

Abstract
Ferroptosis is defined as an iron-dependent, non-apoptotic cell death pathway, with specific morphological phenotypes and biochemical changes. There is a growing realization that ferroptosis has significant implications for several neurodegenerative diseases. Even though ferroptosis is different from other forms of programmed death such as apoptosis and autophagic death, they involve a number of common protein molecules. This review focuses on current research on ferroptosis and summarizes the cross-talk among ferroptosis, apoptosis, and autophagy that are implicated in neurodegenerative diseases. We hope that this information provides new ideas for understanding the mechanisms and searching for potential therapeutic approaches and prevention of neurodegenerative diseases.
Keywords: Ferroptosis, Apoptosis, Autophagic death, Neurodegenerative diseases
Introduction
A new form of programmed death named ferroptosis, which is different from two types of main programmed death known as apoptosis and autophagic death, was first described in 2012. Ferroptosis is caused by the small molecule erastin, which was used for cancer therapy several years before the concept of ferroptosis was proposed. Recent studies have indicated that an increase in iron levels in the brain can activate ferroptosis, which might accelerate the progression of neurodegenerative diseases [1–3]. Apoptosis and autophagic death are common forms of programmed death in the pathogenesis of neurodegenerative diseases. However, the interaction between ferroptosis and these other forms of programmed death is still unclear. What are the different roles of the various forms of programmed neuronal death in the onset and progression of neurodegenerative diseases? Do they work together or independently? Evidence has shown the presence of proteins of the cross-signaling pathways in the regulation of different forms of programmed cell death, such as p53, BRCA1-associated protein 1 (BAP1), Beclin-1, and voltage-dependent anion channels (VDACs). More importantly, these proteins are also involved in the regulation of neurodegenerative diseases [4, 5]. We hypothesize that neurodegenerative diseases are not regulated by a single cell death pathway, but by the joint regulation of multiple processes. Research clarifying the relationship among apoptosis, autophagic death, and ferroptosis might be important for understanding the mechanisms of neurodegenerative diseases.
Ferroptosis
The first description of ferroptosis was by Dixon et al. The preliminary characterization of this pathway demonstrated that erastin inhibits cysteine transport via the binding cystine/glutamate antiporter (system Xc−), which leads to glutathione (GSH) depletion and inactivation of the phospholipid peroxidase glutathione peroxidase 4 (GPX4) induces lipid peroxidation to trigger this specific form of cell death [2, 6]. Ferroptosis is activated by chemical compounds such as erastin and Ras-selective lethal 3 (RSL3), and the occurrence of ferroptosis can be reversed by the iron chelators deferoxamine (DFO) and ferrostatin-1 (Fer-1). Furthermore, ferroptosis is characterized by the depletion of plasma membrane unsaturated fatty acids (PUFAs) and the accumulation of iron-dependent lipid reactive oxygen species (ROS). The regulatory mechanism of ferroptosis is completely different from the other modes of cell death, and can be roughly divided into three parts: iron metabolism, amino-acid metabolism, and lipid metabolism (Fig. 1).
Fig. 1.

Overview of the ferroptosis pathways. Ferroptosis occurs as a result of the production of large amounts of lipid peroxides by the Fenton reaction. Elevated intracellular iron concentration or the disruption of cystine transport leads to a depletion of the antioxidant GSH; the lower level of GPX4 activity causes increased accumulation of ROS that consequently leads to lipid peroxidation and triggers ferroptosis. AA, arachidonic acid; Fe2+, ferrous iron; DMT1, divalent metal transporter 1; Fe3+, ferric iron; Fpn, ferroportin; GPX4, glutathione peroxidase 4; GSH, glutathione; GSSG, glutathione disulfide; NCOA4, nuclear receptor coactivator 4; PUFA, plasma membrane unsaturated fatty acid; ROS, reactive oxygen species; SLC3A2, solute carrier family 3 member 2; SLC7A11, solute carrier family 7 member 11; Tf, transferrin; TfR, transferrin receptor
Iron Metabolism in Ferroptosis
Iron metabolism is a complex process co-regulated by multiple related proteins [7]. These proteins include iron-responsive element-binding proteins (IRPs), transferrin (Tf), transferrin receptors (TfRs), nuclear receptor coactivator 4 (NCOA4), divalent metal transporter 1 (DMT1), ferritin, and ferroportin (Fpn). TfR1, DMT1, ferritin, and Fpn are regulated post-transcriptionally by IRPs/IREs (iron-responsive elements) [8]. Specifically, the IRP/IRE system promotes iron absorption by up-regulating the expression of TfR1 and DMT1 under iron-lacking conditions, while the up-regulation of ferritin and Fpn contributes to the storage and export of iron in cases of iron-overload. In the brain, ferric iron (Fe3+) is imported from extracellular to intracellular through TfR1. Subsequently, Fe3+ is reduced to ferrous iron (Fe2+) via ferric reductases, and delivered from the endosome into the cytoplasm by DMT1. Some of the Fe2+ is stored in the labile iron pool, the excess iron is detoxified to Fe3+ and stored in ferritin, and the remaining part is delivered to the extracellular space via Fpn accompanied by the oxidation of iron [8]. Excessive accumulation of intracellular Fe2+ leads to the Fenton reaction, overproduction of ROS, and the occurrence of lipid peroxidation [2, 7, 9, 10]. Fe2+ accumulation is one of the key factors for ferroptosis, thus an imbalance of iron metabolism may trigger ferroptosis.
In addition, abnormal expression or dysfunction of these proteins involved in iron regulation often results in abnormal iron metabolism in the brain, such as an increase or decrease in the intracellular free iron concentration [8, 9]. Mice carrying a mutation in the iron regulatory protein 2 (IRP2) gene (targeted disruption of the coding gene Ireb2) have misregulated brain iron metabolism, including abnormal accumulation of ferritin in neurons and Fe2+ in the cytosol, which leads to neurodegenerative disease [11]. Tf and TfRs are necessary for ferroptosis owing to their role in importing iron from the extracellular space [12]. It has been reported that TfR1 is overexpressed in brain cancer, promoting ROS formation and iron uptake in cancer cells, and participates in tumor progression [13]. Likewise, ferritinophagy, the degradation of ferritin by selective autophagy, enhances ferroptosis [14, 15]. This process is regulated by the specific cargo receptor NCOA4, which recruits ferritin to autophagosomes and releases intracellular free iron. Overexpression of NCOA4 increases ferritin degradation and promotes ferroptosis [14, 15]. Indeed, decreasing iron overload by iron chelators inhibits ferroptosis, whereas supplying exogenous sources of iron enhances ferroptosis [2, 16]. Thus, the cellular systems involved in the uptake, storage, utilization, and exportation of iron are required for the induction of ferroptosis and the regulation of iron metabolism is a potential point for controlling ferroptosis.
Amino-Acid Metabolism in Ferroptosis
System Xc−, a cystine/glutamate reverse transporter on the cell plasma membrane, is responsible for importing L-cystine into cells and transferring L-glutamate out of cells to maintain redox homeostasis [17]. Intracellularly, cystine is reduced to cysteine by thioredoxin reductase 1 or GSH, soon afterward GSH is synthesized under the action of γ-glutamylcysteine synthase and GSH synthase [18]. As a cofactor of GPX4, GSH has been used to neutralize lipid peroxidase to protect membranes and cells against peroxidation [19]. Due to the strong lipid peroxidation in ferroptosis, system Xc−, GSH, and GPX4 are required for ferroptosis regulation.
The small molecule compounds erastin or sorafenib trigger ferroptosis by blocking the system Xc− and reducing cysteine transport, which results in depletion of GSH and loss of GPX4 activity by reducing intracellular glutamate accumulation and cysteine content [20]. Similarly, deprivation of cysteine or cystine also induces ferroptosis in a serum-dependent manner; this is regulated by transferrin and the glutamine metabolic pathway [12]. In addition, cystine starvation induces non-canonical activity of the glutamate-cysteine ligase catalytic subunit, which causes an accumulation of γ-glutamyl-peptides and maintains glutamate homeostasis, thereby protecting against ferroptosis [21]. Glutamate is an excitatory neurotransmitter, and under normal physiological conditions, low levels of glutamate play a role in synapses. While excessive glutamate bind to specific glutamate receptors present in neuron membranes which could induce oxidative toxicity and trigger ferroptosis [22]. Conversely, in the absence of glutamine or glutaminolysis disorder, the accumulation of ROS, lipid peroxidation, and ferroptosis are inhibited [12].
GPX4 is an intracellular antioxidant selenium enzyme that can reduce potentially toxic lipid peroxides to non-toxic lipid peroxides under the catalysis of GSH. With conditional deletion of GPX4 in forebrain neurons, mice exhibit hippocampal neurodegeneration and significant deficits in spatial learning and memory [23]. Moreover, markers associated with ferroptosis such as elevated lipid peroxidation, extracellular signal-regulated kinases activation, and overt neuroinflammation, have been reported in these mice with conditional gene deletion[23]. The ferroptosis inhibitor liproxstatin-1 (Lip-1) ameliorates these neurodegenerative symptoms [23]. Chemical reagents that deplete GSH or inactivate GPX4 promote ferroptosis; in contrast, up-regulation of GSH or GPX4 expression diminishes ROS-induced ferroptosis [6, 24].
Lipid Metabolism in Ferroptosis
PUFA, a component of the membrane bilayer, is the molecular basis of fluidity and deformation of the cell membrane [25]. However, the carbon-carbon double bonds in PUFAs are unstable and can easily become targets of lipid peroxidation, leading to the cleavage of PUFAs into many products, such as eicosapentaenoic acid and arachidonic acid (AA) [26, 27]. Lipid peroxidation is a process in which oxidants extract an unstable hydrogen atom from the diallyl methylene of PUFAs. In this process, oxidation results in the accumulation of lipid peroxy radicals and hydrogen peroxide [24]. Increasingly, studies have implicated lipid peroxides as a key feature of pathological conditions, including cancer, inflammation, and neurodegenerative diseases [28].
Lipid peroxidation is one of the crucial downstream features of ferroptosis. The results of lipomics have shown that PUFAs are the lipids most vulnerable to peroxidation in the course of ferroptosis [29]. One study has also shown that AA is the most frequently depleted PUFA in cells undergoing ferroptosis [30]. The mechanism of the lipid metabolism pathway involved in ferroptosis induction is as follows: acyl-CoA synthetase, which prefers AA as its main substrate, promotes ferroptosis by producing oxidized phosphatidylethanolamine, controlling membrane phospholipid fatty acid composition [30, 31]. Ferroptosis inducers such as RSL3 inhibit GPX4 by covalently targeting activated selenocysteine, causing the accumulation of PUFAs [29]. Inhibitors of lipid peroxidation such as vitamin E, coenzyme Q10, and DFO, modulate ferroptosis by suppressing lipid peroxidation [2, 5, 32].
The Role of Ferroptosis in Neurodegenerative Diseases
Neurodegenerative disorders are nervous system diseases closely associated with age [33]. The most common neurodegenerative disorders include Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), and stroke. With aging processes, brain iron deposition is more serious in brain areas associated with neurodegenerative disorders than in others [34]. Proteins associated with neurodegeneration, such as α-synuclein (α-syn), tau, and amyloid-β (Aβ), are also involved in molecular crosstalk with iron metabolism proteins [35]. Moreover, new evidence has shown that iron metabolism imbalance, oxidative stress, and glutamate metabolic abnormalities involved in ferroptosis are also linked to neurodegenerative disorders [36–42] (Fig. 2).
Fig. 2.
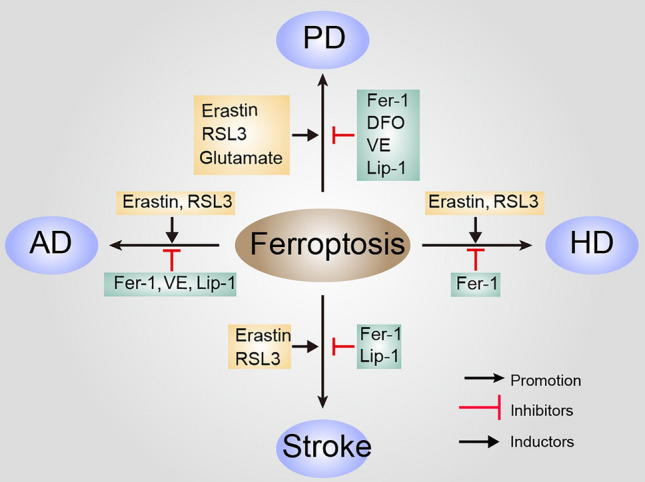
The role of ferroptosis in neurodegenerative diseases. Various inducers and inhibitors of ferroptosis in different neurodegenerative diseases are shown. Inducers of ferroptosis in AD [23, 41], inhibitors of ferroptosis in AD [23]; inducers of ferroptosis in PD [41], inhibitors of ferroptosis in PD [40, 41, 62]; inducers of ferroptosis in HD [42, 69, 70], inhibitors of ferroptosis in HD [69, 70]; inducers of ferroptosis in stroke [22], inhibitors of ferroptosis in stroke [40, 83]. DFO, deferoxamine; Fer-1, ferrostatin-1; RSL3, Ras-selective lethal 3; Lip-1, liproxstatin-1; VE, vitamin E
Ferroptosis in AD
AD is one of the most common and serious neurodegenerative disorders, and its socioeconomic burden is staggering and increasing. The quintessential histopathological characteristics of AD are the deposition of extracellular Aβ in senile plaques and intracellular neurofibrillary tangles formed by the abnormal phosphorylation of tau protein [43–45].
Features related to ferroptosis, such as iron metabolism disorders [46], glutamate excitotoxicity [47], and lipid ROS accumulation, have been detected in the brains of AD patients or mouse models [48]. Brain iron levels are positively correlated with AD progression and cognitive decline [49, 50]. Ayton’s results have revealed that ferritin is closely associated with the level of apolipoprotein E, and the allele ApoE-ɛ4 elevates the risk of AD. Glutamate excitotoxicity is one of the pathogenetic processes of AD. Increased glutamic acid levels in AD patients are associated with abnormal system Xc− function, because system Xc− dysfunction increase the extracellular glutamate concentration and excitotoxicity [51]. Moreover, compared with elderly controls, AD patients demonstrate higher levels of oxidative damage [52]. Iron interacts with Aβ and tau through deposition and takes shape in a peptide hemin complex, participating in producing ROS that may be involved in the ferroptotic death pathway [46]. Recent research has shown that mice with forebrain neuronal conditional GPX4 knockout (GPX4BIKO) have hippocampal neuronal degeneration [23]. Feeding with a vitamin E deficient diet accelerates the neurodegeneration in GPX4BIKO mice, while treatment with Lip-1 or vitamin E mitigates hippocampal neurodegeneration and inflammation [23]. In conclusion, in an AD model, protein misfolding or abnormal amino-acid metabolism aggravates the accumulation of iron in the brain, and then proceeds to the next step to initiate ferroptosis. Although its particular role has not been confirmed, that ferroptosis may be involved in the regulation of AD has attracted increasing attention.
Ferroptosis in PD
Alpha-synuclein (SNCA) gene overexpression causes abnormal aggregation of α-syn protein and leads to neurotoxicity [53]. Excessive accumulation of unfolded or misfolded α-syn perhaps leads cells to autophagic death. In addition, the abnormal accumulation of α-syn also leads to decreased B cell lymphoma-2 (Bcl-2) and increases the Bcl-2-associated X protein (Bax) expression levels, followed by cytochrome C (Cyt C) release and caspase activation [54, 55]. Maintaining a balance between autophagy, apoptosis, or other modes of cell death may be a promising therapeutic strategy.
In the substantia nigra (SN) of PD patients, the iron level is increased [56, 57], GSH levels are decreased [58], and lipid peroxides are increased significantly compared with age-matched controls [59]. And these characteristics coincide with the biochemical characteristics of ferroptosis, suggesting that ferroptosis is involved in the degeneration of dopaminergic neurons. For example, human dopaminergic neuronal precursors the Lund human mesencephalic cells are more sensitive to ferroptosis [60, 61].
The abnormal iron accumulation in PD may well be the result of an imbalance in the iron homeostatic pathway caused by the alterations of iron regulatory proteins [62]. Radioimmunoassays have shown that the level of ferritin in the substantia nigra pars compacta (SNpc) of PD patients declines significantly, indicating that the iron storage capacity decreases [63]. As well, elevated levels of DMT1 in the SNpc of PD patients and several PD mouse models are likely to contribute to increased importation of cellular iron [63]. Ayton et al. found that ceruloplasmin knock-out mice develop parkinsonism, and this is rescued by iron chelation with DFO [64]. The decreased expression of the iron export protein ferroportin1 (FPN1) leads to a decrease in iron translocation, which is also thought to be related to iron deposition in the SN of PD [65]. A new report has shown that ferroptosis is a vital cell death pathway for dopaminergic neurons [27] (Fig. 3). Research workers studied organotypic slice cultures and found that ferroptosis regulates dopaminergic neurons by the protein kinase C (PKC) signaling pathway. Moreover, the ferroptosis inhibitor Fer-1 significantly inhibits the toxicity of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) to dopaminergic neurons [61]. When comparing the expression of ferroptosis-related genes in the brain tissue of normal people and PD patients, it has been found that only the SN region GPX4 and solute carrier family 7 member 11 (SLC7A11) showed significant differences [61]. Exploring the role of ferroptosis in the occurrence of PD would provide a new target for treating the degeneration mechanism of dopaminergic neurons.
Fig. 3.

Relationships among ferroptosis, apoptosis, and autophagic death in PD. Accumulation of iron and misfolded α-syn in SNpc dopaminergic neurons are the hallmark of PD. Although unfolded or misfolded α-syn is partially degraded by the autophagy pathway, its overexpression can result in autophagic death. At the mitochondrial level, α-syn and ROS accumulation induce mitochondrial damage and fragmentation. The increase in the number of damaged mitochondria can trigger apoptosis through the release of Cyt C, degradation of the anti-apoptotic Bcl-2 family, and activation of Bax. In the presence of Fe2+, the most harmful highly-reactive hydroxyl radical is produced by the Fenton reaction, causes oxidative stress, and then triggers ferroptosis. α-syn, α-synuclein; Apaf-1, apoptotic protease activating factor 1; Bak, Bcl-2 homologs antagonist/killer; Bax, Bcl-2-associated X protein; Bcl-2, B cell lymphoma-2; Cyt C, cytochrome C; ROS, reactive oxygen species
Ferroptosis in HD
In the process of HD, many cellular pathways including activation mitochondrial dysfunction and oxidative stress are involved. Mitochondrial DNA (mtDNA) is the main site of oxidative stress lesion and cell models of HD provide convincing evidence that there is significant mtDNA damage in striatum cells expressing mutant huntingtin protein (mHTT), which may contribute to subsequent mitochondrial dysfunction. Previous studies have also shown that mitochondrial dysfunction and oxidative stress are both major events in the pathogenesis of HD [66, 67]. MHTT leads to increased oxidative stress, which increases intracellular ROS levels [68]. The level of lipid peroxidation in the cerebrospinal fluid of HD patients is significantly increased [27]. Treatment with the antioxidant nordihydroguaiaretic acid can restore the mitochondrial membrane potential, improve ATP production in the striatum of the R6/2 mouse model, and prolong the life span of mice by inhibiting lipid peroxidation [69]. Excessive iron accumulation is also a factor in neuronal oxidative stress. In HD patients and mouse models, iron can promote the neurodegenerative process by boosting the accumulation of subcellular oxidative damage, while DFO plays a protective role by inhibiting the redox property of iron [70]. The iron content of the striatum and cortex is increased in the HD transgenic mouse model as well as the elevated expression of iron regulatory protein 1 (IRP1), Tf, ferritin, and TfR; similar results have been reported in mHTT-expressing cells [71]. Magnetic resonance imaging results from HD patients have shown excessive iron deposition in the occipital cortex, globus pallidus, and putamen [72]. Fer-1 improves neuropathy in R6/2 mice as well as significantly inhibiting lipid peroxidation [69]. Compared with healthy participants, HD patients show markedly lower levels of GSH [73]. Intraventricular administration of DFO improves the pathological features and motor phenotype in R6/2 mice [69].
Ferroptosis in Stroke
It is well documented that acutely elevated glutamate levels occur after stroke in animal models and stroke patients [74, 75]. It is worth mentioning that high levels of extracellular iron can activate glutamate receptors, which results in ferroptotic damage by promoting neuronal iron uptake and peroxide overproduction in neurons [76]. Before ferroptosis was defined, clinical studies in mice models had shown that disorder of brain iron homeostasis is associated with acute neural injury after cerebral ischemia, manifested as the accumulation of iron aggregates and neuronal damage during reperfusion. Under such pathological conditions as stroke, the blood-brain barrier (BBB) is destroyed, allowing free iron and ferritin entry into the brain parenchyma, along with a great quantity of oxygen free radicals (oxidants) [77, 78]. Iron overload could aggravate brain damage after stroke by inducing reactive oxygen species, while apotransferrin reduces ischemic neuronal death by preventing the production of ROS and NMDA-induced holotransferrin uptake [79]. On the other hand, in the case of ischemic stroke, the accumulated glutamate inhibits cystine uptake by inhibiting the system Xc− and reducing the generation of the antioxidant GSH [80]. In a hemorrhagic stroke model, the expression of GPX4 is significantly reduced after cerebral hemorrhage, while increasing the level of GPX4 in the brain significantly reduces neuronal dysfunction, BBB damage, and oxidative stress after the hemorrhage [81]. Supplementation with a single dose of selenium drives the expression of GPX4, effectively inhibits GPX4-dependent ferroptosis, and protects neurons [82]. The research results of Liu et al. showed that the ferroptotic death inhibitor Fer-1 and the 5-lipoxygenase (5-LOX) inhibitor zileuton act synergistically to inhibit ferroptosis, protecting neurons against glutamate-induced oxidative stress [83]. Experimental studies in recent years have reported that autophagy may be involved in the regulation of iron accumulation and lipid peroxidation after ischemia, and then promotes ferroptosis. NCOA4 is a selective cargo receptor for ferritinophagy to maintain iron homeostasis by delivering ferritin to lysosomes [84], and a deficiency of NCOA4 affects the degradation of ferritin, causing intracellular iron overload and harmful oxidative stress [85].
The Interaction Between Ferroptosis and Other Classic Forms of Cell Death in Neurodegenerative Diseases
Common Regulatory Factors
Emerging evidence suggests that ferroptosis usually shares a common regulatory factor or pathway with other forms of cell death (Table 1), in spite of being supposed to be genetically and biochemically different from these other forms (Fig. 4).
Table 1.
The expression and function of related proteins in ferroptosis and apoptosis
| Ferroptosis | Apoptosis | References | |||
|---|---|---|---|---|---|
| Expression | Function | Expression | Function | ||
| p53 | p53↑, ferroptosis↑ | p53 enhances ferroptosis through targeting SLC7A11, spermidine/spermine N1-acetyltransferase 1 (SAT1) or glutaminase 2 (GLS2) | p53↑, apoptosis↑ | Cytosolic p53 directly binds to BAX to increase mitochondrial membrane permeabilization and engage the apoptotic program | [102, 103, 155, 156] |
| p53↑, ferroptosis↓ | p53 suppresses ferroptosis by targeting dipeptidyl peptidase 4 (DPP4) or cyclin-dependent kinase inhibitor 1 A (CDKN1A) | Combines with anti-apoptotic mitochondrial proteins (such as Bcl-2 and Bcl-XL) to directly induce apoptosis | |||
| BAP1 | BAP1↑, ferroptosis↑ | BAP1 promotes ferroptosis by repressing the expression of SLC7A11 | BAP1↓, apoptosis↑ | Modulating Ca2+ release from the endoplasmic reticulum into the cytosol and mitochondria, promoting apoptosis | [111–113] |
| Beclin-1 | Beclin-1↑, ferroptosis; Beclin-1↓ ferroptosis↓ | Inhibits system Xc− activity by directly binding to SLC7A11 | Beclin-1-C↑, apoptosis↑ | Caspase-mediated cleavage of Beclin-1 enhances apoptosis by promoting the release of pro-apoptotic factors from mitochondria | [123, 125, 126] |
| VDAC | VDAC2↓, ferroptosis↓ | Erastin induces ferroptosis by binding with VDAC2 | VDAC↑, apoptosis↑ | Dynamic VDAC oligomerization is involved in the release of Cyt C from mitochondria | [131, 132, 134, 157] |
| VDAC1↓, ferroptosis↓ | Lip-1 reduces VDAC1 levels, rescues GPX4 levels, and reduces mitochondrial ROS production | ||||
Fig. 4.

The role of proteins associated with cell death in neurodegenerative diseases. Numerous proteins are multi-functional, regulating ferroptosis, apoptosis, and autophagy. BAP1, BRCA1-associated protein 1; ROS, reactive oxygen species; VDAC, voltage-dependent anion channel
p53
The p53 protein is currently recognized as the most classic and widely-studied tumor suppressor that regulates signaling pathways associated with DNA damage, and is involved in controlling cell survival and division under various stresses [86–88]. P53 has been a focus in genetics and biology for nearly two decades, and previous research mainly focused on its role in programmed cell death and the occurrence of tumors [89]. The expression of p53 is relatively low in normal cells and it is activated when external and internal stress signals are enhanced to induce cell cycle arrest, DNA repair, and survival. On one hand, the activated p53 can directly activate pro-apoptotic members of the Bcl-2 family (such as Bax, Bad, and Bak) to increase mitochondrial membrane permeabilization, and the release of pro-apoptotic factors from the mitochondria can trigger apoptosis. On the other hand, it also combines with anti-apoptotic mitochondrial proteins (such as Bcl-2 and Bcl-XL) to form p53-Bcl-2 complexes to directly induce apoptosis via the mitochondrial p53 pathway [90]. As a commonly used drug to mediate the PD model, rotenone targets the protein deacetylase sirtuin1 (SIRT1) to promote p53 transcription and apoptosis [91]. P53-induced autophagy occurs when cells undergo DNA damage or p53 is reactivated in p53-negative tumor cells [92, 93]. Evidence has shown that p53 can both promote and inhibit autophagy in a different subcellular location: nuclear p53 promotes autophagy, while cytoplasmic p53 inhibits it [94]. Two main pathways are involved in the induction of autophagy: the first pathway is mainly related to the DNA damage-regulated autophagy modulator (DRAM) [95]; the second pathway is implemented primarily by activating the adenosine monophosphate-activated protein kinase (AMPK) signal pathway. DRAM is a lysosomal protein that is not only important for the action of p53 to induce autophagy, but also for its action to induce programmed cell death. DRAM1 was the first protein reported to be directly related to p53 and autophagy [95, 96]. Studies have shown that p53 can directly activate DRAM transcription and expression [96], and overexpression of DRAM also induces apoptosis [95, 97]. Nuclear p53 activates tuberous sclerosis 2 and AMPK, both of which down-regulate mechanistic target of rapamycin (mTOR) activity and indirectly promote autophagy [98]. The activators of AMPK, Sestrins 1 and 2, also up-regulate p53 [98, 99]. Therefore, nuclear p53 induces and regulates the autophagy process by activating its downstream target genes. Besides cytoplasmic p53 binds to Beclin-1 to inhibit autophagy by promoting its ubiquitination and degradation [96].
Recent studies have shown that p53 not only affects apoptosis and autophagy, but also participates in the regulation of ferroptosis. It has been found that p53 plays a specific role in ferroptosis by using an acetylation-deficient p53 mutant in which three lysine residues were replaced by arginine residues [100, 101]. In addition, as a transcription inhibitor of SLC7A11, p53 promotes the occurrence of ferroptosis by inhibiting the intake of cysteine [102]. The mechanism is that ferroptosis is triggered by weakness in cellular antioxidant capacity and an increase in lipid ROS levels, which is led by inhibiting the activity of cystine-dependent GPX4 through down-regulating SLC7A11 expression which suppresses the uptake of cystine by the system Xc− [103].
Not only can p53 eliminate tumor cells through cell-cycle arrest and apoptosis, but also induces ferroptosis in tumor cells under certain conditions [104, 105]. Based on the ability of p53 to regulate oxidative stress and the ability of the metal-organic network (MON) to induce the Fenton reaction, researchers have designed MON encapsulated with p53 plasmid (MON-p53) [106]. Once MON-p53 is internalized, ferric ions induce the Fenton reaction to produce ROS, and excessive intracellular ROS levels result in the lipid peroxidation of biofilms. Studies in vivo and in vitro have shown that MON-p53 kills cancer cells through the ferroptosis/apoptosis hybrid pathway, in which ferroptosis plays a dominant role in regulating cell death [106]. Our previous results indicated that when dopaminergic cells are treated with different concentration gradients of ferric ammonium citrate (FAC), ferroptosis occurs first in a group treated with a relatively low concentration of FAC accompanied by an increased level of lipid peroxidation, while apoptosis occurs with increased doses [16]. Fer-1 eliminates the ferroptosis and apoptosis caused by iron overload. Nevertheless, apoptosis inhibitors do not alleviate ferroptosis. It is worth mentioning that p53 may be involved in the regulation of this link, but the concrete mechanism is unclear [16]. Thus, apoptosis might be transformed into ferroptosis under certain conditions, and ferroptosis would advance the sensitivity of apoptosis. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) initiates an exogenous pathway by binding to its corresponding death receptors (DR4 and DR5) to induce apoptosis [107]. The data show that the ER (endoplasmic reticulum) is stressed by the ferroptosis-inducer erastin, which mediates the expression of DR5 but not DR4 [108]. Some experimental data have suggested that the combination of erastin and TRAIL significantly promotes caspase, especially the activation of caspase-3, to enhance TRAIL-induced apoptosis [109].
BAP1
BRCA1-associated protein1 (BAP1) is a ubiquitin hydrolase that modulates key cellular pathways, inhibiting tumorigenesis by influencing the cell cycle, cellular responses to DNA damage, and cellular proliferation [110]. Similar to p53, BAP1 is also a tumor suppressor; both BAP1 and p53 regulate transcription and DNA damage/repair responses, cell death processes, and cell metabolism. BAP1 is localized in the cytoplasm, binds and deubiquitylates the type-3 inositol-1,4,5-trisphosphate-receptor, and then regulates Ca2+ release from the ER into the cytosol and mitochondria, modulating Ca2+ signaling-mediated cell death [111]. BAP1 encodes the widely-expressed histone H2A deubiquitinase, and the ubiquitin ligase ring finger protein 2 silences genes by mono-nucleating H2A and promotes the apoptosis of BAP1-deficient cells by inhibiting the expression of the pro-survival genes Bcl-2 and Mcl-1 [112]. In general, a decrease in BAP1 causes the accumulation of DNA damage in normal cells and this triggers apoptosis. In addition, BAP1-expressing heterogeneous tumors exhibit the morphological characteristics of ferroptosis by electron microscopic analysis, and BAP1 at least partly promotes lipid peroxidation through repressing SLC7A11 and then triggers ferroptosis in vivo [113]. Cancer-related mutations within the BAP1 gene have been found to disrupt protein structure and induce amyloidogenic aggregation in vitro [114]. Although current research on BAP1 is focused on various cancers, emerging evidence suggests that neurodegenerative diseases show a significant imbalance in the ubiquitination/ deubiquitination process [115], and the role of BAP1 remains to be further revealed.
Beclin-1
Beclin-1, is the mammalian autophagy of yeast autophagy-related 6 (ATG6), which is found in the brains of mice with fatal Sindbis virus encephalitis [116]. It is highly homologous among humans, rats, and mice, and participates in various biological functions [117]. Beclin-1 is a key regulator of autophagy, an indispensable condition for autophagosome formation. A study has shown that Beclin-1 promotes autophagy in human breast carcinoma cells Michigan Cancer Foundation-7 and autophagy-deficient yeast with targeted destruction of APG6/VPS30. The phosphoinositide 3 (PI3) kinase complex is the signaling molecule for autophagosome synthesis and the autophagosome maturation bond lipid, consisting of the core lipid kinase VPS34, the regulatory component VPS15, and Beclin-1 [118]. Active phosphorylated epidermal growth factor receptor (EGFR) interacts with the key autophagy protein Beclin-1 (via activating mutations in EGFR or via ligand-dependent EGFR activation) in human non-small-cell lung carcinoma cells, resulting in the phosphorylation of multiple tyrosine sites, and reduced Beclin-1-associated VPS34 kinase activity suppresses autophagy [119]. BECN1 is significantly lower in AD patients’ brains than in age-matched controls [120, 121]. In the genetically modified AD mouse model that expresses human amyloid precursor protein, the reduction of BECN1 expression increases the accumulation of intraneuronal Aβ and neurodegeneration [122].
Recent research has provided evidence of the interplay between autophagy and apoptosis. Generally speaking, the “BCL-2 homology 3 (BH3)-only members” of the Bcl-2 family can bind and antagonize pro-survival proteins to promote apoptosis [123]. Yet Beclin-1 is unlike other known BH3-only proteins, and does not function as a pro-apoptotic molecule even if it is overexpressed [124]. The overexpression of Beclin-1 does not interfere with the protective effect of Bcl-2 on apoptosis induced by a variety of inducers regardless of the location of Bcl-2 in cells [124]. Caspase-mediated cleavage of Beclin-1 generates the C-terminal fragment Beclin-1-C; this promotes the release of mitochondrial pro-apoptotic factors from mitochondria, increasing Beclin-1 sensitivity to apoptosis [125].
In addition, Beclin-1 plays a non-autophagic role in cells. AMPK-mediated phosphorylation of Beclin-1 promotes ferroptosis by binding to produce the BECN1-SLC7A11 complex, further leading to promote GSH-depletion lipid peroxidation in tumor suppression [126]. Among them, the system Xc− inhibitor erastin or sulfasalazine is able to promote BECN1 binding to SLC7A11, and the expression of BECN1 only affects system Xc− activity, but not SLC7A11 expression [126].
VDAC
The VDACs are also known as mitochondrial proteins, which are located at the mitochondrial outer membrane, forming the interface between the cytosol and mitochondria. The VDAC protein family consists of three isomers (VDAC1, VDAC2, and VDAC3), which participate in regulating the ion and metabolite flux between mitochondria and cytoplasm, and play an intermediate role in metabolism [127–129]. VDACs are considered to be key in mitochondrion-mediated apoptosis since they are recognized to be targets of pro-apoptotic and anti-apoptotic Bcl-2 family proteins.
Moreover, the closure of VDACs can indirectly induce ROS production. The 18-kDa protein TSPO (translocator protein) is located on the outer membrane of mitochondria, and interacts with VDAC1 to regulate both mitochondrial structure and function. The experiment results indicate that when the ratio of TSPO to VDAC1 increases, the production of mitochondrial ATP is limited and the level of ROS is increased [130, 131]. The interaction between VDAC and TSPO is also thought to play a role in apoptosis. ROS production may provide a link between the activation of TSPO and VDAC, and it may be able to release Cyt C from cardiolipins located at the inner mitochondrial membrane, then activates VDAC and allows VDAC-mediated Cyt C release into the cytoplasm. Finally, the apoptotic signaling pathway is activated [131, 132]. In neurodegenerative diseases, the nitration and carbonylation of VDAC1 have been detected in the brain of AD patients, indicating that VDAC1 channel activity contributes to the occurrence and development of AD [133].
Erastin can directly bind to VDAC2/3 to alter the permeability of the mitochondrial outer membrane, which can induce ferroptosis by reducing the oxidation rate of the reduced form of nicotinamide adenine dinucleotide (NADH) [134]. Depletion of VDAC2 or VDAC3 by RNA interference results in significantly increased resistance to erastin compared to a control group. Indeed, suppression of VDAC2 or VDAC3 significantly inhibits erastin-induced ferroptotic events, including lipid ROS production, iron accumulation, GSH depletion, and GSH disulfide generation. Moreover, combined interference with VDAC2 and VDAC3 has a stronger effect on the production of lipid ROS and the accumulation of iron [135]. Down-regulation of neuronal precursor cell-expressed developmentally down-regulated 4 (NEDD4) by short hairpin RNA rescues the VDAC2/3 protein elimination induced by erastin and increases the sensitivity of melanoma cells to erastin but not RSL3 [135]. It has been reported that Lip-1 protects the mouse myocardium against ischemia/reperfusion by reducing the levels of VDAC1 and VDAC1 oligomers, restoring GPX4 protein levels, and decreasing ROS production [136]. VDACs are not only multi-functional channels that allow small molecules to passively cross the outer membrane of mitochondria, but are also active agents that regulate the metabolism of mitochondria and their surrounding environment, as well as controlling cell survival and death.
Protein Degradation
The intracellular protein degradation system is mainly mediated by the ubiquitin-proteasome system (UPS) and the autophagy-lysosome system (referred to here as autophagy) [137]. The UPS regulates ferroptosis by targeting different lipid peroxide-related regulatory factors. For instance, the E3 ligase NEDD4 inhibits ferroptotic cell death by degrading VDACs in mitochondria. NEDD4-like E3 ubiquitin protein ligase binds to the transporter lactotransferrin to inhibit the accumulation of iron and subsequent oxidative damage-mediated intracellular ferroptosis [138, 139]. In addition, UPS dysfunction is associated with neurological diseases [140, 141] characterized by misfolded protein accumulation and mitochondrial dysfunction [142]. Autophagy plays a dual role in cell death [143]. Autophagy usually represents a mechanism that promotes cell survival through catabolism, whereas autophagic cell death may be defined as a process that promotes cell death [144, 145]. Evidence has revealed an interaction between ferroptosis and autophagy, and ferroptosis is a form of autophagy-dependent cell death [146]. Excessive autophagy and lysosome activity can promote ferroptosis through iron accumulation or lipid peroxidation. Mancias’ results have shown that autophagy regulates iron balance through the degradation of ferritin in fibroblasts and cancer cells, and involves the regulation of ROS and ferroptosis [147]. Autophagy-related genes such as ATG5 or ATG7 control the formation and maturation of autophagy-related membrane structures [15]. Moreover, knockdown of ATG5 and ATG7 suppress ferroptosis by preventing iron accumulation and lipid peroxidation after erastin administration [15]. NCOA4 is a selective cargo receptor that plays a critical role by targeting ferritin to autophagosomes [84]. When the intracellular iron is at low levels, NCOA4 bonds to ferritin heavy chain 1 (FTH1) and promotes ferritin degradation to release free iron by transferring autophagosome to lysosomes, which is essential for inducing ferroptosis [147]. Knockdown of NCOA4 inhibits the degradation of ferritin and prevents ferroptosis; on the contrary, NCOA4 over-expression enhances ferritin degradation and promotes ferroptosis [148]. Further, NCOA4 selectively identifies ferritin and guides it to autophagy microbodies, after which free iron is released from ferritin for cellular physiological activities; this process is known as ferritinophagy [14, 84, 149]. This shows that the degradation pathway plays a significant role in the regulation of ferroptosis.
Metabolite Messengers
Previous studies have suggested that apoptosis, as an inert non-cellular process, releases “find-me” signals to recruit phagocytes at the early stage of death [150]. Recently, evidence has demonstrated that apoptotic cells are not “inert corpses” waiting to be swept away [151]. On the contrary, they affect adjacent cells via the release of specific soluble metabolites to inhibit inflammation and cell proliferation [151]. In this process, the caspase-mediated opening of the plasma membrane Pannexin 1 channels facilitates the release of a select subset of the metabolite secretome. Then, certain metabolic pathways continue to remain active during apoptosis with the release of select metabolites [151]. Our results show that iron overload activates ferroptotic death and promotes apoptosis in a PD model, as well as indicating that ferroptosis occurs before apoptosis [16]. The metabolites of AA, a PUFA closely associated with ferroptosis, can act as peroxisome proliferator-activated receptor (PPAR) agonists and decrease the apoptosis of PPAR genes silenced cells [152]. Therefore, we hypothesized that during the ferroptosis process in specific cells, some metabolites may also be produced to affect the physiological state of adjacent cells and accelerate the occurrence of apoptosis. These results all support the close interaction between ferroptosis and other cell death pathways, but their crosstalk mechanisms at the molecular level need further exploration.
Conclusions and Perspectives
With the in-depth study of ferroptosis, its important role in the regulation of neuronal death has been taken seriously. Multiple means of cell death have been discovered, and they may cooperate with ferroptosis to maintain organismal or brain homeostasis. Emerging studies have described the crosstalk between ferroptosis and autophagy, ferroptosis and apoptosis, and autophagy and apoptosis. In this review, we briefly summarized current knowledge about the mechanisms of ferroptosis, and the currently recognized cross-talk of cell death involved in regulating the occurrence and progress of multiple neurodegenerative diseases including AD, PD, HD, and stroke. For example, our previous research indicated that FAC-induced ferroptosis precedes apoptosis [16]. In addition, the ferroptosis inhibitor erastin and TRAIL synergistically induce expression of the pro-apoptotic protein p53 up-regulated modulator of apoptosis without inducing apoptosis [153]. It is worth noting that a study has shown that converting apoptosis into iron death may be a new strategy for the treatment of diseases [106]. A recent study has indicated that simultaneous inhibition of two or more cell death pathways reduces ischemic stroke damage more efficiently than inhibition of a single one [154]. Indeed, other multifarious processes, such as lipid peroxidation, iron metabolism, fatty acid metabolism, and mitochondrial membrane formation, require the co-regulation of mixed types of cell death. Summarizing the results of previous experiments, we suspect that different conditions or external stimuli, such as death inducers, erroneous accumulation of proteins, neuroinflammation, and neuronal damage cause cells to gravitate toward different death pathways. Cell death may be dominated by a single type at a particular stage, while sometimes mixed types of death pathways appear to occur sequentially or simultaneously in neurodegenerative disease. And the mixed types of cell death seem to be more widespread than the “pure” type, because ferroptosis, apoptosis, and autophagic death share common regulators and signaling molecules (as reviewed above). Additional studies are needed to elucidate how “ferroptotic” and other cell death processes work individually or synergistically through multiple molecular mechanisms to improve the prognosis of patients with neurodegenerative diseases. Unanswered questions about the interplay between different cell death pathways remain: (1) are the mixed types of cell death protective or destructive—either pro-survival or pro-death, and (2) how do the mechanisms of multiple signaling molecules proteins work together in the onset and progression of neurodegenerative diseases. Therefore, understanding the mechanisms by which they work together in the onset and progression of neurodegenerative diseases is of great importance for the development of potential therapeutic strategies for various brain disorders.
Acknowledgments
We gratefully acknowledge financial support from the National Natural Science Foundation of China (82071429 and 32171131), Shandong Province Natural Science Foundation (ZR2019ZD31, ZR2020MC072, and ZR2020QH125), and Innovative Research Team of High-level Local Universities in Shanghai, China.
Conflicts of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported. The authors declare that they have no competing interests.
Contributor Information
Qian Jiao, Email: jiaoqian@qdu.edu.cn.
Hong Jiang, Email: hongjiang@qdu.edu.cn.
References
- 1.Yan N, Zhang JJ. Iron metabolism, ferroptosis, and the links with Alzheimer’s disease. Front Neurosci. 2020;13:1443. doi: 10.3389/fnins.2019.01443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weiland A, Wang YM, Wu WH, Lan X, Han XN, Li Q, et al. Ferroptosis and its role in diverse brain diseases. Mol Neurobiol. 2019;56:4880–4893. doi: 10.1007/s12035-018-1403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tang DL, Chen X, Kang R, Kroemer G. Ferroptosis: molecular mechanisms and health implications. Cell Res. 2021;31:107–125. doi: 10.1038/s41422-020-00441-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yan HF, Zou T, Tuo QZ, Xu S, Li H, Belaidi AA, et al. Ferroptosis: mechanisms and links with diseases. Signal Transduct Target Ther. 2021;6:49. doi: 10.1038/s41392-020-00428-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–331. doi: 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Andrews NC, Schmidt PJ. Iron homeostasis. Annu Rev Physiol. 2007;69:69–85. doi: 10.1146/annurev.physiol.69.031905.164337. [DOI] [PubMed] [Google Scholar]
- 8.Jiang H, Wang J, Rogers J, Xie JX. Brain iron metabolism dysfunction in Parkinson’s disease. Mol Neurobiol. 2017;54:3078–3101. doi: 10.1007/s12035-016-9879-1. [DOI] [PubMed] [Google Scholar]
- 9.Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, et al. Ferroptosis: process and function. Cell Death Diff. 2016;23:369–379. doi: 10.1038/cdd.2015.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cao JY, Dixon SJ. Mechanisms of ferroptosis. Cell Mol Life Sci. 2016;73:2195–2209. doi: 10.1007/s00018-016-2194-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.LaVaute T, Smith S, Cooperman S, Iwai K, Land W, Meyron-Holtz E, et al. Targeted deletion of the gene encoding iron regulatory protein-2 causes misregulation of iron metabolism and neurodegenerative disease in mice. Nat Genet. 2001;27:209–214. doi: 10.1038/84859. [DOI] [PubMed] [Google Scholar]
- 12.Gao MH, Monian P, Quadri N, Ramasamy R, Jiang XJ. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell. 2015;59:298–308. doi: 10.1016/j.molcel.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chirasani SR, Markovic DS, Synowitz M, Eichler SA, Wisniewski P, Kaminska B, et al. Transferrin-receptor-mediated iron accumulation controls proliferation and glutamate release in glioma cells. J Mol Med (Berl) 2009;87:153–167. doi: 10.1007/s00109-008-0414-3. [DOI] [PubMed] [Google Scholar]
- 14.Gao MH, Monian P, Pan QH, Zhang W, Xiang J, Jiang XJ. Ferroptosis is an autophagic cell death process. Cell Res. 2016;26:1021–1032. doi: 10.1038/cr.2016.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hou W, Xie YC, Song XX, Sun XF, Lotze MT, Zeh HJ, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12:1425–1428. doi: 10.1080/15548627.2016.1187366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang P, Chen L, Zhao QQ, Du XX, Bi MX, Li Y, et al. Ferroptosis was more initial in cell death caused by iron overload and its underlying mechanism in Parkinson’s disease. Free Radic Biol Med. 2020;152:227–234. doi: 10.1016/j.freeradbiomed.2020.03.015. [DOI] [PubMed] [Google Scholar]
- 17.Sato H, Tamba M, Ishii T, Bannai S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem. 1999;274:11455–11458. doi: 10.1074/jbc.274.17.11455. [DOI] [PubMed] [Google Scholar]
- 18.Gu F, Chauhan V, Chauhan A. Glutathione redox imbalance in brain disorders. Curr Opin Clin Nutr Metab Care. 2015;18:89–95. doi: 10.1097/MCO.0000000000000134. [DOI] [PubMed] [Google Scholar]
- 19.Kaur D, Lee D, Ragapolan S, Andersen JK. Glutathione depletion in immortalized midbrain-derived dopaminergic neurons results in increases in the labile iron pool: Implications for Parkinson’s disease. Free Radic Biol Med. 2009;46:593–598. doi: 10.1016/j.freeradbiomed.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife. 2014;3:2523. doi: 10.7554/eLife.02523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kang YP, Mockabee-Macias A, Jiang C, Falzone A, Prieto-Farigua N, Stone E, et al. Non-canonical glutamate-cysteine ligase activity protects against ferroptosis. Cell Metab. 2021;33:174–189.e7. doi: 10.1016/j.cmet.2020.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Degregorio-Rocasolano N, Martí-Sistac O, Gasull T. Deciphering the iron side of stroke: Neurodegeneration at the crossroads between iron dyshomeostasis, excitotoxicity, and ferroptosis. Front Neurosci. 2019;13:85. doi: 10.3389/fnins.2019.00085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hambright WS, Fonseca RS, Chen LJ, Na R, Ran QT. Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration. Redox Biol. 2017;12:8–17. doi: 10.1016/j.redox.2017.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stockwell BR, Angeli JPF, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171:273–285. doi: 10.1016/j.cell.2017.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yehuda S, Rabinovitz S, Carasso RL, Mostofsky DI. The role of polyunsaturated fatty acids in restoring the aging neuronal membrane. Neurobiol Aging. 2002;23:843–853. doi: 10.1016/S0197-4580(02)00074-X. [DOI] [PubMed] [Google Scholar]
- 26.Cheng ZY, Li YZ. What is responsible for the initiating chemistry of iron-mediated lipid peroxidation: An update. Chem Rev. 2007;107:748–766. doi: 10.1021/cr040077w. [DOI] [PubMed] [Google Scholar]
- 27.From the American Association of Neurological Surgeons (AANS), American Society of Neuroradiology (ASNR), Cardiovascular and Interventional Radiology Society of Europe (CIRSE), Canadian Interventional Radiology Association (CIRA), Congress of Neurological Surgeons (CNS), European Society of Minimally Invasive Neurological Therapy (ESMINT), et al. Multisociety consensus quality improvement revised consensus statement for endovascular therapy of acute ischemic stroke. Int J Stroke 2018, 13: 612–632.
- 28.Gaschler MM, Stockwell BR. Lipid peroxidation in cell death. Biochem Biophys Res Commun. 2017;482:419–425. doi: 10.1016/j.bbrc.2016.10.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci USA. 2016;113:E4966–E4975. doi: 10.1073/pnas.1603244113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kagan VE, Mao GW, Qu F, Angeli JPF, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13:81–90. doi: 10.1038/nchembio.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dixon SJ, Winter GE, Musavi LS, Lee ED, Snijder B, Rebsamen M, et al. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem Biol. 2015;10:1604–1609. doi: 10.1021/acschembio.5b00245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575:693–698. doi: 10.1038/s41586-019-1707-0. [DOI] [PubMed] [Google Scholar]
- 33.Lardenoije R, Pishva E, Lunnon K, van den Hove DL. Neuroepigenetics of aging and age-related neurodegenerative disorders. Prog Mol Biol Transl Sci. 2018;158:49–82. doi: 10.1016/bs.pmbts.2018.04.008. [DOI] [PubMed] [Google Scholar]
- 34.Masaldan S, Bush AI, Devos D, Rolland AS, Moreau C. Striking while the iron is hot: Iron metabolism and ferroptosis in neurodegeneration. Free Radic Biol Med. 2019;133:221–233. doi: 10.1016/j.freeradbiomed.2018.09.033. [DOI] [PubMed] [Google Scholar]
- 35.Caudle WM. Occupational metal exposure and Parkinsonism. Adv Neurobiol. 2017;18:143–158. doi: 10.1007/978-3-319-60189-2_7. [DOI] [PubMed] [Google Scholar]
- 36.Xie AM, Gao J, Xu L, Meng DM. Shared mechanisms of neurodegeneration in Alzheimer’s disease and Parkinson’s disease. Biomed Res Int. 2014;2014:648740. doi: 10.1155/2014/648740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gao HM, Zhou H, Hong JS. NADPH oxidases: Novel therapeutic targets for neurodegenerative diseases. Trends Pharmacol Sci. 2012;33:295–303. doi: 10.1016/j.tips.2012.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brenner S. Parkinson’s disease may be due to failure of melanin in the Substantia Nigra to produce molecular hydrogen from dissociation of water, to protect the brain from oxidative stress. Med Hypotheses. 2014;82:503. doi: 10.1016/j.mehy.2014.01.013. [DOI] [PubMed] [Google Scholar]
- 39.Amalric M. Targeting metabotropic glutamate receptors (mGluRs) in Parkinson’s disease. Curr Opin Pharmacol. 2015;20:29–34. doi: 10.1016/j.coph.2014.11.001. [DOI] [PubMed] [Google Scholar]
- 40.Wu JR, Tuo QZ, Lei P. Ferroptosis, a recent defined form of critical cell death in neurological disorders. J Mol Neurosci. 2018;66:197–206. doi: 10.1007/s12031-018-1155-6. [DOI] [PubMed] [Google Scholar]
- 41.Mahoney-Sánchez L, Bouchaoui H, Ayton S, Devos D, Duce JA, Devedjian JC. Ferroptosis and its potential role in the physiopathology of Parkinson’s Disease. Prog Neurobiol. 2021;196:101890. doi: 10.1016/j.pneurobio.2020.101890. [DOI] [PubMed] [Google Scholar]
- 42.Reichert CO, de Freitas FA, Sampaio-Silva J, Rokita-Rosa L, Barros PL, Levy D, et al. Ferroptosis mechanisms involved in neurodegenerative diseases. Int J Mol Sci. 2020;21:8765. doi: 10.3390/ijms21228765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ballard C, Gauthier S, Corbett A, Brayne C, Aarsland D, Jones E. Alzheimer’s disease. Lancet. 2011;377:1019–1031. doi: 10.1016/S0140-6736(10)61349-9. [DOI] [PubMed] [Google Scholar]
- 44.Lane CA, Hardy J, Schott JM. Alzheimer’s disease. Eur J Neurol. 2018;25:59–70. doi: 10.1111/ene.13439. [DOI] [PubMed] [Google Scholar]
- 45.Lee AK, Khaled H, Chofflet N, Takahashi H. Synaptic organizers in Alzheimer’s disease: A classification based on amyloid-β sensitivity. Front Cell Neurosci. 2020;14:281. doi: 10.3389/fncel.2020.00281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lane DJR, Ayton S, Bush AI. Iron and Alzheimer’s disease: An update on emerging mechanisms. J Alzheimers Dis. 2018;64:S379–S395. doi: 10.3233/JAD-179944. [DOI] [PubMed] [Google Scholar]
- 47.Hynd MR, Scott HL, Dodd PR. Glutamate-mediated excitotoxicity and neurodegeneration in Alzheimer’s disease. Neurochem Int. 2004;45:583–595. doi: 10.1016/j.neuint.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 48.Cheignon C, Tomas M, Bonnefont-Rousselot D, Faller P, Hureau C, Collin F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018;14:450–464. doi: 10.1016/j.redox.2017.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ayton S, Fazlollahi A, Bourgeat P, Raniga P, Ng A, Lim YY, et al. Cerebral quantitative susceptibility mapping predicts amyloid-β-related cognitive decline. Brain. 2017;140:2112–2119. doi: 10.1093/brain/awx137. [DOI] [PubMed] [Google Scholar]
- 50.Ayton S, Wang YM, Diouf I, Schneider JA, Brockman J, Morris MC, et al. Brain iron is associated with accelerated cognitive decline in people with Alzheimer pathology. Mol Psychiatry. 2020;25:2932–2941. doi: 10.1038/s41380-019-0375-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lewerenz J, Maher P. Chronic glutamate toxicity in neurodegenerative diseases-what is the evidence? Front Neurosci. 2015;9:469. doi: 10.3389/fnins.2015.00469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang XL, Wang WZ, Li L, Perry G, Lee HG, Zhu XW. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim Biophys Acta. 2014;1842:1240–1247. doi: 10.1016/j.bbadis.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xilouri M, Brekk OR, Stefanis L. Autophagy and alpha-synuclein: Relevance to Parkinson’s disease and related synucleopathies. Mov Disord. 2016;31:178–192. doi: 10.1002/mds.26477. [DOI] [PubMed] [Google Scholar]
- 54.Seo JH, Rah JC, Choi SH, Shin JK, Min K, Kim HS, et al. Α-Synuclein regulates neuronal survival via Bcl-2 family expression and PI3/Akt kinase pathway. FASEB J. 2002;16:1–20. doi: 10.1096/fj.02-0041fje. [DOI] [PubMed] [Google Scholar]
- 55.Liu J, Liu WJ, Yang H. Balancing apoptosis and autophagy for Parkinson’s disease therapy: Targeting BCL-2. ACS Chem Neurosci. 2019;10:792–802. doi: 10.1021/acschemneuro.8b00356. [DOI] [PubMed] [Google Scholar]
- 56.Dexter DT, Wells FR, Lees AJ, Agid F, Agid Y, Jenner P, et al. Increased nigral iron content and alterations in other metal ions occurring in brain in Parkinson’s disease. J Neurochem. 1989;52:1830–1836. doi: 10.1111/j.1471-4159.1989.tb07264.x. [DOI] [PubMed] [Google Scholar]
- 57.Ayton S, Lei P. Nigral iron elevation is an invariable feature of Parkinson’s disease and is a sufficient cause of neurodegeneration. Biomed Res Int. 2014;2014:581256. doi: 10.1155/2014/581256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sian J, Dexter DT, Lees AJ, Daniel S, Agid Y, Javoy-Agid F, et al. Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Ann Neurol. 1994;36:348–355. doi: 10.1002/ana.410360305. [DOI] [PubMed] [Google Scholar]
- 59.Dexter DT, Carter CJ, Wells FR, Javoy-Agid F, Agid Y, Lees A, et al. Basal lipid peroxidation in substantia nigra is increased in Parkinson’s disease. J Neurochem. 1989;52:381–389. doi: 10.1111/j.1471-4159.1989.tb09133.x. [DOI] [PubMed] [Google Scholar]
- 60.Guiney SJ, Adlard PA, Bush AI, Finkelstein DI, Ayton S. Ferroptosis and cell death mechanisms in Parkinson’s disease. Neurochem Int. 2017;104:34–48. doi: 10.1016/j.neuint.2017.01.004. [DOI] [PubMed] [Google Scholar]
- 61.Van BD, Gouel F, Jonneaux A, Timmerman K, Gelé P, Pétrault M, et al. Ferroptosis, a newly characterized form of cell death in Parkinson’s disease that is regulated by PKC. Neurobiol Dis. 2016;94:169–178. doi: 10.1016/j.nbd.2016.05.011. [DOI] [PubMed] [Google Scholar]
- 62.Jiang H, Song N, Jiao Q, Shi LM, Du XX. Iron pathophysiology in parkinson diseases. Adv Exp Med Biol. 2019;1173:45–66. doi: 10.1007/978-981-13-9589-5_4. [DOI] [PubMed] [Google Scholar]
- 63.Dexter DT, Carayon A, Vidailhet M, Ruberg M, Agid F, Agid Y, et al. Decreased ferritin levels in brain in Parkinson’s disease. J Neurochem. 1990;55:16–20. doi: 10.1111/j.1471-4159.1990.tb08814.x. [DOI] [PubMed] [Google Scholar]
- 64.Ayton S, Lei P, Duce JA, Wong BXW, Sedjahtera A, Adlard PA, et al. Ceruloplasmin dysfunction and therapeutic potential for Parkinson disease. Ann Neurol. 2013;73:554–559. doi: 10.1002/ana.23817. [DOI] [PubMed] [Google Scholar]
- 65.Wang J, Jiang H, Xie JX. Ferroportin1 and hephaestin are involved in the nigral iron accumulation of 6-OHDA-lesioned rats. Eur J Neurosci. 2007;25:2766–2772. doi: 10.1111/j.1460-9568.2007.05515.x. [DOI] [PubMed] [Google Scholar]
- 66.Sandhir R, Sood A, Mehrotra A, Kamboj SS. N-Acetylcysteine reverses mitochondrial dysfunctions and behavioral abnormalities in 3-nitropropionic acid-induced Huntington’s disease. Neurodegener Dis. 2012;9:145–157. doi: 10.1159/000334273. [DOI] [PubMed] [Google Scholar]
- 67.Browne SE, Ferrante RJ, Beal MF. Oxidative stress in Huntington’s disease. Brain Pathol. 1999;9:147–163. doi: 10.1111/j.1750-3639.1999.tb00216.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wyttenbach A, Sauvageot O, Carmichael J, Diaz-Latoud C, Arrigo AP, Rubinsztein DC. Heat shock protein 27 prevents cellular polyglutamine toxicity and suppresses the increase of reactive oxygen species caused by huntingtin. Hum Mol Genet. 2002;11:1137–1151. doi: 10.1093/hmg/11.9.1137. [DOI] [PubMed] [Google Scholar]
- 69.Lee J, Kosaras B, del Signore SJ, Cormier K, McKee A, Ratan RR, et al. Modulation of lipid peroxidation and mitochondrial function improves neuropathology in Huntington’s disease mice. Acta Neuropathol. 2011;121:487–498. doi: 10.1007/s00401-010-0788-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen JF, Marks E, Lai B, Zhang ZJ, Duce JA, Lam LQ, et al. Iron accumulates in Huntington’s disease neurons: Protection by deferoxamine. PLoS One. 2013;8:77023. doi: 10.1371/journal.pone.0077023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Niu L, Ye CF, Sun Y, Peng T, Yang SM, Wang WX, et al. Mutant huntingtin induces iron overload via up-regulating IRP1 in Huntington’s disease. Cell Biosci. 2018;8:41. doi: 10.1186/s13578-018-0239-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rosas HD, Chen YI, Doros G, Salat DH, Chen NK, Kwong KK, et al. Alterations in brain transition metals in Huntington disease: An evolving and intricate story. Arch Neurol. 2012;69:887–893. doi: 10.1001/archneurol.2011.2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Klepac N, Relja M, Klepac R, Hećimović S, Babić T, Trkulja V. Oxidative stress parameters in plasma of Huntington’s disease patients, asymptomatic Huntington’s disease gene carriers and healthy subjects. J Neurol. 2007;254:1676–1683. doi: 10.1007/s00415-007-0611-y. [DOI] [PubMed] [Google Scholar]
- 74.Khanna S, Briggs Z, Rink C. Inducible glutamate oxaloacetate transaminase as a therapeutic target against ischemic stroke. Antioxid Redox Signal. 2015;22:175–186. doi: 10.1089/ars.2014.6106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Takagi K, Ginsberg MD, Globus MY, Dietrich WD, Martinez E, Kraydieh S, et al. Changes in amino acid neurotransmitters and cerebral blood flow in the ischemic penumbral region following middle cerebral artery occlusion in the rat: Correlation with histopathology. J Cereb Blood Flow Metab. 1993;13:575–585. doi: 10.1038/jcbfm.1993.75. [DOI] [PubMed] [Google Scholar]
- 76.Banjac A, Perisic T, Sato H, Seiler A, Bannai S, Weiss N, et al. The cystine/cysteine cycle: A redox cycle regulating susceptibility versus resistance to cell death. Oncogene. 2008;27:1618–1628. doi: 10.1038/sj.onc.1210796. [DOI] [PubMed] [Google Scholar]
- 77.Selim MH, Ratan RR. The role of iron neurotoxicity in ischemic stroke. Ageing Res Rev. 2004;3:345–353. doi: 10.1016/j.arr.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 78.Chen H, Yoshioka H, Kim GS, Jung JE, Okami N, Sakata H, et al. Oxidative stress in ischemic brain damage: Mechanisms of cell death and potential molecular targets for neuroprotection. Antioxid Redox Signal. 2011;14:1505–1517. doi: 10.1089/ars.2010.3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Degregorio-Rocasolano N, Martí-Sistac O, Ponce J, Castelló-Ruiz M, Millán M, Guirao V, et al. Iron-loaded transferrin (Tf) is detrimental whereas iron-free Tf confers protection against brain ischemia by modifying blood Tf saturation and subsequent neuronal damage. Redox Biol. 2018;15:143–158. doi: 10.1016/j.redox.2017.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Domercq M, Sánchez-Gómez MV, Sherwin C, Etxebarria E, Fern R, Matute C. System Xc− and glutamate transporter inhibition mediates microglial toxicity to oligodendrocytes. J Immunol. 2007;178:6549–6556. doi: 10.4049/jimmunol.178.10.6549. [DOI] [PubMed] [Google Scholar]
- 81.Zhang ZW, Wu Y, Yuan S, Zhang P, Zhang JY, Li HY, et al. Glutathione peroxidase 4 participates in secondary brain injury through mediating ferroptosis in a rat model of intracerebral hemorrhage. Brain Res. 2018;1701:112–125. doi: 10.1016/j.brainres.2018.09.012. [DOI] [PubMed] [Google Scholar]
- 82.Alim I, Caulfield JT, Chen YX, Swarup V, Geschwind DH, Ivanova E, et al. Selenium drives a transcriptional adaptive program to block ferroptosis and treat stroke. Cell. 2019;177:1262–1279.e25. doi: 10.1016/j.cell.2019.03.032. [DOI] [PubMed] [Google Scholar]
- 83.Liu Y, Wang W, Li YY, Xiao YQ, Cheng J, Jia J. The 5-lipoxygenase inhibitor zileuton confers neuroprotection against glutamate oxidative damage by inhibiting ferroptosis. Biol Pharm Bull. 2015;38:1234–1239. doi: 10.1248/bpb.b15-00048. [DOI] [PubMed] [Google Scholar]
- 84.Mancias JD, Wang XX, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature. 2014;509:105–109. doi: 10.1038/nature13148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Quiles del Rey M, Mancias JD. NCOA4-mediated ferritinophagy: A potential link to neurodegeneration. Front Neurosci. 2019;13:238. doi: 10.3389/fnins.2019.00238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Levine AJ, Oren M. The first 30 years of p53: Growing ever more complex. Nat Rev Cancer. 2009;9:749–758. doi: 10.1038/nrc2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kastenhuber ER, Lowe SW. Putting p53 in context. Cell. 2017;170:1062–1078. doi: 10.1016/j.cell.2017.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mello SS, Attardi LD. Deciphering p53 signaling in tumor suppression. Curr Opin Cell Biol. 2018;51:65–72. doi: 10.1016/j.ceb.2017.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chen JD. The cell-cycle arrest and apoptotic functions of p53 in tumor initiation and progression. Cold Spring Harb Perspect Med. 2016;6:26104. doi: 10.1101/cshperspect.a026104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tomita Y, Marchenko N, Erster S, Nemajerova A, Dehner A, Klein C, et al. WT p53, but not tumor-derived mutants, bind to Bcl2 via the DNA binding domain and induce mitochondrial permeabilization. J Biol Chem. 2006;281:8600–8606. doi: 10.1074/jbc.M507611200. [DOI] [PubMed] [Google Scholar]
- 91.Feng Y, Liu T, Dong SY, Guo YJ, Jankovic J, Xu HX, et al. Rotenone affects p53 transcriptional activity and apoptosis via targeting SIRT1 and H3K9 acetylation in SH-SY5Y cells. J Neurochem. 2015;134:668–676. doi: 10.1111/jnc.13172. [DOI] [PubMed] [Google Scholar]
- 92.Abida WM, Gu W. p53-dependent and p53-independent activation of autophagy by ARF. Cancer Res. 2008;68:352–357. doi: 10.1158/0008-5472.CAN-07-2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Verma N, Manna SK. Advanced glycation end products (AGE) potentiates cell death in p53 negative cells via upregulaion of NF-kappa B and impairment of autophagy. J Cell Physiol. 2017;232:3598–3610. doi: 10.1002/jcp.25828. [DOI] [PubMed] [Google Scholar]
- 94.Zhang XF, Cheng Q, Yin HJ, Yang G. Regulation of autophagy and EMT by the interplay between p53 and RAS during cancer progression (Review) Int J Oncol. 2017;51:18–24. doi: 10.3892/ijo.2017.4025. [DOI] [PubMed] [Google Scholar]
- 95.Mrschtik M, Ryan KM. Another DRAM involved in autophagy and cell death. Autophagy. 2016;12:603–605. doi: 10.1080/15548627.2015.1137412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Crighton D, Wilkinson S, Ryan KM. DRAM links autophagy to p53 and programmed cell death. Autophagy. 2007;3:72–74. doi: 10.4161/auto.3438. [DOI] [PubMed] [Google Scholar]
- 97.Crighton D, Wilkinson S, O’Prey J, Syed N, Smith P, Harrison PR, et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126:121–134. doi: 10.1016/j.cell.2006.05.034. [DOI] [PubMed] [Google Scholar]
- 98.Villanueva-Paz M, Cotán D, Garrido-Maraver J, Oropesa-Ávila M, de la Mata M, Delgado-Pavón A, et al. AMPK regulation of cell growth, apoptosis, autophagy, and bioenergetics. Exp Suppl. 2016;107:45–71. doi: 10.1007/978-3-319-43589-3_3. [DOI] [PubMed] [Google Scholar]
- 99.Ma X, Zhang S, He L, Rong YG, Brier LW, Sun QM, et al. MTORC1-mediated NRBF2 phosphorylation functions as a switch for the class III PtdIns3K and autophagy. Autophagy. 2017;13:592–607. doi: 10.1080/15548627.2016.1269988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Li TY, Kon N, Jiang L, Tan MJ, Ludwig T, Zhao YM, et al. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell. 2012;149:1269–1283. doi: 10.1016/j.cell.2012.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wang SJ, Li DW, Ou Y, Jiang L, Chen Y, Zhao YM, et al. Acetylation is crucial for p53-mediated ferroptosis and tumor suppression. Cell Rep. 2016;17:366–373. doi: 10.1016/j.celrep.2016.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jiang L, Kon N, Li TY, Wang SJ, Su T, Hibshoosh H, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57–62. doi: 10.1038/nature14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Thomasova D, Bruns HA, Kretschmer V, Ebrahim M, Romoli S, Liapis H, et al. Murine double minute-2 prevents p53-overactivation-related cell death (podoptosis) of podocytes. J Am Soc Nephrol. 2015;26:1513–1523. doi: 10.1681/ASN.2014040345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mou YH, Wang J, Wu JC, He D, Zhang CF, Duan CJ, et al. Ferroptosis, a new form of cell death: Opportunities and challenges in cancer. J Hematol Oncol. 2019;12:34. doi: 10.1186/s13045-019-0720-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yu HT, Guo PY, Xie XZ, Wang Y, Chen G. Ferroptosis, a new form of cell death, and its relationships with tumourous diseases. J Cell Mol Med. 2017;21:648–657. doi: 10.1111/jcmm.13008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zheng DW, Lei Q, Zhu JY, Fan JX, Li CX, Li C, et al. Switching apoptosis to ferroptosis: Metal-organic network for high-efficiency anticancer therapy. Nano Lett. 2017;17:284–291. doi: 10.1021/acs.nanolett.6b04060. [DOI] [PubMed] [Google Scholar]
- 107.Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lee YS, Lee DH, Jeong SY, Park SH, Oh SC, Park YS, et al. Ferroptosis-inducing agents enhance TRAIL-induced apoptosis through up-regulation of death receptor 5. J Cell Biochem. 2019;120:928–939. doi: 10.1002/jcb.27456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lee YS, Kalimuthu K, Park YS, Luo X, Choudry MHA, Bartlett DL, et al. BAX-dependent mitochondrial pathway mediates the crosstalk between ferroptosis and apoptosis. Apoptosis. 2020;25:625–631. doi: 10.1007/s10495-020-01627-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Carbone M, Yang HN, Pass HI, Krausz T, Testa JR, Gaudino G. BAP1 and cancer. Nat Rev Cancer. 2013;13:153–159. doi: 10.1038/nrc3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bononi A, Giorgi C, Patergnani S, Larson D, Verbruggen K, Tanji MK, et al. BAP1 regulates IP3R3-mediated Ca2+ flux to mitochondria suppressing cell transformation. Nature. 2017;546:549–553. doi: 10.1038/nature22798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.He M, Chaurushiya MS, Webster JD, Kummerfeld S, Reja R, Chaudhuri S, et al. Intrinsic apoptosis shapes the tumor spectrum linked to inactivation of the deubiquitinase BAP1. Science. 2019;364:283–285. doi: 10.1126/science.aav4902. [DOI] [PubMed] [Google Scholar]
- 113.Zhang YL, Zhuang L, Gan BY. BAP1 suppresses tumor development by inducing ferroptosis upon SLC7A11 repression. Mol Cell Oncol. 2018;6:1536845. doi: 10.1080/23723556.2018.1536845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bhattacharya S, Hanpude P, Maiti TK. Cancer associated missense mutations in BAP1 catalytic domain induce amyloidogenic aggregation: A new insight in enzymatic inactivation. Sci Rep. 2015;5:18462. doi: 10.1038/srep18462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sharma A, Liu HD, Tobar-Tosse F, Chand Dakal T, Ludwig M, Holz FG, et al. Ubiquitin carboxyl-terminal hydrolases (UCHs): Potential mediators for cancer and neurodegeneration. Int J Mol Sci. 2020;21:3910. doi: 10.3390/ijms21113910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Liang XH, Kleeman LK, Jiang HH, Gordon G, Goldman JE, Berry G, et al. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J Virol. 1998;72:8586–8596. doi: 10.1128/JVI.72.11.8586-8596.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Huang WJ, Choi W, Hu WQ, Mi N, Guo Q, Ma MS, et al. Crystal structure and biochemical analyses reveal Beclin 1 as a novel membrane binding protein. Cell Res. 2012;22:473–489. doi: 10.1038/cr.2012.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Furuya N, Yu J, Byfield M, Pattingre S, Levine B. The evolutionarily conserved domain of Beclin 1 is required for Vps34 binding, autophagy and tumor suppressor function. Autophagy. 2005;1:46–52. doi: 10.4161/auto.1.1.1542. [DOI] [PubMed] [Google Scholar]
- 119.Wei YJ, Zou ZJ, Becker N, Anderson M, Sumpter R, Xiao GH, et al. EGFR-mediated beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell. 2013;154:1269–1284. doi: 10.1016/j.cell.2013.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Salminen A, Kaarniranta K, Kauppinen A, Ojala J, Haapasalo A, Soininen H, et al. Impaired autophagy and APP processing in Alzheimer’s disease: The potential role of Beclin 1 interactome. Prog Neurobiol. 2013;106(107):33–54. doi: 10.1016/j.pneurobio.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 121.Jaeger PA, Pickford F, Sun CH, Lucin KM, Masliah E, Wyss-Coray T. Regulation of amyloid precursor protein processing by the Beclin 1 complex. PLoS One. 2010;5:e11102. doi: 10.1371/journal.pone.0011102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest. 2008;118:2190–2199. doi: 10.1172/JCI33585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Maiuri MC, le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P, et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 2007;26:2527–2539. doi: 10.1038/sj.emboj.7601689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ciechomska IA, Goemans GC, Skepper JN, Tolkovsky AM. Bcl-2 complexed with Beclin-1 maintains full anti-apoptotic function. Oncogene. 2009;28:2128–2141. doi: 10.1038/onc.2009.60. [DOI] [PubMed] [Google Scholar]
- 125.Wirawan E, Vande Walle L, Kersse K, Cornelis S, Claerhout S, Vanoverberghe I, et al. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis. 2010;1:e18. doi: 10.1038/cddis.2009.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Song XX, Zhu S, Chen P, Hou W, Wen QR, Liu J, et al. AMPK-mediated BECN1 phosphorylation promotes ferroptosis by directly blocking system X c- activity. Curr Biol. 2018;28:2388–2399.e5. doi: 10.1016/j.cub.2018.05.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Báthori G, Sahin-Tóth M, Fonyó A, Ligeti E. Transport properties and inhibitor sensitivity of isolated and reconstituted porin differ from those of intact mitochondria. Biochim Biophys Acta BBA Biomembr. 1993;1145:168–176. doi: 10.1016/0005-2736(93)90394-F. [DOI] [PubMed] [Google Scholar]
- 128.Lee AC, Xu X, Blachly-Dyson E, Forte M, Colombini M. The role of yeast VDAC genes on the permeability of the mitochondrial outer membrane. J Membr Biol. 1998;161:173–181. doi: 10.1007/s002329900324. [DOI] [PubMed] [Google Scholar]
- 129.Colombini M. Structure and mode of action of a voltage dependent anion-selective channel (VDAC) located in the outer mitochondrial membrane. Ann N Y Acad Sci. 1980;341:552–563. doi: 10.1111/j.1749-6632.1980.tb47198.x. [DOI] [PubMed] [Google Scholar]
- 130.Gatliff J, Campanella M. The 18 kDa translocator protein (TSPO): A new perspective in mitochondrial biology. Curr Mol Med. 2012;12:356–368. doi: 10.2174/1566524011207040356. [DOI] [PubMed] [Google Scholar]
- 131.Veenman L, Shandalov Y, Gavish M. VDAC activation by the 18 kDa translocator protein (TSPO), implications for apoptosis. J Bioenerg Biomembr. 2008;40:199–205. doi: 10.1007/s10863-008-9142-1. [DOI] [PubMed] [Google Scholar]
- 132.Veenman L, Papadopoulos V, Gavish M. Channel-like functions of the 18-kDa translocator protein (TSPO): Regulation of apoptosis and steroidogenesis as part of the host-defense response. Curr Pharm Des. 2007;13:2385–2405. doi: 10.2174/138161207781368710. [DOI] [PubMed] [Google Scholar]
- 133.Lovell MA, Xie C, Markesbery WR. Acrolein is increased in Alzheimer’s disease brain and is toxic to primary hippocampal cultures. Neurobiol Aging. 2001;22:187–194. doi: 10.1016/S0197-4580(00)00235-9. [DOI] [PubMed] [Google Scholar]
- 134.Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature. 2007;447:864–868. doi: 10.1038/nature05859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Yang YF, Luo MY, Zhang KX, Zhang J, Gao TT, Connell DO, et al. Nedd4 ubiquitylates VDAC2/3 to suppress erastin-induced ferroptosis in melanoma. Nat Commun. 2020;11:433. doi: 10.1038/s41467-020-14324-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Feng YS, Madungwe NB, Imam Aliagan AD, Tombo N, Bopassa JC. Liproxstatin-1 protects the mouse myocardium against ischemia/reperfusion injury by decreasing VDAC1 levels and restoring GPX4 levels. Biochem Biophys Res Commun. 2019;520:606–611. doi: 10.1016/j.bbrc.2019.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ji CH, Kwon YT. Crosstalk and interplay between the ubiquitin-proteasome system and autophagy. Mol Cells. 2017;40:441–449. doi: 10.14348/molcells.2017.0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wang Y, Liu Y, Liu J, Kang R, Tang DL. NEDD4L-mediated LTF protein degradation limits ferroptosis. Biochem Biophys Res Commun. 2020;531:581–587. doi: 10.1016/j.bbrc.2020.07.032. [DOI] [PubMed] [Google Scholar]
- 139.Chen X, Yu CH, Kang R, Kroemer G, Tang DL. Cellular degradation systems in ferroptosis. Cell Death Differ. 2021;28:1135–1148. doi: 10.1038/s41418-020-00728-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.van Leeuwen FW, Hol EM, Fischer DF. Frameshift proteins in Alzheimer’s disease and in other conformational disorders: Time for the ubiquitin-proteasome system. J Alzheimers Dis. 2006;9:319–325. doi: 10.3233/JAD-2006-9S336. [DOI] [PubMed] [Google Scholar]
- 141.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 142.Gong B, Radulovic M, Figueiredo-Pereira ME, Cardozo C. The ubiquitin-proteasome system: Potential therapeutic targets for Alzheimer’s disease and spinal cord injury. Front Mol Neurosci. 2016;9:4. doi: 10.3389/fnmol.2016.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kimura T, Jia JY, Claude-Taupin A, Kumar S, Choi SW, Gu YX, et al. Cellular and molecular mechanism for secretory autophagy. Autophagy. 2017;13:1084–1085. doi: 10.1080/15548627.2017.1307486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Tsujimoto Y, Shimizu S. Another way to die: Autophagic programmed cell death. Cell Death Differ. 2005;12:1528–1534. doi: 10.1038/sj.cdd.4401777. [DOI] [PubMed] [Google Scholar]
- 145.Kroemer G, Levine B. Autophagic cell death: The story of a misnomer. Nat Rev Mol Cell Biol. 2008;9:1004–1010. doi: 10.1038/nrm2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zhou BR, Liu J, Kang R, Klionsky DJ, Kroemer G, Tang DL. Ferroptosis is a type of autophagy-dependent cell death. Semin Cancer Biol. 2020;66:89–100. doi: 10.1016/j.semcancer.2019.03.002. [DOI] [PubMed] [Google Scholar]
- 147.Ma SM, Dielschneider RF, Henson ES, Xiao WY, Choquette TR, Blankstein AR, et al. Ferroptosis and autophagy induced cell death occur independently after siramesine and lapatinib treatment in breast cancer cells. PLoS One. 2017;12:e0182921. doi: 10.1371/journal.pone.0182921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Lu B, Chen XB, Ying MD, He QJ, Cao J, Yang B. The role of ferroptosis in cancer development and treatment response. Front Pharmacol. 2018;8:992. doi: 10.3389/fphar.2017.00992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Torii S, Shintoku R, Kubota C, Yaegashi M, Torii R, Sasaki M, et al. An essential role for functional lysosomes in ferroptosis of cancer cells. Biochem J. 2016;473:769–777. doi: 10.1042/BJ20150658. [DOI] [PubMed] [Google Scholar]
- 150.Chekeni FB, Elliott MR, Sandilos JK, Walk SF, Kinchen JM, Lazarowski ER, et al. Pannexin 1 channels mediate “find-me” signal release and membrane permeability during apoptosis. Nature. 2010;467:863–867. doi: 10.1038/nature09413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Medina CB, Mehrotra P, Arandjelovic S, Perry JSA, Guo YZ, Morioka S, et al. Metabolites released from apoptotic cells act as tissue messengers. Nature. 2020;580:130–135. doi: 10.1038/s41586-020-2121-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Szczepańska AA, Łupicka M, Korzekwa AJ. Do arachidonic acid metabolites affect apoptosis in bovine endometrial cells with silenced PPAR genes? Prostaglandins Other Lipid Mediat 2019, 143: 106336. [DOI] [PubMed]
- 153.Hong SH, Lee DH, Lee YS, Jo MJ, Jeong YA, Kwon WT, et al. Molecular crosstalk between ferroptosis and apoptosis: Emerging role of ER stress-induced p53-independent PUMA expression. Oncotarget. 2017;8:115164–115178. doi: 10.18632/oncotarget.23046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Tian J, Guo S, Chen H, Peng JJ, Jia MM, Li NS, et al. Combination of emricasan with ponatinib synergistically reduces ischemia/reperfusion injury in rat brain through simultaneous prevention of apoptosis and necroptosis. Transl Stroke Res. 2018;9:382–392. doi: 10.1007/s12975-017-0581-z. [DOI] [PubMed] [Google Scholar]
- 155.Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M, et al. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science. 2004;303:1010–1014. doi: 10.1126/science.1092734. [DOI] [PubMed] [Google Scholar]
- 156.Green DR, Kroemer G. Cytoplasmic functions of the tumour suppressor p53. Nature. 2009;458:1127–1130. doi: 10.1038/nature07986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Tan WZ, Colombini M. VDAC closure increases calcium ion flux. Biochim Biophys Acta. 2007;1768:2510–2515. doi: 10.1016/j.bbamem.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]