

Abstract
A dipyrrin-supported nickel catalyst (AdFL)Ni(py) (AdFL: 1,9-di(1-adamantyl)-5-perfluorophenyldipyrrin; py: pyridine) displays productive intramolecular C−H bond amination to afford N−heterocyclic products using aliphatic azide substrates. The catalytic amination conditions are mild, requiring 0.1−2% catalyst loading and operational at room temperature. The scope of C−H bond substrates was explored and benzylic, tertiary, secondary, and primary C−H bonds are successfully aminated. The amination chemoselectivity was examined using substrates featuring multiple activatable C−H bonds. Uniformly, the catalyst showcases high chemoselectivity favoring C–H bonds with lower bond dissociation energy as well as a wide range of functional group tolerance (e.g., ethers, halides, thioetheres, esters, etc.). Sequential cyclization of substrates with ester groups could be achieved, providing facile preparation of indolizidine framework that is commonly found in a variety of alkaloids. The amination cyclization reaction mechanism was examined employing nuclear magnetic resonance (NMR) spectroscopy to determine the reaction kinetic profile. A large, primary intermolecular kinetic isotope effect (KIE = 31.9 ± 1.0) suggests H−atom abstraction (HAA) is the rate determining step, indicative of H−atom tunneling being operative. The reaction rate has first order dependence in the catalyst and zeroth order in substrate, consistent with the resting state of the catalyst as the corresponding nickel iminyl radical. The presence of the nickel iminyl was determined by multi-nuclear NMR spectroscopy observed during catalysis. The activation parameters (ΔH≠ = 13.4 ± 0.5 kcal/mol; ΔS≠ = −24.3 ± 1.7 cal/mol·K) were measured using Eyring analysis, implying a highly ordered transition state during the HAA step. The proposed mechanism of rapid iminyl formation, rate-determining HAA, and subsequent radical recombination was corroborated by intramolecular isotope labeling experiments and theoretical calculations.
Keywords: If you are submitting your paper to a journal that requires keywords, provide
1. INTRODUCTION
Saturated N-heterocycles constitute a critically important class of molecules found in bioactive alkaloids,1 pharmaceutical agents,2–3 and as chiral elements in asymmetric catalysis.4 Indeed, over half of the top twenty best-selling drugs contain an N-heterocycle in the last six years.5–6 Cyclic amine architectures form heterocyclic cores in medicines used to treat dementia (solanidine),6 gastrointestinal dysmotility (SC-53116),7 mantle cell lymphoma (acalabrutinib),8 and hepatitis C.9 Given the ubiquity and importance of N-heterocycles in medicine, streamlined syntheses for these architectures is a high priority.
Traditional strategies to synthesize N-heterocyclic cores include functional group exchange via nucleophilic substitution, rearrangements, cycloadditions, and reductive amination.10 However, the utility of forming heterocyclic skeletons via direct C–H bond functionalization is becoming a more prevalent strategy. In general, the introduction of diverse chemical functionality into unactivated C–H bonds is an evolving, but not yet solved problem in synthetic chemistry.11–18 Rendering aliphatic C–H bonds into an interchangeable functional group would greatly expand methodology. Indeed, streamlined syntheses for functionalized products while minimizing waste generation would have tremendous impact on the synthesis of fine chemicals and pharmaceuticals. For examples, over 84% of pharmaceuticals feature at least one N atom,19 and nearly one in six reactions involve formation of C–N bonds.20 Thus, direct C–H bond amination methods could provide a streamlined protocol and may serve to enhance the scope of conventional syntheses.
The current state of the art in C–H bond functionalization largely depend on late transition metal catalysts that template the C–H bond activation followed by a C–heteroatom bond forming step.21 Previously, we reported the synthesis of N-heterocycles using an iron catalyzed, intramolecular amination of unactivated aliphatic C–H bonds.22 The dipyrrin-iron catalyst served to extrude dinitrogen from the azide substrate, leaving an activated iminyl radical23–25 that could functionalize intramolecular C–H bonds (benzylic, allylic, tertiary, secondary, primary) leading to the formation of cyclic amines. Several limitations with the methodology included high catalyst loadings and the requirement for product sequestration by in situ protection of the secondary amine products. Several groups have adapted this protocol using different catalyst systems, including iron porphyrins,26–27 cobalt corroles,28 iron bound within redox-active pincer ligands,29 and iron beta-diketiminate metal organic frameworks.30 Each of these systems have improved upon the catalyst performance with regards to decreasing catalyst loading, yet each require elevated reaction temperatures and product sequestration to retard product inhibition. Aryl azide substrates which give rise to cyclic aniline products (far less potent Lewis bases) have been synthesized that bypass this requirement.31–33 More recently, non-base-metal catalyzed variants have been discovered that employ dirhodium catalysts to effect the amination of aliphatic bonds to generate free amine products following deprotection.34 While this strategy exhibits remarkable diastereoselectivity, the Rh2-nitrenoid proposed cannot access primary C−H bond substrates that the high-spin iron catalyzed22 system can (albeit poorly). Collectively, these strategies highlight how direct, ring-closing amination can generate heterocyclic cores without requiring activating groups12, 35 and demonstrating modest functional group tolerance.19, 36–38 Despite these advances, improvements on the state of the art catalysis known include enhancing catalytic performance, increasing functional group tolerance (many of the cited examples feature sparse functional group presence), and an understanding of how different electronic structures give rise to different catalytic capabilities.13
Recently we reported that nickel dipyrrinato complexes are capable of stabilizing reactive iminyl functionalities potent for intermolecular functionalization of benzylic C–H bonds.39 Preparation and characterization of the reactive intermediate suggest an iminyl (2NAr coupled to a triple NiII)or imido (1NR, NiIII) electronic configuration, depending on the N-functional group giving rise to the doublet spin configurations observed. Reaction of the NiI complex with 4-(azidobutyl)benzene exclusively formed 4-phenylbutanenitrile. We report herein that alkylation of the azide-bearing methylene unit mitigates nitrile formation, yielding the corresponding N-heterocyclic products. Furthermore, we find the nickel dipyrrin catalyst is effective for amination of benzylic, tertiary, secondary, and primary C−H bonds with high turn-over numbers, mild reaction conditions, and excellent chemoselectivity. A detailed mechanistic study is presented resulting from NMR kinetic profiling of the reaction, corroborated with intramolecular isotope labelling experiments and density functional theory (DFT) computations.
2. RESULTS AND DISCUSSION
2.1. Catalytic intramolecular C–H amination optimization.
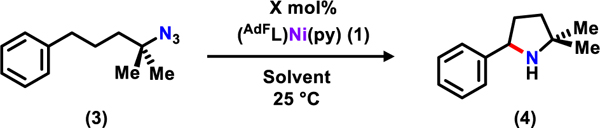
In our previous study, (AdFL)Ni(py) (1) rapidly consumes tertiary azides to afford the corresponding iminyl complexes (e.g., (AdFL)Ni(NAd) (2)) and rapidly converts 4-(azidobutyl)benzene to 4-phenylbutanenitrile via β-hydride elimination (Scheme 1). To inhibit nitrile generation, gem-dimethyl groups were introduced to the azide substrate to promote C–H bond amination. As an initial test, 10 mol% of (AdFL)Ni(py) (1) was subjected to (4-azido-4-methylpentyl)benzene (3) in C6D6 (Table 1, entry 1). An immediate color change from dark brown to deep pink was observed along with intense effervescence. The 19F NMR spectra obtained during the reaction has similar features to that of 2 (Supplementary Information, Figure S5). Bubbling ceased within 10 min with the solution color reverted to dark brown. Both 1H and 19F NMR spectra of the reaction mixture showed complete regeneration of 1 as well as quantitative conversion of 3 into 2,2-dimethyl-5-phenylpyrrolidine (4).
Scheme. 1.

Table 1.
Optimization of Amination Conditionsa
| ||||
|---|---|---|---|---|
| Entry | Catalyst Loading (mol%) | Solvent | Time | Yield (%)d |
| 1 | 10b | C6D6 | 10 min | > 99 |
| 2 | 5b | C6D6 | 20 min | > 99 |
| 3 | 2b | C6D6 | 40 min | 97 |
| 4 | 1b | C6D6 | 90 min | 96 |
| 5 | 0.1 | C6D6 | 48 h | 73 |
| 6 | 1b | Hexanes | 90 min | 95 |
| 7 | 1b | Et2O | 90 min | 96 |
| 8 | 1b | (d8-)Toluenee | 90 min | 95 |
| 9 | 1b | d8-THF | 90 min | 94 |
| 10 | 10 | d6-DMSO | N/Ac | 0 |
| 11 | 10 | d4-Methanol | N/Ac | 0 |
| 12 | 10 | DCM | N/A | 0 |
Reaction were conducted on 0.2 mmol scale, see SI for details of the experiments.
Regeneration of 1 by 19F NMR spectroscopy.
Catalyst decomposition.
Yields were determined by 1H NMR spectroscopy using tetrakis(trimethylsilyl)silane as internal standard.
Both proteo- and deutero-toluene were tested as solvent, and no difference was observed.
Given the full regeneration of 1, the catalyst loading was further lowered to 1 mol % without a significant loss in yield (96%). When 0.1 mol% catalyst loading was used, the reaction took two days at room temperature until no further product formation was observed, providing a final NMR yield of 73% (Table 1, entry 1–5; TONmax = 730). To compare to dipyrrin iron and cobalt systems, the observed reaction rate of 1.4 min−1 (25° C) for 1 is superior to the (AdL)FeCl22 and (ArL)Co(py)40 systems (require heating > 50 °C) or (TrL)Co (0.063 min−1),41 indicating 1 has a much lower activation barrier. The performance and rate of catalysis do not vary significantly with solvent polarity, e.g. from hexanes to THF (Table 1, entry 6–9). Tetrahydrofuran is a catalyst inhibitor for related iron-dipyrrin amination catalysts.22 Notably, even though the iminyl complex (AdFL)Ni(NAd) (2) can abstract the benzylic hydrogen from toluene,39 the intramolecular reaction is sufficiently faster than intermolecular C–H bond amination that the cyclization reaction can be run in toluene (Table 1, entry 8) without diminishing the yield (95%). However, rapid catalyst decomposition via demetallation was observed in protic (MeOH) and redox-active (DMSO) solvents (Table 1, entry 10–11). When dichloromethane is used as solvent, 1 undergoes instantaneous halogen abstraction, generating the corresponding divalent nickel chloride complex (AdFL)NiCl based on the 19F NMR features of the mixture (Table 1, entry 12).39 Based on these results, the optimal conditions were chosen to be 1 mol% catalyst loading in C6D6 for the ease of reaction monitoring.
2.2. The bond dissociation energy limit of activatable C–H bonds.
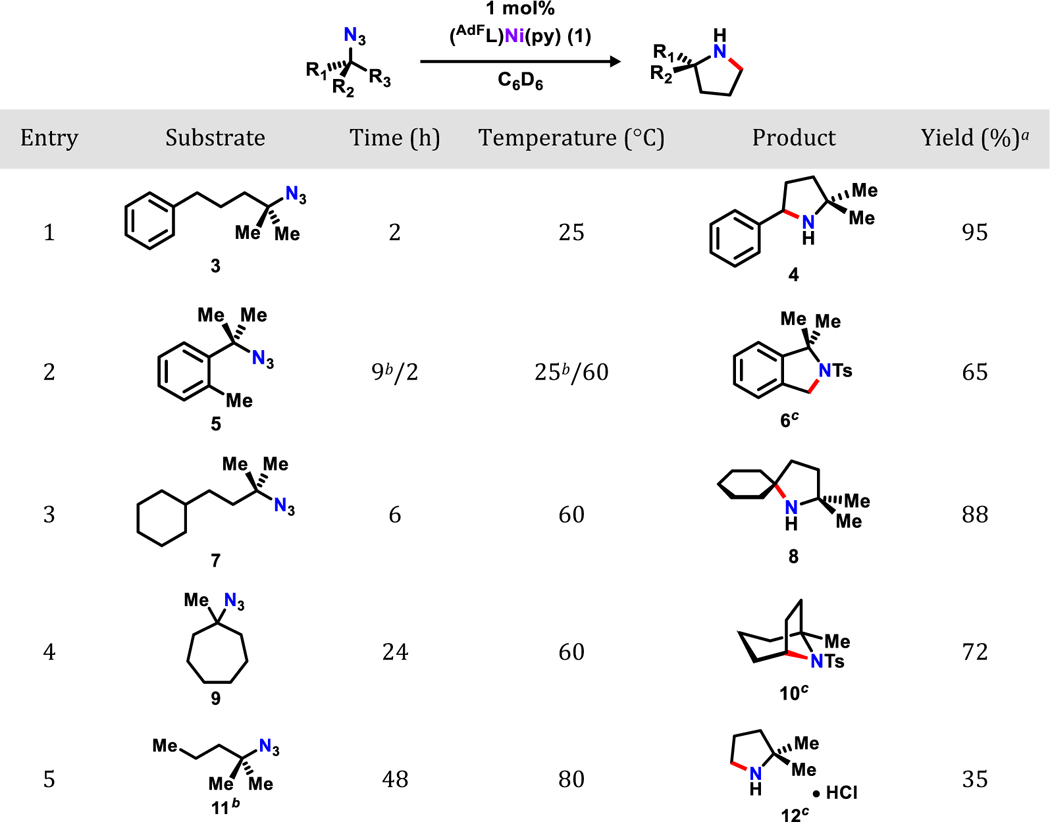
To test the bond strength limit for activatable C–H bonds of this cyclization reaction, a range of substrates with C–H bonds of various bond dissociation energies was examined (Table 2). Substrate 5 features a primary benzylic C–H bond (BDE: 89.7 ± 1.2 kcal/mol)42 requires 9 h at room temperature with 10 mol% catalyst loading to be fully converted to the corresponding pyrrolidine product 6. The same reaction can be completed in only 2 h with 1 mol% catalyst loading heated to 60 °C (65 %). Isolation of the pyrrolidine product 6 was achieved either by an acid-base aqueous extraction or via N-tolsylation (addition TsCl, NEt3, 40 °C, 2 h) followed by column chromatography. Worth noting, the rate difference is likely caused by a combination of an entropically unfavored transition state due to the incorporation of a rigid phenyl group as well as the increased C–H bond dissociation energy in 5 (as compared to 3). Substrates with tertiary and secondary aliphatic C–H bonds can also be readily functionalized in good yields using 1 mol% catalyst loading at 60 °C in 6 and 24 h, respectively (7 and 9). A higher catalyst loading of 10 mol% was required to aminate a substrate featuring a primary C–H bond (11) even at elevated temperature (80 °C, 48 h). Due to the volatility of the product, 12 is separated as an ammonium chloride salt by vacuum transferring into an HCl-diethyl ether solution, leading to a final isolated yield of 35%.
Table 2.
Substrates with various bond dissociation energiesa
|
Isolated yields;
10 mol% of (AdFL)Ni(py) (1) was used;
Derivatized for ease of isolation. Combined yield for two steps.
2.3. Chemoselectivity between C–H bonds with different bond dissociation energies.
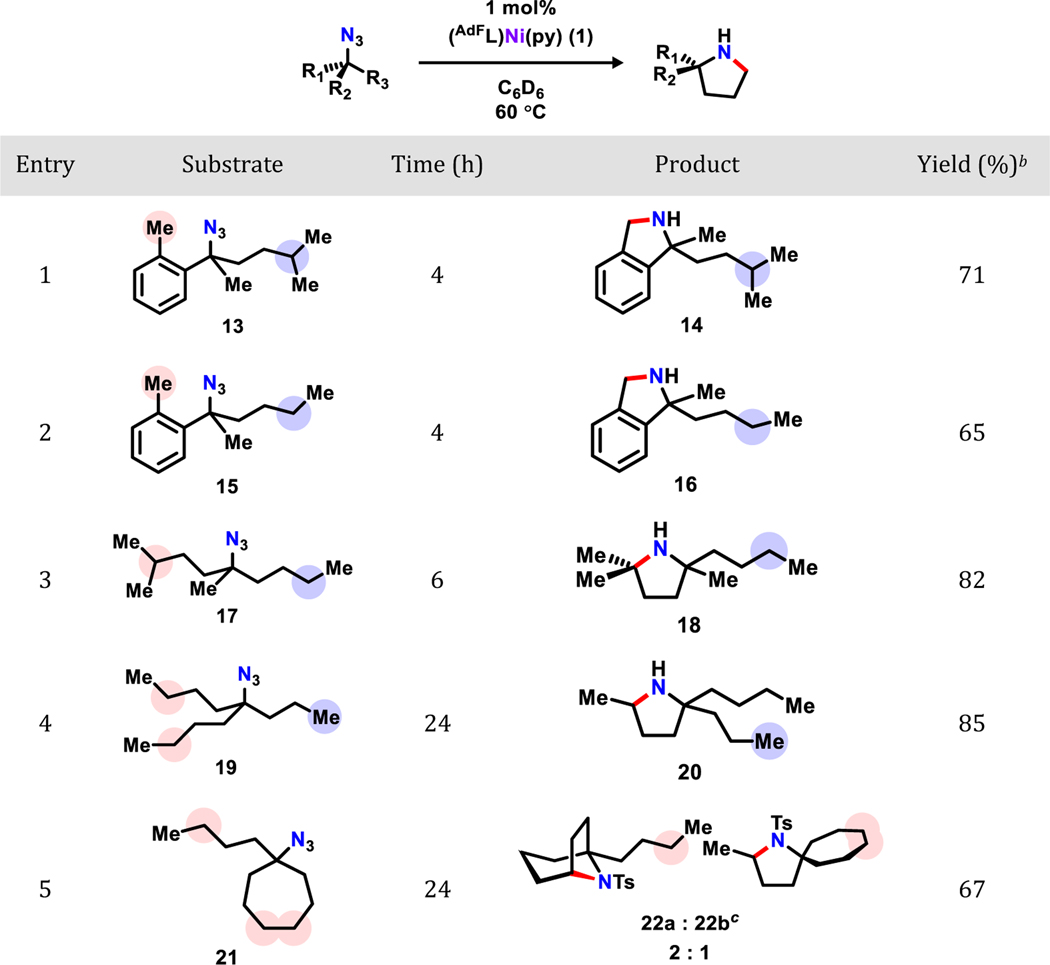
To evaluate the chemoselectivity of this cyclization reaction, azide substrates featuring C–H bonds with different bond dissociation energies were examined under the optimized conditions (entry 1–4; Table 3). Gratifyingly, high levels of chemoselectivity (d.r. > 99:1) favoring C–H bonds with lower bond dissociation energies were observed in every substrate tested. Product corresponding to the stronger C–H bonds was undetectable by 1H NMR spectroscopy. Moreover, when different C–H bonds with similar bond dissociation energy were present in the same substrate in a 2:1 ratio, the expected statistical product mixture (2:1) was obtained (22a-b).
Table 3.
Substrates with competitive C–H bonds of different bond dissociation energiesa
|
Within each substrate, the more activated C–H bonds are noted with red circles, and the less activated ones are labeled with blue circles;
Isolated yields;
Derivatized for ease of isolation. Combined yield for two steps.
2.4. Functional group tolerance.
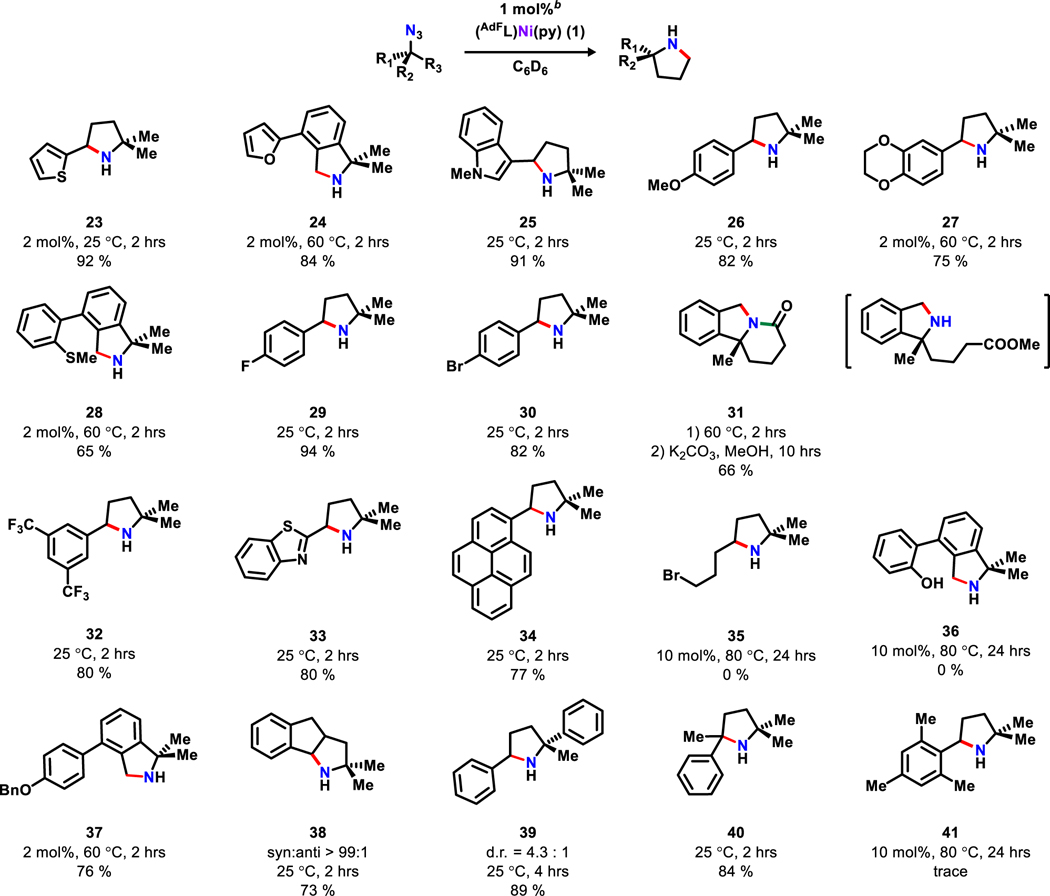
The catalytic amination cyclization reaction tolerates a wide range of functional groups (Table 4): electron rich groups such as thiophene (23), furan (24), N-methylindole (25), phenoxides (26-27) and thioether (28); as well as electron withdrawing groups like aryl halides (29-30), ester (31), and trifluoro methyl groups (32). Notably, when the ester group is positioned appropriately, tandem cyclization can be accomplished, leading to indolizidine (31) framework that is commonly found in a variety of alkaloids.43 Substrates with coordinating functional groups such as benzo[d]thiazole (33) readily undergoes cyclization in good isolated yield (80%). To examine if large, aromatic substituents alpha to the aminated C−H bond would impede catalysis, a pyrene terminated substrate (34) was examined and did not impact catalysis.
Table 4.
Functional group tolerancea
|
All yields noted are isolated yields;
Default catalyst loading (1 mol %) unless noted otherwise.
An aliphatic azide featuring a pendant alkyl bromide (35) leads to rapid catalyst decomposition in a similar way that dichloromethane deactivates the catalyst via halide abstraction (Table 1, entry 12). Acidic functional groups such as phenol (36) also leads to rapid catalyst decomposition likely via proto-demetallation (free (AdFL)H observed by 1H NMR spectroscopy). However, substrates containing protected phenol such as benzyloxy group can be readily cyclized (37).
When the cyclization leads to a fused ring structure (38), high level of diastereoselectivity is observed (syn:anti > 99:1).44 The minor anti isomer is not detectable by 1H NMR spectroscopy. If different α-methylene substituents are introduced at the gem position (Ph/Me), the reaction exhibits a moderate level of diatereoselectivity (39, d.r. = 4.3:1; 83%).45 Although the pyrene substituent in 34 and sterically encumbered tertiary benzylic C–H bond (40) undergo cyclization smoothly, the mesityl substituent in 41 only led to trace amounts of the cyclized product, likely owing to the ortho-phenyl substituents that can sterically impede activation.
2.5. Mechanistic study.
To elucidate the mechanism of the amination cyclization, the concentrations of azide 3 and product 4 during the catalytic reaction with 8 mol% of 1 at room temperature were measured as a function of time (Figure 1a) using 1H NMR spectroscopy. Linear relationships were observed for product generation and azide consumption over time (3𝜏1/2, 14 min), indicating a zeroth order dependence of the reaction rate on the substrate concentration. Similar experiments were carried out using various catalyst loadings from 2−10 mol%. Plotting the slopes obtained from first three half-lives of each run (kobs) versus catalyst loadings reveals a linear relationship, indicating a first order dependence of the reaction rate on the catalyst loading (Figure 1b). Each kinetic experiment is reproduced in triplicate, and error bars are plotted using the standard deviations. The 1H and 19F NMR spectra during catalysis indicate complete conversion of 1 into a paramagnetic species with similar spectroscopic features to the previously reported (AdFL)Ni(NAd) (2) (Figure S5). Notably, the concentration of the new species remains constant over the course of the reaction, suggesting the resting state of the catalyst is the corresponding nickel(II) iminyl species. Complete regeneration of catalyst (AdFL)Ni(py) (1) was observed after reaction completion by 19F NMR spectroscopy.
Figure 1.

(a) Conversion of substrate 3 into 4 as a function of time using 8 mol% of 1. The black dashed line represents the linear fit over the first three half-lives (3𝜏1/2) used to extracted kobs values. (b) Observed rate constants as a function of catalyst loadings with every measurement done in triplicate. Error bars represent standard deviations. Inset: observed rate constants as a function of exogenous pyridine concentrations using 8 mol% of 1. Every measurement is done in triplicate, and error bars represent standard deviations. (c) Eyring analysis with every measurement done in triplicate. Error bars represent standard deviations. Parameters extracted from the Eyring plot: ΔH≠ = 13.4 ± 0.5 kcal/mol; ΔS≠ = −24.3 ± 1.7 cal/mol·K; ΔG≠(25°C) = 20.6 ± 0.5 kcal/mol.
Considering our recently reported cobalt dipyrrin catalytic system whose rate depends on concentration of exogenous L-type ligand such as pyridine, we sought to examine if a similar effect exists for the nickel system. As the isolated nickel adamantyl iminyl complex 2 does not have pyridine bound, several catalytic runs with 8 mol% catalyst loading were carried out with pyridine ranging from 0–30 mol% added. Based on the kobs values extracted from the slopes over first three half-lives of the trial runs, the reaction rate does not vary significantly with exogenous pyridine concentrations (Figure 1b), possibly due to the increased electronegativity of nickel compared to cobalt.40–41 Notably, at high pyridine concentration (30 mol%), even though the product generation follows a linear relationship over the first three half-lives, the correlation deviates from linearity at low substrate concertation, indicating a competitive binding equilibrium between the substrate 3 and pyridine.
We have shown in our previous Fe- and Co-based catalytic systems by measuring the kinetic isotope effects that H-atom abstraction contributes to the rate determining step of the amination.22, 40–41, 45 Similarly, the bis-deutero substituted substrate (4-azido-4-methylpentyl-1,1-d2)benzene (3D2) was prepared and treated with 8 mol% of 1. By measuring concentration of the product 4D over time (Figure S4), an intermolecular kinetic isotope effect of 31.9 ± 1.0 was determined. The large KIE value suggests the C–H bond activation goes through a radical-based H−atom abstraction with significant tunneling being operative rather than a concerted C−H bond insertion mechanism.46 Given the configurational restriction by tunneling effect, a large, negative activation entropy is expected. Indeed, by conducting the same kinetic experiment at various temperatures and performing Eyring analysis, activation enthalpy of 13.4 ± 0.5 kcal/mol and activation entropy of −24.3 ± 1.7 cal/mol·K were obtained, consistent with other previously reported kinetic measurements on similar 1,5-HAA reactions (Figure 1).47–48
Based on the above kinetic measurements, a catalytic cycle is proposed (Figure 2). Substrate 3 displaces pyridine from 1, followed by release of dinitrogen giving rise to the corresponding nickel iminyl species (e.g., NiII(2NR)) as the resting state of the catalyst.39 The next step is the rate-determining hydrogen atom abstraction via tunneling followed by radical recombination to furnish the bound cyclized product. Product displacement is aided by ligand substitution with pyridine or substrate 3, releasing the pyrrolidine product 4. Since 3 enters the cycle before the resting state, the reaction rate is not dependent on its concentration.46
Figure 2.

Proposed mechanism for C–H amination.
2.6. Theoretical Modelling of the Reaction Profile.
Density functional theory (DFT) calculations were employed in order to provide support for the proposed mechanism in Figure 3, and to gain insight into geometric factors that may influence substrate turnover during catalysis. The calculations were performed with the B3LYP functional and the def2-SVP (C, H) and def2-TZVP (Ni, N, F) basis sets on an untruncated system.49–52 The full computational methodology is described in detail in the Supporting Information. We first optimized the minima along the reaction coordinate corresponding to the resting state alkyl iminyl species B and the ring-closed, pyrrolidine bound D. The DFT optimized resting state B has similar coordination bond lengths (Ni−Nim = 1.668 Å) to the crystallographically characterized adamantyl imido we previously described (Ni−Nim = 1.642(7) Å), whereas the product bound D has a significantly longer Ni−Nim bond, (Ni–Nim = 2.126 Å) more in line with the Ni(I) catalyst (1) (Ni–Npyr = 1.9566(19) Å). Product-bound D is 30.4 kcal/mol lower in energy relative to the catalyst resting state. In order to locate transition states corresponding to hydrogen-atom abstraction (HAA – TS1) and pyrrolidine ring closure (TS2) as a result of alkyl radical rebound, a relaxed potential energy surface scan along the C4–Nim bond vector (20×0.1 Å steps) was conducted starting from the product bound D and searching for an energy maximum. The corresponding maximum energy geometry was then subjected to a transition state optimization, resulting in a species with a single imaginary frequency (νim = −297 cm−1) located 32.6 kcal/mol above the ring closed product, corresponding to TS2, ring closure between C4 and Nim as a result of alkyl radical rebound. Connectivity between TS2 and D was established via an intrinsic reaction coordinate (IRC) calculation, during which the Ni–Nim bond length lengthens substantially (1.877 Å in TS2 to 2.126 Å in D) while moving along the reaction coordinate. Furthermore, C4 is planarized in TS2, suggesting radical character at that position.
Figure 3.

DFT calculated reaction pathway for the cyclization of 3 into 2-phenyl-5,5-dimethylpyrrolidine (4) mediated by (AdFL)Ni(py) (1). Energies are reported in kcal/mol relative to the (AdFL)Ni(•NR) catalyst resting state. [B3LYP/ def2-SVP (C, H) and def2-TZVP (Ni, N, F)]49–52
We then endeavored to locate the transition state corresponding to H−atom abstraction by moving the proton from Nim back to C4 and performing an optimization, resulting in a local minimum immediately preceding HAA (see the Supporting Information for geometry and coordinates, Figure S7–S13, Table S2–S8). A relaxed potential energy surface scan along the Nim−H vector located a maximum energy structure which was optimized to a transition state corresponding to HAA with a single imaginary frequency (νim = −1693 cm−1) located 23.3 kcal/mol above the catalyst resting state. While this is in excellent agreement with the barrier determined experimentally (20.6 ± 0.5 kcal/mol), the intermediate C following HAA and preceding radical rebound was found as confirmation of HAA as rate determining. The reaction coordinate was followed in the forward direction from TS1 to locate a local minimum structure preceding radical rebound, located 5.3 kcal/mol below TS2. This modest barrier to radical rebound agrees with HAA as the rate determining step by both experimental and theoretical means.
2.7. Intramolecular isotope labelling experiment.
In order to gain kinetic information on the rate of radical recombination step, a pair of isotopically labelled enantiopure substrates (S-3HD and R-3HD) were prepared.55 Subjecting this pair of substrates to standard catalytic conditions could lead to four potential products: S-4H, R-4H, S-4D and R-4D (Figure 4). The two pairs of enantiomers (S-4H, R-4H; S-4D, R-4D) are resolved using Mosher analysis53–54 by condensing the isolated mixture of 4H and 4D with (S)-(+)-α-Methoxy-α-(trifluoromethyl)phenylacetyl chloride (S-Mosher acyl chloride). The product distributions are obtained from 1H NMR spectrum of the final mixture. Since catalyst 1 is achiral and the pair of substrates (S-3HD and R-3HD) are enantiomers, the same product distribution with inverted chirality should be expected from both reactions. Indeed, the same distribution was observed with a large ratio between 4D and 4H in both cases, suggesting the H−atom abstraction is favored over its deuterium counterpart, yielding an intramolecular KIE of 18.1, much larger than the values observed in our previously published iron and cobalt dipyrrin systems.22, 40
Figure 4.

Product distribution using enantiopure mono D-labeled substrates S-3HD and R-3HD. The two pairs of enantiomers (S-4H and R-4H; S-4D and R-4D) are resolved using Mosher analysis53–54 and the ratios are obtained using 1H-NMR spectroscopy. All reactions were carried out using standard catalytic conditions at room temperature.
Once the H/D-atom is abstracted, two processes take place in competition: the planar, carbon-based benzyl radical rotation to expose either the Re or Si face near the iminyl N, and radical rebound to form the C–N bond. Given the substrates S-3HD and R-3HD are enantiopure, if the rate of radical trapping process exceeds that of the carboradical rotation, the corresponding enantiopure pyrrolidines (4H or 4D) should be expected. Otherwise, if the radical rebound process is sufficiently slow that the carboradical rotation reaches thermo-equilibrium first, completely racemic mixtures would be observed instead. The experimental results, however, revealed an intermediate ratio of 4.6 (average from two reactions) between the major and minor isomers. The intermediate value suggests that radical is trapped before its rotation could reach thermo-equilibrium, suggesting that the two processes happen on a comparable time scale. A similar strategy to probe the rate of radical rebound has been used to examine the mechanism of C–H hydroxylation using cytochrome P450,56 intramolecular C–H insertion using iron-carbene complexes,57 allylic C–H abstraction by singlet oxygen,58 and more recently enantioselective carbene C–H insertion catalyzed by chiral cobalt porphyrin complexes.59
Based on these results, two potential strategies toward enantioselective C–H amination catalysis can be proposed depending on the relative rate between radical recombination and rotation.59 If the radical rebound is much faster, the enantioselectivity is solely determined by the H−atom abstraction step. Therefore, the catalyst should have its chiral elements proximal to the iminyl nitrogen. If, however, carboradical rotation reaches thermo-equilibrium first, the reaction will be under Curtin-Hammett control.46, 59 The enantioselectivity is then dictated by the relative barrier height of the radical rebound process with Re or Si face of the carbon-based radical. In this case, the catalyst should be designed to better accommodate one prochiral face of the radical versus the other. The enantioselective C–H amination catalysis is currently under study.
3. CONCLUSION
In conclusion, we have developed an efficient nickel dipyrrinato catalyst capable of aminating strong, aliphatic C–H bonds with simple ligand synthesis, low catalyst loading, mild reaction conditions, wide substrate scope and high chemoselectivity. A detailed mechanistic study was also carried out to provide insights into further improvements of the system. Theoretical studies closely reproduced experimentally measured kinetics barriers, corroborating proposed mechanistic cycle. Intramolecular isotope labelling experiment suggests that the radical recombination step happens on the same time scale as the radical rotation. Based on these results, two potential strategies for enantioselective amination are proposed, and catalysts competent for such enantioselective catalysis are currently under study.
Supplementary Material
ACKNOWLEDGMENT
This work was generously supported by a grant from the NIH (GM-115815), the Dreyfus Foundation (Teacher-Scholar Award to T.A.B.), and Harvard University. R.M.C. gratefully acknowledges an NSERC Postdoctoral Fellowship.
Footnotes
ASSOCIATED CONTENT
The Supporting Information is available free of charge on the
ACS Publications website.
General experimental considerations and procedures, multinuclear NMR data(PDF)
The authors declare no competing financial interest.
REFERENCES
- 1.Modern alkaloids . Structure, isolation, synthesis, and biology. Wiley-VCH Verlag GmbH & Co.: Weinheim, 2012. [Google Scholar]
- 2.Vitaku E; Smith DT; Njardarson JT, Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J. Med. Chem 2014, 57, 10257. [DOI] [PubMed] [Google Scholar]
- 3.Taylor RD; MacCoss M; Lawson ADG, Rings in drugs. J. Med. Chem 2014, 57, 5845. [DOI] [PubMed] [Google Scholar]
- 4.Banik SM; Levina A; Hyde AM; Jacobsen EN, Lewis acid enhancement by hydrogen-bond donors for asymmetric catalysis. Science 2017, 358, 761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vitaku E; Smith BR; Smith DT; Njardarson JT, Top US pharmaceutical products of 2013. 2014. [Google Scholar]
- 6.Race NJ; Hazelden IR; Flaulkner A; Bower JF, Recent developments in the use of aza-Heck cyclizations for the synthesis of chiral N-heterocycles. Chem. Sci 2017, 8, 5248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Flynn DL; Zabrowski DL; Becker DP; Nosal R; Villamil CI; Gullikson GW; Moummi C; Yang DC, SC-53116: the first selective agonist at the newly identified serotonin 5-HT4 receptor subtype. J. Med. Chem 1992, 35, 1486. [DOI] [PubMed] [Google Scholar]
- 8.Wang M; Rule S; Zinzani PL; Goy A; Casasnovas O; Smith SD; Damaj G; Doorduijn J; Lamy T; Morschhauser F; Panizo C; Shah B; Davies A; Eek R; Dupuis J; Jacobsen E; Kater AP; Le Gouill S; Oberic L; Robak T; Covey T; Dua R; Hamdy A; Huang X; Izumi R; Patel P; Rothbaum W; Slatter JG; Jurczak W, Acalabrutinib in relapsed or refratory matle cell lymphoma (ACE-LY-004): a single-arm, multicentre, phase 2 trial. Lancet 2018, 391, 659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Poordad F; Felizarta F; Asatryan A; Sulkowski MS; Reindollar RW; Landis CS; Gordon SC; Flamm SL; W FM; Bernstein DE; Lin CW; Liu R; Lovell SS; I NT; Kort J; Mensa FJ, Glecarprevir and pirbrentasvir for 12 weeks for hepatitis C virus gentype 1 infection and prior direct-acting antiviral treatment. Hepatology 2017, 66, 389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Candeias NR; Branco LC; Gois PMP; Afonso CAM; Trindade AF, More sustainable approaches for the synthsis of N-based heterocycles. Chem. Rev 2009, 109, 2703. [DOI] [PubMed] [Google Scholar]
- 11.Labinger JA; Bercaw JE, Understanding and Exploiting C–H Bond Activation. Nature 2002, 417, 507. [DOI] [PubMed] [Google Scholar]
- 12.Dick AR; Sanford MS, Transition metal catalyzed oxidative functionalization of carbon–hydrogen bonds. Tetrahedron 2006, 62, 2439. [Google Scholar]
- 13.Goswami M; Lyaskovskyy V; Domingos SR; Buma WJ; Woutersen S; Troeppner O; Ivanović-Burmazović I; Lu H; Cui X; Zhang XP; Reijerse EJ; DeBeer S; van Schooneveld MM; Pfaff FF; Ray K; de Bruin B, Characterization of Porphyrin-Co(III)-’Nitrene Radical’ Species Relevant in Catalytic Nitrene Transfer Reactions. J. Am. Chem. Soc 2015, 137, 5468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li C; Lang K; Lu H; Hu Y; Cui X; Wojtas L; Zhang XP, Catalytic Radical Process for Enantioselective Amination of C(sp3)−H Bonds. Angew. Chem. Int. Ed 2018, 57, 16837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu H; Hu Y; Jiang H; Wojtas L; Zhang XP, Stereoselective Radical Amination of Electron-Deficient C(sp3)–H Bonds by Co(II)-Based Metalloradical Catalysis: Direct Synthesis of α-Amino Acid Derivatives via α-C–H Amination. Org. Lett 2012, 14, 5158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu H; Jiang H; Hu Y; Wojtas L; Zhang XP, Chemoselective intramolecular allylic C–H amination versus C=C aziridination through Co(ii)-based metalloradical catalysis. Chem. Sci 2011, 2, 2361. [Google Scholar]
- 17.Lu H; Lang K; Jiang H; Wojtas L; Zhang XP, Intramolecular 1,5-C(sp3)–H radical amination via Co(ii)-based metalloradical catalysis for five-membered cyclic sulfamides. Chem. Sci 2016, 7, 6934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu H; Li C; Jiang H; Lizardi CL; Zhang XP, Chemoselective Amination of Propargylic C(sp3)–H Bonds by Cobalt(II)-Based Metalloradical Catalysis. Angew. Chem. Int. Ed 2014, 53, 7028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vitaku E; Smith DT; Njardarson JT, Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J. Med. Chem 2014, 57, 10257. [DOI] [PubMed] [Google Scholar]
- 20.Roughley SD; Jordan AM, The medicinal chemist’s toolbox: an analysis of reactions used in the pursuit of drug candidates. J. Med. Chem 2011, 54, 3451. [DOI] [PubMed] [Google Scholar]
- 21.Nakamura I; Yamamoto Y, Transition-metal-catalyzed reactions in heterocyclic synthesis. Chem. Rev 2004, 104, 2127. [DOI] [PubMed] [Google Scholar]
- 22.Hennessy ET; Betley TA, Complex N-Heterocycle synthesis via iron-catalyzed, direct C-H bond amination. Science 2013, 340, 591. [DOI] [PubMed] [Google Scholar]
- 23.King ER; Hennessy ET; Betley TA, Catalytic C-H bond amination from high-spin iron imide complexes. J. Am. Chem. Soc 2011, 133, 4917. [DOI] [PubMed] [Google Scholar]
- 24.Wilding MJT; Iovan DA; Wrobel AT; Luken JT; MacMillan SN; Lancaster KM; Betley TA, Direct comparison of C–H bond amination efficacy through manipulation of nitrogen-valance centered redox: imido versus iminyl. J. Am. Chem. Soc 2017, 139, 14757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iovan DA; Betley TA, Characterization of iron–imido species relevant for N–group transfer chemistry. J. Am. Chem. Soc 2016, 138, 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Du Y-D; Xu Z-J; Zhou C-Y; Che C-M, An effective [FeIII(TF4DMAP)Cl] catalyst for C-H bond amination with aryl and alkyl azides. Org. Lett 2019, 21, 895. [DOI] [PubMed] [Google Scholar]
- 27.Shing K-P; Liu Y; Cao B; Chang X-Y; You T; Che C-M, N-heterocyclic carbene iron(III) porphyrin-catalyzed intramolecular C(sp3)-H amination of alkyl azides. Angew. Chem. Int. Ed 2018, 57, 11947. [DOI] [PubMed] [Google Scholar]
- 28.Goswami M; Geuijen P; Reek JNH; de Bruin B, Application of [Co(Corrole)]- complexes in ring-closing C-H amination of aliphatic azides via nitrene radical intermediates. Eur. J. Inorg. Chem 2018, 617. [Google Scholar]
- 29.Bagh B; Broere DLJ; Sinha V; Kuijpers PF; van Leest NP; de Bruin B; Demeshko S; Siegler MA; van der Vlugt JI, Catalytic synthesis of N-heterocycles via direct C(sp3)–H amination using an air-stable iron(III) species with a redox-active ligand. J. Am. Chem. Soc 2017, 139, 5117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thacker NC; Lin Z; Zhang T; Gilhula JC; Abney CW; Lin W, Robust and porous β-diketiminate-functionalized metal–organic frameworks for earth-abundant-metal-catalyzed C–H amination and hydrogenation. J. Am. Chem. Soc 2016, 138, 3501. [DOI] [PubMed] [Google Scholar]
- 31.Alt IT; Guttroff C; Plietker B, Iron-catalyzed intramolecular aminations of C(sp3)-H bonds in alkylaryl azides. Angew. Chem. Int. Ed 2017, 56, 10582. [DOI] [PubMed] [Google Scholar]
- 32.Liu Y; Wei J; Che C-M, [Fe(F20TPP)Cl] catalyzed intramolecular C-N bond formation for alkaloid synthesis using aryl azides as nitrogen source. Chem. Commun 2010, 46, 6926. [DOI] [PubMed] [Google Scholar]
- 33.Villanueva O; Weldy NM; Blakey SB; MacBeth CE, Cobalt catalyzed sp3 C-H amination utilizing aryl azides. Chem. Sci 2015, 6, 6672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Munnuri S; Adebesin AM; Paudyal MP; Yousufuddin M; Dalipe A; Falck JR, Catalyst-controlled diastereoselective synthesis of cyclic amines via C-H functionalization. J. Am. Chem. Soc 2017, 139, 18288. [DOI] [PubMed] [Google Scholar]
- 35.Hinman A; Du Bois J, A Stereoselective Synthesis of (−)-Tetrodotoxin. J. Am. Chem. Soc 2003, 125, 11510. [DOI] [PubMed] [Google Scholar]
- 36.Hartwig JF, Organtransition Metal Chemistry: From Bonding to Catalysis. University Science Books: Mill Valley, CA, 2010. [Google Scholar]
- 37.Hartwig JF, Catalyst-controlled site-selective bond activation. Acc. Chem. Res 2017, 50, 549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hartwig JF, Borylation and silylation of C-H bonds: a platform for diverse C-H bond functionalizations. Acc. Chem. Res 2012, 45, 864. [DOI] [PubMed] [Google Scholar]
- 39.Dong Y; Lukens JT; Clarke RM; Zheng SL; Lancaster KM; Betley TA, Synthesis, characterization and C-H amination reactivity of nickel iminyl complexes. Chem. Sci 2020, 11, 1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baek Y; Betley TA, Catalytic C–H Amination Mediated by Dipyrrin Cobalt Imidos. J. Am. Chem. Soc 2019, 141, 7797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baek Y; Hennessy ET; Betley TA, Direct manipulation of metal imido geometry: key principles to enhance C-H amination efficacy. J. Am. Chem. Soc 2019, 141, 16944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Luo Y-R, Comprehensive Handbook of Chemical Bond Energies. 1st ed.; CRC Press: Boca Raton, 2007. [Google Scholar]
- 43.Howard AS; Michael JP, Chapter 3 Simple Indolizidine and Quinolizidine Alkaloids. In The Alkaloids: Chemistry and Pharmacology, Brossi A, Ed. Academic Press: 1986; Vol. 28, pp 183. [Google Scholar]
- 44.Wang H-M; Zhou H; Xu Q-S; Liu T-S; Zhuang C-L; Shen M-H; Xu H-D, Copper-Catalyzed Borylative Cyclization of Substituted N-(2-Vinylaryl)benzaldimines. Org. Lett 2018, 20, 1777. [DOI] [PubMed] [Google Scholar]
- 45.Iovan DA; Wilding MJT; Baek Y; Hennessy ET; Betley TA, Diastereoselective C−H Bond Amination for Disubstituted Pyrrolidines. Angew. Chem. Int. Ed 2017, 56, 15599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Anslyn EV; Dougherty DA, Modern Physical Organic Chemistry. University Science Books: 2006. [Google Scholar]
- 47.Chiba S; Chen H, sp3 C–H oxidation by remote H-radical shift with oxygen- and nitrogen-radicals: a recent update. Org. Biomol. Chem 2014, 12, 4051. [DOI] [PubMed] [Google Scholar]
- 48.Wang Y; Olankitwanit A; Rajca S; Rajca A, Intramolecular Hydrogen Atom Transfer in Aminyl Radical at Room Temperature with Large Kinetic Isotope Effect. J. Am. Chem. Soc 2017, 139, 7144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schäfer A; Horn H; Ahlrichs R, Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys 1992, 97, 2571. [Google Scholar]
- 50.Becke AD, Density‐functional thermochemistry. III. The role of exact exchange. J. Chem. Phys 1993, 98, 5648. [Google Scholar]
- 51.Becke AD, A new mixing of Hartree–Fock and local density‐functional theories. J. Chem. Phys 1993, 98, 1372. [Google Scholar]
- 52.Schäfer A; Huber C; Ahlrichs R, Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys 1994, 100, 5829. [Google Scholar]
- 53.Dale JA; Dull DL; Mosher HS, .alpha.-Methoxy-.alpha.-trifluoromethylphenylacetic acid, a versatile reagent for the determination of enantiomeric composition of alcohols and amines. The Journal of Organic Chemistry 1969, 34, 2543. [Google Scholar]
- 54.Dale JA; Mosher HS, Nuclear magnetic resonance enantiomer regents. Configurational correlations via nuclear magnetic resonance chemical shifts of diastereomeric mandelate, O-methylmandelate, and .alpha.-methoxy-.alpha.-trifluoromethylphenylacetate (MTPA) esters. J. Am. Chem. Soc 1973, 95, 512. [Google Scholar]
- 55.Elsenbaumer RL; Mosher HS, Enantiomerically pure (R)-(+)-2-phenylethanol-2-d and −1,1,2-d3, and (S)-(+)-1-phenylethane-1-d, −1,2,-d2, −1,2,2-d3, and −1,2,2,2-d4. The Journal of Organic Chemistry 1979, 44, 600. [Google Scholar]
- 56.White RE; Miller JP; Favreau LV; Bhattacharyya A, Stereochemical dynamics of aliphatic hydroxylation by cytochrome P-450. J. Am. Chem. Soc 1986, 108, 6024. [DOI] [PubMed] [Google Scholar]
- 57.Ishii S; Zhao S; Helquist P, Stereochemical Probes of Intramolecular C−H Insertion Reactions of Iron-Carbene Complexes. J. Am. Chem. Soc 2000, 122, 5897. [Google Scholar]
- 58.Alberti MN; Vassilikogiannakis G; Orfanopoulos M, Stereochemistry of the Singlet Oxygenation of Simple Alkenes: A Stereospecific Transformation. Org. Lett 2008, 10, 3997. [DOI] [PubMed] [Google Scholar]
- 59.Lang K; Torker S; Wojtas L; Zhang XP, Asymmetric Induction and Enantiodivergence in Catalytic Radical C–H Amination via Enantiodifferentiative H-Atom Abstraction and Stereoretentive Radical Substitution. J. Am. Chem. Soc 2019, 141, 12388. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.