to target HSF1 protein (Figure 7C). On the other hand, metformin entered the active site of AMPK target protein through hydrophobic and van der chemistry, and interacted with VAL11, LEU18, GLY19, LYS31, SEP108, VAL113, and other amino acid residues at the active site of the protein. The binding conformation of metformin and AMPK target protein acted on the active site of the protein in a non-covalent manner. The hydrogen atom of the ligand could form a hydrogen bond with the oxygen on the GLY19 amino acid, the hydrogen bond length was 2.1 A; it formed two hydrogen bonds with SEP108, the bond length was 1.8 and 2.0 A. The formation of hydrogen bonds enhanced the ability of metformin to target AMPK protein (Figure 7D). This might be the molecular basis explaining that metformin could activate HSF1 and AMPK proteins.
For real-time detection of Surface Plasmon Resonance (SPR) equipment, a set of concentration gradients (200 nM, 400 nM, 800 nM, 1,600 nM, 3,200 nM) of AMPK and HSF1 proteins were distributed in the experiment. We drew the specific binding curve (Figures 7E and 7F) of the interaction concentration gradient between the two proteins of different concentrations and the stationary phase metformin. We fitted the interaction kinetic curve and output the affinity parameters, and measured that metformin has a strong interaction with the two proteins, and the binding signal of metformin-HSF1 was stronger than metformin-AMPK (Figures 7G and 7H). We calculated the binding kinetics and affinity data of metformin and HSF1 and AMPK (Table 2). The above results suggested that HSF1 and AMPK proteins could interact strongly with metformin. The qualitative relationship of its strength was HSF1>AMPK.
Table 2.
Analyze the affinity data of metformin with two proteins by SPR
| No. | Mobile phase | Stationary phase | Avg kon (1/Ms) | Avg koff (1/s) | Avg KD (M) | Int. intensity level | ABS (tr_KD) |
|---|---|---|---|---|---|---|---|
| 1 | HSF1 | Metformin | 5.38 × 103 | 1.86 × 10−4 | 3.46 × 10−8 | Strong | 24.787 |
| 1-BK | HSF1 | DMSO | 1.25E+00 | 5.93 × 10−1 | 4.73 × 10−1 | VW/None | 1.080 |
| 2 | AMPK | Metformin | 7.78 × 102 | 2.91 × 10−2 | 3.74 × 10−5 | Strong | 14.708 |
| 2-BK | AMPK | DMSO | 1.72E+00 | 5.79 × 10−1 | 3.37 × 10−1 | VW/None | 1.569 |
| NC | PBS | Metformin | 1.05E+00 | 8.38 × 10−1 | 8.00 × 10−1 | VW/None | 0.321 |
| NC-BK | PBS | DMSO | 1.09E+00 | 6.91 × 10−1 | 6.37 × 10−1 | VW/None | 0.651 |
Conclusion
In this study, we found that HSF1 was a key target for metformin to exert its efficacy. We further found that there was a regulatory relationship between HSF1 and the AMPK/mTOR signaling pathway. Importantly, metformin preferentially targeted HSF1 compared with AMPK (as shown in Figure 8). Our findings provide new clues for a comprehensive and in-depth understanding of the specific pharmacological mechanism of metformin.
Figure 8.

Mechanistic diagram of metformin (via HSF1 and AMPK) protecting mouse myocardium
Discussion
Among patients admitted to hospital due to acute MI, the prevalence of type 2 diabetes is as high as 20% to 30%.16 It was found that metformin might be a useful drug for secondary prevention in diabetic patients.17 An average follow-up study found that metformin could reduce the risk of all-cause death, cardiovascular disease, infection, or acidosis in diabetic patients.18 In addition, overweight type 2 diabetes patients treated by metformin had a lower risk of MI than conventional treatment (mainly diet therapy).19 In terms of the comprehensive endpoints of macrovascular disease (MI, sudden death, angina pectoris, stroke, and peripheral diseases), metformin had a lower risk than the traditional treatment.19
Metformin is a widely used drug that has obvious benefits for glucose metabolism and diabetes-related complications. In this study, the protective effect of metformin on the infarcted myocardium was confirmed. It could effectively improve the tissue surrounding the infarcted myocardium and prevent the infarct area from further expanding. At the same time, it could stabilize blood pressure and improve the stress response after MI. On the other hand, metformin could inhibit the basal apoptotic rate of isolated cardiomyocytes. However, the mechanism behind its benefits has not yet been fully elucidated. The currently known important pharmacological molecular mechanism of metformin is the activation of AMPK. It reduces the activity of AMPK substrate acetyl-CoA carboxylase (ACC), increases fatty acid oxidation, inhibits fat synthesis, and promotes glucose uptake in vascular smooth muscle.20 However, how metformin activates AMPK remains controversial. Another study showed that metformin could activate AMPK by phosphorylating LKB1 at serine 428 through PKC.21 In fact, neither LKB1 nor AMPK is a direct target of metformin.22 At present, people have shifted from the understanding of the simple mechanism of action of metformin through AMPK activation to improve blood glucose to the exploration of more complex mechanisms of action reflecting its multiple modes of action.23 To truly understand the mechanism of action of this drug in its target population, more work needs to be done.
The HSR is a widespread biological effect in all life characterized by the induction of molecular chaperones following a sudden increase in temperature. In eukaryotes, HSRs are regulated by a series of conserved HSFs.24 Yeast and invertebrates have a single HSF - Hsf1, while vertebrates have four Hsf genes, Hsf1-4. HSF1 is considered to be a master regulator of the HSR and is the only homolog of the HSF gene in invertebrates. HSF2 is a ubiquitously expressed HSF1 paralog that interacts with HSF1 during HSR and participates in developmental pathways. HSF3 and HSF4 show tissue-restricted expression and their roles in HSR remain to be explored.25 Among them, HSF1 is the "master regulator" of HSR, which controls a subset of heat-shock-inducible genes by increasing RNA polymerase II release from promoter-proximal pause.26
HSF1 drives distinct transcriptional programs in physical development and inflammation, in animal models of obesity, and in highly malignant cancer cells, and is able to reduce the sensitivity of the aged heart to stress.27, 28, 29, 30 In addition to protease inhibitors, HSF1 also controls cell cycle and metabolic genes, and promotes tumorigenesis in a variety of tumor types.29 HSF1 is also active in cancer-related fibroblasts in the matrix surrounding tumors, and drives transcriptional programs in these cells to promote tumor growth through paracrine signals.31 At the same time, HSF1 has also been shown to be phosphorylated in a variety of eukaryotes, and phosphorylation is essential for the activation of HSF1.32
We explored the importance of HSF1 for metformin in cardioprotection. Existing reports have shown that HSF1 plays a prominent role in protecting cells from stressful environments in mammals.33 Due to the emergence of emergency conditions of ischemia and hypoxia in the myocardial tissue, the activation of HSF1 in the tissue around infarcted myocardium was inhibited. Metformin treatment reduced the myocardial damage and increased the translocation of phosphorylation of HSF1 into the nucleus. Other study showed that HSF1 expression increased after heart injury to prevent inactivation of cardiomyocytes.9 In addition, its expression was compensatorily increased in some heart diseases such as heart failure. HSF1 could promote angiogenesis, alleviated cardiomyocyte hypertrophy, and ultimately lowered the risk of heart failure.13
So far, there are few research reports that are focused on metformin-targeted autophagy. However, autophagy may be an important part of metformin’s pharmacological effects.34 Autophagy plays a dual role in different cells of type 2 diabetic patients treated with metformin.35 Metformin can not only improve some of the biochemical characteristics of diabetes, but also inhibit the increase of autophagy in white blood cells, and restore the reduction of autophagy in the blood monocytes of patients with type 2 diabetes.36 At the same time, activated AMPK is also involved in the occurrence of autophagy.37 We therefore believe that metformin can protect and control the infarct focus by activating autophagy in the surrounding tissues of the infarcted myocardium.
We found that the AMPK/mTOR pathway is a key link for metformin to protect the heart. AMPK is an important molecular energy sensor that can be activated by increasing the ratio of adenosine monophosphate (AMP) to adenosine triphosphate (ATP) and/or the ratio of adenosine diphosphate (ADP) to ATP.37 Metformin can significantly activate AMPKα1 and α2 in isolated rat muscles, inhibit mitochondrial oxidative phosphorylation, reduce ATP synthesis in cells, and increase the ratio of AMP/ATP in cells to activate AMPK.38 Many studies have suggested that metformin exerts various biological activities by targeting AMPK.38, 39, 40 However, in this study, we propose that metformin may become an activating molecule of HSF1. Therefore, we used AMPK as a positive control to compare the affinity of HSF1 and AMPK (known targets of metformin) for metformin, respectively. Through computer analysis and in vitro experimental analysis and comparison, we have proved that HSF1 had a stronger affinity for metformin than AMPK, thus HSF1 may be the effective target of metformin.
The present study additionally demonstrates that HSF1 can inhibit the induction of SGs by metformin. It is generally believed that the production of SGs is a self-protection mechanism of cells.41 Upon adverse environmental stimuli, eukaryotic cells may form SGs in the cytoplasmic matrix to prevent damage and promote cell survival. The initiation, mRNA and protein composition, distribution, and degradation of SGs are affected by multiple intracellular post-translational modifications and signaling pathways in response to stress injury. Abnormal and/or persistent intracellular SG and aggregation of SG-related proteins may lead to various diseases when stress persists.42 This is similar to the protective role that autophagy plays in living organisms. For mouse cardiomyocytes, the formation of SGs and the increase in autophagy levels both reflect the protective effects of metformin. Unfortunately, the cross-talk mechanism between SGs and autophagic phenotype has not been explored in this study. Given that the beneficial manifestations of metformin in many diseases and its complex molecular mechanisms of action are not well understood, this study provides only a limited exploration of its effects on infarcted heart.
Materials and methods
HSF1 knockout and WT mice
In this study, 8-week-old SPF male C57BL/6 mice weighing about 25 g, Hsf+/+ and Hsf−/− mice were kindly provided by Dr. lvor J. Benjamin (Froedtert & Medical College of Wisconsin, Milwaukee, WI, USA). The mouse tail genotype rapid identification kit (Beyotime, Shanghai, China) was used to identify knockout mice. The specific operation procedures refer to the instructions attached to the kit. The study was carried out in accordance with the Laboratory Animal-Guideline for Ethical Review of Animal Welfare. The experimental protocol was approved by the Laboratory Animal Welfare and Ethical Committee of Central South University.
Animal modeling and metformin treatment
Mice were anesthetized by ether inhalation. A microsurgical approach was used to ligate the left anterior descending (LAD) coronary artery. The sham-operated mice underwent the same surgical procedure without the LAD suture. Experimental procedures should be as gentle as possible. The operation should be successful once, to avoid repeated operations to cause animal suffering. After the ligation of the LAD surgical model was successfully established, they were fed normally. On the seventh day, the mice were randomly divided into groups, and the administration group received intraperitoneal injection of metformin (Sigma-Aldrich, St Louis, MO, USA, 100 mg/kg, once). The untreated group was injected intraperitoneally with the same volume of DMSO. Finally, the mice were killed on the seventh day after the administration (Figure 1A). The heart was then removed, and Masson staining was performed after fixation. Masson-stained pictures were used to calculate the percentage of the area of infarcted myocardium to the area of left ventricular muscle.
Histological analysis
For histological analysis, the heart from the mouse was fixed with 4% paraformaldehyde and embedded in paraffin and cross-cut into 5-μm sections. Heart sections were stained with Masson to assess the area of infarcted myocardium.
The effect of metformin on myocardial tissue adjacent to infarcted myocardium was observed under an electron microscope. Microsurgery instruments were used to carefully remove the infarcted myocardial tissue. The myocardial tissue was cut 2 mm from the edge of the infarcted myocardium with microscissors. After fixation, the microstructure of myocardial fibers was observed by electron microscope.
Isolation and culture of primary cardiomyocytes
Primary cardiomyocytes were taken from mice at 3 days after birth. In short, the heart was placed on a plate containing D-Hanks solution, and the remaining blood clots were removed. The heart was cut into small pieces and incubated with 0.25% trypsin. The digested suspension was centrifuged to resuspend and placed in a 5% CO2 incubator at 37°C. After 24 h, beating cardiomyocytes could be observed. For cell experiments, metformin was administered at a concentration of 100 μM, and the medium was changed after 24 h.
RNA extraction and qRT-PCR
The RNA was extracted using the Trizol method. The RNA reverse transcription procedure was carried out according to the instructions of the TaKaRa reverse transcription kit (TaKaRa, Beijing, China). The qPCR primer sequences of related genes were designed and synthesized by Shanghai Shenggong Biological Engineering Co., Ltd. All genes have been compared with Actb as an internal reference gene. The primer sequence is shown in Table S1.
Cell fractionation
Cells were fractionated to nuclear and cytoplasmic fractions using the nuclear cytoplasmic proteins separation kit (Biotechnology, China). The cytoplasm and nuclear protein were respectively mixed with SDS denaturation buffer and boiled. Histone H3 was used as the internal reference for nuclear proteins, and β-actin was used as the internal reference for cytoplasmic proteins.
SGs detection
Neonatal mouse primary cardiomyocytes were cultured in normal medium containing 10% serum. The administration group was treated with 100 μM metformin. The corresponding control group was treated with DMSO at the same time. The knockdown of HSF1 was achieved by small interfering RNA (siRNA) transfection (Jima Pharmaceutical Technology, Shanghai, China). Subsequently, immunofluorescence was used to detect different markers to determine the formation of SG. Anti-FMRP, anti-G3BP, anti-FXR1, and DDX3 antibodies were purchased from Abcam, Inc. (Abcam, Cambridge, MA, USA).
ImageJ software was used to set the exposure threshold of fluorescence images, and the number of cells in the unit field of view was counted. Further, by increasing the threshold of fluorescence intensity, the number of cells with obvious granule-like fluorescence in the cytoplasm was counted. Finally, the percentage of SGs was calculated and published as follows: SGs% = (number of granule-like fluorescent positive cells/total number of cells) × 100%.
Western blot
Western blot was used to evaluate the level of protein expression. The protein concentration was determined with BCA kit (Beyotime, Beijing, China), and 25 to 30 μ of protein was taken from each sample for SDS-PAGE separation experiment. According to standard procedures, the protein bands were transferred to PVDF membranes, blocked with 5% nonfat dry milk, and then incubated with primary and secondary antibodies in sequence. Finally, it was developed by enhanced chemiluminescence kit (Beyotime, Beijing, China), and the gray value of the band was analyzed by ImageJ software, and the protein expression was quantitatively analyzed. The primary antibodies used were anti-HSF1 and p-HSF1Ser326 (Cell Signaling Technology, USA), anti-β-actin (Cell Signaling Technology, USA), anti-LC3 (Cell Signaling Technology, USA), anti-p62 (Cell Signaling Technology, USA), Histone H3 (Cell Signaling Technology, USA), anti-mTOR and anti-p-mTORSer2448 (Abcam, USA), anti-AMPK and anti-p-AMPKThr172 (Abcam, USA).
Autophagy analysis
For observation of the autophagosomes, the cardiomyocytes were transfected with of GFP-LC3-expressing adenovirus for 24 h (HANBIO, Shanghai, China). The primary mouse cardiomyocytes were inoculated into a six-well plate, and 50 MOI of virus was added to each well. After 24 h of transfection, the expression of GFP was observed under a microscope. The number of fluorescent spots in each field of view was calculated. The level of autophagy was compared by the number of spots. Labeled autophagic vesicles were measured using ImageJ software and shown as a percentage of the total number of GFP-LC3 dots.
Immunofluorescence
The isolated cardiomyocytes were placed in a culture plate, and then treated in different groups. After the treatment, the cells was fixed for immunofluorescence staining. In this experiment, goat serum was used to block at room temperature, and then a sufficient amount of diluted primary antibody was added dropwise and placed in a wet box, and incubated overnight at 4°C. The diluted fluorescent secondary antibody was incubated in a humidified box at room temperature. All operation steps should be carried out in a dark place as much as possible. DAPI was added dropwise to counterstain the cell nucleus, and then the image was collected by observation under a fluorescence microscope.
For observing the subcellular localization of HSF1, the isolated cardiomyocytes were cultured in groups. They were treated with nutrient deprivation culture and metformin after nutrient deprivation. In the control group, primary cardiomyocytes were cultured under serum-free conditions for 48 h. In the experimental group, cells cultured under nutrient deprivation conditions were treated with metformin for 12 h.
Apoptosis detection
After reselection, the cells were collected into a 10-mL centrifuge tube, the number of cells in each sample was 5 × 106/mL, 500 to 1,000 r/min, centrifuged for 5 min, and the medium was discarded. After washing with buffer solution, 100 μL of cell suspension was added to a 5-mL culture tube, and 5 μL of fluorescein isothiocyanate (FITC)-labeled Annexin-V and 5 μL of propidium iodide (PI) were added (MultiSciences Biotech, Zhejiang, China). After mixing, the suspension was incubated for 30 min at room temperature in the dark, and 400 μL of 1× binding buffer was added. The entire procedure was performed gently to avoid damaging cells as much as possible.
Finally, cells were detected by flow cytometry. The excitation wavelength of the flow cytometer was 488 nm, a 515 nm passband filter was used to detect FITC fluorescence, and another filter with a wavelength greater than 560 nm was used to detect PI. Single positive controls and a negative control were used to set a threshold for measuring apoptosis, and the proportions of early and late apoptotic cells were summed and compared with different treatment groups.
siRNA cell transfection
The siRNA and its corresponding control sequence were designed and synthesized by Shanghai Jima Pharmaceutical Technology Co., Ltd. The knockdown effect was tested by qPCR and western blot. Cells were digested into single cell suspensions with 0.25% trypsin. After the cells were counted, the cell concentration was adjusted to 0.5 × 106/mL, and the cells were seeded into low-adsorption six-well plates. The diluted siRNA and Lipo reagent were mixed, and kept at room temperature for 10 min, and then it was added to the cell well plate of the corresponding group, and transferred to the cell incubator. After 8 h of culture, the culture medium was replaced with serum-containing culture medium. Transfection efficiency can be detected after 24 h. The siRNA sequence information is exhibited in Table S2.
Adenovirus-mediated genes expression in vitro
The HSF1 and AMPKα2 overexpression adenovirus vectors and their corresponding control adenovirus vectors were designed and synthesized by Shanghai Jima Biotechnology Co., Ltd. After cell adhesion, virus was added into each well, MOI = 50. The cells was cultured at 37°C with 5% CO2 incubator for 4 h. After transfection, the cells were taken out and RNA was extracted. The overexpression efficiencies were detected by qPCR and western blot.
Luciferase reporter of AMPKα2 promoter region
In brief, the target DNA fragments were obtained by extraction of mouse cardiomyocyte DNA and PCR technology, and the fragment was inserted into the pGL-4.1 reporter gene vector. The AMPKα2-promoter-pGL-4.1 promoter recombinant plasmid and HSF1 overexpression vector were co-transfected into WT HEK293 cells. Subsequently, the intensity of the luciferase reaction was detected by a microplate reader under dark conditions. Bioinformatics analysis predicted the possible binding sites of HSF1 in the AMPKα2 promoter region. A site-directed mutagenesis kit (Biotechnology, China) was used to carry out multiple adjacent base mutations to the predicted binding site to construct the AMPKα2 promoter site-directed mutation reporter gene. The binding of HSF1 to target DNA was determined by comparing the ratio of the reaction intensity of firefly luciferase and Renilla luciferase.
ChIP analysis of HSF1 and AMPKα2 promoter region binding
After DNA fixation, the long-strand DNA was broken into 200 to 1,000 base pair DNA fragments. The concentration of the system was adjusted, and HSF1 antibody and IgG antibody were added. Then, the unbound DNA fragment was discarded, and the solution buffer was added to elute the antibody in the complex. The target DNA fragment was detected by PCR.
Lentiviral vector in vivo
The AMPKα2 gene-silencing lentivirus, the HSF1 gene overexpression lentivirus, and their corresponding control sequence lentiviral vector were designed and synthesized by Shanghai Jima Biotechnology Co., Ltd. Surgical model mice were randomly selected for grouping. Lentivirus was injected via the tail vein, and the dose of lentivirus injection was 2 × 108 U/mouse. Two weeks later, the efficiencies were tested.
Molecular docking
In this study, ChemDraw software was used to construct a molecular model of metformin. The protein crystal model of HSF1 and AMPK were constructed by homology modeling, and the protein structures were processed with Mgtools 1.5.6. The protein structure database (Protein DataBank, PDB; http://www.rcsb.org/) was used as the data source for the crystal structure of HSF1 (5JCT-HSF1) and AMPK (4CFF-AMPK) proteins that were obtained by crystal X-ray diffraction.
According to the change of binding energy of receptor and ligand molecules and the principle of the lowest binding energy, a reasonable binding model was found. With the active site of the protein as the center, a 32∗32∗40 box was constructed, and the GLG file was generated through autogrid4.exe operation. Using Lamarckian genetic algorithm, the DLG file was generated by autodock4.exe. According to the reasonable docking model, the docking energy and amino acid residues were calculated.
SPR technology analyzes the binding of metformin to HSF1 and AMPK proteins
The kinetic parameters and affinity parameters of the interaction between metformin and the two proteins were compared by SPR to further analyze the binding ability of metformin and the two proteins. The metformin reagent and two recombinant protein samples used in the experiment were purchased from Abcam Company.
The metformin working fluid was printed on the three-dimensional light cross-linked chip by the Biodot AD1520 chip array printer. The printed chip was dried and placed in a photo-cross-linking instrument for photo-cross-linking reaction. The protein sample was diluted into five concentration gradients: 200 nM, 400 nM, 800 nM, 1,600 nM, 3,200 nM. PBST was used as a flow carrier throughout the experiment. The experiment was carried out in strict accordance with the relevant standard operating procedure process. The experimental method design and the parameters involved are shown in Table S3.
Statistical analysis
Statistical analysis was performed using SPSS 17.0 (SPSS Inc., Chicago, IL, USA) software. The data are expressed in the form of mean ± SD. For the comparison of two samples, when the condition of homogeneity of variance is met, the independent sample t test is used. When the condition of homogeneity of variance is not satisfied, the approximate t test or the non-parametric Mann-Whitney U test is used. One-way ANOVA was used to compare samples of more than two groups, and the Tukey method was used for comparison between groups. p < 0.05 indicates that the difference is statistically significant.
Data availability
All data are available from the authors upon reasonable request.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (Grant No. 81470408); the Natural Science Foundation of Hunan Province, China (Grant No. 2020JJ4774; 2022JJ40818; 2022JJ30786); Science and Technology Planning Project of Changsha (Grant No. kq2004080). The authors thank BioRender software for creation of the schematic diagram.
Author contributions
Conceived and performed the study: K.W., M.W., and N.W. Designed the experiments: K.W. and M.W. Wrote the manuscript: M.W. All authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2022.07.009.
Contributor Information
Nian Wang, Email: wangnian@csu.edu.cn.
Kangkai Wang, Email: wangkangkai@csu.edu.cn.
Supplemental information
References
- 1.Cui H., Liu C., Esworthy T., Huang Y., Yu Z.X., Zhou X., San H., Lee S.J., Hann S.Y., Boehm M., et al. 4D physiologically adaptable cardiac patch: a 4-month in vivo study for the treatment of myocardial infarction. Sci. Adv. 2020;6:b5067. doi: 10.1126/sciadv.abb5067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kalogeris T., Baines C.P., Krenz M., Korthuis R.J. Cell biology of ischemia/reperfusion injury. Int. Rev. Cell Mol. Biol. 2012;298:229–317. doi: 10.1016/B978-0-12-394309-5.00006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gulati R., Behfar A., Narula J., Kanwar A., Lerman A., Cooper L., Singh M. Acute myocardial infarction in young individuals. Mayo Clin. Proc. 2020;95:136–156. doi: 10.1016/j.mayocp.2019.05.001. [DOI] [PubMed] [Google Scholar]
- 4.Zhu J., Yu X., Zheng Y., Li J., Wang Y., Lin Y., He Z., Zhao W., Chen C., Qiu K., Wu J. Association of glucose-lowering medications with cardiovascular outcomes: an umbrella review and evidence map. Lancet Diabetes Endocrinol. 2020;8:192–205. doi: 10.1016/S2213-8587(19)30422-X. [DOI] [PubMed] [Google Scholar]
- 5.Bromage D.I., Godec T.R., Pujades-Rodriguez M., Gonzalez-Izquierdo A., Denaxas S., Hemingway H., Yellon D.M. Metformin use and cardiovascular outcomes after acute myocardial infarction in patients with type 2 diabetes: a cohort study. Cardiovasc. Diabetol. 2019;18:168. doi: 10.1186/s12933-019-0972-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Inzucchi S.E., Masoudi F.A., Wang Y., Kosiborod M., Foody J.M., Setaro J.F., Havranek E.P., Krumholz H.M. Insulin-sensitizing antihyperglycemic drugs and mortality after acute myocardial infarction: insights from the National Heart Care Project. Diabetes Care. 2005;28:1680–1689. doi: 10.2337/diacare.28.7.1680. [DOI] [PubMed] [Google Scholar]
- 7.Wang Y., Huang Y., Lam K.S.L., Li Y., Wong W.T., Ye H., Lau C.W., Vanhoutte P.M., Xu A. Berberine prevents hyperglycemia-induced endothelial injury and enhances vasodilatation via adenosine monophosphate-activated protein kinase and endothelial nitric oxide synthase. Cardiovasc. Res. 2009;82:484–492. doi: 10.1093/cvr/cvp078. [DOI] [PubMed] [Google Scholar]
- 8.Huang C.Y., Kuo W.W., Yeh Y.L., Ho T.J., Lin J.Y., Lin D.Y., Chu C.H., Tsai F.J., Tsai C.H., Huang C.Y. ANG II promotes IGF-IIR expression and cardiomyocyte apoptosis by inhibiting HSF1 via JNK activation and SIRT1 degradation. Cell Death Differ. 2014;21:1262–1274. doi: 10.1038/cdd.2014.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou N., Ye Y., Wang X., Ma B., Wu J., Li L., Wang L., Wang D.W., Zou Y. Heat shock transcription factor 1 protects against pressure overload-induced cardiac fibrosis via Smad3. J. Mol. Med. 2017;95:445–460. doi: 10.1007/s00109-016-1504-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Björk J.K., Åkerfelt M., Joutsen J., Puustinen M.C., Cheng F., Sistonen L., Nees M. Heat-shock factor 2 is a suppressor of prostate cancer invasion. Oncogene. 2016;35:1770–1784. doi: 10.1038/onc.2015.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pincus D. Regulation of Hsf1 and the heat shock response. Adv. Exp. Med. Biol. 2020;1243:41–50. doi: 10.1007/978-3-030-40204-4_3. [DOI] [PubMed] [Google Scholar]
- 12.The E., Du P., Chang Y., Dai F., Wei C., Li J. Role of HSF1-upregulated AC6 in ameliorating heart failure in mice. Environ. Toxicol. Pharmacol. 2016;47:79–85. doi: 10.1016/j.etap.2016.08.015. [DOI] [PubMed] [Google Scholar]
- 13.Wang S., Wu J., You J., Shi H., Xue X., Huang J., Xu L., Jiang G., Yuan L., Gong X., et al. HSF1 deficiency accelerates the transition from pressure overload-induced cardiac hypertrophy to heart failure through endothelial miR-195a-3p-mediated impairment of cardiac angiogenesis. J. Mol. Cell. Cardiol. 2018;118:193–207. doi: 10.1016/j.yjmcc.2018.03.017. [DOI] [PubMed] [Google Scholar]
- 14.Wang Y., Morgan W.D. Cooperative interaction of human HSF1 heat shock transcription factor with promoter DNA. Nucleic Acids Res. 1994;22:3113–3118. doi: 10.1093/nar/22.15.3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee A.K., Klein J., Fon Tacer K., Lord T., Oatley M.J., Oatley J.M., Porter S.N., Pruett-Miller S.M., Tikhonova E.B., Karamyshev A.L., et al. Translational repression of G3BP in cancer and germ cells suppresses stress granules and enhances stress tolerance. Mol. Cell. 2020;79:645–659.e9. doi: 10.1016/j.molcel.2020.06.037. [DOI] [PubMed] [Google Scholar]
- 16.Norhammar A., Lindbäck J., Rydén L., Wallentin L., Stenestrand U., Register of Information and Knowledge about Swedish Heart Intensive Care Admission RIKS-HIA Improved but still high short- and long-term mortality rates after myocardial infarction in patients with diabetes mellitus: a time-trend report from the Swedish Register of Information and Knowledge about Swedish Heart Intensive Care Admission. Heart. 2007;93:1577–1583. doi: 10.1136/hrt.2006.097956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roussel R., Travert F., Pasquet B., Wilson P.W.F., Smith S.C., Jr., Goto S., Ravaud P., Marre M., Porath A., Bhatt D.L., et al. Metformin use and mortality among patients with diabetes and atherothrombosis. Arch. Intern. Med. 2010;170:1892–1899. doi: 10.1001/archinternmed.2010.409. [DOI] [PubMed] [Google Scholar]
- 18.Ekström N., Schiöler L., Svensson A.M., Gudbjörnsdottir S., Eeg-Olofsson K., Eeg-Olofsson K., Miao Jonasson J., Zethelius B., Cederholm J., Eliasson B. Effectiveness and safety of metformin in 51 675 patients with type 2 diabetes and different levels of renal function: a cohort study from the Swedish National Diabetes Register. BMJ Open. 2012;2:e001076. doi: 10.1136/bmjopen-2012-001076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.UK Prospective Diabetes Study (UKPDS) Group Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34) Lancet. 1998;352:854–865. [PubMed] [Google Scholar]
- 20.Li S., Shi Y., Liu P., Song Y., Liu Y., Ying L., Quan K., Yu G., Fan Z., Zhu W. Metformin inhibits intracranial aneurysm formation and progression by regulating vascular smooth muscle cell phenotype switching via the AMPK/ACC pathway. J. Neuroinflammation. 2020;17:191. doi: 10.1186/s12974-020-01868-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xie Z., Dong Y., Scholz R., Neumann D., Zou M.H. Phosphorylation of LKB1 at serine 428 by protein kinase C-zeta is required for metformin-enhanced activation of the AMP-activated protein kinase in endothelial cells. Circulation. 2008;117:952–962. doi: 10.1161/CIRCULATIONAHA.107.744490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hardie D.G. Neither LKB1 nor AMPK are the direct targets of metformin. Gastroenterology. 2006;131:973. doi: 10.1053/j.gastro.2006.07.032. [DOI] [PubMed] [Google Scholar]
- 23.Rena G., Hardie D.G., Pearson E.R. The mechanisms of action of metformin. Diabetologia. 2017;60:1577–1585. doi: 10.1007/s00125-017-4342-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Parker C.S., Topol J. A Drosophila RNA polymerase II transcription factor binds to the regulatory site of an hsp 70 gene. Cell. 1984;37:273–283. doi: 10.1016/0092-8674(84)90323-4. [DOI] [PubMed] [Google Scholar]
- 25.Akerfelt M., Morimoto R.I., Sistonen L. Heat shock factors: integrators of cell stress, development and lifespan. Nat. Rev. Mol. Cell Biol. 2010;11:545–555. doi: 10.1038/nrm2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mahat D.B., Salamanca H.H., Duarte F.M., Danko C.G., Lis J.T. Mammalian heat shock response and mechanisms Underlying its genome-wide transcriptional regulation. Mol. Cell. 2016;62:63–78. doi: 10.1016/j.molcel.2016.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ali A., Biswas A., Pal M. HSF1 mediated TNF-alpha production during proteotoxic stress response pioneers proinflammatory signal in human cells. Faseb. J. 2019;33:2621–2635. doi: 10.1096/fj.201801482R. [DOI] [PubMed] [Google Scholar]
- 28.Li J., Chauve L., Phelps G., Brielmann R.M., Morimoto R.I. E2F coregulates an essential HSF developmental program that is distinct from the heat-shock response. Genes Dev. 2016;30:2062–2075. doi: 10.1101/gad.283317.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mendillo M.L., Santagata S., Koeva M., Bell G.W., Hu R., Tamimi R.M., Fraenkel E., Ince T.A., Whitesell L., Lindquist S. HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell. 2012;150:549–562. doi: 10.1016/j.cell.2012.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Locke M., Tanguay R.M. Diminished heat shock response in the aged myocardium. Cell Stress Chaperones. 1996;1:251–260. doi: 10.1379/1466-1268(1996)001<0251:dhsrit>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scherz-Shouval R., Santagata S., Mendillo M.L., Sholl L.M., Ben-Aharon I., Beck A.H., Dias-Santagata D., Koeva M., Stemmer S.M., Whitesell L., Lindquist S. The reprogramming of tumor stroma by HSF1 is a potent enabler of malignancy. Cell. 2014;158:564–578. doi: 10.1016/j.cell.2014.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anckar J., Sistonen L. Regulation of HSF1 function in the heat stress response: implications in aging and disease. Annu. Rev. Biochem. 2011;80:1089–1115. doi: 10.1146/annurev-biochem-060809-095203. [DOI] [PubMed] [Google Scholar]
- 33.Lindquist S. Protein folding sculpting evolutionary change. Cold Spring Harb. Symp. Quant. Biol. 2009;74:103–108. doi: 10.1101/sqb.2009.74.043. [DOI] [PubMed] [Google Scholar]
- 34.Lu G., Wu Z., Shang J., Xie Z., Chen C., Zhang C. The effects of metformin on autophagy. Biomed. Pharmacother. 2021;137:111286. doi: 10.1016/j.biopha.2021.111286. [DOI] [PubMed] [Google Scholar]
- 35.Bhansali S., Bhansali A., Dutta P., Walia R., Dhawan V. Metformin upregulates mitophagy in patients with T2DM: a randomized placebo-controlled study. J. Cell Mol. Med. 2020;24:2832–2846. doi: 10.1111/jcmm.14834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Diaz-Morales N., Iannantuoni F., Escribano-Lopez I., Bañuls C., Rovira-Llopis S., Sola E., Rocha M., Hernandez-Mijares A., Victor V.M. Does metformin modulate endoplasmic reticulum stress and autophagy in type 2 diabetic peripheral blood mononuclear cells? Antioxidants Redox Signal. 2018;28:1562–1569. doi: 10.1089/ars.2017.7409. [DOI] [PubMed] [Google Scholar]
- 37.Li Y., Chen Y. AMPK and autophagy. Adv. Exp. Med. Biol. 2019;1206:85–108. doi: 10.1007/978-981-15-0602-4_4. [DOI] [PubMed] [Google Scholar]
- 38.Foretz M., Guigas B., Bertrand L., Pollak M., Viollet B. Metformin: from mechanisms of action to therapies. Cell Metabol. 2014;20:953–966. doi: 10.1016/j.cmet.2014.09.018. [DOI] [PubMed] [Google Scholar]
- 39.Zhang C.S., Li M., Ma T., Zong Y., Cui J., Feng J.W., Wu Y.Q., Lin S.Y., Lin S.C. Metformin activates AMPK through the lysosomal pathway. Cell Metabol. 2016;24:521–522. doi: 10.1016/j.cmet.2016.09.003. [DOI] [PubMed] [Google Scholar]
- 40.Wang Y., An H., Liu T., Qin C., Sesaki H., Guo S., Radovick S., Hussain M., Maheshwari A., Wondisford F.E., et al. Metformin improves mitochondrial respiratory activity through activation of AMPK. Cell Rep. 2019;29:1511–1523.e5. doi: 10.1016/j.celrep.2019.09.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Verma A., Sumi S., Seervi M. Heat shock proteins-driven stress granule dynamics: yet another avenue for cell survival. Apoptosis. 2021;26:371–384. doi: 10.1007/s10495-021-01678-w. [DOI] [PubMed] [Google Scholar]
- 42.Wang J., Gan Y., Cao J., Dong X., Ouyang W. Pathophysiology of stress granules: an emerging link to diseases (Review) Int. J. Mol. Med. 2022;49:44. doi: 10.3892/ijmm.2022.5099. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available from the authors upon reasonable request.