

Abstract
Chronic stress is generally accepted as the main risk factor in the development of cognitive decline; however, the underlying mechanisms remain unclear. Previous data have demonstrated that the levels of homocysteine (Hcy) are significantly elevated in the plasma of stressed animals, which suggests that Hcy is associated with stress and cognitive decline. To test this hypothesis, we analyzed the cognitive function, plasma concentrations of Hcy, and brain-derived neurotropic factor (BDNF) levels in rats undergoing chronic unpredicted mild stress (CUMS). The results showed that decreased cognitive behavioral performance and decreased BDNF transcription and protein expression were correlated with hyperhomocysteinemia (HHcy) levels in stressed rats. Diet-induced HHcy mimicked the cognitive decline and BDNF downregulation in the same manner as CUMS, while Hcy reduction (by means of vitamin B complex supplements) alleviated the cognitive deficits and BDNF reduction in CUMS rats. Furthermore, we also found that both stress and HHcy disturbed the DNA methylation process in the brain and induced DNA hypermethylation in the BDNF promoter. In contrast, control of Hcy blocked BDNF promoter methylation and upregulated BDNF levels in the brain. These results imply the possibility of a causal role of Hcy in stress-induced cognitive decline. We also used ten-eleven translocation (TET1), an enzyme that induces DNA demethylation, to verify the involvement of Hcy and DNA methylation in the regulation of BDNF expression and the development of stress-related cognitive decline. The data showed that TET1-expressing viral injection into the hippocampus inhibited BDNF promoter methylation and significantly mitigated the cognitive decline in HHcy rats. Taken together, novel evidence from the present study suggests that Hcy is likely involved in chronic stress-induced BDNF reduction and related cognitive deficits. In addition, the negative side-effects of HHcy may be associated with Hcy-induced DNA hypermethylation in the BDNF promoter. The results also suggest the possibility of Hcy as a target for therapy and the potential value of vitamin B intake in preventing stress-induced cognitive decline.
Supplementary Information
The online version contains supplementary material available at 10.1007/s12264-022-00852-7.
Keywords: Stress-associated cognitive decline, Homocysteine, DNA methylation, BDNF
Introduction
According to current research, stress is a nonspecific systemic response of the organism to stimuli from internal and external environments [1, 2]. Although stress of appropriate intensity helps the organism cope with adverse environmental effects, prolonged high-intensity stress has a negative impact on human functioning [3, 4]. Epidemiological studies have shown that PTSD occurs in 7–8% of the population [5] and even in 20% of the veteran population [6]. Thus, stress has received increasing research attention in recent years.
The hippocampus is a part of the brain with great importance for the stress response given that it is rich in stress hormone targets [7] and is primarily responsible for memory and learning [8]. At present, chronic stress is recognized as an important risk factor leading to cognitive impairment and even dementia [9–11]. Early studies in rodents have shown that stress leads to structural and functional alterations in corresponding brain regions by reducing brain weight [12], mediating hippocampal atrophy, and affecting neurogenesis [13], thereby ultimately inducing the onset of cognitive impairment. However, the underlying molecular mechanisms leading to cognitive decline have not been fully defined. Therefore, there is a need to clarify the molecular mechanisms underlying the development of stress-related cognitive impairment.
Methylation is a type of epigenetic modification, and DNA modification by 5mC methylation is one of the most prominent types. Erin L. Kinnally et al. reported elevated plasmatic 5-HTT genes as well as genome-wide methylation in bonnet macaques that had experienced early-life stress [14]. In addition, it has been shown that elevated TACR2 expression in rat brain induced by chronic stress is associated with elevated methylation of the TACR2 gene [15]. Previous studies have shown that the effects of stress on methylation are widespread, but differ with different kinds of stress. In addition, it has been suggested that 5mC methylation plays an important role in several diseases with clinical manifestations of cognitive impairment (e.g., AD, MCI, and depression). For example, Yang et al. suggested that elevated SORL methylation in blood is closely associated with reduced cognitive function as reflected by MOCA scores [16]. In addition, DNA methylation in the NCAPH2/LMF2 promoter region has been implicated in hippocampal atrophy through apoptosis [17], suggesting that stress is closely related to methylation and that methylation may be involved in cognitive regulation.
Proteins play important roles in the maintenance of cognitive function in animals. Brain-derived neurotrophic factor (BDNF) plays an important role in a variety of neuropsychiatric disorders [18]. According to McEwen et al., BDNF is one of the important proteins in the stress-activated brain [19]. Furthermore, several studies have found that various stress patterns, such as PTSD, prenatal stress, and negative early-life events, affect BDNF expression in the rat hippocampus. All of these processes are closely associated with methylation [20, 21]. However, whether BDNF is regulated by methylation during chronic stress needs further study.
Homocysteine (Hcy), a sulfur-containing amino-acid, is an important product of methionine metabolism and an important intermediate of energy metabolism and methylation dependence in the body [22]. According to our previous study, chronic stress increases Hcy levels by affecting the expression of Hcy metabolic enzymes in the liver [23], and the consequent Hcy accumulation induces cognitive impairment [24]. In addition, a retrospective study on a large population has shown that timely detection of the early elevation of Hcy is an effective measure to prevent cognitive impairment in patients [25]. Considering that James et al. found that Hcy metabolites affect methylation [26], it has been suggested that Hcy is closely related to methylation modifications [27].
Thus, the focus of our study was to unravel whether Hcy induces cognitive impairment in rats by affecting methylation during chronic stress. This study used the classical chronic unpredictable mild stress (CUMS) model as an animal model of chronic stress [28]. We found that chronic stress: (1) affects the transcription and protein expression of hippocampal BDNF by increasing plasma Hcy concentration and changing methylation levels in the promoter region of hippocampal BDNF in rats, and (2) induces cognitive decline. This study provides a possible biological mechanism for further understanding stress-induced cognitive disorders.
Materials and Methods
Animals and Ethical Approval
Adult male Sprague-Dawley rats (6 weeks old and weighing 80–120 g) purchased from Laboratory Animal Center of the Academy of Military Sciences (AMS) were randomly assigned to different groups. Rats were exposed to a 12-h/12-h light/dark cycle and had free access to pure water and food except during the stress intervention and behavioral experiments. All of the experiments were approved by the Institutional Animal Care and Use Committee of the AMS (Permit No: IACUC-DWZX-2020-670) and were in accordance with the NIH Guide for the Care and Use of Laboratory Animals (NIH Publications No. 8023) and the ARRIVE guidelines from NC3Rs. We made all efforts to minimize animal suffering.
Model and Behavioral Testing
Chronic Unpredictable Mild Stress
CUMS is a common paradigm used to mimic long-term negative stress both physically and psychologically [28]. In this study, CUMS was used to simulate the effects of long-term negative stimulation in rats according to previous studies [24, 29]. The stimulation procedures were as follows: physical restriction for 2 h; food and water deprivation for 24 h; cage tilting at 45° for 24 h; wet bedding for 24 h; forced swimming in 4 °C water for 3 min, after which they were towel-dried; and continuous lighting for 24 h. The stimulation methods were selected randomly and were always different from those used on the previous day. Schematic diagrams of CUMS rats from 2 weeks to 8 weeks are shown in Fig. S1A.
Open Field Test (OFT)
The OFT was applied as in our previous study [27]. The area of the black square open field was 1 m2, and the bottom of the field was divided into 25 squares (each 0.04 m2). At the beginning of the test, the rats were placed in the corner of the bottom. The open field score was used to evaluate locomotor activity and exploratory behavior.
Morris Water Maze Test (MWM)
The water maze was 1.2 m in diameter and 75 cm in depth, and the temperature of the water was kept at 24 °C. Rats were trained to find a hidden platform 2 cm underwater in five consecutive half-days, one training session per half-day for 3 min. If they did not find the platform within 3 min, they were manually directed to the platform and allowed to stay there for 10 s so that they could remember its location. Final tests were performed 24 h after the last training session; they consisted of two 3-min free swims, one with and one without a platform. The latency to find the hidden platform (platform latency) and the number of entries to the area of the platform (platform passes) were recorded to evaluate spatial memory.
Step-Down Test
This test was conducted in a passive avoidance device. Briefly, the bottom of the device was an electrically-conductive grid with an insulated cylindrical table 8 cm in diameter and height, which allowed the rats to avoid an electric shock when the grid was energized. The test was divided into two parts: training and testing. Before training, the grid current was set to 0.8 mA, and a rat was then placed in the device and familiarized with it for 30 min; 24 h later, the rat was placed on the insulated table in the device with the bottom voltage set at 0.8 mA. The time needed to jump from the insulated table to the electrical fence was recorded as the step-down latency, which reflected the memory performance of the rats.
Novel Object Recognition Test
The apparatus consisted of a square open field box (65 cm × 65 cm × 45 cm) with black-painted surroundings. The test was divided into two steps: adaptation and testing. On the day before the experiment, rats were placed in the open field to adapt for 10 min; 24 h later, they were placed in the same cognitive box containing two identical white cubes (5 cm × 5 cm × 5 cm) and allowed to explore for a total of 30 s, with total duration up to 10 min. After 1 h, one block was replaced with a 5-cm-high glass bottle, and the time spent by the rats exploring (including limb contact and whisker exploration) the new (Tnew) and the old (Told) objects within 10 min were measured separately. Cognitive index was calculated as follows: Cognitive index = (Tnew − Told)/(Tnew + Told).
Homocysteine and Diet Intervention
To study the effects of Hcy on their cognitive function, rats were fed a diet containing 1% methionine to simulate the HHcy status in vivo (Met diet group) [27]. The homocysteine intervention group was given vitamin B intragastrically at 09:00 every day during the stress process (CUMS+VBco group). The total volume of gavage was 1.5 mL, and the content of each component was as follows: VB6 24 mg/kg body weight per day; VB12 20 μg/kg body weight per day; folic acid 10 mg/kg body weight per day [24]. The experimental schematic is shown in Fig. S1B, C.
BDNF Intervention
In the CUMS+BDNF AAV group, AAV overexpressing BDNF (synthesized by Gene Chem) was injected into the hippocampal DG region (coordinates: − 3.2 mm caudal to bregma, ± 1.9 mm from midline, − 3.0 mm deep from the skull) by stereotactic localization before stress was administered. The AAV injection dose was 3 × 109 vg (dissolved in 1 μL sterile saline). In the CUMS + Ctrl AAV group, AAV without any protein expression was injected into the same brain region. AAV injected into the DG region spread to the surrounding hippocampus. Intracranial injections were administered under isoflurane anesthesia, and penicillin was given intramuscularly (80,000 units per 100 g body weight postoperatively). The experimental schematic is shown in Fig. S1D.
Methylation Intervention
The CUMS+TET1 AAV group was set to study the effects of hippocampal DNA methylation on the cognitive function of stressed rats. Briefly, AAV overexpressing TET1 (1 μL of AAV at a dose of 3 × 109 vg, synthesized by Hanbio) was stereotactically injected into the hippocampus DG area. The experimental schematic is also shown in Fig. S1D.
Sample Preparation for Biochemical Analysis
After behavioral experiments, blood was collected from the heart when the rat was sacrificed under sodium pentobarbital anesthesia (1%, 50 mg/kg, i.p.) was placed in a blood collection tube containing sodium citrate, and centrifuged at 3,500 r/min at 4 °C for 10 min. The supernatant was frozen at − 80 °C for subsequent Hcy measurement. Rat brains were then quickly stripped out and placed on an ice table. For Golgi staining, the whole brain was immersed in sterile saline. For DNA, RNA, and protein extraction, the hippocampus was carefully dissected from the rest of the brain, post-processed in accordance with the requirements of the RIPA lysate (Solarbio, R0010), DNA extraction kit (Qiagen, 69504), and TRIzol (Sigma, T9424) extraction kit.
Immunohistochemical Analysis
After induction of deep anesthesia with sodium pentobarbital (1%, 50 mg/kg, i.p.), the rats were rapidly sequentially perfused with 120 mL of 37 °C saline and 37 °C 4% paraformaldehyde. The brain was subsequently removed and post-fixed in 4% paraformaldehyde at 4 °C for 24 h. Before immunoreaction, brain sections (20 μm thick cut on a Leica CM 1950) were mounted on glass slides, washed in 1× PBS for 15 min, then washed again in 1× TBS with 0.3% Triton for 2 h at room temperature (RT). Then, the tissue was blocked in goat serum overnight at 4 °C and incubated with primary antibody diluted in 1× TBS with 1% BSA for 24 h at 4 °C. The primary antibody used was polyclonal rabbit anti-BDNF (1:500, Santa Cruz, sc-20981). After incubation, the tissue was washed with 1× TBS with 0.025% Triton three times for 10 min each. The tissue was incubated with the secondary antibody diluted in 1× TBS with 1% BSA for 2 h at RT in the dark. The secondary antibody was TRITC-labeled goat anti rabbit IgG (1:200, Boster, BA1090). The tissue was washed with 1× TBS for 10 min and incubated with DAPI (Solarbio, C0065) solution for 5 min at RT. After incubation, the tissue was washed thoroughly with 1× PBS. Finally, the tissue was sealed with anti-fluorescence mounting medium. Images were captured using an Olympus IX-71 fluorescence microscope with CellSence Standard.
Western Blotting
Before electrophoresis, protein extracted from the hippocampus was diluted by SDS-PAGE loading buffer (Biorad, 1610747). The mixture was boiled for 5 min until denaturation, then centrifuged at 12,000 r/min for 3 min. Liquid supernatant was extracted for subsequent use. The sample was electrophoresed in 8% SDS-PAGE gel and 12% SDS-PAGE gel then transferred onto a PVDF membrane (Millipore, IPVH00010) using a wet transfer system (Bio-Rad, Power Pac HC). Membranes were blocked in 5% skim milk with 1× PBS containing 0.5% Tween-20 at RT for 1 h. After washing three times in 1× PBS, membranes were incubated with primary antibodies diluted in 5% skim milk with 1× PBS overnight at 4 °C. The primary antibodies used were rabbit anti-BDNF (1:1000, Santa Cruz, sc-20981) and rabbit anti-TET1 (1:1000, Abcam, ab191698), mouse anti- DNA methyl-transferase 1 (DNMT1, 1:1000, Santa Cruz, sc-271729), mouse anti-DNMT3a (1:1000, Santa Cruz, sc-365769), mouse anti-DNMT3b (1:1000, Santa Cruz, sc-376043), and mouse anti-GAPDH (1:5000, Proteintech, 60004-1-Ig). After washing three times in 1× PBS, membranes were incubated with secondary antibodies labeled with HRP for 2 h at RT. The secondary antibodies used were HRP-labeled goat anti-rabbit IgG (1:5000, ZSJQ, ZB2301) and HRP-labeled goat anti-mouse IgG (1:5000, ZSJQ, ZB-2306). After incubation, the membranes were developed with an ECL western blotting substrate kit (Thermo Scientific, 34095). Images were captured and analyzed using GE Image Quant LAS 4000.
Quantitative Polymerase Chain Reaction (q-PCR)
After extraction using TRI reagent, RNA from the hippocampus was measured using a NanoDrop nucleic acid quantitative instrument. Reverse transcription was conducted using ABScript Master Mix (Abclonal, RK20402), and final q-PCR was carried out in accordance with the Taq enzyme (Takara, DRR820A) system. All measurements were repeated three times. Primers are shown in Fig. S2A. The ΔΔCq calculation method was used.
Methyl-Specific PCR and Sequenom Mass Spectrometry (MS)
DNA extracted from the hippocampus was transformed as described above. Based on the sequence of the target gene, primers were designed as shown in Fig. S2B and diluted to 100 μmol/L with DEPC water. After that, a 2× PCR reagent (Tiagen, KT201) was used, and the reaction system was applied according to the manufacturer’s instructions. Amplification products were analyzed by an agarose gel electrophoresis system (Bio-Rad) and a fluorescence chemical imager (GE LAS4000).
Sequenom MS information is shown in Fig. S3. Briefly, 4 μg hippocampal DNA was extracted as noted above; PCR amplification reactions were performed after treatment with NaHSO3; SAP reaction and T-cut/RNase A digestion were performed sequentially after ensuring that the amplified products were electrophoretically clear and with no breaks. The purified samples were finally loaded onto a SpectroCHIP chip and analyzed using a MassARRAY Nano dispenser RS1000 spotter. Sequenom MS was completed in cooperation with Shanghai OE Biotech.
Plasma Hcy Analysis
Total plasma Hcy was measured by high performance liquid chromatography (HPLC) with fluorimetric detection and isocratic elution [30]. HPLC containing a Waters LC2695 instrument, Waters 2475 fluorescence detector, and a Symmetry Shield RP18 column of C18 model (3.9 mm i.d. × 150 mm, 5 μm microparticles) was used for chromatographic separation. The volume of the tested plasma was 200 μL and the final conditions of excitation at 390 nm and emission at 470 nm were used to detect the compounds in the liquid column. Known concentrations of Hcy and N-acetyl-L-cysteine were also used as calibration curves to calculate the Hcy content.
Enzyme-Linked Immunosorbent Assay (ELISA)
ELISA kits were used to detect S-adenosyl methionine (SAM, Arthus Bio, IK00201), S-adenosyl homocysteine (SAH, Arthus Bio, IK00301), and DNA total 5mC (Epigentek, P-1035-96) in the hippocampus homogenate. Briefly, the samples were diluted with antibody at an appropriate ratio, and antibody was added to the standard and samples. The samples were incubated at 37 °C for 1 h. After washing, chromogenic solution was added to each well, and the concentrations of SAM, SAH, and DNA total 5mC were assessed by the microplate reader after incubation and termination solution were added.
Golgi Staining
This step strictly complied with the FD rapid Golgi stain kit (FD Neuro Technologies). Brains were removed quickly after deep sodium pentobarbital anesthesia (1%, 50 mg/kg, i.p.), and the brain surface was rinsed with double-distilled water to remove blood. Brain tissue was then placed in pre-configured A/B solution in the dark for 2 weeks, replaced with new solution on the next day, immersed in solution C in the dark for 4 days, and replaced with a new solution on the next day. After soaking, 100 μm-thick coronal slices were cut on a vibrating slicer (Leica, VT1200) then mounted onto slides. The slices were then rinsed with distilled water and placed in D/E solution for 10 min. After rinsing thoroughly with distilled water, the slices were dehydrated in an alcohol gradient and were cleared with xylene. Finally, the slices were sealed with neutral gum. The density of dendritic spines was counted using ImageJ.
Data Analysis
All of the data are listed as the mean ± SEM. For comparisons between two independent groups, we used an unpaired t-test; for comparisons of more than two groups, we used one-way ANOVA followed by Fisher’s protected least significant difference post-hoc analysis. All analyses were performed using the GraphPad Prism 8.0 statistical package, and P < 0.05 was considered significantly different.
Results
Stress Induces Cognitive Decline by Elevating Plasma Hcy Levels in Rats
Behavioral tests in CUMS and Ctrl rats were conducted at different time points from 0 week to 8 weeks to assess the effect of stress on cognitive function. Compared with the Ctrl group at the same time points, we found a time-dependent decrease in cognitive function in the CUMS group, with significant differences in open field score (unpaired t-test, t = 5.018, P = 0.0037) (Fig. 1A), cognitive index (unpaired t-test, t = 4.176, P = 0.0087) (Fig. 1B), and platform latency (unpaired t-test, t = 3.445, P = 0.0183) (Fig. 1E) from week 4. Significant differences were found in the platform passes (unpaired t-test, t = 7.997, P = 0.0005) (Fig. 1C) and step-down latency (unpaired t-test, t = 6.929, P = 0.0010) (Fig. 1D) from week 6. Plasma Hcy level also increased, showing significant differences from week 4 [ANOVA, F (4, 25) =94.2, P = 0.038] (Fig. 1F). In addition, we found that the open field score (r2 = 0.4287, P < 0.0001) (Fig. 1G), cognitive index (r2 = 0.6929, P < 0.0001) (Fig. 1H), step-down latency (r2 = 0.2213, P = 0.0087) (Fig. 1I), and platform latency (r2 = 0.6058, P < 0.0001) (Fig. 1J) showed significant correlation with plasma Hcy levels. These findings suggest that Hcy is closely associated with the reduction of cognitive functions in rats during chronic stress. Thus, it was particularly important to investigate whether Hcy is involved in the development of cognitive decline. In this part of the study, CUMS rats were gavaged with a vitamin B complex to reduce Hcy, while a high methionine diet was used to elevate Hcy.
Fig. 1.

Stress induces cognitive decline by elevating plasma Hcy levels in rats. A–E Open field score, cognitive index, platform passes, step down latency, and platform latency of Ctrl and CUMS groups at different time points (0 week–8 weeks). Data shown are the mean ± SEM, unpaired t-test, n = 6, **P < 0.01, *P < 0.05 vs Ctrl. F Plasma Hcy levels of the two groups above at different time points (0–8 weeks). F (4, 25) = 94.20, n = 6. G–J Correlation between plasma Hcy levels and behavioral tests. n = 30. K Plasma Hcy levels of Ctrl and CUMS groups as well as Hcy-regulated groups. F (3, 32) = 35.04, n = 9. L–O Open field score, cognitive index, step down latency, and platform latency of the four groups above. l: F (3, 32) = 22.07, m: F (3, 32) = 8.427, n: F (3, 32) = 13.34, o: F (3, 32) = 17.6, n = 9. Data shown are the mean ± SEM, One-way ANOVA, *P < 0.05, **P < 0.01 vs Ctrl, #P < 0.05, ##P < 0.01 vs CUMS.
Plasma Hcy was measured to confirm the effect of the Hcy intervention. We found that VBco gavage significantly reversed the elevated plasma Hcy levels in CUMS rats [ANOVA, F (3, 32) = 35.04, P < 0.0001]; in contrast, the methionine diet significantly the elevated Hcy level, thereby mimicking the effect of stress on Hcy (ANOVA, F (3, 32) = 8.437, P < 0.0001) (Fig. 1K). Behavioral tests showed that the decreases in open field score [ANOVA, F (3, 32) = 22.07, P = 0.004] (Fig. 1L), cognitive index [ANOVA, F (3, 32) = 8.427, P = 0.0021] (Fig. 1M), step-down latency [ANOVA, F (3, 32) = 13.34, P = 0.0003] (Fig. 1N), and elevated platform latency [ANOVA, F (3, 32) = 17.6, P = 0.0001] (Fig. 1O) caused by CUMS were significantly reversed by VBco gavage; in contrast, the high-methionine diet led to behavioral test results similar to those caused by CUMS.
Moreover, plasma Hcy measurement and behavioral tests were also conducted after VBco gavage controls in order to clarify the effect of the gavage procedure. Compared with the Ctrl group, there were no significant differences in plasma Hcy level [ANOVA, F (4, 20) = 41.7, P = 0.5354] (Fig. S4A) and behavioral test results [e.g., open field score (ANOVA, F (4, 20) = 28.5, P = 0.5354) (Fig. S4B) and cognitive index (ANOVA, F (4, 20) = 15.54, P = 0.9230) (Fig. S4C)] after VBco gavage. These data indicate that gavage itself did not affect the Hcy level and behavioral scores. In summary, our findings suggest that Hcy may be one of the causes of stress-related cognitive decline in rats.
Downregulation of BDNF in Hippocampus is Involved in the Development of Cognitive Decline in Stressed Rats
The hippocampal mRNA and protein levels of BDNF, a gene closely related to cognitive function, were measured at different time points (week 0 to week 8). We found that the transcription and expression levels of BDNF decreased with the duration of increased stress, with significant differences in both transcription [ANOVA, F (4, 10) = 9.614, P = 0.0278] (Fig. 2A) and expression levels [ANOVA, F (4, 10) = 26.68, P = 0.0022] (Fig. 2B) from week 6. By week 8, the mRNA and protein levels decreased up to 50% and 70%, respectively. There was a significant negative correlation between plasma Hcy and BDNF transcription levels in the hippocampus (r2 = 0.8058, P < 0.0001) (Fig. 2C). Our results suggest that reduced BDNF levels during stress are closely associated with elevated Hcy. To further reveal whether BDNF is regulated by Hcy, we measured the BDNF transcription and expression levels in the hippocampus after Hcy modulation. We found that, compared with the CUMS, VBco gavage significantly elevated the BDNF transcription [ANOVA, F (3, 8) = 10.39, P = 0.0088] and expression levels [ANOVA, F (3, 8) = 13.73, P = 0.0164], almost to those of the Ctrl group. In contrast, similar to stress, a high-methionine diet in rats significantly reduced the hippocampal BDNF transcription [ANOVA, F (3, 8) = 10.39, P = 0.0289] and expression [ANOVA, F (3, 8) = 13.73, P = 0.0069] levels (Fig. 2D–E). In addition, we used immunofluorescence to visualize the expression of BDNF protein in the brain. We showed that BDNF was abundantly expressed in the hippocampus (CA1, 2, 3, and DG region), and the fluorescence intensity decreased after stress and high-methionine intervention. In contrast, the fluorescence intensity increased after VBco gavage treatment compared with the stress group, which was consistent with the findings from qPCR and WB (left upper two lines of Fig. 2I). Given that neural structures, especially neuronal structures, are the basis of cognition [31], the dendritic structure of hippocampal neurons in different groups of rats was analyzed using Golgi staining. Significant reductions of the hippocampal neuronal dendritic spine density were identified by skeletonization analysis in stress rats [ANOVA, F (5, 17) = 9.987, P = 0.0130]. Similar to stress, a high-methionine diet significantly reduced hippocampal dendritic spine density [ANOVA, F (5, 17) = 9.987, P = 0.0046]. However, the stress-related decrease in dendritic spine density was significantly reversed with VBco treatment, yielding values similar to the Ctrl group [ANOVA, F (5, 17) = 9.987, P = 0.0065] (2F, left lower two lines of Fig. 2I). Taken together, hippocampal BDNF and dendritic spine density may be negatively regulated by Hcy during stress.
Fig. 2.

Downregulation of BDNF in the hippocampus is involved in the development of cognitive decline in stressed rats. A, B Hippocampal BDNF transcription and expression of Ctrl and CUMS groups at different time points (0 week–8 weeks). A: F(4, 10) = 9.614, B: F(4, 10) = 26.68, n = 3. C Correlation between plasma Hcy levels and BDNF transcription in the hippocampus. n = 15. D, E BDNF transcription and expression levels of Ctrl and CUMS groups as well as Hcy-regulated groups. D: F(3, 8) = 10.39, E: F(3, 8) = 13.73, n = 3. F Spine density of hippocampal neurons of Ctrl and CUMS groups as well as Hcy- and BDNF-regulated groups, F(5, 17) = 9.987, CUMS + BDNF AAV: n = 3, other groups: n = 4. G BDNF expression in the hippocampus of Ctrl and CUMS groups as well as BDNF-regulated groups measured by WB. H Plasma Hcy levels of the four groups above, F(3, 16) = 5.887, n = 5. I Upper two lines: distribution of BDNF in the hippocampus measured by immunofluorescence as well as co-labeled image of BDNF and DAPI (× 40 and partial × 200). Lower two lines: neuronal dendritic spines measured by Golgi staining (× 200) and skeletonized images. J–N Open field score, cognitive index, platform passes, step-down latency, and platform latency of the four groups above. J: F (3, 16) = 23.95, K: F (3, 16) = 8.317, L: F (3, 16) = 14.74, M: F (3, 16) = 11.22, N: F (3, 16) = 29.02, n = 3. Data shown are the mean ± SEM, one-way ANOVA, *P < 0.05, **P < 0.01 vs Ctrl; #P < 0.05, ##P < 0.01 vs CUMS.
Given that BDNF is closely associated with cognitive function during stress, to further test whether BDNF supplementation improves cognitive function in stressed rats, we subsequently performed a series of assays after overexpression of BDNF during stress in the hippocampus with the aid of an AAV vector labeled with green fluorescent protein (GFP). Immunofluorescence was assessed to verify successful infection. We showed good co-labeling performance of GFP with BDNF in the hippocampus, which indicated successful infection by AAV (Fig. S4A).
Subsequent results showed that rats infected with AAV-BDNF had significantly elevated BDNF protein levels despite the stress intervention compared with the CUMS group and even the Ctrl group [CUMS+BDNF AAV vs CUMS: ANOVA, F (3, 8) = 108.6, P = 0.0001; CUMS+BDNF AAV vs Ctrl: ANOVA, F (3, 8) = 108.6, P = 0.0014] (Figs. 2G and S4C). This was similar to the results of immunofluorescence (right upper two lines of Fig. 2I). In addition, the density of neuronal dendritic spines in the hippocampus after infection with AAV-BDNF was significantly higher than that of the stress group [ANOVA, F (5, 17) = 9.987, P = 0.0195] and was not significantly different from that of the Ctrl group (Fig. 2F). Simultaneously, the behavioral test results, such as the open field score [ANOVA, F (3, 16) = 23.95, P < 0.0001] (Fig. 2J), cognitive index [ANOVA, F (3, 16) = 8.317, P = 0.0083] (Fig. 2K), platform passes [ANOVA, F (3, 16) = 14.74, P = 0.0003] (Fig. 2L), step-down latency [ANOVA, F (3, 16) =11.22, P = 0.0072] (Fig. 2M), and platform latency [ANOVA, F (3, 16) = 29.02, P = 0.0003] (Fig. 2N) in AAV-BDNF-infected rats showed significant improvements compared with those in the CUMS group, and there were no significant differences from those in the Ctrl group. However, the plasma Hcy level in AAV-BDNF-infected rats remained high and significantly differed from that in the Ctrl group [ANOVA, F (3, 16) = 5.887, P = 0.8226] (Fig. 2H). Our findings indicate that BDNF has a protective effect on the cognitive function of rats during stress and that Hcy may impact the cognitive function of rats by affecting BDNF in the hippocampus.
Hcy Affects DNA Methylation in the Hippocampus and Facilitates 5mC Methylation in Specific BDNF Promoter Sites
To further investigate the molecular mechanisms affecting the transcription and expression of BDNF during stress, follow-up studies related to methylation were conducted. The ratio of SAM to SAH, which is the methylation potential, is an important indicator of the methylation process in an organism [27]. Thus, the changes in methylation potential after stress and Hcy regulation were examined. We found that the hippocampal methylation potential decreased after stress [ANOVA, F (3, 12) = 27.87, P = 0.0036] and a high-methionine diet [ANOVA, F (3, 12) = 27.87, P = 0.0004]; in contrast, the methylation potential was reversed after VBco supplementation, yielding levels similar to the Ctrl group [ANOVA, F (3, 12) = 27.87, P = 0.1036] (Fig. 3A). Next, we examined the extent of total 5mC methylation modification in the hippocampus. Similar to the trend of altered methylation potential, stress [ANOVA, F (3, 12) = 60.96, P = 0.0003] and a high-methionine diet [ANOVA, F (3, 12) = 60.96, P = 0.0003] led to a decrease in hippocampal total 5mC methylation levels, whereas VBco gavage reversed the stress-induced hypomethylation status [ANOVA, F (3, 12) = 60.96, P < 0.0001] (Fig. 3B).
Fig. 3.
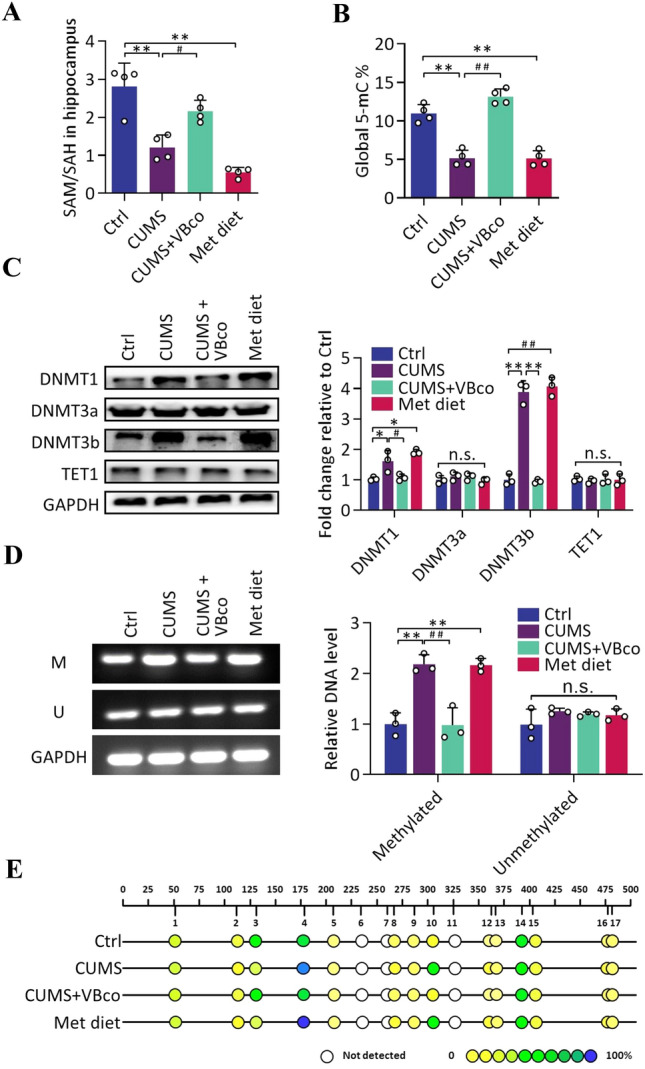
Hcy affects DNA methylation in the hippocampus and facilitates 5mC methylation at specific BDNF promoter sites. A, B The ratio of SAM and SAH (methylation potential) and DNA global 5mC methylation in the hippocampus of Ctrl and CUMS groups as well as Hcy-regulated groups. A: F (3, 12) = 27.87, B: F (3, 12) = 60.96, n = 4. C Expression of methyltransferases and demethylase measured by WB in the hippocampus, and corresponding gray-scale analysis. DNMT1: F (3, 8) = 15.19, DNMT3a: F (3, 8) = 1.201, DNMT3b: F (3, 8) = 136.5, TET1: F (3, 8) = 0.1302, n = 3. D Methylation levels in the BDNF promoter region measured by MSP, and corresponding trans-gray-scale analysis. Methylated: F (3, 8) = 25.87, Unmethylated: F (3, 8) = 1.362, n = 3. E 5mC methylation of CpG islands in the BDNF promoter region detected by Sequenom sequencing. Each dot represents a “GC” locus (color ranging from yellow to blue indicates the average degree of methylation from low to high), n = 6. Data shown are the mean ± SEM, one-way ANOVA, *P < 0.05, **P < 0.01 vs Ctrl; #P < 0.05, ##P < 0.01 vs CUMS.
Since methylation depends on the involvement of methyltransferases and demethylases, the expression levels of methylation-related enzymes were further examined. The results showed that stress [DNMT1: ANOVA, F (3, 8) = 15.19, P = 0.0453; DNMT3b: ANOVA, F (3, 8) = 136.5, P = 0.0003] and a high-methionine diet [DNMT1: ANOVA, F (3, 8) = 15.19, P = 0.0002; DNMT3b: ANOVA, F (3, 8) = 136.5, P = 0.0001] resulted in significantly elevated expression of DNMT1 and DNMT3b in the hippocampus. In contrast, VBco supplementation effectively reversed the stress-induced elevation in the expression levels of DNMT1 and DNMT3b [DNMT1: ANOVA, F (3, 8) = 15.19, P = 0.0415; DNMT3b: ANOVA, F (3, 8) = 136.5, P = 0.0002], resulting in expression levels similar to those in the control group (Fig. 3c). Given that the 5mC methylation regulatory site is often located in the CpG island of the gene promoter region [32], and that the transcription of specific genes is often regulated by the level of methylation in their promoter regions, we next measured the methylation levels of the CpG island within the BDNF promoter. Methyl-specific PCR (MSP) results showed that hippocampal BDNF promoter 5mC methylation significantly increased after stress [ANOVA, F (3, 8) = 25.87, P = 0.0021] and a high-methionine diet [ANOVA, F (3, 8) = 25.87, P = 0.0015], while VBco supplementation was able to reverse the methylation levels to normal [ANOVA, F (3, 8) = 25.87, P = 0.0057] (Fig. 3D). To further measure the methylation of the BDNF promoter region accurately, a follow-up study using Sequenom sequencing was conducted. Seventeen CpG regions with possible 5mC methylation modifications were identified in the selected sequences. Site 4 and site 10 showed increased levels of methylation after stress and a high-methionine diet, whereas VBco supplementation was able to reverse the stress-induced hypermethylation status. Site 3 showed the opposite trend (Fig. 3E). These results suggest that the patterns of methylation modification changes at different loci are not identical and that MSP can reflect a combination of methylation changes at each site.
Hippocampal TET-1 Transfection Decreases Methylation in the BDNF Promoter and Mitigates Cognitive Decline in Stressed Rats
TET1, an enzyme with active demethylation, has been used in recent years [33]. Considering that we found that stress and a high-methionine diet led to elevated methylation in the hippocampal BDNF promoter region in rats, a series of studies using intracranial infection with an AAV that highly expressed TET1 were then performed.
Immunofluorescence was used to confirm the successful infection by AAV. Green fluorescence overlapped with TET1 expression in the hippocampus, showing successful AAV infection (Fig. S4B). WB and qPCR results showed a significant increase in TET1 expression in the hippocampus after TET1 overexpression compared with the stressed group [ANOVA, F (3, 8) = 19.14, P = 0.0036], as well as a significant increase in BDNF transcription [ANOVA, F (3, 8) = 10.97, P = 0.0165] and expression levels [ANOVA, F (3, 8) = 11.37, P = 0.0098] compared to control and virus-infected controls (Figs. 4A–B and S4D). Similar to WB, immunofluorescence showed that rats with TET1 overexpression showed higher BDNF expression in the hippocampus despite the stress intervention (upper two lines of Fig. 4D). In addition, Golgi staining showed a significant increase in hippocampal dendritic spine density after overexpression of TET1 in the hippocampus [ANOVA, F (3, 12) = 11.48, P = 0.0119], which may be associated with the elevated BDNF levels (Fig. 4E and lower two lines of Fig. 4D). We then examined the methylation of the BDNF promoter region and showed that TET1 overexpression reversed the stress-induced hypermethylation [ANOVA, F (3, 8) = 34.77, P = 0.0022], while the methylation level of the control virus-transfected group remained high and did not differ from that of the stressed group [ANOVA, F (3, 8) = 34.77, P = 0.5399] (Fig. 4F). However, plasma Hcy remained at a high level after overexpression of TET1 [ANOVA, F (3, 8) = 30.02, P = 0.0004] and stress intervention [ANOVA, F (3, 8) = 30.02, P = 0.0018] (Fig. 4C). Finally, the behavioral effects of TET1 overexpression were examined. Compared with the control group and virus-infected control group, rats overexpressing TET1 had a significantly higher open field score [ANOVA, F (3, 16) = 51.46, P < 0.0001] (Fig. 4G), cognitive index [ANOVA, F (3, 16) = 22.75, P < 0.0001] (Fig. 4H), and step-down latency [ANOVA, F (3, 16) = 8.354, P = 0.0054] (Fig. 4I), and a significantly shorter platform latency [ANOVA, F (3, 16) = 17.97, P = 0.0010] (Fig. 4J). These results suggest that the decrease in hippocampal BDNF during stress may be due to the increased level of promoter methylation.
Fig. 4.
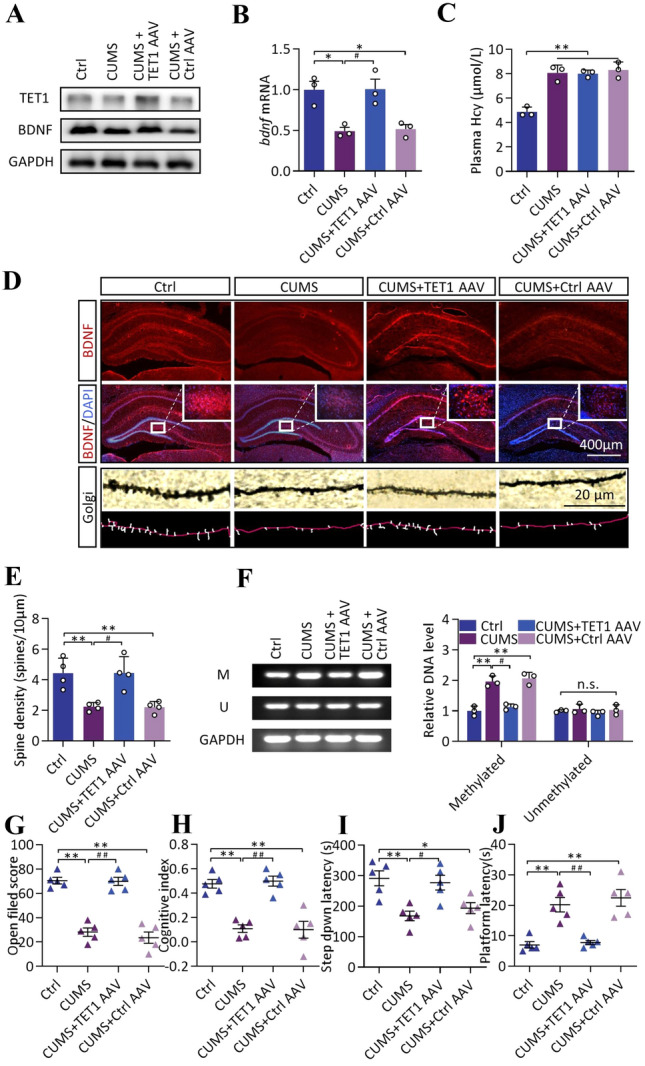
Hippocampal TET-1 transfection decreases methylation in the BDNF promoter and mitigates cognitive decline in stressed rats. A Expression of TET1 and BDNF measured by WB in the hippocampus of Ctrl and CUMS groups as well as TET1-regulated groups. B Transcription of BDNF measured by q-PCR in the hippocampus of above rats, F (3, 8) = 10.97, n = 3. C Plasma Hcy levels in the above rats, F (3, 8) = 30.02, n = 3. D Upper two lines: distribution of BDNF in the hippocampus measured by immunofluorescence as well as co-labeled image of BDNF and DAPI (× 40 and partial × 200). Lower two lines: neuronal dendritic spines measured by Golgi staining (× 200) and skeletonized images. E Spine density of neurons in the hippocampus of the above rats, F (3, 12) = 11.48, n = 4. F Methylation levels in the BDNF promoter region measured by MSP, and corresponding trans-gray-scale analysis. Methylated: F (3, 8) = 34.77, Unmethylated: F (3, 8) = 0.666, n = 3. G–I Open field score, cognitive index, step down latency, and platform latency of the four groups above. G: F (3, 16) = 51.46, H: F (3, 16) = 22.75, I: F (3, 16) = 8.354, J: F (3, 16) = 17.97, n = 5. Data shown are the mean ± SEM, one-way ANOVA, *P < 0.05, **P < 0.01 vs Ctrl; #P < 0.05, ##P < 0.01 vs CUMS.
Taken together, our findings suggest that BDNF promoter hypermethylation results in lower transcription and expression levels. In addition, TET1 downregulates the degree of BDNF promoter methylation, thereby promoting BDNF transcription and expression and contributing to recovery from stress-induced cognitive impairment.
Discussion
As a response to stimulation by internal and external environmental factors, stress involves systemic reactions in several organ systems, such as the nervous, endocrine, and immune systems [34]. Our study revealed that BDNF transcriptional repression in the hippocampus of rats is regulated by 5mC methylation modifications in response to stressful stimuli, which ultimately causes abnormal cognitive function. The above process may be caused by the stress-related elevation in Hcy levels.
There has been a longstanding interest in the effects of stress on cognitive function [35]. Our present results show that a longer duration of stress is associated with decreasing trends in learning memory for novelty and spatial location and the decreased levels were more stable at 6 weeks–8 weeks. These data are consistent with a previous study that showed that 6 weeks of CUMS induces cognitive decline as measured by behavioral tests such as OFT and MWM [36, 37]. However, 3 weeks of chronic stress does not affect spatial learning memory [38], which highlights the time-dependence of the effects of stress on cognitive function. According to epidemiological studies, stress is also an important cause of several neuropsychiatric disorders, such as depression, Alzheimer's disease (AD), and even schizophrenia [39], and cognitive impairment is one of the possible clinical manifestations of these diseases. The above illustrates the importance of the effects of chronic stress on cognition.
Our study also highlights the role of Hcy, a small amino-acid involved in vital processes such as energy metabolism and methylation [40], in the process of stress-induced cognitive dysfunction in rats. Epidemiological studies have suggested that Hcy is one of the critical causes of cognitive impairment. Hcy is considered to be a risk factor in the occurrence of neurodegenerative diseases [41] such as AD [42], and supplementation with B vitamins can improve some of the cognitive symptoms of AD patients [43]. Studies have also found a negative correlation between plasma Hcy level and Cambridge cognitive score in the elderly [44]. Recent studies suggest that the effect of Hcy on cerebrovascular injury may be one of the important reasons for the occurrence of cognitive impairment induced by Hcy. Taking cerebral small vascular disease (CSVD) as an example, vascular endothelial dysfunction is an important factor in its occurrence [45, 46]. Clinical studies have found that the plasma Hcy level is strongly correlated with decreased brain volume in patients with CSVD [47]. In addition, enhanced Hcy reduction strategies may be necessary to reduce the risk and progression of CSVD [48]. This indicates that vascular injury might be one of the reasons for the role of Hcy in cognitive impairment. However, the role of Hcy cannot be fully revealed from the perspective of vascular injury alone. Zhou et al. found that increased Hcy is associated with decreased autonomous activity in CUMS rats, and folic acid supplementation alleviates these symptoms [49]. In addition, increased Hcy in CUMS may be closely related to 5-HT metabolism in related brain regions, and lack of a methyl donor may also lead to stress-like effects [50]. These studies suggest that methylation may be a potential mechanism by which Hcy functions.
Neurotrophic factors are essential for well-maintained memory in mammals [51]. As a neurotrophic factor, BDNF plays an important role in maintaining normal brain function, such as the formation of synapses, growth, and the differentiation and migration of neurons [52]. Our study showed that both stress and high levels of Hcy caused a decrease in BDNF transcription and expression in the hippocampus, reduced dendritic spine density, and induced abnormal cognitive function. However, overexpression of BDNF in the hippocampus of rats increased the density of dendritic spines and improved the cognitive function of rats without affecting the high Hcy state caused by stress. These findings suggest that BDNF may be a downstream target molecule of Hcy in the process of stress-induced cognitive decline. In addition, the inhibition of BDNF transcription results from chronic isolation stress [53], chronic social defeat stress [54], and chronic mild stress, [55] suggesting that negative regulation of BDNF by stress is widespread in a variety of chronic stress processes. Moreover, interference with BDNF in the dentate gyrus triggers depressive-like behavior and affects neuronal regeneration in rats [56], and knockdown of BDNF in mice inhibits neuronal plasticity and induces the onset of psychiatric symptoms such as anxiety in mice [57]. These findings indicate that BDNF is a major factor in the pathogenesis of mental disorders. Ma et al. reported that the expression of BDNF could be regulated by methylation [58], while another study showed that the administration of methyl donors could block the negative effects of Hcy during stress [49]. Accordingly, we have focused to determine whether Hcy affects BDNF transcription through methylation.
The Hcy metabolite SAM is a universal methyl donor, and SAH is an important methyltransferase inhibitor [59]. The ratio of SAM to SAH, also known as methylation potential, is an important indicator of the methylation process [27]. We found that CUMS and a high-methionine diet led to a reduced methylation potential and total methylation levels in the rat hippocampus, in conjunction with elevated DNMT1 and DNMT3b expression and concomitant elevated methylation in the BDNF promoter region. Other reports have also pointed out that maternal stress and unpredictable stress lead to increased methylation in the BDNF promoter and decreased expression in the hippocampus and mPFC of the offspring rats, respectively [21, 60]. Moreover, elevated dorsal hippocampal methylation levels have also been reported in PTSD rats [21]. In addition, stressful events are associated with elevated BDNF promoter methylation levels in the blood of patients with depression [61]. The above study suggests that methylation may be a target of stress-regulated gene expression. Moreover, it has been pointed out that Hcy is closely associated with methylation modifications and that Hcy clearance helps to protect the maintenance of methylation [59]. Hcy also elevates the methylation levels of the GPX4 promoter and affect its transcription during oxidative stress [62], which indicates that methylation is a mechanism by which Hcy affects gene transcription. Taken together, methylation may be one of the key targets of Hcy in regulating BDNF transcription during stress.
However, the inconsistency between overall methylation and specific gene (BDNF) promoter methylation levels in stress and with high-methionine diets is intriguing. We propose the following hypothesis: stress and a high-methionine diet lead to elevated expression levels of methyltransferases (DNMTs); DNMTs promote SAM transmethylation, leading to its depletion, and SAH accumulates in large amounts as a product of the transmethylation reaction; SAH inhibits the methylation reaction, which eventually leads to a lower methylation potential and a lower total methylation level. Given that B vitamins are important coenzymes for Hcy isomerization metabolism, providing B vitamins during stress can reduce the accumulation of Hcy and methionine, thereby decreasing the level of DNMTs and returning the methylation potential to normal. DNA DNMTs are 5mC methyltransferases that maintain the environment of the CpG site [63]. Maternal separation stress has been reported to increase DNMT activity in the hippocampus, while intrahippocampal DNMT inhibition induces antidepressant-like behavior [64]. This indicates that stress may be a factor in the elevation of DNMT and may be involved in emotion neural function regulation. In addition, some studies have focused on the effect of Hcy on DNMT. Rats with elevated Hcy levels due to a high-methionine diet have elevated levels of DNMT3a transcripts [65]. Moreover, reducing the methionine levels in cervical cancer cells results in reduced levels of DNMT3a and 3b in cells [66]. In addition, in an atherosclerosis study, Hcy has been found to increase DNMT levels in mice [67]. These studies suggest that Hcy may be a factor in the upregulation of DNMTs. On the other hand, HHcy may interfere with DNA methylation by affecting SAH/SAM levels. In animal models, HHcy have been associated with high SAH and low SAM/SAH ratios, but changes in SAM are not consistent in different models [68, 69]. Taken together with our results, stress and methionine lead to elevated DNMT, a reduced methylation potential, and lower overall methylation. However, the specific mechanism needs to be further investigated. Of note, it has been found that the overall methylation and specific gene methylation trends under the same conditions are not equivalent, and the genome-wide hypomethylation status can be accompanied by the hypermethylation status of some genes [70]. This is similar to our results, suggesting that epigenetic modification of BDNF is selective and not consistent with the overall degree of methylation.
Since DNMT3b and DNMT1 knockdown in the hippocampus have detrimental effects on synaptic function and recognition memory in animals [71, 72], overexpression of TET1 was chosen for the subsequent modulation of methylation. TET1 is an enzyme that acts in the mammalian brain; it is involved in the regulation of cognitive functions and exerts 5mC demethylation [73, 74]. We found that overexpression of TET1 in the hippocampus successfully downregulated the BDNF promoter methylation levels in stressed rats without affecting Hcy; this was accompanied by increased BDNF transcription and expression levels and improved behavioral performance. These results suggest that methylation is a downstream target of Hcy during stress, and intervention by TET1 may be a potential breakthrough to control stress-induced cognitive impairment. However, as a widespread demethylase of 5mC methylation, TET1 may also act on other genes while affecting BDNF methylation. The regulation of methylation by TET1 is still wide and nonspecific, and we cannot exclude the possibility that Hcy affects BDNF transcriptional regulation by influencing the methylation of other genes. Further studies should attempt specific interference with BDNF promoter methylation to provide more accurate results about the mechanism of Hcy in BDNF transcriptional regulation. In addition, the Sequenom technology used in our study has the advantage of high accuracy, but at the same time, due to its narrow coverage, it cannot fully reflect the changes of the BDNF promoter. Methods with higher coverage should be considered in subsequent studies.
In summary, we found that chronic stress increases Hcy levels in rats, inhibits BDNF transcription and protein expression by upregulating methylation in the hippocampal BDNF promoter region, and leads to cognitive decline. These findings provide new insights into the mechanisms underlying cognitive decline caused by stress. In addition, we propose that intervention in methylation may be a new target for controlling stress-induced cognitive impairment. However, the specific intervention methods need to be further studied.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
This work was supported by Grants from the National Natural Science Foundation of China (31771290 and 81702454), the Beijing National Science Foundation (5222033), the Military Logistics Scientific Research Foundation of China (BWS17J027) and the National Basic Scientific Research Program of China (JCKY2019548B001).
Conflict of interest
The authors listed in this article have no known competing financial interests or personal relationships that may affect the reported work.
Footnotes
Shi-Da Wang and Xue Wang contributed equally to this work.
Contributor Information
Fang Xie, Email: vancoxie@sina.com.
Ling-Jia Qian, Email: newjia@vip.sina.com.
References
- 1.de Kloet ER, Joëls M, Holsboer F. Stress and the brain: From adaptation to disease. Nat Rev Neurosci. 2005;6:463–475. doi: 10.1038/nrn1683. [DOI] [PubMed] [Google Scholar]
- 2.Sandi C, Haller J. Stress and the social brain: Behavioural effects and neurobiological mechanisms. Nat Rev Neurosci. 2015;16:290–304. doi: 10.1038/nrn3918. [DOI] [PubMed] [Google Scholar]
- 3.Sapolsky RM. Stress and the brain: Individual variability and the inverted-U. Nat Neurosci. 2015;18:1344–1346. doi: 10.1038/nn.4109. [DOI] [PubMed] [Google Scholar]
- 4.Liu QJ, Liu Y, Leng XC, Han JF, Xia F, Chen H. Impact of chronic stress on attention control: Evidence from behavioral and event-related potential analyses. Neurosci Bull. 2020;36:1395–1410. doi: 10.1007/s12264-020-00549-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kessler RC, Birnbaum H, Demler O, Falloon IRH, Gagnon E, Guyer M, et al. The prevalence and correlates of nonaffective psychosis in the national comorbidity survey replication (NCS-R) Biol Psychiatry. 2005;58:668–676. doi: 10.1016/j.biopsych.2005.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Edmondson D, von Känel R. Post-traumatic stress disorder and cardiovascular disease. Lancet Psychiatry. 2017;4:320–329. doi: 10.1016/S2215-0366(16)30377-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McEwen BS, Weiss JM, Schwartz LS. Selective retention of corticosterone by limbic structures in rat brain. Nature. 1968;220:911–912. doi: 10.1038/220911a0. [DOI] [PubMed] [Google Scholar]
- 8.McEwen BS. Stress and hippocampal plasticity. Annu Rev Neurosci. 1999;22:105–122. doi: 10.1146/annurev.neuro.22.1.105. [DOI] [PubMed] [Google Scholar]
- 9.Greenberg MS, Tanev K, Marin MF, Pitman RK. Stress, PTSD, and dementia. Alzheimers Dement. 2014;10:S155–S165. doi: 10.1016/j.jalz.2014.04.008. [DOI] [PubMed] [Google Scholar]
- 10.Machado A, Herrera AJ, de Pablos RM, Espinosa-Oliva AM, Sarmiento M, Ayala A, et al. Chronic stress as a risk factor for Alzheimer's disease. Rev Neurosci. 2014;25:785–804. doi: 10.1515/revneuro-2014-0035. [DOI] [PubMed] [Google Scholar]
- 11.Polis B, Samson AO. Neurogenesis versus neurodegeneration: The broken balance in Alzheimer's disease. Neural Regen Res. 2021;16:496–497. doi: 10.4103/1673-5374.293138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sarahian N, Sahraei H, Zardooz H, Alibeik H, Sadeghi B. Effect of memantine administration within the nucleus accumbens on changes in weight and volume of the brain and adrenal gland during chronic stress in female mice. Modares J Med Sci Pathobiol. 2014;17:71–82. [Google Scholar]
- 13.Lupien SJ, Lepage M. Stress, memory, and the Hippocampus: Can't live with it, can't live without it. Behav Brain Res. 2001;127:137–158. doi: 10.1016/S0166-4328(01)00361-8. [DOI] [PubMed] [Google Scholar]
- 14.Kinnally EL, Feinberg C, Kim D, Ferguson K, Leibel R, Coplan JD, et al. DNA methylation as a risk factor in the effects of early life stress. Brain Behav Immun. 2011;25:1548–1553. doi: 10.1016/j.bbi.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xiang D, Xiao JW, Fu LY, Yao LH, Wan QR, Xiao L, et al. DNA methylation of the Tacr2 gene in a CUMS model of depression. Behav Brain Res. 2019;365:103–109. doi: 10.1016/j.bbr.2019.01.059. [DOI] [PubMed] [Google Scholar]
- 16.Yu Y, Wang MJ, Yang XH, Sui M, Zhang T, Liang J, et al. Association between DNA methylation of SORL1 5′-flanking region and mild cognitive impairment in type 2 diabetes mellitus. Ann Endocrinol (Paris) 2016;77:625–632. doi: 10.1016/j.ando.2016.02.008. [DOI] [PubMed] [Google Scholar]
- 17.Shinagawa S, Kobayashi N, Nagata T, Kusaka A, Yamada H, Kondo K, et al. DNA methylation in the NCAPH2/LMF2 promoter region is associated with hippocampal atrophy in Alzheimer's disease and amnesic mild cognitive impairment patients. Neurosci Lett. 2016;629:33–37. doi: 10.1016/j.neulet.2016.06.055. [DOI] [PubMed] [Google Scholar]
- 18.Notaras M, van den Buuse M. Neurobiology of BDNF in fear memory, sensitivity to stress, and stress-related disorders. Mol Psychiatry. 2020;25:2251–2274. doi: 10.1038/s41380-019-0639-2. [DOI] [PubMed] [Google Scholar]
- 19.McEwen BS. Physiology and neurobiology of stress and adaptation: Central role of the brain. Physiol Rev. 2007;87:873–904. doi: 10.1152/physrev.00041.2006. [DOI] [PubMed] [Google Scholar]
- 20.Boersma GJ, Lee RS, Cordner ZA, Ewald ER, Purcell RH, Moghadam AA, et al. Prenatal stress decreases Bdnf expression and increases methylation of Bdnf exon IV in rats. Epigenetics. 2014;9:437–447. doi: 10.4161/epi.27558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roth TL, Zoladz PR, Sweatt JD, Diamond DM. Epigenetic modification of hippocampal Bdnf DNA in adult rats in an animal model of post-traumatic stress disorder. J Psychiatr Res. 2011;45:919–926. doi: 10.1016/j.jpsychires.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller AL. The methionine-homocysteine cycle and its effects on cognitive diseases. Altern Med Rev. 2003;8:7–19. [PubMed] [Google Scholar]
- 23.Zhao Y, Wu SQ, Gao XJ, Zhang ZQ, Gong JB, Zhan R, et al. Inhibition of cystathionine β-synthase is associated with glucocorticoids over-secretion in psychological stress-induced hyperhomocystinemia rat liver. Cell Stress Chaperones. 2013;18:631–641. doi: 10.1007/s12192-013-0416-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xie F, Zhao Y, Ma J, Gong JB, Wang SD, Zhang L, et al. The involvement of homocysteine in stress-induced Aβ precursor protein misprocessing and related cognitive decline in rats. Cell Stress Chaperones. 2016;21:915–926. doi: 10.1007/s12192-016-0718-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Setién-Suero E, Suárez-Pinilla M, Suárez-Pinilla P, Crespo-Facorro B, Ayesa-Arriola R. Homocysteine and cognition: A systematic review of 111 studies. Neurosci Biobehav Rev. 2016;69:280–298. doi: 10.1016/j.neubiorev.2016.08.014. [DOI] [PubMed] [Google Scholar]
- 26.James SJ, Melnyk S, Pogribna M, Pogribny IP, Caudill MA. Elevation in S-adenosylhomocysteine and DNA hypomethylation: Potential epigenetic mechanism for homocysteine-related pathology. J Nutr. 2002;132:2361S–2366S. doi: 10.1093/jn/132.8.2361S. [DOI] [PubMed] [Google Scholar]
- 27.Mandaviya PR, Stolk L, Heil SG. Homocysteine and DNA methylation: A review of animal and human literature. Mol Genet Metab. 2014;113:243–252. doi: 10.1016/j.ymgme.2014.10.006. [DOI] [PubMed] [Google Scholar]
- 28.Qiao H, Li MX, Xu C, Chen HB, An SC, Ma XM. Dendritic spines in depression: What we learned from animal models. Neural Plast. 2016;2016:8056370. doi: 10.1155/2016/8056370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu J, Wang HT, Chen YR, Yan LY, Han YY, Liu LY, et al. The Joubert syndrome gene arl13b is critical for early cerebellar development in zebrafish. Neurosci Bull. 2020;36:1023–1034. doi: 10.1007/s12264-020-00554-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang XX, Liu W, Wu L, Zhan R, Jin BY, Qian LJ. A neuroendocrine mechanism of co-morbidity of depression-like behavior and myocardial injury in rats. PLoS One. 2014;9:e88427. doi: 10.1371/journal.pone.0088427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Forrest MP, Parnell E, Penzes P. Dendritic structural plasticity and neuropsychiatric disease. Nat Rev Neurosci. 2018;19:215–234. doi: 10.1038/nrn.2018.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y, et al. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302:890–893. doi: 10.1126/science.1090842. [DOI] [PubMed] [Google Scholar]
- 33.Wu SC, Zhang Y. Active DNA demethylation: Many roads lead to Rome. Nat Rev Mol Cell Biol. 2010;11:607–620. doi: 10.1038/nrm2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Joëls M, Baram TZ. The neuro-symphony of stress. Nat Rev Neurosci. 2009;10:459–466. doi: 10.1038/nrn2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chrousos GP, Gold PW. The concepts of stress and stress system disorders. Overview of physical and behavioral homeostasis. JAMA. 1992;267:1244–1252. doi: 10.1001/jama.1992.03480090092034. [DOI] [PubMed] [Google Scholar]
- 36.Song AQ, Gao B, Fan JJ, Zhu YJ, Zhou J, Wang YL, et al. NLRP1 inflammasome contributes to chronic stress-induced depressive-like behaviors in mice. J Neuroinflammation. 2020;17:178. doi: 10.1186/s12974-020-01848-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cortese A, Delgado-Morales R, Almeida OFX, Romberg C. The Arctic/Swedish APP mutation alters the impact of chronic stress on cognition in mice. Eur J Neurosci. 2019;50:2773–2785. doi: 10.1111/ejn.14500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zain MA, Pandy V, Majeed ABA, Wong WF, Mohamed Z. Chronic restraint stress impairs sociability but not social recognition and spatial memoryin C57BL/6J mice. Exp Anim. 2019;68:113–124. doi: 10.1538/expanim.18-0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Atrooz F, Liu HS, Salim S. Stress, psychiatric disorders, molecular targets, and more. Prog Mol Biol Transl Sci. 2019;167:77–105. doi: 10.1016/bs.pmbts.2019.06.006. [DOI] [PubMed] [Google Scholar]
- 40.Bosevski M, Zlatanovikj N, Petkoska D, Gjorgievski A, Lazarova E, Stojanovska L. Plasma homocysteine in patients with coronary and carotid artery disease: A case control study. Pril (Makedon Akad Nauk Umet Odd Med Nauki) 2020;41:15–22. doi: 10.2478/prilozi-2020-0019. [DOI] [PubMed] [Google Scholar]
- 41.Kaplan P, Tatarkova Z, Sivonova MK, Racay P, Lehotsky J. Homocysteine and mitochondria in cardiovascular and cerebrovascular systems. Int J Mol Sci. 2020;21:E7698. doi: 10.3390/ijms21207698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen CS, Chou MC, Yeh YC, Yang YH, Lai CL, Yen CF, et al. Plasma homocysteine levels and major depressive disorders in Alzheimer disease. Am J Geriatr Psychiatry. 2010;18:1045–1048. doi: 10.1097/JGP.0b013e3181dba6f1. [DOI] [PubMed] [Google Scholar]
- 43.Gariballa S. Testing homocysteine-induced neurotransmitter deficiency, and depression of mood hypothesis in clinical practice. Age Ageing. 2011;40:702–705. doi: 10.1093/ageing/afr086. [DOI] [PubMed] [Google Scholar]
- 44.Seshadri S, Beiser A, Selhub J, Jacques PF, Rosenberg IH, D'Agostino RB, et al. Plasma homocysteine as a risk factor for dementia and Alzheimer's disease. N Engl J Med. 2002;346:476–483. doi: 10.1056/NEJMoa011613. [DOI] [PubMed] [Google Scholar]
- 45.Zhang CE, Wong SM, van de Haar HJ, Staals J, Jansen JFA, Jeukens CRLPN, et al. Blood-brain barrier leakage is more widespread in patients with cerebral small vessel disease. Neurology. 2017;88:426–432. doi: 10.1212/WNL.0000000000003556. [DOI] [PubMed] [Google Scholar]
- 46.Schreiber S, Bueche CZ, Garz C, Braun H. Blood brain barrier breakdown as the starting point of cerebral small vessel disease? - New insights from a rat model. Exp Transl Stroke Med. 2013;5:4. doi: 10.1186/2040-7378-5-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ji YF, Li XY, Teng ZJ, Li XS, Jin W, Lv PY. Homocysteine is associated with the development of cerebral small vessel disease: Retrospective analyses from neuroimaging and cognitive outcomes. J Stroke Cerebrovasc Dis. 2020;29:105393. doi: 10.1016/j.jstrokecerebrovasdis.2020.105393. [DOI] [PubMed] [Google Scholar]
- 48.Cao YZ, Su N, Zhang DD, Zhou LX, Yao M, Zhang SY, et al. Correlation between total homocysteine and cerebral small vessel disease: A Mendelian randomization study. Eur J Neurol. 2021;28:1931–1938. doi: 10.1111/ene.14708. [DOI] [PubMed] [Google Scholar]
- 49.Zhou Y, Cong Y, Liu H. Folic acid ameliorates depression-like behaviour in a rat model of chronic unpredictable mild stress. BMC Neurosci. 2020;21:1. doi: 10.1186/s12868-020-0551-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Javelot H, Messaoudi M, Jacquelin C, Bisson JF, Rozan P, Nejdi A, et al. Behavioral and neurochemical effects of dietary methyl donor deficiency combined with unpredictable chronic mild stress in rats. Behav Brain Res. 2014;261:8–16. doi: 10.1016/j.bbr.2013.11.047. [DOI] [PubMed] [Google Scholar]
- 51.Allen SJ, Watson JJ, Shoemark DK, Barua NU, Patel NK. GDNF, NGF and BDNF as therapeutic options for neurodegeneration. Pharmacol Ther. 2013;138:155–175. doi: 10.1016/j.pharmthera.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 52.Tapia-Arancibia L, Aliaga E, Silhol M, Arancibia S. New insights into brain BDNF function in normal aging and Alzheimer disease. Brain Res Rev. 2008;59:201–220. doi: 10.1016/j.brainresrev.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 53.Zaletel I, Filipović D, Puškaš N. Hippocampal BDNF in physiological conditions and social isolation. Rev Neurosci. 2017;28:675–692. doi: 10.1515/revneuro-2016-0072. [DOI] [PubMed] [Google Scholar]
- 54.Wook Koo J, Labonté B, Engmann O, Calipari ES, Juarez B, Lorsch Z, et al. Essential role of mesolimbic brain-derived neurotrophic factor in chronic social stress-induced depressive behaviors. Biol Psychiatry. 2016;80:469–478. doi: 10.1016/j.biopsych.2015.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tornese P, Sala N, Bonini D, Bonifacino T, la Via L, Milanese M, et al. Chronic mild stress induces anhedonic behavior and changes in glutamate release, BDNF trafficking and dendrite morphology only in stress vulnerable rats. The rapid restorative action of ketamine. Neurobiol Stress. 2019;10:100160. doi: 10.1016/j.ynstr.2019.100160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Taliaz D, Stall N, Dar DE, Zangen A. Knockdown of brain-derived neurotrophic factor in specific brain sites precipitates behaviors associated with depression and reduces neurogenesis. Mol Psychiatry. 2010;15:80–92. doi: 10.1038/mp.2009.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhu GB, Sun XF, Yang Y, Du Y, Lin YH, Xiang JM, et al. Reduction of BDNF results in GABAergic neuroplasticity dysfunction and contributes to late-life anxiety disorder. Behav Neurosci. 2019;133:212–224. doi: 10.1037/bne0000301. [DOI] [PubMed] [Google Scholar]
- 58.Ma DK, Jang MH, Guo JU, Kitabatake Y, Chang ML, Pow-Anpongkul N, et al. Neuronal activity-induced Gadd45b promotes epigenetic DNA demethylation and adult neurogenesis. Science. 2009;323:1074–1077. doi: 10.1126/science.1166859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shen W, Gao C, Cueto R, Liu L, Fu HF, Shao Y, et al. Homocysteine-methionine cycle is a metabolic sensor system controlling methylation-regulated pathological signaling. Redox Biol. 2020;28:101322. doi: 10.1016/j.redox.2019.101322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Niknazar S, Nahavandi A, Peyvandi AA, Peyvandi H, Zare Mehrjerdi F, Karimi M. Effect of maternal stress prior to conception on hippocampal BDNF signaling in rat offspring. Mol Neurobiol. 2017;54:6436–6445. doi: 10.1007/s12035-016-0143-5. [DOI] [PubMed] [Google Scholar]
- 61.Song YX, Miyaki K, Suzuki T, Sasaki Y, Tsutsumi A, Kawakami N, et al. Altered DNA methylation status of human brain derived neurotrophis factor gene could be useful as biomarker of depression. Am J Med Genet B Neuropsychiatr Genet. 2014;165B:357–364. doi: 10.1002/ajmg.b.32238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen C, Liu L, Shu YQ, Jing P, Lu Y, Zhang XX, et al. Blockade of HCN2 channels provides neuroprotection against ischemic injury via accelerating autophagic degradation in hippocampal neurons. Neurosci Bull. 2020;36:875–894. doi: 10.1007/s12264-020-00513-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang Y, Wu HD, Sun ZS. The biological basis of sexual orientation: How hormonal, genetic, and environmental factors influence to whom we are sexually attracted. Front Neuroendocrinol. 2019;55:100798. doi: 10.1016/j.yfrne.2019.100798. [DOI] [PubMed] [Google Scholar]
- 64.Uchida S, Hara K, Kobayashi A, Otsuki K, Yamagata H, Hobara T, et al. Epigenetic status of gdnf in the ventral striatum determines susceptibility and adaptation to daily stressful events. Neuron. 2011;69:359–372. doi: 10.1016/j.neuron.2010.12.023. [DOI] [PubMed] [Google Scholar]
- 65.Jiang YD, Sun T, Xiong JT, Cao J, Li GZ, Wang SR. Hyperhomocysteinemia-mediated DNA hypomethylation and its potential epigenetic role in rats. Acta Biochim Biophys Sin (Shanghai) 2007;39:657–667. doi: 10.1111/j.1745-7270.2007.00327.x. [DOI] [PubMed] [Google Scholar]
- 66.Poomipark N, Flatley JE, Hill MH, Mangnall B, Azar E, Grabowski P, et al. Methyl donor status influences DNMT expression and global DNA methylation in cervical cancer cells. Asian Pac J Cancer Prev. 2016;17:3213–3222. [PubMed] [Google Scholar]
- 67.Ma XF, Jiang ZS, Wang Z, Zhang ZH. Administration of metformin alleviates atherosclerosis by promoting H2S production via regulating CSE expression. J Cell Physiol. 2020;235:2102–2112. doi: 10.1002/jcp.29112. [DOI] [PubMed] [Google Scholar]
- 68.Esse R, Imbard A, Florindo C, Gupta S, Quinlivan EP, Davids M, et al. Protein arginine hypomethylation in a mouse model of cystathionine β-synthase deficiency. FASEB J. 2014;28:2686–2695. doi: 10.1096/fj.13-246579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Perła-Kaján J, Jakubowski H. Dysregulation of epigenetic mechanisms of gene expression in the pathologies of hyperhomocysteinemia. Int J Mol Sci. 2019;20:3140. doi: 10.3390/ijms20133140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li M, Du W, Shao F, Wang WW. Cognitive dysfunction and epigenetic alterations of the BDNF gene are induced by social isolation during early adolescence. Behav Brain Res. 2016;313:177–183. doi: 10.1016/j.bbr.2016.07.025. [DOI] [PubMed] [Google Scholar]
- 71.Sun W, Kong QN, Zhang M, Mi X, Sun XM, Yu M, et al. Virus-mediated Dnmt1 and Dnmt3a deletion disrupts excitatory synaptogenesis and synaptic function in primary cultured hippocampal neurons. Biochem Biophys Res Commun. 2020;526:361–367. doi: 10.1016/j.bbrc.2020.03.094. [DOI] [PubMed] [Google Scholar]
- 72.Kong QN, Yu M, Zhang M, Wei C, Gu HT, Yu SY, et al. Conditional Dnmt3b deletion in hippocampal dCA1 impairs recognition memory. Mol Brain. 2020;13:42. doi: 10.1186/s13041-020-00574-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liu XS, Wu H, Ji X, Stelzer Y, Wu XB, Czauderna S, et al. Editing DNA methylation in the mammalian genome. Cell. 2016;167:233–247.e17. doi: 10.1016/j.cell.2016.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Guo JU, Su YJ, Zhong C, Ming GL, Song HJ. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell. 2011;145:423–434. doi: 10.1016/j.cell.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.