

Abstract
Diverse host factors/phenotypes may exacerbate or diminish biological responses induced by air pollutant exposure. We lack an understanding of biological indicators of environmental exposures which culminate in a physiological response versus those which lead to adversity. Variations in response phenotype might arise centrally and/or at the local tissue-level. In addition to genetic differences, the current evidence supports the roles of preexisting cardiopulmonary diseases, diabetes, diet, adverse prenatal environments, neurobehavioral disorders, childhood infections, microbiome, sex and psychosocial stressors in modifying the susceptibility to air pollutant exposures. Animal models of human diseases, obesity, nutritional inadequacies, and neurobehavioral conditions have been compared with healthy controls to understand the causes of variations in susceptibility. While psychosocial stressors have been associated with increased susceptibility to air pollutant effects, the contribution of neuroendocrine stress pathways in mediating these effects are just emerging. The new findings of neuroendocrine activation leading to systemic metabolic and immunological effects of air pollutants, and the potential contribution to allostatic load, emphasizes the consideration of these mechanisms into susceptibility. Variations in susceptibility to air pollution health effects are likely to underlie host genetic and physiological conditions in concert with disrupted neuroendocrine circuitry that alter physiological stability under the influence of stressors.
Keywords: susceptibility variations, air pollution, neuroendocrine, metabolic diseases, allostatic load, animal models
Introduction
The World Health Organization report of May 2018 shows that ambient (outdoor) and household air pollution causes nearly 7 million annual deaths worldwide. Air pollution accounts for nearly 70% of all deaths attributed to environmental causes.1 However, for decades we have recognized that not every individual that is exposed to air pollutants or other environmental stressors will die or develop diseases.2–4 For example, only about 20% of smokers go on to develop COPD.5 Likewise, variations in susceptibilities are likely to predispose some individuals to granulomatous diseases after exposure to silica or asbestos. Individuals from the same genetic background will show relatively similar susceptibility for a given disease.6–7 When discussing susceptibility variations, one also must keep in mind the impact of longevity and the intensity of environmental exposures. This becomes important as chronic exposures to some pollutants may rarely result in active disease. Understanding the causes of susceptibility variations are critical in developing mitigation strategies and therapeutic interventions. In the context of the current paper, “susceptibility” is defined as any unhealthy host condition/s that modify the biological response to environmental or other stressors. Here we focus on experimental studies of animal models of selected human susceptibilities and how they modify risk of adverse outcomes caused by air pollution.
Targeted hypothesis driven research has led us to identify and understand some of the biological pathways and key genetic differences or physiological conditions that make one more susceptible to pollutants.8–11 In the past two decades, the application of a systems approach with high throughput global gene expression, proteomic and metabolomic techniques has accelerated the understanding of variations in susceptibilities but has also exposed the complexities of biological networks, cross talks between multiple processes within a cell, dynamics of these processes, and the communication between organ systems. At what step in the biological network and in what organ system the normal homeostatic processes activated by air pollutants are interrupted due to abnormal host conditions becomes a critical determinant of the type and the degree of variation in response. Perturbation in any one of the homeostatic processes either due to a genetic defect or from chronic or overstimulated responses, will likely lead to cumulative or organ-specific impact after exposure to environmental pollutants. In this paper, I will first briefly provide a historical perspective on what we have learned using animal models with defined susceptibilities/diseases by giving examples of air pollutant exposures in these models. Recent work12 on the role of neuroendocrine stress pathways in mediating response to air pollutants will highlight how these new mechanisms will be critical in identifying variations in individual susceptibilities. Based on this body of work, a new mechanistic paradigm is proposed that will be useful when determining causes of variations in susceptibility to air pollutants and the importance of allostatic load. Experimental studies that provide insights into the biological mechanisms of susceptibility variations (non-communicable; non-cancer effects) will be discussed in this paper but will not include models of infections, allergies, or developmental stresses.
Historical perspective on susceptibility variations in air pollution research
A variety of host conditions can modify the response to air pollution exposure that can impact health (Figure 1). It is conceivable that one who is susceptible to developing a disease from unknown or known causes is likely to have increased susceptibility to subsequent air pollutant exposure. Likewise, when an individual is exposed chronically to a given air pollutant, will likely develop exposure-related disease over time. However, the mechanisms by which air pollutants may cause disease or simply exacerbate the preexisting disease condition may depend on each individual’s collective and individually-tailored response for survival. The understanding of variations in each individual’s response is further complicated by the fact that in ambient-environmental conditions, individuals are generally exposed to rather low levels of a diverse range of pollutants that produce changes in homeostatic processes through different mechanisms.
Figure 1.

Host susceptibility factors that interactively produce variations in individual response to air pollution health effects.
Most of the earlier research on air pollution nearly 50 years ago focused on occupational exposures to source-enriched pollutants at rather high concentrations. Because of the lack of exposure controls, workers who were exposed to specific source pollutants developed many types of pulmonary diseases based on the nature of inhalants. Due to the dominance of pulmonary effects, the acute and subtle effects on extra-pulmonary organs likely went unnoticed. For example, exposure to silica in miners caused acute and chronic silicosis13; coal miners’ exposures were associated with coal worker’s pneumoconiosis14; asbestos mining caused fibrotic lung disease referred to as asbestosis, adenoma, and also pleural diseases with plaques of fibrous tissue, calcification, and mesothelioma15; and exposure of farm dust caused hypersensitivity pneumonitis often referred to as farmers lung.16 Some of these pollutants were retained in the lungs with little clearance and the diseases occurred decades later, for example, mesothelioma occurs in susceptible individuals decades after asbestos exposure.17 The mechanisms of the delay in expression of disease still puzzles researchers and new insights continue to emerge. It was believed that air pollutants affected primarily the lung while the knowledge of peripheral effects was limited or these effects were considered non-specific, and therefore of lower significance when examining pulmonary diseases. Thus, the experimental studies mainly included lung pathologies and functional impairment. Likewise, animal models of susceptibility used for research were primarily of those with lung diseases.18–21
The October 1948 Donora Pennsylvania smog incident and the December 1952 Great London Smog incident raised the awareness that ambient level air pollution can cause mortality and morbidity and influenced the creation of ambient air quality standards for criteria air pollutants.22 These episodes also prompted epidemiological and toxicological studies to determine what type of pollutants were responsible for adverse health effects and especially who was susceptible. Human mortality and hospital admissions were primarily associated with pulmonary complications of bronchitis and exacerbation of chronic obstructive pulmonary disease (COPD). Based on these observations, and along with the awareness of cigarette smoke-related, allergic and infectious lung diseases in the population, the emphasis for research was placed on the animal models of bronchitis, COPD, pneumonia and asthma.4, 23–29 The research experiments included understanding of biological factors/host conditions that may increase pulmonary susceptibility to inflammation and obstructive lung diseases.
Host genetics as the contributor to variations in air pollution health effects
The contribution of genetic differences to the variations in susceptibility to air pollution health effects continues to be examined experimentally.20, 30–33 Mouse models of different genetic backgrounds have shown differential susceptibilities to pulmonary inflammation after exposure to ozone and other pollutants.34–37 Linkage analysis studies have provided insights into the contribution of candidate genes within specific chromosomal loci influencing response to air pollutants. For example, in studies involving genetically diverse mouse strains, the DNA sequences on chromosomes 4, 11 and 17 spanning Toll-like receptor-4 and many inflammatory cytokines were linked to increased ozone-induced lung injury and inflammation.38 The mutations in Toll-like receptor-4 and its contributions to increased allergen-induced lung injury and inflammation were also identified in mouse models as well as in humans.9 Although these studies provide insights for the gene targets associated with specific pathological conditions, the understanding of collective contribution of gene networks and the causal factors to observed susceptibility differences remains sparse. The limited genetic diversity of laboratory rodents limits the scope of investigations identifying mechanisms of biological variations among genetically diverse humans.
More recent studies have provided insights into the complex interplay of genetic networks that might govern phenotypic expression of disease or adverse health effects in each strain/species of animals.39–40 The individual strain/species variability in orchestrating transcriptional response will likely be a major contributing factor to producing variability in ultimate disease phenotype. The newer approach of collaborative cross of mouse models to create highly variable genetic backgrounds, that better represent the diversity seen in the human population will provide some insights. Their systematic assessment using high throughput platforms of genetic contribution to each disease phenotype has already begun to provide breakthroughs in understanding how individual genes influence the variability in susceptibility to not only air pollutants but all to other types of environmental stressors.33, 41–42
Use of animal models of human cardiovascular diseases in air pollution susceptibility studies
The robust epidemiological evidence from the 1990’s expanding air pollution effects beyond the lung to the cardiovascular system and how these effects might be important in human mortality and morbidity43 led to research interest in cardiovascular effects and cardiopulmonary interactions. Likewise, the focus of susceptibility variations expanded from lung disease models to using animal models of cardiovascular diseases for understanding how those with preexisting hypertension or other cardiovascular complications might have exacerbated responses to air pollutants.4, 44–47 Guided by the epidemiological associations between air pollution and cardiovascular diseases, our introduction of the use of a spontaneously hypertensive (SH) rat model in examining their susceptibility to air pollutants has led to use of these rats in many air pollution studies.48 These rats are now used world-wide in understanding of the key susceptibility attributes associated with hypertensive disorder in exacerbating pulmonary and cardiovascular effects of air pollutants.49–54
We have shown that relative to healthy normotensive rats, SH rats have greater pulmonary injury and inflammation, especially when air pollution exposures occur at high concentrations.48–49, 55 They have compromised ability to increase lung lining antioxidants when exposed to residual oil fly ash particles.48 SH rats are not able to respond to inhaled combustion source particles by increasing expression of genes involved in inflammation in the lung and the heart as baseline expression of some of these genes is already high in these rats.48 When global gene expression in the heart of normotensive control Wistar Kyoto (WKY) rats was compared to SH rats following diesel exhaust inhalation, it was apparent that WKY rats showed remarkable changes in gene clusters reflecting cardiovascular disease and these expression changes mimicked the expression pattern of the same genes at baseline in SH rats.56 Diesel exhaust inhalation, however, was not able to further exacerbate the transcriptome expression in SH rats, implying that their ability to induce changes in response to pollutant exposure has been compromised due to disease.56 When hypertensive rats were made normotensive by treating them with hydralazine, diesel exhaust inhalation-induced lung injury, inflammation and vascular effects were diminished.10 These data clearly demonstrated the role of hypertension in exacerbating the effects of inhaled pollutants. However, what brings about hypertension-associated physiological changes becomes an important determinant of the susceptibility of SH rats to pollutant-induced cardiopulmonary effects. These findings led us to understand how susceptibility variations may depend on the phenotypic outcome being selected for in a given genetic background of SH rats and how underlying host conditions might, in a complex and interactive manner, impact the response to air pollution stress. In epidemiological studies air pollution has been variably linked to increases in blood pressure among healthy and susceptible individuals.57–58
Numerous epidemiological studies have linked air pollution to exacerbation of cardiovascular diseases. Since autonomic imbalance can mediate cardiovascular effects of air pollutants59–60, the individuals with underlying cardiovascular diseases might be impacted through effects on sensory and central autonomic pathways. Using WKY, SH, and 6 other healthy and cardiovascular and/or metabolically compromised rat models46,61, we obtained insights into how underlying host genetics and disease may influence the response to inhaled ozone. Measures of breathing62, injury and inflammation varied greatly even between healthy strains.63 Furthermore, alveolar lining levels of antioxidants, namely ascorbate and glutathione, at baseline64 were correlated with lung physiological changes, protein leakage and inflammation when the theoretical ozone dose was plotted against levels of ascorbate in the lung lavage fluid.11 It was apparent that strains with highest levels of ascorbate in the lavage fluid were minimally affected by ozone in terms of injury and inflammation.63 For example, obese and insulin resistant James C. Russell (JCR) rats, which have a propensity to develop vascular disease, with low ozone effective dose (based on theoretical calculation) and high levels of ascorbate at baseline in the lung lining fluid, were least affected by ozone-induced lung vascular leakage and inflammation. On the contrary, the stroke-prone SH rats, which had the lowest level of lung lining ascorbate and received the highest effective ozone dose, were highly susceptible to lung injury and inflammation from ozone exposure.11, 63 These data collectively demonstrated that ascorbate and glutathione, the first line of defense in the lung lining65, are at least one of the critical determinants of susceptibility variation in how ozone may induce lung injury and inflammation. However, the causal mechanisms responsible for achieving differential lung lining antioxidant levels, and the connectivity to other cellular physiological processes require further investigations.
When examining the global gene expression patterns of the lung in 5 genetically-related strains which included healthy WKY, SH, stroke-prone SH, obese spontaneously hypertensive heart failure (SHHF) and obese insulin resistant JCR rats with vascular impairment61, we observed marked strain-related differences in transcriptome at baseline. We noted that the baseline pulmonary expression patterns of genetically-closer strains (SH, SH-stroke-prone and SHHF), which were cardiovascular compromised, were similar and reflected more severe cardiac complications.40 However, the lung gene expression pattern of JCR rats (genetically somewhat distinct than other strains; derived by crossing LA/N-cp and SH obese rats46) was generally, the opposite of the other strains, implying a strain-specific variation in the transcriptome. We noted that 2 obese strains due to their leptin receptor mutation (JCR and SHHF) had opposing patterns of expression for most gene clusters in the lung including the genes involved in metabolic processes.39 When exposed to ozone, these 5 strains shifted their lung expression in the same direction, albeit at different levels.39 These changes in different rat strains suggest that while inhaled ozone induces similar effects on the lung, there are quantitative differences in expression of specific gene clusters in each strain based on their underlying condition.39 Ozone effects on genes in the lung were primarily noted in SHR and stoke-prone SHR but not in severely compromised SHHF and JCR.39 The whole transcriptome analysis of WKY and SH after ozone exposure39, unlike few inflammatory gene changes examined in these strains after fly ash particle exposure48, provided a different picture and insights into greater effects in SH rats of ozone than in WKY rats.
As in the lung, when baseline expression of genes in the heart tissue was determined, it was further apparent that JCR and SHHF had opposing expression profiles whereas those genetically closer strains SHR, stoke-prone SHR and SHHF showed similar expression of genes.40 These data clearly emphasized that the phenotypic expression of disease might be similar in two strains of rats, however, the mechanisms that bring about these phenotypes depend on the underlying genetic differences and individual variability in the transcriptional response of regulatory networks. Because of these complexities, the therapeutic interventions targeted for a given disease phenotype might not be similarly effective in all individuals. These studies emphasize that the susceptibility variations are complex and a systems-based approach with continued use of high throughput technologies and identification of patterns of changes in gene expression, epigenetic regulators and phenotypes collectively will provide increased understanding of mechanisms and guide personalized therapeutic interventions that will be effective.
Use of rodent models of metabolic alterations in air pollution susceptibility studies
Research on air pollution further expanded to include metabolic diseases as a risk factors for exacerbated air pollution effects as new epidemiological evidence began to emerge linking air pollution to these diseases (i.e. diabetes, metabolic syndrome, obesity).66–67 The interest in how diet could modify health effects of pollutants prompted studies that included unhealthy diets as modifiers of air pollution health effects. Since air pollution was linked to wide array of systemic effects, the quest to identify circulating mediators that produce systemic effects was also apparent.68–70 Likewise, the choice of animal models used for understanding mechanisms of susceptibility variations began to include diet-induced predisposition to metabolic diseases, such as obesity and diabetes. A number of studies have been published examining how ozone-induced inflammation may be associated with greater production of IL-33 in obese mice and the modulation through IL-17A, and the influence of gut microbiota in exacerbating ozone-induced airway hyperresponsiveness.71–73 While these studies have shown increases in ozone-induced inflammatory response in obese mice, our use of two rat models of obesity with genetic predisposition to cardiovascular diseases showed no exacerbation of net ozone-induced injury and inflammation relative to healthy rats63. This suggests that multiple host factors including genetic background might be critical in the exacerbation of the inflammatory response in obese models. Moreover, the regulatory mechanisms that control obesity might influence susceptibility to lung injury and inflammation differently in individuals.
Diet supplementations and nutrient deficiencies have been examined as modifying factors of air pollution-induced cardiopulmonary health impairments.74–77 Feeding of a high fat diet in healthy rats has been linked to increased metabolic impairment and insulin resistance after repeated particulate matter instillation.78 In a mouse model, high fat diet has been shown to increase insulin resistance after repeated exposure to particulate matter, which correlated with increased plasma to tissue ratio of HSP-70 that has been shown to be a biomarker of systemic changes associated with type 2 diabetes.79 High fat diet in a mouse model has also been shown to exacerbate changes in blood brain barrier integrity after traffic-related motor vehicle exhaust exposures as evidenced by increased expression of oxidized-LDL signaling in the vasculature.80 High fat or high carbohydrate/fructose diets in male and female Brown Norway rats, which are resistant to developing diet-induced obesity, showed increased body fat percentage relative to lean mass in males81, but when exposed to ozone, diet did not significantly exacerbate lung injury or inflammation. These studies imply that that diet-induced susceptibility differences are dependent on model selection and biological endpoint.
Individuals consuming healthy dietary fats might have reduced responsiveness to air pollutants. Based on the health benefits of omega-6 and omega-3 fatty acids in a clinical study showing decreases in particulate air pollution-induced vasoconstriction82, we demonstrated that rats fed an omega-3-rich diet, similar to humans had attenuated vascular contractility response to ozone.75 However, these rats developed foamy macrophage accumulation in the lung with impaired lipid transporters expression, suggesting inhibition of lipid surfactant clearance.75 Although it is likely that the type and the concentration of dietary supplements might play a role in this adverse effect, lipid-rich diets with several unsaturated binding sites are likely to have impact on the lung as lipids can accumulate in the form of surfactant. And constant exposure to oxygen and oxidant pollutants also make the lipids more susceptible to oxidation within the lung. Thus, not only host factors but also the type of intervention can impact the effects of air pollutants.
Air pollution and neurobehavioral impact: use of animal models of neural diseases
In the past decade, significant epidemiological and experimental evidence has been accumulated pertaining to the effects of air pollutants on various brain areas and neurobehavioral alterations.83 In epidemiological studies, exposure to air pollutants has been linked to Alzheimer’s disease84, depression and suicide85, cognitive dysfunction86, Parkinson’s disease87, autism spectrum disorder88, and neurodegenerative diseases.89 Recently, short term exposure to air pollution has also been associated with increases in criminal activities.90 However, to-date only limited experimental studies have used animal models of brain disorders to determine if and how air pollutants interactively exacerbate the disease phenotype. In one study, exposure to diesel exhaust for 5–13 weeks in female 5X Familial AD (5XFAD) mice prone to Alzheimer’s disease, has been shown to accelerate amyloid-beta plaque formation in the brain without having significant systemic inflammation.91 In a genetic stress sensitive rat model of depression, the Flinders Sensitive Line (FSL) rat, repeated ozone exposure is associated with exacerbated behavioral alterations and oxidative stress in the hippocampus and frontal cortex, relative to control animals.92 These studies are beginning emphasize the role of neural regulation in health effects of air pollutants and how central mechanisms may contribute to human susceptibility variations.
Evidence of the role of neuroendocrine system in mediating air pollutant health effects and its implications in susceptibility variations
It is well documented that when a stress is encountered, a highly conserved host response will involve the activation of two major survival mechanisms, i.e. 1) mobilization of energy sources from storage sites and channeling of metabolite precursors to organs where they are needed the most, and 2) recognizing the location of stress and mobilizing immune cells to the site of injury to fight invading organisms or creating an innate immune response that removes unwanted or damaged tissues/cells to initiate healing process in case of an injury or tissue damage.93 The degree of host response to a stressor will depend on the underlying physiological condition and genetic variability. The peripheral autonomic sensory nerves recognize stress in the body and relay the information to the brain centers through the activation of where it is processed.94 The sympathetic and parasympathetic nervous system are activated. This leads to the activation of neuroendocrine stress response pathways involving sympathetic-adrenal-medullary (SAM), and hypothalamus-pituitary-adrenal (HPA) axes, which release adrenal-derived stress hormones, such as epinephrine and corticosterone/cortisol, respectively.95–96 These are the hormones, which through their receptors in various tissues, mediate cellular metabolic and immune changes to protect the host from stress. When stress is no longer a threat, these same hormones reverse acute stressor-induced effects through feedback inhibition.97 Because of selective distribution of the stress hormone receptor subtypes in the brain centers, these hormones also regulate learning and memory that is likely linked to an adaptation/habituation response.95, 98–99 These host survival mechanisms, well characterized for bodily injuries and infections, are tailored to the type of stressor encountered and the location of the injury in the body (Figure 2). Any variations in these mechanisms100 might impact how individuals differ in their susceptibility to stressors.
Figure 2. The proposed schematic of the mechanisms by which chemical and non-chemical stressors affect multiple organs through the activation/suppression of neuroendocrine axes.
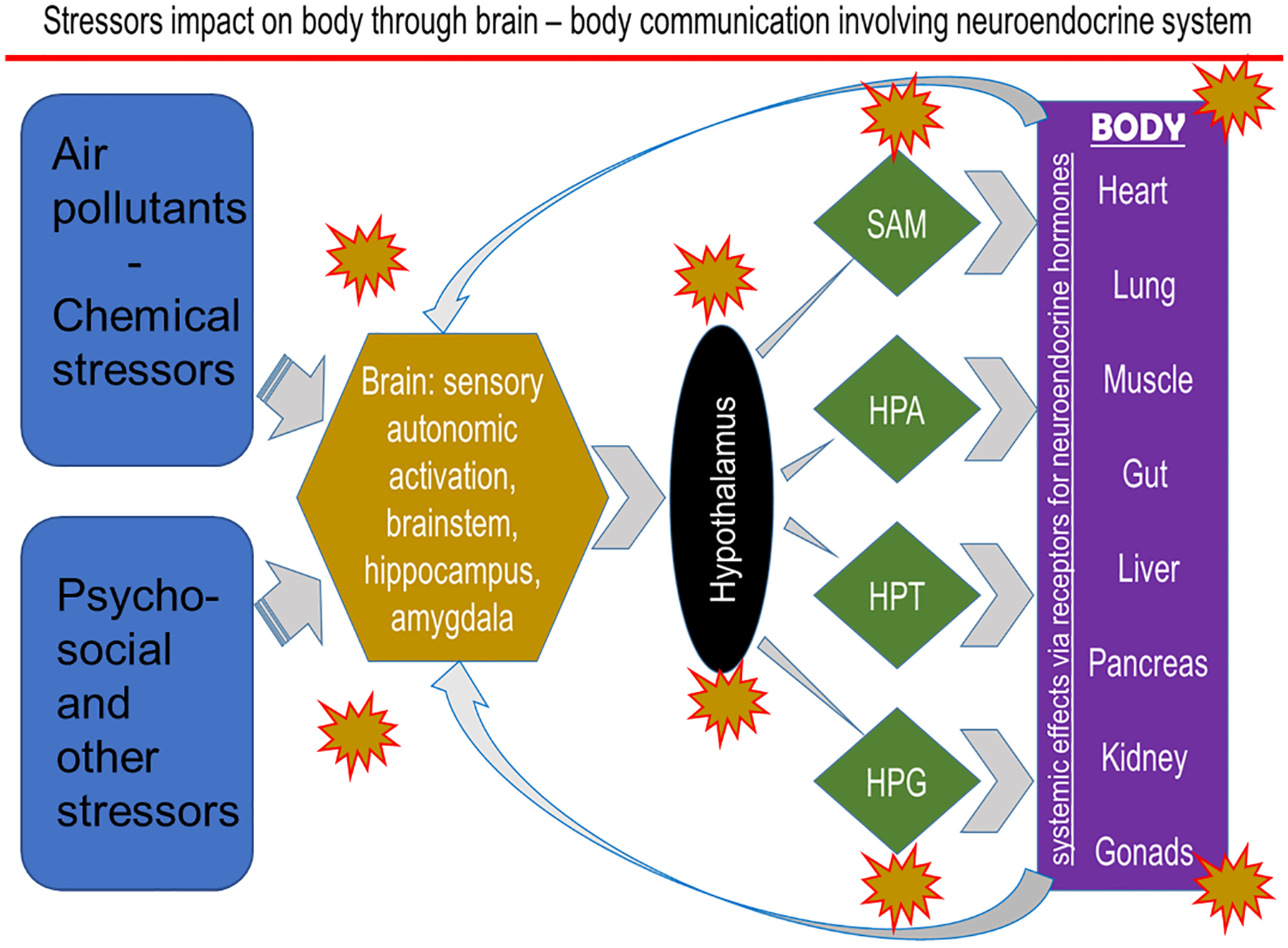
SAM, sympathetic-adrenal-medullary; HPA, hypothalamic-pituitary-adrenal; HPT, hypothalamus-pituitary-thyroid; HPG, hypothalamus-pituitary gonadal axes.
Although the neuroendocrine-mediated activation of SAM and HPA stress axes in maintaining homeostatic balance are well characterized, prior air pollution studies neither systematically examined their involvement in peripheral and local health effects, nor assessed their potential significance in disease causation or susceptibility variations. In inhalation toxicology studies, these acute reversible physiological changes have been often recognized but not characterized for their systemic and pulmonary consequences.101 Air pollution effects on autonomic nervous system, and sympathetic and parasympathetic reflex cardiopulmonary changes, however, have been well characterized102–103 such that air pollution regulations, in part, rely on evidence of autonomically-mediated cardiovascular changes in heart rate variability.104–105
The research effort for identifying circulating mediators responsible for extrapulmonary effects of air pollutants prompted us to examine a wide array of circulating cytokines, acute phase proteins, and metabolic hormones in our experimental studies. The lack of increases in circulating cytokines, in general, but striking increases in leptin and acute phase proteins in many of our studies106–108, led us to consider neural effects and subsequent peripheral changes. The Gackiere et al109 paper of 2011 showing the activation of hypothalamic stress responsive regions in rats acutely exposed to ozone served as a catalyst for us to consider neural mechanisms, especially the neuroendocrine stress pathways in examining air pollution health effects and how this may be important in susceptibility variations.
Linking peripheral metabolic and immune effects of irritant air pollutants to neuroendocrine stress pathways
Our first few studies on neuroendocrine pathways focused on circulating stress hormones and characterization of resultant peripheral effects. We demonstrated that ozone exposure increased circuiting epinephrine, corticosterone /cortisol and acute phase response proteins.70, 107–108 A single exposure to ozone also resulted in a series of metabolic effects together with depletion of circulating lymphocytes in a reversible manner. These effects were not associated with the release of cytokines from the lung into the circulation as metabolic effects occurred before detectable lung inflammation.107–108 These effects were characterized by the release of numerous free fatty acids into the circulation, likely through ozone-induced adipose lipolysis, increases in circulating branched-chain amino acids indicating muscle protein catabolism, increased gluconeogenesis, inhibition of lipid synthesis as indicated by downregulated gene expressions in the liver, and inhibition of bile acids release into the circulation with increased cholesterol, indicating inhibition of cholesterol breakdown.108 Ozone exposure produced hyperglycemia, glucose intolerance, and diminished glucose-mediated insulin secretion from pancreatic beta cells.70, 107–108 Subsequently published epidemiological studies incorporating metabolic homeostasis markers have reported association between air pollution and indices of altered glucose homeostasis.110–111
Animal studies involving exposure to another irritant air pollutant, acrolein, which deposits primarily in nasal airways, also exhibited similar peripheral metabolic changes.112 Acrolein-induced nasal injury/inflammation and also lung inflammation were associated with increases in circulating stress hormones, with diabetic rats being more susceptible to metabolic effects.112 In a human clinical study where ozone exposure occurred during intermittent exercise, similar increases in a variety of free fatty acids in the circulation and metabolites of membrane lipid breakdown together with increases in cortisol/corticosterone were shown.113 Virtually all pathways associated with lipid metabolism were altered as evident by changes in circulating lipid metabolites in humans exposed to ozone.113 Thus, we demonstrated that this stress response to ozone was coherent between species. There are a few other recent studies that have also examined the activation of the neuroendocrine system, especially the HPA in mediating ozone effects.114–116 One earlier study examining the effects of concentrated ambient particles in rats with allergic airways disease showed changes in the stress response pathway.117 All these effects observed in rat models and in humans indicated that SAM and HPA axes were stimulated by ozone and acrolein exposures and caused peripheral effects.118 This fight-or-flight response, induced by other host stressors or injuries, likely contribute to variations in susceptibility to effects of air pollution. Epidemiological studies have begun examining the biomarkers of HPA activation and subsequent associations with circulating stress hormones as well as metabolic alterations after exposure to increased levels of air pollutants.119
The confirmation of the role of circulating stress hormones in mediating systemic and pulmonary effects of ozone was initially made using the intervention of adrenalectomy, since adrenals are the main source of adrenaline and glucocorticoids. As expected, bilateral total adrenalectomy depleted circulating corticosterone and epinephrine nearly completely, but nor-epinephrine levels did not change as it is produced by nerve terminals and acts locally in a paracrine manner.120 To our surprise, adrenalectomy resulted in near elimination of all ozone-mediated systemic metabolic, immunological and pulmonary effects emphasizing the role of circulating adrenal-derived stress hormones in mediating these effects.120–121 In follow-up studies, we demonstrated that if the receptors of these stress hormones (i.e. adrenergic and glucocorticoid) are selectively blocked using pharmacological treatments, ozone-induced lung injury and inflammation were inhibited in an antagonist-specific manner.122 It was evident that the effect of blocking glucocorticoid receptors was more central and reduced all metabolic effects and systemic immuno suppression effects of ozone (i.e. depletion of circulating lymphocytes). On the other hand, as beta adrenergic receptors are densely distributed in the lung and regulate respiratory muscle tone123, the blockade of these receptors was associated with reduced local lung vascular leakage and extravasation of neutrophils at the site of stress.122 These data implied that the tissue distribution for adrenergic and glucocorticoid receptors is a critical determinant of the organ-specific effects of air pollution-induced stress response. The roles of beta adrenergic and glucocorticoid receptors in mediating ozone-induced lung injury/inflammation and systemic effects was further established by a gain of function experiment where adrenalectomized rats were treated with beta adrenergic and glucocorticoid receptor agonists and exposed to ozone.124 Thus, any variations on these receptors distribution in specific organs or the alterations of receptors sensitivity can contribute to variability in host response to environmental stressors in peripheral organs.
The autonomic activation by air pollutants likely involves stimulation of sensory vagus nerves in the lung to activate brain regions including brainstem and hypothalamus109, leading to the activation of SAM and HPA axes. The vagus nerve innervating the lung originates from jugular and nodose ganglion, which receives projections from the nucleus tractus solitarius (NTS) of the brainstem.125 The neural connection from NTS to hypothalamus involves glutaminergic and GLP-1 neurons.126 Although ozone has been shown to activate these stress responsive regions, it is still not clear if ozone-induced changes in the lung initiates changes in the brain through vagal sensory nerves or if there are circulating mediators which reach and cross the blood-brain barrier to induce discrete effects on different brain regions including the hypothalamus. The key initiating event for systemic changes is currently being actively investigated in the field of air pollution health effects.127
Our recent study has shown that acute ozone exposure induces a wide array of gene expression changes in the brainstem and hypothalamus that are similar to those induced during hypoxia (i.e. interferon signaling and the activation of stress pathways).128 These ozone-induced gene expression changes in rats are associated with increases in adrenaline and corticosterone while depleting prolactin, luteinizing hormone and also thyroid stimulating hormone, suggesting the concomitant inhibition of hypothalamus-pituitary-gonadal (HPG) and hypothalamus-pituitary-thyroid (HPT) axes. The activation of SAM and HPA and associated inhibition of HPG and HPT axes have been reported after application of other stressors.129 These data suggest that air pollutants, especially those that induce lung irritation, impact all neuroendocrine axes leading to suppression of thyroid and reproductive pathways along with the activation of adrenal axes (Figure 2). Importantly, most of the ozone-induced changes in hormones are not evident in adrenalectomized rats, implying the contribution of circulating adrenal-derived stress hormones in regulating brain effects of ozone. Circulating stress hormones are needed to produce lung effects of ozone and if no lung effects occur due to lack of stress hormones, no changes in brain regions are noted.128 Glucocorticoids in conjunction with mineralocorticoids and catecholamines have been shown to interactively regulate the process of adaptation and habituation centrally.99 The regulatory roles of adrenergic and glucocorticoid receptors systems and feedback inhibition need to be further examined in relation to health effects of acute and chronic air pollution exposures and more importantly how neuroendocrine response variations may lead to variations in human susceptibility to air pollution health effects.
The potential contribution of stress hormones in adaptation/plasticity and susceptibility variations
Once released into the circulation, stress hormones exert their cellular effects through adrenergic and glucocorticoid receptors.130–131 Subtypes of adrenergic receptors regulate cardiovascular functionality, smooth muscle tone, and other homeostatic processes.130 Impaired adrenergic receptor function in the brain has been shown to contribute to chronic stress in the central and peripheral organs.132–135 Glucocorticoid receptors are important in homeostatic cellular metabolic and immunological response of peripheral organs and also in regulating stress, memory, learning and habituation centrally.98, 136 Impaired regulation of adrenergic and glucocorticoid receptors in the brain has been linked to chronic stress and a number of neurological disorders including depression and PTSD. For example, glucocorticoid resistance has been noted in a variety of neuropathies and inflammatory conditions.131, 137 There is evidence that the selective distribution of mineralocorticoid and glucocorticoids receptors in limbic areas, which regulate stress response, are involved in habituation and learning.99 However, the impacts of air pollutants on these receptor pathways have been poorly examined.
Reversibility of air pollution-induced changes and the loss of effects upon repeated daily exposure (often referred to as adaptation, habituation or tolerance) to some pollutants, specifically ozone, has been the area of research interest with limited understanding of the mechanisms.138–139 Based on the role of adrenergic and glucocorticoid regulation of habituation, learning and memory99, 130–131, and the activation of SAM and HPA axes after air pollution exposure, it is conceivable that the adaptation that occurs upon repeated exposure to ozone will involve the loss of neuroendocrine activation through adrenergic and glucocorticoid mechanisms. The importance of the presence of catecholamines and glucocorticoids in the peripheral circulation, and their increases following stress, in mediating systemic, pulmonary and even brain effects of air pollutants120–121, 124, 128 emphasizes their role in regulation of adaptive, homeostatic, and pathological processes.131–132 It has been shown that after applying psychological stress, the changes in peripheral stress hormones is necessary for inducing inflammatory changes in the brain135, implying the regulatory role of peripheral stress hormones in orchestrating brain response. Our recent study showing the lack of ozone effects on brain in adrenalectomized rats corroborates with these findings.128 Thus, the potential interactions between underlying host neurobehavioral disorders and air pollutant modification of susceptibility through neuroendocrine system are likely (Figure 3). The role of developmental reprogramming of neuroendocrine pathways has been proposed to increase susceptibility of children to allergic respiratory diseases in epidemiological studies.140–141 The use of animal models with impaired activation of neuroendocrine stress response will prove useful in understanding how loss of adaptation can impact chronic diseases after exposure to air pollutants.
Figure 3. A flowchart of interactive effects of air pollutants and non-chemical stressors.
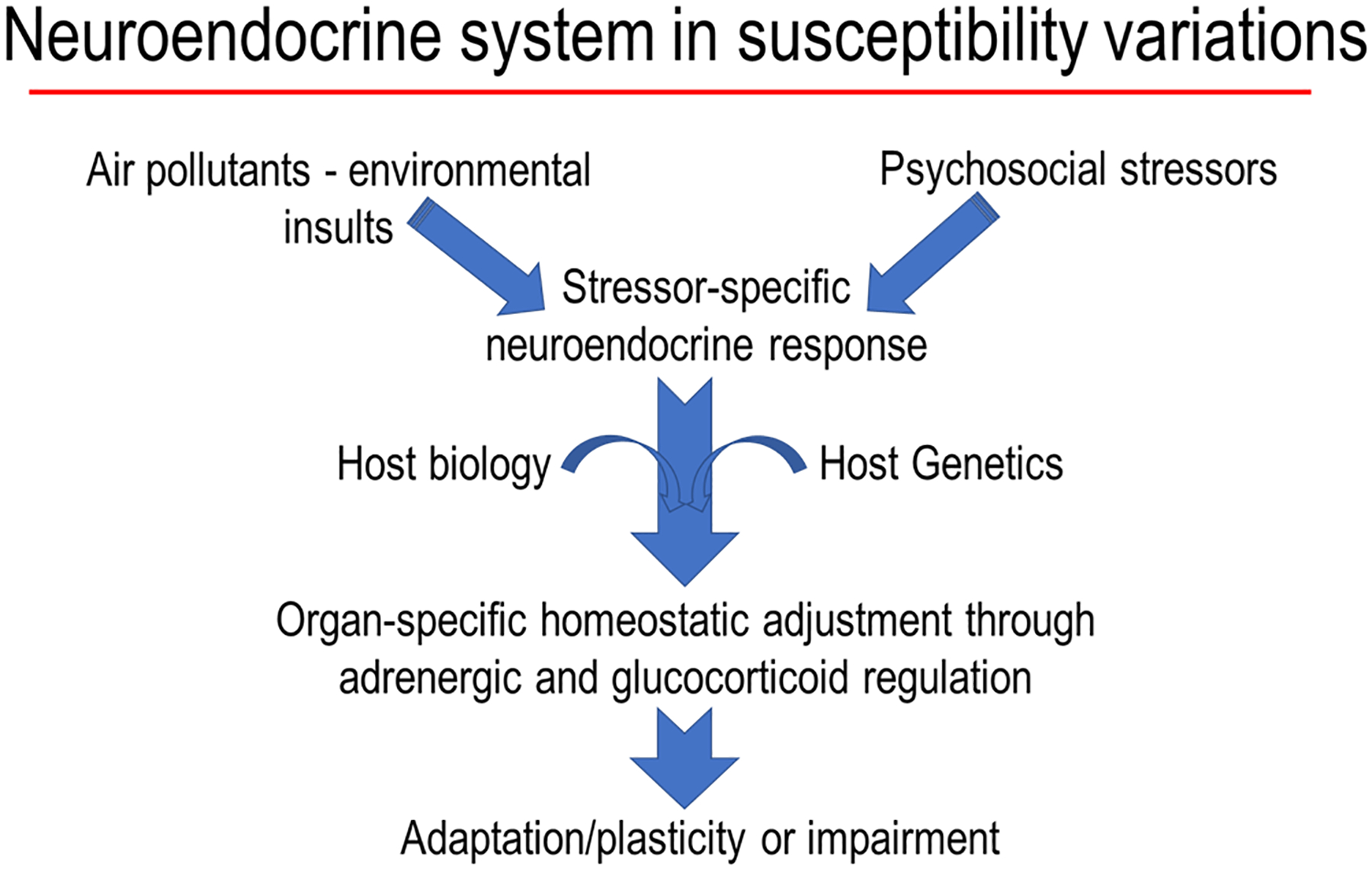
Air pollutant-induced neuroendocrine activation producing a stress response, which when combined with the impact of other stressors and host susceptibilities may still maintain plasticity and lead to adaptation through adrenergic and glucocorticoid receptors or produce an impairment.
Neuroendocrine activation and allostatic load in susceptibility variations
The concepts of neuroendocrine-mediated allostasis, habituation/adaptation, homeostasis as well as allostatic load are not new and have been examined for decades in relation to the plasticity or the lack thereof of neuroendocrine system to accelerate disease.95, 126, 142–145 How poor socioeconomic conditions, diets, environmental exposures, and psychosocial stressors together with genetic predisposition can push the homeostatic system to lose plasticity and increase the susceptibility to chronic diseases, including acceleration of aging, have been studied and incorporated in modeling approaches to quantify health burden from stressful life conditions. However, this has not been done for air pollution as an environmental stressor. The new epidemiological119, 146 and experimental12 evidence of the involvement of neuroendocrine system in mediating air pollution effects necessitates its incorporation in allostatic load modeling and assessing the contribution to human mortality and morbidity.
Air pollutants are physicochemically diverse and therefore, their deposition, solubility and retention can influence the extent to which, acute neuroendocrine stress response is produced after inhalation. When exposures occur repeatedly over a long period of time or when chronic exposure leads to retention of inhaled pollutants, local lung irritation can result in induction of fibrotic processes ultimately causing proliferative changes or even cancer with some exposures (Figure 4). The pollutants which activate central stress responsive regions and the neuroendocrine axes will, based on host susceptibilities or the presence of other stressors, induce systemic metabolic and immunological changes to counteract stress. When overwhelmed by repeated encounters of stressors or cumulative impacts, the adrenergic and glucocorticoid functions can be impaired. This can contribute to allostatic load, causing neural dysregulation, chronic inflammation and peripheral metabolic disorders based on the preexisting conditions (Figure 4). Thus, when examining susceptibility variations from exposure to air pollutants, multiple host conditions, the contribution of neuroendocrine pathways as well as chronicity of air pollution exposure should be incorporated into study designs.
Figure 4. A proposed mechanistic paradigm of how air pollutants with other stressors contribute to individual variations in disease susceptibility.
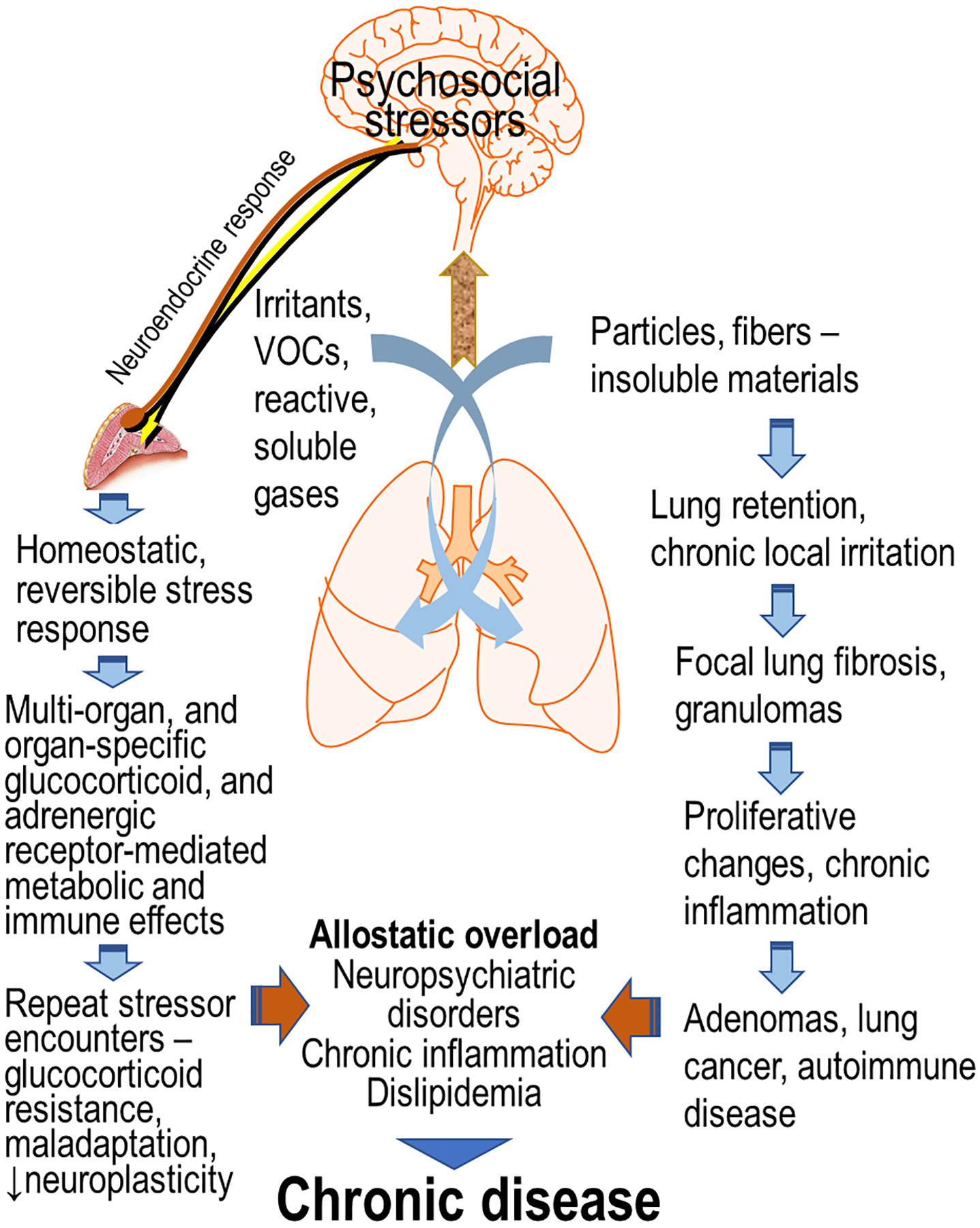
Acute exposure to all air pollutants may produce reversible homeostatic systemic metabolic and immune effects in a stressor-specific manner through neuroendocrine activation. Upon repeated encounter of stressors, however, there is likely a loss of adaptation/plasticity adding to allostatic load which can exacerbate disease. The pollutants which accumulate in the lung over time and produce low chronic irritation in lung epithelial cells additionally can have local proliferative changes that can ultimately lead to active granuloma/fibrosis and/or lung cancer. This can have added contribution to the allostatic load and contribute to variation in susceptibility.
Future perspective
The epidemiological studies have provided associations between air pollution and biological as well as physiological indicators of chronic pulmonary and cardiovascular diseases57–58, metabolic diseases (including obesity)66–67, 146, neuropsychiatric disorders and even low birth weight outcomes147 suggesting a mechanistic link. Given the evidence that the air pollutant effects are mediated through changes in endocrine activities, that the impairment of neuroendocrine function is linked to each disease and also air pollution, and that the neurobehavioral disorders and chronic peripheral diseases co-exist, necessitates more focused studies along neuroendocrine axes but using systems approach. Epidemiological associations, such as those showing increases in blood glucose148 and increases in pulmonary inflammation in children who are receiving bronchodilators149, can be linked to air pollution-induced activation of neuroendocrine response and release of stress hormones in the circulation.12, 119, 146 Thus, the role of neuroendocrine system in air pollution and chronic disease susceptibility is mechanistically plausible, however, clear understanding of stressor-specific outcomes and interactive role of neural regulation in susceptibility variations remains an area needing further research. While adrenergic and glucocorticoid receptor systems have been widely manipulated for currently used therapeutic interventions to treat chronic diseases150–151, further understanding of the role of central neuroendocrine mechanisms in air pollution health effects offers opportunities to identify new mechanistic pathways for therapeutic interventions. Other well-recognized interventions that could improve health of susceptible individuals include access to healthy living conditions, healthy diets and reduced psychosocial stressors that are linked to health disparities.
Impaired neuroendocrine regulation of homeostasis can contribute to host susceptibility in the presence of other stressors, such as chronic diseases, obesity, psychosocial pressures, poor diets, genetic predisposition and air pollution. Epidemiological association studies have begun incorporate the allostatic load concept in air pollution studies.152–153 The concepts of chemical and non-chemical stressors, external and internal exposomes, and one health have already emerged.154 Given the role of neuroendocrine system in processing all perceived stress signals centrally and producing stressor-specific metabolic and immune responses through coordination of all organ systems, one can presume that its malfunction can lead to abnormal stress response and increase in host susceptibility. Assessing the biological plausibility for interactive influence of each of these risk factors and identifying the biological indicators or group of indicators of stressor-induced susceptibilities with or without air pollutants will contribute to mechanistic understanding of the overall health outcomes.145, 155–156 As modeling approaches have been applied to assess the risk of cumulative stressors on overall wellbeing, health and longevity, more emphasis is needed to understand molecular mechanisms with systems concepts in mind. New insights have also emerged in the biology of developmental processes, and therefore the research on understanding how early life stresses may predispose one to adulthood diseases has grown157 (not covered in this paper). The incorporation of the neuroendocrine homeostatic mechanisms and dynamicity of response with consideration of all life stressors, host physiology as well as genetics, with or without incorporating air pollutant exposure will be useful in understanding the mechanisms of susceptibility variations.
Acknowledgements:
The authors thank Drs. Michael Madden, Colette Miller and Ian Gilmour of the US EPA for their critical review of the manuscript. Most of the previously published work presented in the second half of the paper is performed at the National Health and Environmental Effects Laboratory of the US Environmental Protection Agency, Research Triangle Park, NC, USA by my three excellent trainees, Drs Andres Henriquez, Samantha Snow and Desinia Miller.
Footnotes
Disclaimer: The research described in this article has been reviewed by the National Health and Environmental Effects Research Laboratory, U.S. Environmental Protection Agency, and approved for publication. Approval does not signify that the contents necessarily reflect the views and policies of the Agency, nor does the mention of trade names of commercial products constitute endorsement or recommendation for use.
Declaration of Conflicting Interests: Author declares no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- 1.Landrigan PJ, Fuller R, Acosta NJR, et al. The Lancet Commission on pollution and health. Lancet. 2018;391(10119):462–512. [DOI] [PubMed] [Google Scholar]
- 2.Brain JD, Valberg PA, Mensah GA. Species differences. In: Variations in Susceptibility to Inhaled Pollutants. Identification, Mechanisms, and Policy Implications (Brain JD, Beck BD, Warren AJ, Shaikh RA, eds). Baltimore: The Johns Hopkins University Press, 1988;89–103. [Google Scholar]
- 3.Sweeney TD, Brain JD, Godleski JJ. Preexisting disease. In: Variations in Susceptibility to Inhaled Pollutants. Identification, Mechanisms, and Policy Implications (Brain JD, Beck BD, Warren AJ, Shaikh RA, eds). Baltimore: The Johns Hopkins University Press, 1988;142–158. [Google Scholar]
- 4.Kodavanti UP, Costa DL, Bromberg PA. Rodent models of cardiopulmonary disease: their potential applicability in studies of air pollutant susceptibility. Environ Health Perspect. 1998;106 Suppl 1:111–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Siafakas NM, Tzortzaki EG. Few smokers develop COPD. Why? Respir Med. 2002;96(8):615–624. [DOI] [PubMed] [Google Scholar]
- 6.Yang IV, Schwartz DA. Epigenetic mechanisms and the development of asthma. J Allergy Clin Immunol. 2012;130(6):1243–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yauk CL, Lucas Argueso J, Auerbach SS, et al. Harnessing genomics to identify environmental determinants of heritable disease. Mutat Res. 2013;752(1):6–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arbour NC, Lorenz E, Schutte BC, et al. TLR4 mutations are associated with endotoxin hyporesponsiveness in humans. Nat Genet. 2000;25(2):187–191. [DOI] [PubMed] [Google Scholar]
- 9.Hollingsworth JW 2nd, Cook DN, Brass DM, et al. The role of Toll-like receptor 4 in environmental airway injury in mice. Am J Respir Crit Care Med. 2004;170(2):126–132. [DOI] [PubMed] [Google Scholar]
- 10.Kodavanti UP, Thomas RF, Ledbetter AD, et al. Diesel exhaust induced pulmonary and cardiovascular impairment: the role of hypertension intervention. Toxicol Appl Pharmacol. 2013;268(2):232–240. [DOI] [PubMed] [Google Scholar]
- 11.Dye JA, Costa DL, Kodavanti UP. Executive Summary: variation in susceptibility to ozone-induced health effects in rodent models of cardiometabolic disease. Inhal Toxicol. 2015;27 Suppl 1:105–15. [DOI] [PubMed] [Google Scholar]
- 12.Snow SJ, Henriquez AR, Costa DL, Kodavanti UP. Neuroendocrine Regulation of Air Pollution Health Effects: Emerging Insights. Toxicol Sci. 2018;164(1):9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ding M, Chen F, Shi X, Yucesoy B, Mossman B, Vallyathan V. Diseases caused by silica: mechanisms of injury and disease development. Int Immunopharmacol. 2002;2(2–3):173–182. [DOI] [PubMed] [Google Scholar]
- 14.Castranova V, Vallyathan V. Silicosis and coal workers’ pneumoconiosis. Environ Health Perspect. 2000;108 Suppl 4:675–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang SX, Jaurand MC, Kamp DW, Whysner J, Hei TK. Role of mutagenicity in asbestos fiber-induced carcinogenicity and other diseases. J Toxicol Environ Health B Crit Rev. 2011;14(1–4):179–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.May S, Romberger DJ, Poole JA. Respiratory health effects of large animal farming environments. J Toxicol Environ Health B Crit Rev. 2012;15(8):524–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Donaldson K, Murphy FA, Duffin R, Poland CA. Asbestos, carbon nanotubes and the pleural mesothelium: a review of the hypothesis regarding the role of long fibre retention in the parietal pleura, inflammation and mesothelioma. Part Fibre Toxicol. 2010;7:5. doi: 10.1186/1743-8977-7-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tryka AF, Sweeney TD, Brain JD, Godleski JJ. Short-term regional clearance of an inhaled submicrometric aerosol in pulmonary fibrosis. Am Rev Respir Dis. 1985;132(3):606–611. [DOI] [PubMed] [Google Scholar]
- 19.Adamson IY, Hedgecock C. Patterns of particle deposition and retention after instillation to mouse lung during acute injury and fibrotic repair. Exp Lung Res. 1995;21(5):695–709. [DOI] [PubMed] [Google Scholar]
- 20.Kodavanti UP, Jaskot RH, Su WY, Costa DL, Ghio AJ, Dreher KL. Genetic variability in combustion particle-induced chronic lung injury. Am J Physiol. 1997;272(3 Pt 1):L521–L532. [DOI] [PubMed] [Google Scholar]
- 21.Kodavanti UP, Jackson MC, Ledbetter AD, et al. Lung injury from intratracheal and inhalation exposures to residual oil fly ash in a rat model of monocrotaline-induced pulmonary hypertension. J Toxicol Environ Health A. 1999;57(8):543–563. [DOI] [PubMed] [Google Scholar]
- 22.Shy CM. Epidemiologic evidence and the United States air quality standards. Am J Epidemiol. 1979;110(6):661–671. [DOI] [PubMed] [Google Scholar]
- 23.Drazen JM, Takebayashi T, Long NC, De Sanctis GT, Shore SA. Animal models of asthma and chronic bronchitis. Clin Exp Allergy. 1999;29 Suppl 2:37–47. [DOI] [PubMed] [Google Scholar]
- 24.Bice DE, Seagrave J, Green FH. Animal models of asthma: potential usefulness for studying health effects of inhaled particles. Inhal Toxicol. 2000;12(9):829–862. [DOI] [PubMed] [Google Scholar]
- 25.Kodavanti UP, Mebane R, Ledbetter A, et al. Variable pulmonary responses from exposure to concentrated ambient air particles in a rat model of bronchitis. Toxicol Sci. 2000;54(2):441–451. [DOI] [PubMed] [Google Scholar]
- 26.Kodavanti UP, Schladweiler MC, Ledbetter AD, et al. The spontaneously hypertensive rat: an experimental model of sulfur dioxide-induced airways disease. Toxicol Sci. 2006;94(1):193–205. [DOI] [PubMed] [Google Scholar]
- 27.Nikula KJ, Green FH. Animal models of chronic bronchitis and their relevance to studies of particle-induced disease. Inhal Toxicol. 2000;12 Suppl 4:123–153. [DOI] [PubMed] [Google Scholar]
- 28.Chapman RW. Canine models of asthma and COPD. Pulm Pharmacol Ther. 2008;21(5):731–742. [DOI] [PubMed] [Google Scholar]
- 29.Wright JL, Cosio M, Churg A. Animal models of chronic obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol. 2008;295(1):L1–L15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gilmour PS, Schladweiler MC, Richards JH, Ledbetter AD, Kodavanti UP. Hypertensive rats are susceptible to TLR4-mediated signaling following exposure to combustion source particulate matter. Inhal Toxicol. 2004;16 Suppl 1:5–18. [DOI] [PubMed] [Google Scholar]
- 31.Romieu I, Moreno-Macias H, London SJ. Gene by environment interaction and ambient air pollution. Proc Am Thorac Soc. 2010;7(2):116–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scoville DK, Botta D, Galdanes K, et al. Genetic determinants of susceptibility to silver nanoparticle-induced acute lung inflammation in mice. FASEB J. 2017;31(10):4600–4611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saul MC, Philip VM, Reinholdt LG; Center for Systems Neurogenetics of Addiction, Chesler EJ. High-Diversity Mouse Populations for Complex Traits. Trends Genet. 2019;35(7):501–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Savov JD, Whitehead GS, Wang J, et al. Ozone-induced acute pulmonary injury in inbred mouse strains. Am J Respir Cell Mol Biol. 2004;31(1):69–77. [DOI] [PubMed] [Google Scholar]
- 35.Kleeberger SR. Genetic aspects of pulmonary responses to inhaled pollutants. Exp Toxicol Pathol. 2005;57 Suppl 1:147–53. [DOI] [PubMed] [Google Scholar]
- 36.Bauer AK, Kleeberger SR. Genetic mechanisms of susceptibility to ozone-induced lung disease. Ann N Y Acad Sci. 2010;1203:113–119. [DOI] [PubMed] [Google Scholar]
- 37.Gershwin LJ. Comparative immunology of allergic responses. Annu Rev Anim Biosci. 2015;3:327–346. [DOI] [PubMed] [Google Scholar]
- 38.Kleeberger SR. Genetic aspects of susceptibility to air pollution. Eur Respir J Suppl. 2003;40:52s–56s. [DOI] [PubMed] [Google Scholar]
- 39.Ward WO, Kodavanti UP. Pulmonary transcriptional response to ozone in healthy and cardiovascular compromised rat models. Inhal Toxicol. 2015;27 Suppl 1:93–104. [DOI] [PubMed] [Google Scholar]
- 40.Ward WO, Kodavanti UP. Left ventricular gene expression profile of healthy and cardiovascular compromised rat models used in air pollution studies. Inhal Toxicol. 2015;27 Suppl 1:63–79. [DOI] [PubMed] [Google Scholar]
- 41.Smith CM, Sassetti CM. Modeling Diversity: Do Homogeneous Laboratory Strains Limit Discovery? Trends Microbiol. 2018;26(11):892–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Salimova E, Nowak KJ, Estrada AC, et al. Variable outcomes of human heart attack recapitulated in genetically diverse mice. NPJ Regen Med. 2019;4:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dockery DW, Pope CA 3rd, Xu X, Spengler JD, Ware JH, Fay ME, Ferris BG Jr, Speizer FE. An association between air pollution and mortality in six U.S. cities. N Engl J Med. 1993;329(24):1753–1759. [DOI] [PubMed] [Google Scholar]
- 44.Wallenborn JG, Schladweiler MC, Nyska A, et al. Cardiopulmonary responses of Wistar Kyoto, spontaneously hypertensive, and stroke-prone spontaneously hypertensive rats to particulate matter (PM) exposure. J Toxicol Environ Health A. 2007;70(22):1912–1922. [DOI] [PubMed] [Google Scholar]
- 45.Kodavanti UP, Schladweiler MC, Ledbetter AD, et al. Consistent pulmonary and systemic responses from inhalation of fine concentrated ambient particles: roles of rat strains used and physicochemical properties. Environ Health Perspect. 2005;113(11):1561–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kodavanti UP, Russell JC, Costa DL. Rat models of cardiometabolic diseases: baseline clinical chemistries, and rationale for their use in examining air pollution health effects. Inhal Toxicol. 2015;27 Suppl 1:2–13. [DOI] [PubMed] [Google Scholar]
- 47.Costa DL, Kodavanti UP. Toxic responses of the lung to inhaled pollutants: benefits and limitations of lung-disease models. Toxicol Lett. 2003;140–141:195–203. [DOI] [PubMed] [Google Scholar]
- 48.Kodavanti UP, Schladweiler MC, Ledbetter AD, et al. The spontaneously hypertensive rat as a model of human cardiovascular disease: evidence of exacerbated cardiopulmonary injury and oxidative stress from inhaled emission particulate matter. Toxicol Appl Pharmacol. 2000;164(3):250–263. [DOI] [PubMed] [Google Scholar]
- 49.Kodavanti UP, Schladweiler MC, Ledbetter AD, et al. Temporal association between pulmonary and systemic effects of particulate matter in healthy and cardiovascular compromised rats. J Toxicol Environ Health A. 2002;65(20):1545–1569. [DOI] [PubMed] [Google Scholar]
- 50.Saxena RK, Gilmour MI, Schladweiler MC, McClure M, Hays M, Kodavanti UP. Differential pulmonary retention of diesel exhaust particles in Wistar Kyoto and spontaneously hypertensive rats. Toxicol Sci. 2009;111(2):392–401. [DOI] [PubMed] [Google Scholar]
- 51.Shen YH, Pham AK, Davis B, et al. Sex and strain-based inflammatory response to repeated tobacco smoke exposure in spontaneously hypertensive and Wistar Kyoto rats. Inhal Toxicol. 2016;28(14):677–685. [DOI] [PubMed] [Google Scholar]
- 52.Mumaw CL, Surace M, Levesque S, et al. Atypical microglial response to biodiesel exhaust in healthy and hypertensive rats. Neurotoxicology. 2017;59:155–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tavera Busso I, Mateos AC, Juncos LI, Canals N, Carreras HA. Kidney damage induced by sub-chronic fine particulate matter exposure. Environ Int. 2018;121(Pt 1):635–642. [DOI] [PubMed] [Google Scholar]
- 54.Wong EM, Walby WF, Wilson DW, Tablin F, Schelegle ES. Ultrafine Particulate Matter Combined With Ozone Exacerbates Lung Injury in Mature Adult Rats With Cardiovascular Disease. Toxicol Sci. 2018;163(1):140–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shannahan JH, Nyska A, Cesta M, et al. Subchronic pulmonary pathology, iron overload, and transcriptional activity after Libby amphibole exposure in rat models of cardiovascular disease. Environ Health Perspect. 2012;120(1):85–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gottipolu RR, Wallenborn JG, Karoly ED, et al. One-month diesel exhaust inhalation produces hypertensive gene expression pattern in healthy rats. Environ Health Perspect. 2009;117(1):38–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bind MA, Peters A, Koutrakis P, Coull B, Vokonas P, Schwartz J. Quantile Regression Analysis of the Distributional Effects of Air Pollution on Blood Pressure, Heart Rate Variability, Blood Lipids, and Biomarkers of Inflammation in Elderly American Men: The Normative Aging Study. Environ Health Perspect. 2016; 124(8):1189–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Adar SD, Chen YH, D’Souza JC, O’Neill MS, Szpiro AA, Auchincloss AH, Park SK, Daviglus ML, Diez Roux AV, Kaufman JD. Longitudinal Analysis of Long-Term Air Pollution Levels and Blood Pressure: A Cautionary Tale from the Multi-Ethnic Study of Atherosclerosis. Environ Health Perspect. 2018; 126(10):107003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dabass A, Talbott EO, Rager JR, Marsh GM, Venkat A, Holguin F, Sharma RK. Systemic inflammatory markers associated with cardiovascular disease and acute and chronic exposure to fine particulate matter air pollution (PM2.5) among US NHANES adults with metabolic syndrome. Environ Res. 2018; 161:485–491. [DOI] [PubMed] [Google Scholar]
- 60.Raza A, Dahlquist M, Lind T, Ljungman PLS. Susceptibility to short-term ozone exposure and cardiovascular and respiratory mortality by previous hospitalizations. Environ Health. 2018; 17(1):37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ramot Y, Kodavanti UP, Kissling GE, Ledbetter AD, Nyska A. Clinical and pathological manifestations of cardiovascular disease in rat models: the influence of acute ozone exposure. Inhal Toxicol. 2015;27 Suppl 1:26–38. [DOI] [PubMed] [Google Scholar]
- 62.Dye JA, Ledbetter AD, Schladweiler MC, Costa DL, Kodavanti UP. Whole body plethysmography reveals differential ventilatory responses to ozone in rat models of cardiovascular disease. Inhal Toxicol. 2015;27 Suppl 1:14–25. [DOI] [PubMed] [Google Scholar]
- 63.Kodavanti UP, Ledbetter AD, Thomas RF, et al. Variability in ozone-induced pulmonary injury and inflammation in healthy and cardiovascular-compromised rat models. Inhal Toxicol. 2015;27 Suppl 1:39–53. [DOI] [PubMed] [Google Scholar]
- 64.Hatch GE, Crissman K, Schmid J, et al. Strain differences in antioxidants in rat models of cardiovascular disease exposed to ozone. Inhal Toxicol. 2015;27 Suppl 1:54–62. [DOI] [PubMed] [Google Scholar]
- 65.Jain A, Mårtensson J, Mehta T, Krauss AN, Auld PA, Meister A. Ascorbic acid prevents oxidative stress in glutathione-deficient mice: effects on lung type 2 cell lamellar bodies, lung surfactant, and skeletal muscle. Proc Natl Acad Sci U S A. 1992;89(11):5093–5097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rajagopalan S, Brook RD. Air pollution and type 2 diabetes: mechanistic insights. Diabetes. 2012; 61(12):3037–3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Thiering E, Heinrich J. Epidemiology of air pollution and diabetes. Trends Endocrinol Metab. 2015; 26(7):384–394. [DOI] [PubMed] [Google Scholar]
- 68.Brook RD, Sun Z, Brook JR, et al. Extreme Air Pollution Conditions Adversely Affect Blood Pressure and Insulin Resistance: The Air Pollution and Cardiometabolic Disease Study. Hypertension. 2016;67(1):77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen Z, Salam MT, Toledo-Corral C, et al. Ambient Air Pollutants Have Adverse Effects on Insulin and Glucose Homeostasis in Mexican Americans. Diabetes Care. 2016;39(4):547–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Miller DB, Snow SJ, Henriquez A, et al. Systemic metabolic derangement, pulmonary effects, and insulin insufficiency following subchronic ozone exposure in rats. Toxicol Appl Pharmacol. 2016;306:47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shore SA. Mechanistic Basis for Obesity-related Increases in Ozone-induced Airway Hyperresponsiveness in Mice. Ann Am Thorac Soc. 2017;14(Supplement_5):S357–S362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mathews JA, Krishnamoorthy N, Kasahara DI, et al. Augmented Responses to Ozone in Obese Mice Require IL-17A and Gastrin-Releasing Peptide. Am J Respir Cell Mol Biol. 2018;58(3):341–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tashiro H, Cho Y, Kasahara DI, et al. Microbiota contribute to obesity-related increases in the pulmonary response to ozone. Am J Respir Cell Mol Biol. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Moon KY, Park MK, Leikauf GD, Park CS, Jang AS. Diesel exhaust particle-induced airway responses are augmented in obese rats. Int J Toxicol. 2014;33(1):21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Snow SJ, Cheng WY, Henriquez A, et al. Ozone-Induced Vascular Contractility and Pulmonary Injury Are Differentially Impacted by Diets Enriched With Coconut Oil, Fish Oil, and Olive Oil. Toxicol Sci. 2018;163(1):57–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wesselink AK, Hatch EE, Wise LA. Invited Commentary: Interaction Between Diet and Chemical Exposures. Am J Epidemiol. 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Miller CN, Rayalam S. The role of micronutrients in the response to ambient air pollutants: Potential mechanisms and suggestions for research design. J Toxicol Environ Health B Crit Rev. 2017; 20(1):38–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yan YH, Chou CC, Lee CT, Liu JY, Cheng TJ. Enhanced insulin resistance in diet-induced obese rats exposed to fine particles by instillation. Inhal Toxicol. 2011;23(9):507–519. [DOI] [PubMed] [Google Scholar]
- 79.Goettems-Fiorin PB, Grochanke BS, Baldissera FG, et al. Fine particulate matter potentiates type 2 diabetes development in high-fat diet-treated mice: stress response and extracellular to intracellular HSP70 ratio analysis. J Physiol Biochem. 2016;72(4):643–656. [DOI] [PubMed] [Google Scholar]
- 80.Suwannasual U, Lucero J, McDonald JD, Lund AK. Exposure to traffic-generated air pollutants mediates alterations in brain microvascular integrity in wildtype mice on a high-fat diet. Environ Res. 2018;160:449–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gordon CJ, Phillips PM, Johnstone AF, et al. Effect of high-fructose and high-fat diets on pulmonary sensitivity, motor activity, and body composition of brown Norway rats exposed to ozone. Inhal Toxicol. 2016;28(5):203–215. [DOI] [PubMed] [Google Scholar]
- 82.Brigham EP, Woo H, McCormack M, et al. Omega-3 and Omega-6 Intake Modifies Asthma Severity and Response to Indoor Air Pollution in Children. Am J Respir Crit Care Med. 2019;199(12):1478–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Russ TC, Reis S, van Tongeren M. Air pollution and brain health: defining the research agenda. Curr Opin Psychiatry. 2019;32(2):97–104. [DOI] [PubMed] [Google Scholar]
- 84.Calderón-Garcidueñas L, González-Maciel A, Kulesza RJ, et al. Air Pollution, Combustion and Friction Derived Nanoparticles, and Alzheimer’s Disease in Urban Children and Young Adults. J Alzheimers Dis. 2019;doi: 10.3233/JAD-190331. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 85.Gładka A, Rymaszewska J, Zatoński T. Impact of air pollution on depression and suicide. Int J Occup Med Environ Health. 2018;31(6):711–721. [DOI] [PubMed] [Google Scholar]
- 86.Cipriani G, Danti S, Carlesi C, Borin G. Danger in the Air: Air Pollution and Cognitive Dysfunction. Am J Alzheimers Dis Other Demen. 2018;33(6):333–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Palacios N, Fitzgerald KC, Hart JE, et al. Air Pollution and Risk of Parkinson’s Disease in a Large Prospective Study of Men. Environ Health Perspect. 2017;125(8):087011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lam J, Sutton P, Kalkbrenner A, et al. A Systematic Review and Meta-Analysis of Multiple Airborne Pollutants and Autism Spectrum Disorder. PLoS One. 2016;11(9):e0161851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jayaraj RL, Rodriguez EA, Wang Y, Block ML. Outdoor Ambient Air Pollution and Neurodegenerative Diseases: the Neuroinflammation Hypothesis. Curr Environ Health Rep. 2017;4(2):166–179. [DOI] [PubMed] [Google Scholar]
- 90.Berman JD, Burkhardt J, Bayham J, Carter E, Wilson A. Acute air pollution exposure and the risk of violent behavior in the United States. Epidemiology. 2019; doi: 10.1097/EDE.0000000000001085. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 91.Hullmann M, Albrecht C, van Berlo D, et al. Diesel engine exhaust accelerates plaque formation in a mouse model of Alzheimer’s disease. Part Fibre Toxicol. 2017;14(1):35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mokoena ML, Harvey BH, Viljoen F, Ellis SM, Brink CB. Ozone exposure of Flinders Sensitive Line rats is a rodent translational model of neurobiological oxidative stress with relevance for depression and antidepressant response. Psychopharmacology (Berl). 2015;232(16):2921–2938. [DOI] [PubMed] [Google Scholar]
- 93.Russell G, Lightman S. The human stress response. Nat Rev Endocrinol. 2019; doi: 10.1038/s41574-019-0228-0. [Epub ahead of print] Review. [DOI] [PubMed] [Google Scholar]
- 94.Waxenbaum JA, Varacallo M. Anatomy, Autonomic Nervous System. StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2019. [PubMed] [Google Scholar]
- 95.McEwen BS. From molecules to mind. Stress, individual differences, and the social environment. Ann N Y Acad Sci. 2001;935:42–49. [PubMed] [Google Scholar]
- 96.Herman JP, McKlveen JM, Ghosal S, et al. Regulation of the Hypothalamic-Pituitary-Adrenocortical Stress Response. Compr Physiol. 2016;6(2):603–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.McEwen BS. In pursuit of resilience: stress, epigenetics, and brain plasticity. Ann N Y Acad Sci. 2016;1373(1):56–64. [DOI] [PubMed] [Google Scholar]
- 98.Viho EMG, Buurstede JC, Mahfouz A, et al. Corticosteroid action in the brain: the potential of selective receptor modulation. Neuroendocrinology. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.de Kloet ER, de Kloet SF, de Kloet CS, de Kloet AD. Top-down and bottom-up control of stress-coping. J Neuroendocrinol. 2019;31(3):e12675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Levran O, Peles E, Randesi M, et al. Genetic variations in genes of the stress response pathway are associated with prolonged abstinence from heroin. Pharmacogenomics. 2018;19(4):333–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pauluhn J Inhalation toxicology: methodological and regulatory challenges. Exp Toxicol Pathol. 2008;60(2–3):111–124. [DOI] [PubMed] [Google Scholar]
- 102.Evans JM, Jenkins RA, Ilgner RH, Knapp CF, Zhang Q, Patwardhan AR. Acute cardiovascular autonomic responses to inhaled particulates. Eur J Appl Physiol. 2015;115(2):257–268. [DOI] [PubMed] [Google Scholar]
- 103.Perez CM, Hazari MS, Farraj AK. Role of autonomic reflex arcs in cardiovascular responses to air pollution exposure. Cardiovasc Toxicol. 2015;15(1):69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Park SK, Auchincloss AH, O’Neill MS, et al. Particulate air pollution, metabolic syndrome, and heart rate variability: the multi-ethnic study of atherosclerosis (MESA). Environ Health Perspect. 2010;118(10):1406–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ljungman PLS, Li W, Rice MB, et al. Long- and short-term air pollution exposure and measures of arterial stiffness in the Framingham Heart Study. Environ Int. 2018;121(Pt 1):139–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Shannahan JH, Alzate O, Winnik WM, et al. Acute phase response, inflammation and metabolic syndrome biomarkers of Libby asbestos exposure. Toxicol Appl Pharmacol. 2012;260(2):105–114. [DOI] [PubMed] [Google Scholar]
- 107.Bass V, Gordon CJ, Jarema KA, et al. Ozone induces glucose intolerance and systemic metabolic effects in young and aged Brown Norway rats. Toxicol Appl Pharmacol. 2013;273(3):551–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Miller DB, Karoly ED, Jones JC, et al. Inhaled ozone (O3)-induces changes in serum metabolomic and liver transcriptomic profiles in rats. Toxicol Appl Pharmacol. 2015;286(2):65–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gackière F, Saliba L, Baude A, Bosler O, Strube C. Ozone inhalation activates stress-responsive regions of the CNS. J Neurochem. 2011;117(6):961–972. [DOI] [PubMed] [Google Scholar]
- 110.Li W, Dorans KS, Wilker EH, Rice MB, Kloog I, Schwartz JD, Koutrakis P, Coull BA, Gold DR, Meigs JB, Fox CS, Mittleman MA. Ambient air pollution, adipokines, and glucose homeostasis: The Framingham Heart Study. Environ Int. 2018; 111:14–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yitshak Sade M, Kloog I, Liberty IF, Schwartz J, Novack V. The Association Between Air Pollution Exposure and Glucose and Lipids Levels. J Clin Endocrinol Metab. 2016; 101(6):2460–2467. [DOI] [PubMed] [Google Scholar]
- 112.Snow SJ, McGee MA, Henriquez A, et al. Respiratory Effects and Systemic Stress Response Following Acute Acrolein Inhalation in Rats.Toxicol Sci. 2017;158(2):454–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Miller DB, Ghio AJ, Karoly ED, et al. Ozone Exposure Increases Circulating Stress Hormones and Lipid Metabolites in Humans. Am J Respir Crit Care Med. 2016;193(12):1382–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Thomson EM, Vladisavljevic D, Mohottalage S, Kumarathasan P, Vincent R. Mapping acute systemic effects of inhaled particulate matter and ozone: multiorgan gene expression and glucocorticoid activity. Toxicol Sci. 2013;135(1):169–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Thomson EM, Pal S, Guénette J, et al. Ozone Inhalation Provokes Glucocorticoid-Dependent and -Independent Effects on Inflammatory and Metabolic Pathways. Toxicol Sci. 2016;152(1):17–28. [DOI] [PubMed] [Google Scholar]
- 116.Thomas J, Guénette J, Thomson EM. Stress axis variability is associated with differential ozone-induced lung inflammatory signaling and injury biomarker response. Environ Res. 2018;167:751–758. [DOI] [PubMed] [Google Scholar]
- 117.Sirivelu MP, MohanKumar SM, Wagner JG, Harkema JR, MohanKumar PS. Activation of the stress axis and neurochemical alterations in specific brain areas by concentrated ambient particle exposure with concomitant allergic airway disease. Environ Health Perspect. 2006.;114(6):870–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kodavanti UP. Stretching the stress boundary: Linking air pollution health effects to a neurohormonal stress response. Biochim Biophys Acta. 2016;1860(12):2880–2890. [DOI] [PubMed] [Google Scholar]
- 119.Li H, Cai J, Chen R, Zhao Z, Ying Z, Wang L, Chen J, Hao K, Kinney PL, Chen H, Kan H. Particulate Matter Exposure and Stress Hormone Levels: A Randomized, Double-Blind, Crossover Trial of Air Purification. Circulation. 2017; 136(7):618–627. [DOI] [PubMed] [Google Scholar]
- 120.Miller DB, Snow SJ, Schladweiler MC, et al. Acute Ozone-Induced Pulmonary and Systemic Metabolic Effects Are Diminished in Adrenalectomized Rats. Toxicol Sci. 2016;150(2):312–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Henriquez A, House J, Miller DB, et al. Adrenal-derived stress hormones modulate ozone-induced lung injury and inflammation. Toxicol Appl Pharmacol. 2017;329:249–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Henriquez AR, Snow SJ, Schladweiler MC, et al. Adrenergic and glucocorticoid receptor antagonists reduce ozone-induced lung injury and inflammation. Toxicol Appl Pharmacol. 2018;339:161–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Barnes PJ Distribution of receptor targets in the lung. Proc Am Thorac Soc. 2004;1(4):345–351. [DOI] [PubMed] [Google Scholar]
- 124.Henriquez AR, Snow SJ, Schladweiler MC, et al. Beta-2 Adrenergic and Glucocorticoid Receptor Agonists Modulate Ozone-Induced Pulmonary Protein Leakage and Inflammation in Healthy and Adrenalectomized Rats. Toxicol Sci. 2018;166(2):288–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Mazzone SB, Undem BJ Vagal afferent innervation of the airways in health and disease. Physiol. Rev 2016; 96, 975–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Herman JP Regulation of hypothalamo-pituitary-adrenocortical responses to stressors by the nucleus of the solitary tract/dorsal vagal complex. Cell. Mol. Neurobiol 2018;38, 25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Aragon MJ, Topper L, Tyler CR, et al. Serum-borne bioactivity caused by pulmonary multiwalled carbon nanotubes induces neuroinflammation via blood-brain barrier impairment. Proc Natl Acad Sci U S A. 2017;114(10):E1968–E1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Henriquez AR, House JS, Snow SJ, et al. Ozone-induced dysregulation of neuroendocrine axes requires adrenal-derived stress hormones. Toxicol Sci, 2019; pii: kfz182. doi: 10.1093/toxsci/kfz182. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Acevedo-Rodriguez A, Kauffman AS, Cherrington BD, Borges CS, Roepke TA, Laconi M. Emerging insights into hypothalamic-pituitary-gonadal axis regulation and interaction with stress signalling. J Neuroendocrinol. 2018;30(10):e12590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Tank AW, Lee Wong D. Peripheral and central effects of circulating catecholamines. Compr Physiol. 2015;5(1):1–15. [DOI] [PubMed] [Google Scholar]
- 131.Merkulov VM, Merkulova TI, Bondar NP. Mechanisms of Brain Glucocorticoid Resistance in Stress-Induced Psychopathologies. Biochemistry (Mosc). 2017;82(3):351–365. [DOI] [PubMed] [Google Scholar]
- 132.Mravec B Role of catecholamine-induced activation of vagal afferent pathways in regulation of sympathoadrenal system activity: negative feedback loop of stress response. Endocr Regul. 2011;45(1):37–41. [PubMed] [Google Scholar]
- 133.Goldstein DS, Kopin IJ. Linking Stress, Catecholamine Autotoxicity, and Allostatic Load with Neurodegenerative Diseases: A Focused Review in Memory of Richard Kvetnansky. Cell Mol Neurobiol. 2018;38(1):13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Nakai A, Suzuki K. Adrenergic control of lymphocyte trafficking and adaptive immune responses. Neurochem Int. 2018. Oct 19. [DOI] [PubMed] [Google Scholar]
- 135.Johnson JD, Barnard DF, Kulp AC, Mehta DM. Neuroendocrine Regulation of Brain Cytokines After Psychological Stress. J Endocr Soc. 2019;3(7):1302–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Baker JD, Ozsan I, Rodriguez Ospina S, Gulick D, Blair LJ. Hsp90 Heterocomplexes Regulate Steroid Hormone Receptors: From Stress Response to Psychiatric Disease. Int J Mol Sci. 2018;20(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ingawale DK, Mandlik SK, Patel SS. An emphasis on molecular mechanisms of anti-inflammatory effects and glucocorticoid resistance. J Complement Integr Med. 2015;12(1):1–13. [DOI] [PubMed] [Google Scholar]
- 138.Chen CY, Bonham AC, Plopper CG, Joad JP. Neuroplasticity in nucleus tractus solitarius neurons after episodic ozone exposure in infant primates. J Appl Physiol (1985). 2003;94(2):819–827. [DOI] [PubMed] [Google Scholar]
- 139.Jang AS, Choi IS, Lee JH, Park CS, Park CS. Prolonged ozone exposure in an allergic airway disease model: adaptation of airway responsiveness and airway remodeling. Respir Res. 2006;7:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Deng Q, Deng L, Lu C, Li Y, Norbäck D. Parental stress and air pollution increase childhood asthma in China. Environ Res. 2018; 165:23–31. [DOI] [PubMed] [Google Scholar]
- 141.Bandoli G, von Ehrenstein O, Ghosh JK, Ritz B. Synergistic effects of air pollution and psychosocial stressors on adolescent lung function. J Allergy Clin Immunol. 2016; 138(3):918–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.McEwen BS, Stellar E. Stress and the individual. Mechanisms leading to disease. Arch Intern Med. 1993;153(18):2093–2101. [PubMed] [Google Scholar]
- 143.McEwen BS. Hormones as regulators of brain development: life-long effects related to health and disease. Acta Paediatr Suppl. 1997;422:41–44. [DOI] [PubMed] [Google Scholar]
- 144.Coste SC, Murray SE, Stenzel-Poore MP. Animal models of CRH excess and CRH receptor deficiency display altered adaptations to stress. Peptides. 2001;22(5):733–741. [DOI] [PubMed] [Google Scholar]
- 145.Mair CA, Cutchin MP, Kristen Peek M. Allostatic load in an environmental riskscape: the role of stressors and gender. Health Place. 2011;17(4):978–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hajat A, Diez Roux AV, Castro-Diehl C, Cosselman K, Golden SH, Hazlehurst MF, Szpiro A, Vedal S, Kaufman JD. The Association between Long-Term Air Pollution and Urinary Catecholamines: Evidence from the Multi-Ethnic Study of Atherosclerosis. Environ Health Perspect. 2019; 127(5):57007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Nobles CJ, Grantz KL, Liu D, Williams A, Ouidir M, Seeni I, Sherman S, Mendola P. Ambient air pollution and fetal growth restriction: Physician diagnosis of fetal growth restriction versus population-based small-for-gestational age. Sci Total Environ. 2019; 650(Pt 2):2641–2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Lucht SA, Hennig F, Matthiessen C, Ohlwein S, Icks A, Moebus S, Jöckel KH, Jakobs H, Hoffmann B. Air Pollution and Glucose Metabolism: An Analysis in Non-Diabetic Participants of the Heinz Nixdorf Recall Study. Environ Health Perspect. 2018; 126(4):047001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Qian Z, Lin HM, Chinchilli VM, Lehman EB, Duan Y, Craig TJ, Wilson WE, Liao D, Lazarus SC, Bascom R. Interaction of ambient air pollution with asthma medication on exhaled nitric oxide among asthmatics. Arch Environ Occup Health. 2009; 64(3):168–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Dézsi CA, Szentes V. The Real Role of β-Blockers in Daily Cardiovascular Therapy. Am J Cardiovasc Drugs. 2017; 17(5):361–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Singh D, Fabbri LM, Vezzoli S, Petruzzelli S, Papi A. Extrafine triple therapy delays COPD clinically important deterioration vs ICS/LABA, LAMA, or LABA/LAMA. Int J Chron Obstruct Pulmon Dis. 2019; 14:531–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Egorov AI, Griffin SM, Converse RR, et al. Vegetated land cover near residence is associated with reduced allostatic load and improved biomarkers of neuroendocrine, metabolic and immune functions. Environ Res. 2017;158:508–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Thomson Errol M. Air Pollution, Stress, and Allostatic Load: Linking Systemic and Central Nervous System Impacts JOURNAL OF ALZHEIMERS DISEASE. 2019; 69(3):597–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Burwell-Naney K, Wilson SM, Whitlock ST, Puett R. Hybrid Resiliency-Stressor Conceptual Framework for Informing Decision Support Tools and Addressing Environmental Injustice and Health Inequities. Int J Environ Res Public Health. 2019;16(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Logan AC, Prescott SL, Haahtela T, Katz DL. The importance of the exposome and allostatic load in the planetary health paradigm. J Physiol Anthropol. 2018;37(1):15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Crews DE, Kawa NC, Cohen JH, Ulmer GL, Edes AN. Climate change, uncertainty and allostatic load. Ann Hum Biol. 2019;46(1):3–16. [DOI] [PubMed] [Google Scholar]
- 157.Morello-Frosch R, Shenassa ED. The environmental “riskscape” and social inequality: implications for explaining maternal and child health disparities. Environ Health Perspect. 2006;114(8):1150–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]