

Abstract
Characterization of a nitrite reductase-negative Staphylococcus carnosus Tn917 mutant led to the identification of the nir operon, which encodes NirBD, the dissimilatory NADH-dependent nitrite reductase; SirA, the putative oxidase and chelatase, and SirB, the uroporphyrinogen III methylase, both of which are necessary for biosynthesis of the siroheme prosthetic group; and NirR, which revealed no convincing similarity to proteins with known functions. We suggest that NirR is essential for nir promoter activity. In the absence of NirR, a weak promoter upstream of sirA seems to drive transcription of sirA, nirB, nirD, and sirB in the stationary-growth phase. In primer extension experiments one predominant and several weaker transcription start sites were identified in the nir promoter region. Northern blot analyses indicated that anaerobiosis and nitrite are induction factors of the nir operon: cells grown aerobically with nitrite revealed small amounts of full-length transcript whereas cells grown anaerobically with or without nitrite showed large amounts of full-length transcript. Although a transcript is detectable, no nitrite reduction occurs in cells grown aerobically with nitrite, indicating an additional oxygen-controlled step at the level of translation, enzyme folding, assembly, or insertion of prosthetic groups. The nitrite-reducing activity expressed during anaerobiosis is switched off reversibly when the oxygen tension increases, most likely due to competition for electrons with the aerobic respiratory chain. Another gene, nirC, is located upstream of the nir operon. nirC encodes a putative integral membrane-spanning protein of unknown function. A nirC mutant showed no distinct phenotype.
Two major pathways of nitrite reduction to ammonia in members of the family Enterobacteriaceae have been described (reviewed by Cole [6]). The main nitrite reductase activity in Escherichia coli is contributed by the cytoplasmic NADH-nitrite reductase, which detoxifies the nitrite formed as the product of nitrate reduction. The less active pathway in most E. coli strains is the electrogenic, formate-dependent nitrite reductase (Nrf pathway; reviewed in references 6 and 19). Both enzymes in E. coli are induced by anaerobiosis and nitrite and fulfill a dissimilatory rather than an assimilatory role (5).
The nir operon in E. coli encodes the NADH-dependent nitrite reductase and consists of five open reading frames (ORFs): nirB, nirD, nirE, nirC, and cysG (21). Essential and sufficient for NADH-dependent nitrite reduction are NirB and NirD, the two subunits of the enzyme (11), and CysG, which is essential for biosynthesis of the siroheme prosthetic group (12, 27, 32). Expression of the nir operon is totally dependent on fumarate and nitrate reductase regulation (FNR) (28), the global E. coli transcription regulator of anaerobically controlled genes (10). FNR-dependent transcription is modulated by two two-component regulatory systems (NarX-NarL and NarQ-NarP), which appear to coordinate transcriptional responses to nitrate and nitrite (7, 22, 33).
High concentrations of nitrate and nitrite in foodstuffs and drinking water can cause severe health problems. A potential application of microorganisms for denitrification has directed attention to the food-grade organism Staphylococcus carnosus, which has been used for a long time in the production of raw fermented sausages due to its ability to reduce nitrate to nitrite, which is essential for development of the typical red color. The nitrate- and nitrite-reducing system of the organism has been characterized physiologically and may have dissimilatory functions (17). Recently, Pantel et al. (20) identified the narGHJI operon, which encodes the dissimilatory nitrate reductase of S. carnosus. The enzyme shows similarity to the corresponding E. coli enzyme with respect to sequence characteristics, induction factors (anaerobiosis, nitrate, and nitrite), and energy gain. Nitrite is converted to ammonia in S. carnosus (17) only in the absence of oxygen and nitrate and if the initial nitrite concentration does not exceed 10 mM (17).
In this paper, we identify and characterize the S. carnosus nitrite reductase operon, which is comprised of five genes: nirR, sirA, nirB, nirD, and sirB. In accordance with the physiological results, the sequence characteristics of the nitrite reductase NirBD resembles the NADH-dependent nitrite reductase of E. coli. SirA and SirB appear to be necessary for biosynthesis of the siroheme prosthetic group. The protein NirR shows no convincing similarity to proteins with known functions, and we attempted to determine its potential function in the enzymatic process or in regulation. Furthermore, we analyzed the intriguing regulation of the nitrite-reducing system in response to oxygen and nitrite.
MATERIALS AND METHODS
Media and culture conditions.
The bacterial strains were grown at 37°C in basic medium (BM) (9) or in modified BM (17). Aerobic cultures were incubated on a rotary shaker at 160 rpm. Anaerobic cultures were incubated in screw-cap bottles with slow stirring (100 rpm). The medium was supplemented with Oxyrase (20 ml/liter of medium; Oxyrase Inc., Mansfield, Ohio) to create anoxic conditions.
Bacterial strains, plasmids, and plasmid constructions.
S. carnosus TM300 (24) was used as the wild-type strain for transposon mutagenesis and also as a control strain and cloning host. E. coli SURE (Stratagene) served as the cloning host for vectors constructed for sequencing and for pRB473 derivatives. This shuttle vector, a derivative of pRB373 (4), was used to construct complementation vectors. The E. coli vectors pUC18 (29) and pBluescriptII KS(+) (Stratagene) were used for subcloning DNA fragments for sequencing.
For complementation of mutant nir1 (see Results and Discussion), pUCnir1-1 (this study) was digested with HindIII/SphI and HindIII/XmaI and the obtained 3- and 5-kb fragments, respectively, were cloned into pRB473, resulting in plasmids pR-AB and pR-ABDB, respectively (Fig. 1). For complementation of mutant M12, plasmids pR-PNO, pR-PABDB, and pR-PBDB were constructed (Fig. 1). Plasmid pR-PNO contains the complete nir operon and 371 nucleotides upstream of the predicted start codon as the promoter region. The nir promoter region together with the 5′ end of nirR was amplified by PCR with the primers MutnirC2, 5′-GAAGGCTTGTCTAAAGCTTTCTTTATCGC-3′, and MutnirC1, 5′-GCATGTGCAATCGTCATATCTGAGTTGCCC-3′. The obtained PCR product was cleaved with HindIII and cloned into HindIII-digested pR-ABDB. For construction of pR-PABDB and pR-PBDB, the nir promoter region was cloned into plasmids pR-ABDB and pR-BDB, respectively. For construction of pR-BDB, in which the genes nirB, nirD, and sirB are cloned with their ribosome-binding-site sequences but without the nir promoter, the beginning of nirB was amplified by PCR with the primers nir1*, 5′-GTATTTACAGGAAGCTTATGGGAGGGAATAACGG-3′, and nirB-int, 5′-CACGCAGCATATCTCCTGCTTGACGG-3′, digested with HindIII and AvaI, and ligated together with the 3.6-kb AvaI fragment of pUCnir1-1 into HindIII- and AvaI-digested pRB473. The nir promoter was amplified with the primers MutnirC2 (see above) and PNOP1: 5′-GCGACATGATGTGTATCAAGCTTTAATTGCATTGTACC-3′. After HindIII cleavage, the PCR product was ligated into the HindIII-digested vectors pR-BDB and pR-ABDB, resulting in pR-PBDB and pR-PABDB, respectively. Underlined sequences in the primers are the introduced restriction sites. Plasmid pRBnirC, used for complementation of mutant MC11, was obtained by EcoRI cleavage of pRBC-Δ12-B (see below) and religation of the 7.1-kb fragment.
FIG. 1.

Map of the locus comprising genes involved in nitrite reduction in S. carnosus. The DNA fragments isolated from partial gene libraries or by inverse PCR and used for sequencing are shown above the gene map together with the names of the plasmids in which these fragments were cloned. Please note that plasmid pRBnirC comprises only the 1.56-kb HindIII/EcoRI fragment. Below the gene map, the different fragments cloned in pRB473 are depicted together with the names of the respective plasmid constructs that were used for complementation analysis and for homologous recombination. The restriction sites of enzymes indicated in parentheses belong to the vector pRB473. Restriction sites with a subscript “i” are inserted by means of PCR. The location of the transposon Tn917 in the transposon mutant nir1 is marked in the gene map (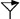 ). The promoter (Pnir) in front of the nir operon was mapped by primer extension analysis (Fig. 3), while the promoters upstream of sirA (Pstat, stationary-phase promoter) and nirC (PnirC) are postulated. Putative transcription terminors (
). The promoter (Pnir) in front of the nir operon was mapped by primer extension analysis (Fig. 3), while the promoters upstream of sirA (Pstat, stationary-phase promoter) and nirC (PnirC) are postulated. Putative transcription terminors (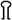 ) are indicated.
) are indicated.
Transposon mutagenesis and screening of mutants affected in nitrite reduction.
S. carnosus TM300 harboring pTV1ts, which contains the transposon Tn917 (33), was grown in BM containing chloramphenicol (20 μg/ml) and erythromycin (20 μg/ml) at 30°C to an optical density at 578 nm (OD578) of 1. The culture was then diluted 100-fold into BM supplemented with erythromycin (2.5 μg/ml) and cultivated for 16 h at 42°C. After two further cycles of cultivation at 42°C (first cycle, 500-fold dilution, 8 h of cultivation; second cycle, 100-fold dilution, 16 h of cultivation), the cells were spread on BM agar plates containing erythromycin (4 μg/ml) and incubated for 1 day at 37°C. Erythromycin-resistant and chloramphenicol-sensitive (Emr Cms) mutants were screened for their ability to reduce nitrite in microtiter plates containing modified BM supplemented with nitrite (1 mM). To decrease the oxygen tension, the medium was overlaid with paraffin oil. The inoculated microtiter plates were incubated at 37°C with shaking (120 rpm). The presence or absence of nitrite was analyzed after 20 h of incubation.
Nitrite reductase activity.
Nitrite reductase activity in intact cells was assayed with glucose as the electron donor, as described by Neubauer and Götz (17). Nitrite reductase activity in cell extracts was monitored as nitrite-specific NADH oxidation as described previously (17). For reconstitution of nitrite reductase activity, cells of strains M12 (pRP-ABDB) and nir1 were grown anaerobically with nitrite, harvested in the mid-exponential phase, washed twice with buffer (50 mM MOPS [morpholinepropanesulfonic acid], 150 mM NaCl, pH 7.0), and then lysed with lysostaphin at 37°C in equal volumes either separately or together. The lysates were incubated for up to 40 min. After subsequent centrifugation, the supernatants comprising mainly the cytosolic fractions were used for the determination of NADH-dependent nitrite reductase activity.
Nitrite reduction after shift from aerobiosis to anaerobiosis.
Cells were grown aerobically in modified BM supplemented either without or with nitrate (20 mM) or nitrite (2 mM). At an OD578 of 3, chloramphenicol (200 μM) or rifampin (200 μM) was added to one-third of each culture and the aerobic incubation was continued for 20 min. Subsequently, the nine different cell batches were washed twice with modified BM with or without antibiotic to remove nitrate and nitrite. All washing steps were performed at 4°C to minimize protein synthesis or enzyme activation at this stage. The cells were then resuspended to an OD578 of 2.8 in modified BM with or without chloramphenicol and switched to anaerobiosis by the addition of Oxyrase, and the tubes were transferred to a 37°C water bath. The experiment was started by addition of nitrite (600 μM without antibiotics, 100 μM with antibiotic). The nitrite concentrations in the different batches were monitored for up to 75 min.
DNA preparation, transformation, and molecular techniques.
Chromosomal DNAs from staphylococci were isolated according to the method of Marmur (15). E. coli plasmid DNA was prepared with a Qiafilter plasmid midi kit 100 (Qiagen, Hilden, Germany) according to the protocol supplied by the manufacturer. S. carnosus plasmid DNA was prepared essentially as described for E. coli, with the exception that lysostaphin (Sigma) was added to the lysis buffer (to 12.5 μg/ml). S. carnosus was transformed either by protoplast transformation (9) or by electroporation (2). Other molecular techniques followed established protocols (23).
DNA amplification by PCR, and DNA sequencing and analysis.
Vent polymerase (New England BioLabs, Schwalbach, Germany) was used for PCRs. rTrh (recombinant Thermus thermophilus) DNA polymerase XL and an X-large PCR kit were used for inverse PCR according to the protocol supplied by the manufacturer (Perkin-Elmer). When the PCR product was used for sequencing, the sequence was verified by direct sequencing of chromosomal DNA. Double-stranded plasmid and chromosomal DNAs were sequenced by using the dideoxy procedure, a Thermo Sequenase Fluorescent labeled primer cycle sequencing kit (Amersham), and a Li-Cor DNA Sequencer (Lincoln Cooperation). The program Gapped BLAST (1) was used for protein similarity searches.
Isolation of the nir promoter region.
A 1.9-kb EcoRI fragment (Fig. 1) from the wild-type genome adjacent to the SnaBI fragment was amplified by inverse PCR with ligated chromosomal EcoRI fragments as the template. The primers PraenirPCR1, 5′-GCCCCTTTGTTATCATCCAGCAC-3′, and PraenirPCR4, 5′-GCAAAGGCACGATGGTAAAATAATGCTCGCC-3′, bind to either of the two sides of the 0.2-kb overlapping region in opposite directions.
Homologous recombination.
For insertional inactivation, major parts of nirR and sirA were replaced by a 1.5-kb ermB fragment (3). For this, ermB from plasmid pEC2 (3), isolated by cleavage with EcoRI and PstI, with flanking regions of 1.3 kb upstream and 2.7 kb downstream of the disrupted genes was cloned in pRB473, yielding pRBCΔ12 (Fig. 1). The 1.3-kb fragment upstream of nirR was amplified by inverse PCR with the primers MutORF1, 5′-GCTCGCCTTCTGCTGAATTCTTACGTATAACTTGCGG-3′, and PraenirL2, 5′-GCCCCTTTGTTATCATCCAGCAC-3′, and with chromosomal DNA of wild-type S. carnosus as the template after EcoRI digestion and ligation. The introduced EcoRI site and a naturally occurring HindIII site were used for cloning. The downstream region was cloned in two fragments. The 0.8-kb fragment directly downstream was amplified with the primers MutORF2, 5′-GATATAGCAGCTGCAGATGTTGTGATTATCGCGACGG-3′, and NirBint, 5′-CACGCAGCATATCTCCTGCTTGACGG-3′, and digested with AvaI and PstI. The adjacent downstream fragment was obtained by AvaI cleavage of pR-AB. The four different fragments were ligated into HindIII- and AvaI-digested pRB473, cloned in E. coli SURE, and transformed into wild-type S. carnosus.
A similar strategy was used to construct the nirC mutant strain. Plasmid pRBΔC-RAB contained ermB together with upstream (0.7-kb) and downstream (4.0-kb) flanking regions of nirC (Fig. 1). The upstream fragment was amplified by inverse PCR as described above with the primers PraenirL2 (see above) and MutnirC3, 5′-GCCCCTAATAAGTTAATGGATCCTTCATTGATACCG-3′, and cleaved with EcoRI and BamHI. The downstream region was cloned in two fragments. With the primers MutnirC2, 5′-GAAGGCTTGTCTAAAGCTTTCTTTATCGC-3′, and MutnirC1, 5′-GCATGTGCAATCGTCATATCTGAGTTGCCC-3′, a 1-kb fragment directly downstream was amplified and digested with HindIII. The remaining 3.1-kb fragment further downstream was obtained by cleaving pR-AB with HindIII and PstI. Together with the BamHI- and HindIII-digested ermB, the three fragments were ligated into EcoRI- and PstI-digested pRB473, cloned in E. coli SURE, and transformed into wild-type S. carnosus.
Both insertional inactivation constructs were transferred to the chromosome by double crossover as described by Brückner (3). Plasmid loss and recombination occurred at frequencies of 1 and 74% for pRBCΔ12 and pRBΔC-RAB, respectively.
mRNA manipulations.
Total RNA was isolated as described by Sizemore et al. (26) from S. carnosus cells grown aerobically or anaerobically without or with nitrite (2 mM). IRD 800-labeled oligonucleotides (obtained from MWG-Biotech) complementary to positions +108 to +79 of the nirR gene relative to the transcription start site were used for primer extension reactions according to the method of Wagner et al. (30). For Northern blot analyses 15 μg of total RNA was separated according to size by electrophoresis through a denaturing agarose gel (final formaldehyde concentration, 1.06 M) and then transferred to a positively charged nylon membrane (Boehringer Mannheim). A DIG RNA labeling kit (SP6/T7) (Boehringer Mannheim) was used for preparing an RNA probe. As a probe, an 870-bp EcoRV fragment of the nir operon comprising the entire sirA gene, approximately 300 bp of the 5′ end of nirB, and approximately 100 bp of the 3′ end of nirR was chosen. Hybridization was carried out overnight at 68°C with 100 ng of RNA probe per ml of DIG Easy Hyb (Boehringer Mannheim). The subsequent washing steps and detection with CSPD [disodium 3-(4-methoxyspiro{1,2-dioxetane-3,2′-(5′-chloro)tricyclo[3.3.1.13,7]decan}-4-yl)phenyl phosphate] (Boehringer Mannheim) were carried out as recommended by the manufacturers. Digoxigenin-labeled RNA molecular weight marker I (Boehringer Mannheim) was used as a standard.
Analytical determinations.
The nitrite concentration was measured colorimetrically by the method of Nicholas and Nason (18) as modified by Showe and DeMoss (25). The protein concentration was determined as described by Lowry et al. (13) in the presence of sodium dodecyl sulfate (8) with bovine serum albumin as the standard.
Nucleotide sequence accession number.
The sequence of the genes involved in nitrite reduction has been assigned GenBank accession no. AF029224.
RESULTS AND DISCUSSION
The nitrite reductase operon. (i) Isolation of nitrite reductase-negative Tn917 insertion mutants of S. carnosus.
In order to identify genes involved in nitrite reduction in S. carnosus TM300, transposon mutagenesis was performed and 530 of the obtained mutants were analyzed in a rapid screening for their ability to reduce nitrite during anaerobic growth. Twenty-seven mutants (nir1 to nir27) were unable to reduce nitrite, which was confirmed by anaerobic cultivation with different concentrations of nitrite (0.1, 1, and 10 mM). Nitrite did not decrease during 20 h of incubation. It is unlikely that the nitrite reductase-negative phenotype is due to a general defect in anaerobic metabolism since the nitrate reductase activity of the mutants was unaffected.
(ii) Molecular characterization of the mutants.
The transposons in the genomes of mutants were localized by digesting chromosomal DNAs with various restriction endonucleases and probing the fragments with Tn917-specific DNA. The restriction patterns of the mutants were similar, indicating that the transposon hit the same chromosomal DNA region. Being representative of the various mutants, the transposon and flanking DNA of nir1 was cloned in pUC18 on a 4.5-kb SalI/EcoRI fragment from a partial library enriched for 3- to 6-kb fragments; the insert was identified by Southern hybridization. A similar strategy was used to isolate the corresponding intact DNA region from wild-type S. carnosus. After Southern hybridization with a 1.7-kb AvaI/EcoRI fragment flanking Tn917 in nir1, a 5.1-kb SnaBI fragment was cloned into the HincII site of pUC18, yielding pUCnir1-1, which was identified by Southern hybridization and restriction analysis. The SnaBI fragment was sequenced, and four ORFs transcribed in the same direction (Fig. 1) were identified. Potential ribosome-binding-site sequences were found at appropriate distances from the predicted start codons.
(iii) Sequence analysis.
In a computer-aided similarity search, the deduced amino acid sequences of the proteins revealed similarity to those of proteins involved in nitrite reduction (Table 1). The second and third ORFs encode proteins similar to NirB and NirD, respectively, the subunits of the dissimilatory NADH-dependent nitrite reductase of E. coli. The presence of NirBD is in agreement with our physiological results (17). The first ORF encodes a protein similar in sequence to the N terminus of E. coli CysG (residues 5 to 153), and the fourth ORF encodes a protein similar in sequence to the C-terminal portion of CysG (residues 216 to 450); the ORFs were termed sirA and sirB, respectively, to indicate that the enzyme activities of the trifunctional CysG might be attributed to two enzymes in S. carnosus. In E. coli, the C terminus of CysG catalyzes the S-adenosylmethionine-dependent methylation of uroporphyrinogen III to dihydrosirohydrochlorin, and the N terminus is important for pyridine-dinucleotide-dependent dehydrogenation of dihydrosirohydrochlorin to sirohydrochlorin and for the subsequent insertion of Fe2+ (31). It has been speculated that the E. coli enzyme arose by a gene fusion between a uroporphyrinogen III methylase and the oxidase and chelatase enzyme (31). Thus, the presence of the two separate enzymes in S. carnosus further supports this theory.
TABLE 1.
Sequence similarities of the proteins involved in nitrite reduction in S. carnosus to other known proteins
| S. carnosus protein (no. of aaa) | Identity (%) | Similarity (%) | No. of aa that overlap | Similar protein (no. of aa) |
|---|---|---|---|---|
| NirC (276) | 24.6 | 49.2 | 240 | E. coli NirC (268) |
| 26.7 | 50.5 | 206 | B. subtilis YrhG (266) | |
| NirR (240) | 22.1 | 42.3 | 253 | B. subtilis YlnE (261) |
| SirA (151) | 27.0 | 52.6 | 152 | B. subtilis YlnF (162) |
| 24.1 | 47.0 | 149 | E. coli CysG (457) | |
| NirB (801) | 52.0 | 87.7 | 796 | B. subtilis NasD (805) |
| 34.9 | 75.0 | 816 | E. coli NirB (847) | |
| NirD (104) | 49.5 | 83.2 | 101 | B. subtilis NasE (106) |
| 30.0 | 68.0 | 100 | E. coli NirD (102) | |
| SirB (317) | 37.2 | 56.1 | 285 | B. subtilis NasF (483) |
| 41.2 | 58.4 | 250 | B. subtilis YlnD (257) | |
| 37.0 | 74.1 | 243 | E. coli CysG (457) |
aa, amino acids.
The genomic organization of sirA, nirB, nirD, and sirB (some overlapping and some separated by short intergenic regions) suggests a transcriptional coupling and operon structure, and the operon was termed the nitrite reductase (nir) operon. A putative promoter region was suggested upstream of sirA. The transposon insertion site in nir1 was mapped to the beginning of nirB (Fig. 1). A predicted stem-loop structure as a potential rho-independent transcription terminator is located downstream of sirB (ΔG = −16.7 kcal/mol).
(iv) Complementation studies.
When nir1 was complemented with the genes sirA, nirB, nirD, and sirB together with a putative promoter region of 209 nucleotides (plasmid pR-ABDB) (Fig. 1), the rate of nitrite reduction during anaerobic growth was only 30% of that of the wild type and nitrite reduction started at the beginning of the stationary phase. Therefore, we speculated that the plasmid lacks the primary promoter region. The delayed nitrite reduction might be due to an additional weak promoter upstream of sirA or nirB that is switched on in the stationary phase. In nir1 complemented with only sirA and nirB (plasmid pR-AB) (Fig. 1) the rate of nitrite reduction was even lower (6%), suggesting that the transposon has a polar effect on nirD and sirB transcription and that no additional internal promoters are present upstream of nirD or sirB.
(v) Identification of the nir promoter region.
A potential ORF upstream of sirA that was incomplete on the SnaBI fragment was identified. An EcoRI chromosomal fragment that overlaps the SnaBI fragment was amplified by inverse PCR (see Materials and Methods), cloned, and sequenced. The ORF upstream of sirA was identified as the first gene of the nir operon. The gene product, NirR, displays low similarity to the deduced amino acid sequence of Bacillus subtilis ylnE (Table 1). Interestingly, ylnE is located between two genes, ylnD and ylnF, which are similar to sirB and sirA of S. carnosus, respectively (Table 1). Interestingly, ylnE is located between two genes, ylnD and ylnF, which are similar to sirB and sirA of S. carnosus, respectively (Table 1). Computer analyses predict that NirR is a cytoplasmic protein with an estimated molecular weight of 28,035. A region upstream of nirR contains the putative nir operon promoter, which was confirmed by primer extension analysis (see below, Fig. 3).
FIG. 3.

Primer extension analysis of the regulation of the nir promoter. The figure shows the reverse transcripts obtained with an IRD 800-labeled nir-specific primer (0.5 pmol) and 20 μg of total RNA (1 of 3 μl was loaded on the gel) isolated from mid-exponential-growth-phase S. carnosus wild-type cells grown in modified BM under aerobic (Ae) or anaerobic (An) conditions and in the absence or presence of nitrite (2 mM). Lanes A, C, G, and T contained the respective sequencing reaction mixtures. The arrow indicates the predominant +1 site.
(vi) Construction and analysis of a nirR sirA mutant.
To analyze the role of nirR and sirA in nitrite reduction, a mutant strain (M12) was constructed by homologous recombination in which the majority of the two genes was replaced by an erythromycin resistance cassette (plasmid pRBCΔ12-B) (Fig. 1). The inserted ermB gene was transcribed divergently from the nitrite reductase genes (Fig. 1). Proper recombination was checked by Southern blot analysis (data not shown). As expected, nitrite reduction in mutant M12 was completely abolished.
We constructed different plasmids in which all five genes (pR-PNO); the four genes sirA, nirB, nirD, and sirB (pR-PABDB); or only the genes nirB, nirD, and sirB (pR-PBDB) were placed under the control of the nir promoter (Fig. 1). Mutant M12 was transformed with each of the constructs, and the resulting strains were tested for nitrite reduction during anaerobic growth. In addition to nitrite, nitrate was added to the medium to determine whether nitrate reduction or the interaction of nitrite and nitrate reduction was affected in one of the strains. In all cell batches, nitrate was reduced prior to nitrite (Fig. 2B) at similar rates. The strains behaved differently only after nitrate had been reduced. Nitrite reduction was fully restored in cells containing plasmid pR-PNO, but no nitrite reduction occurred in cells containing pR-PBDB (Fig. 2B). In cells containing plasmid pR-PABDB, nitrite reduction began only after the cells reached the stationary phase and the rate was significantly lower than that of the wild type (Fig. 2B). These results indicate that NirR is important for nitrite reduction.
FIG. 2.

Levels of nitrate and nitrite reduction during anaerobic growth of wild-type S. carnosus (filled circles) and of the complementation clones M12 (pR-PBDB), M12 (pR-PABDB), and M12 (pR-PNO) (open triangles, filled squares, and open circles, respectively) in comparison to levels in the nirR sirA mutant M12 (open squares). The cells were cultivated anaerobically in modified BM supplemented with 500 μM nitrite and 400 μM nitrate. During growth, the OD578 (A) and levels of reduction in nitrate (monitored as nitrite increase) and nitrite (monitored as nitrite decrease) (B) were determined as described in Materials and Methods.
(vii) Functional analysis of NirR.
The amino acid sequence of NirR revealed no obvious DNA-binding motif and no similarity to any family of proteins in the PROSITE database. However, it should be mentioned that the N-terminal part of NirR (residues 22 to 90) reveals some identity (28%) to parts of the central domain of the transcriptional activator NifA of Bradyrhizobium japonicum (residues 349 to 419). The central domain is involved in interaction with ς54 (16).
NirR may have a regulatory function or an enzymatic function or be involved in enzyme assembly, e.g., in the insertion of siroheme into the apoenzyme. In the latter cases, it should be possible to reconstitute enzyme activity by combining cell extracts of a NirR− strain and a NirR+ strain. Induced cells of M12 (pR-PABDB) (a NirR− strain) and nir1 (a nitrite-reductase-negative but NirR+ strain) were harvested in the mid-exponential-growth phase and were lysed either separately or together. The cytosolic fractions of the two strains separately had no NADH-dependent nitrite reductase activity. Likewise, the combination of cytosolic fractions of the strains had no activity, not even after prolonged incubation (up to 40 min) of the extracts at 37°C. A gradual increase in activity would have been expected for an enzyme-catalyzed reaction. Therefore, we have no evidence of a role for NirR in enzyme reaction or in assembly.
We then determined whether NirR is important for transcription from the nir promoter. Nitrite reduction was analyzed in the nirR sirA mutant M12 complemented with sirA, nirB, nirD, and sirB with or without the nir promoter (plasmid pR-PABDB or pR-ABDB, respectively) (Fig. 1). As discussed above, M12 (pR-PABDB) showed reduced nitrite reductase activity, which was expressed only at the beginning of the stationary phase (Fig. 2). Surprisingly, the same result was obtained when the nir promoter was absent, i.e., with M12 (pR-ABDB) (data not shown). With the nir promoter and nirR (together with sirA, nirB, nirD, and sirB; plasmid pRPNO) full complementation was achieved (see above). These results suggest that the nir promoter (Pnir) is not functional in the absence of NirR. As already speculated above, the observed nitrite reduction is most likely due to an additional weak promoter upstream of sirA (Pstat) (Fig. 1). NirR may be involved in expression of nitrate reductase and nitrite reductase activity. However, in the nirR mutant M12, nitrate reductase activity after anaerobic growth with nitrite was similar to that of the wild type. Therefore, it is unlikely that NirR is involved in expression of nitrate reductase.
Regulation of nitrite reduction.
Aerobically growing cells of S. carnosus are unable to reduce nitrite, and nitrite was unable to induce nitrite reductase activity in the presence of oxygen (17), suggesting that induction of nitrite reductase is strictly coupled to anaerobiosis. In the absence of oxygen, nitrite further increases nitrite reductase activity (17). To analyze this induction process, we investigated the expression of nitrite reductase in more detail.
(i) Transcription initiation at the nir promoter in response to oxygen and nitrite.
Primer extension experiments with RNA isolated from cells grown anaerobically with nitrite revealed a predominant start site 17 nucleotides upstream of the predicted start codon of nirR (Fig. 3). Besides this predominant site, also other weaker transcription initiation sites were visible further upstream. For the predominant transcription initiation site, the sequences of the corresponding deduced −10 region (TACAAT) and −35 region (TTCACA) differ from the optimal consensus sequence for ς70-dependent promoters by one nucleotide each (boldface). The −35 region is located in an inverted repeat centered at −38.5 (TGTGAATXXATTCACA). Whether this palindrome is a binding site for a regulatory protein is not known.
To study the influence of anaerobiosis and nitrite at the transcriptional level, primer extension analyses were performed with RNA preparations from cells grown aerobically or anaerobically without or with nitrite (2 mM). The results depicted in Fig. 3 indicate that the amount of transcript is maximal during anaerobic growth with nitrite and minimal during aerobic and anaerobic growth without nitrite. Induction by nitrite was apparent during aerobic and during anaerobic growth. The presence of transcript in cells grown aerobically with or without nitrite was surprising, since no nitrite reductase activity was detectable under these conditions. In addition, anaerobically grown cells showed relatively low transcription initiation although they expressed relatively high enzyme activity. This result raised the question of whether a high rate of transcription initiation results in large amounts of full-length transcript.
(ii) Northern blot analyses.
Coinciding with the results from the primer extension studies, Northern blot analyses revealed small amounts of full-length transcript of the nir operon (approximately 5 kb) in cells grown aerobically with nitrite but no full-length transcript in cells grown aerobically without nitrite (Fig. 4). In cells grown anaerobically, large amounts of full-length transcript were detectable and the amount of full-length transcript seemed to be slightly greater when nitrite was present. The amount of full-length transcript in anaerobically grown cells does not reflect the rate of transcription initiation from the predominant start site. We speculate that this result might be due to transcription initiation from different start sites and/or that another unknown factor(s) favors large amounts of full-length transcript during anaerobic growth.
FIG. 4.

Northern blot analyses of the nir operon. Northern hybridization was performed as described in Materials and Methods. Total RNA (15 μg) isolated from cells grown aerobically (Ae) or anaerobically (An) with (+) or without (−) nitrite were transferred to a positively charged nylon membrane (Boehringer Mannheim). Northern hybridization was carried out with a digoxigenin-labeled RNA probe (comprising the entire sirA gene, approximately 300 bp of the 5′ end of nirB, and approximately 100 bp of the 3′ end of nirR). For chemiluminescence detection, CSPD (Boehringer Mannheim) was used. The full-length transcript of the nir operon (approximately 5 kb) is indicated by an arrow. As a standard, 90 ng of digoxigenin-labeled RNA molecular weight marker I (Boehringer Mannheim) was used.
(iii) Other oxygen-regulated steps are involved in nir expression and Nir activity.
Both primer extension studies and Northern blot analyses confirmed the presence of transcript in cells grown aerobically with nitrite which clearly lack nitrite reductase activity. This suggests that an additional mechanism(s) controls induction of nitrite reductase activity. Several steps are conceivable: (i) reversible or irreversible inhibition of the nitrite-reducing system by oxygen, (ii) competition for electrons with the aerobic electron transfer chain, (iii) oxygen-controlled regulation of translation, and (iv) oxygen-controlled enzyme folding and/or assembly.
Washed cells, grown anaerobically with nitrite, rapidly reduced nitrite when they were incubated anaerobically but decreased nitrite reduction immediately upon an increase in oxygen tension (Fig. 5). To test for inactivation of the induced nitrite-reducing system by oxygen, the induced cells were incubated with oxygen for up to 60 min. After this preincubation, the conditions were switched to anaerobiosis and the ability to reduce nitrite was detected. As a control, induced cells were incubated anaerobically for 60 min, which showed that even after extensive aerobic incubation nitrite reduction was fully restored after the switch to anoxic conditions (Fig. 5). This finding indicates that nitrite reduction, once the system is expressed, is reversibly inhibited by oxygen.
FIG. 5.

Reversible inhibition of nitrite reductase activity by oxygen. Induced cells (grown anaerobically with 2 mM nitrite) were incubated aerobically (for 10 min [filled circles], and for 60 min [filled triangles]) or anaerobically (for 60 min [open triangles]). After this preincubation, the conditions were switched to anaerobiosis and the ability of the cells to reduce nitrite was measured as nitrite decrease. In parallel, nitrite decrease was detected in the induced cells incubated aerobically (filled squares).
In a second experiment we analyzed the involvement of posttranscriptional regulatory mechanisms. Cells grown aerobically either with or without nitrite were switched to anaerobic growth with nitrite, and no antibiotic or rifampin or chloramphenicol was added. The development of nitrite reductase activity was monitored as nitrite decreases. When translation was blocked (by chloramphenicol), the nitrite concentrations remained constant (data not shown). This indicates that no activation occurs without protein biosynthesis, i.e., the simple lack of oxygen is not sufficient for activation. When transcription was inhibited (by rifampin), a low level of nitrite reductase activity developed (Fig. 6). This result correlates well with the amounts of transcript detected in the Northern blot (Fig. 4). Without antibiotics a high level of nitrite reductase activity was observed (Fig. 6). In accordance with the rifampin data, the induction process was accelerated when nitrite had been present in the aerobic preculture. A certain period is needed to create anaerobic conditions (by Oxyrase) and to allow the cells to perform energy-consuming processes such as protein biosynthesis after the washing steps at 4°C. This might explain the delayed development of enzyme activity in these experiments.
FIG. 6.

Influence of rifampin on induction of nitrite reductase activity after a shift from aerobiosis to anaerobiosis. After aerobic precultivation without (circles) or with (triangles) nitrite, no antibiotic or 200 μM rifampin was added to equal parts of each culture and the incubation was continued for 15 min. Subsequently, the remaining nitrite was removed by centrifugation and two washing steps. Cells were resuspended in modified BM with or without rifampin and incubated anaerobically at 37°C. After addition of nitrite, samples were taken at different time points and the amount of nitrite was monitored.
Taken together, our results suggest that a full-length transcript is present under aerobic conditions and that it is sufficiently stable. Either translation is inhibited during aerobic growth or protein folding or assembly is oxygen regulated. Considering that NADH-dependent nitrite reductase requires several prosthetic groups for activity (NADH, flavin adenine dinucleotide, non-heme iron-sulfur centers, siroheme), the assembly of the iron-sulfur clusters, for example, may be an oxygen-controlled step in the biogenesis of the enzyme. Although such a step is unfavorable from an energetic point of view, an incorrect assembly might lead to protein degradation.
The transporter NirC.
Upstream of the nir operon, we identified another ORF, which encodes a putative integral transmembrane protein of 276 amino acids (estimated molecular weight, 30,800). From hydrophobicity plots, it was estimated to have six membrane-spanning segments. The protein shows similarity to the putative E. coli nitrite transporter NirC (6) (Table 1). The E. coli nirC gene is located in the nir operon (21). A putative transcription termination structure was identified downstream of S. carnosus nirC (ΔG = −30.2 kcal/mol).
Construction and analysis of a nirC mutant.
By homologous recombination of plasmid pRBΔC-RAB onto the chromosome (Fig. 1), we constructed a mutant strain (MC11) in which nirC was replaced by an erythromycin resistance cassette transcribed in the direction opposite to that of the nitrite reductase genes. Proper recombination was confirmed by Southern blot analysis (data not shown). Nitrite reduction during anaerobic growth and growth at high concentrations of nitrite (100 mM) in this mutant were unaffected. To analyze a function in nitrite uptake, nitrite reduction in the nirC mutant was analyzed with increasing pH. Nitrite uptake at physiological pH might take place by passive diffusion of nitrous acid (14). Since the concentration of available nitrous acid decreases (pKa, 3.4) as the pH of the medium is raised, the contribution of active transport might be expected only at alkaline pH. However, levels of nitrite reduction at pH 8.6 and 9.6 were similar in the nirC mutant and in the wild type (80% of the activity at pH 7.2). Thus, NirC is not responsible for nitrite uptake at alkaline pH values.
A function of NirC in nitrite export is unlikely since levels of nitrate reduction and nitrite accumulation were similar to those in the wild type. The involvement of NirC in ammonia excretion was analyzed by measuring nitrite reduction with decreasing pH. It is conceivable that with decreasing pH, when the amount of protonated ammonia increases, passive diffusion is inefficient and that export becomes dependent on a transport system. In the wild type, levels of nitrite reductase activity were similar at pH 7.1 and 6.5 but decreased to about 20% of the former level at pH 5.6. However, this pattern was similar to that of the nirC mutant. Therefore, NirC probably does not influence ammonia excretion.
Also a higher nirC gene dosage, obtained by cloning nirC, including the putative promoter region, into the high-copy-number plasmid pRB473 (plasmid pRBnirC) (Fig. 1) in mutant MC11, did not lead to a recognizable change in the phenotype. Finally, there is still no evidence that S. carnosus nirC encodes a nitrite efflux or uptake system. This is in agreement with the results obtained with the homologous E. coli protein (6).
Our experiments indicate that the regulation of nitrite reduction occurs at various levels. The primary regulation is at the transcription level (Fig. 7). At least one regulatory protein mediates the presence of the effectors nitrite and oxygen. The induction in response to each effector can be further modulated by the presence of another. The first gene product of the nir operon, NirR, is obviously important for expression of nitrite reductase activity. Other steps also appear to be regulated by oxygen. Translation and protein folding and/or assembly may be sensitive to oxygen (Fig. 7), or the insertion of prosthetic groups (e.g., siroheme) might be a critical step. Once nitrite reductase is expressed (under oxygen starvation), the enzyme is active in the absence of oxygen but inactive in the presence of oxygen. Reversible mechanisms such as competition for electrons are probably responsible for this. Further investigations are needed to understand the whole complexity of the nitrite-reducing system of S. carnosus.
FIG. 7.

Model of nir expression and Nir activity. Three different oxygen-regulated steps are postulated and indicated by arrows. Whether the second regulated step is at the level of translation, protein folding, assembly, or insertion of prosthetic groups is not known. The detailed function of NirR for promoter activity is unclear.
As already suggested from the physiological characterization (17), the regulatory features together with the sequence characteristics speak in favor of a dissimilatory nitrite reductase. The characterization of the nitrite reductase-negative mutants nir1 (Tn917::nirB) and M12 (nirR sirA mutant) suggests that the enzyme described here is the only system present in S. carnosus under the chosen growth conditions.
ACKNOWLEDGMENTS
We are indebted to Reinhold Brückner, University of Tübingen, for providing plasmid pRB473. We thank Regine Stemmler, Silke Egner, and Vera Augsburger for expert technical assistance and Karen A. Brune for editing the manuscript.
This study was supported by grants from Nestlé Inc.
REFERENCES
- 1.Altschul S F, Madden T L, Schäffer A A, Zhang J, Zhang Z, Miller W, Lipman D J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Augustin J, Götz F. Transformation of Staphylococcus epidermidis and other staphylococcal species with plasmid DNA by electroporation. FEMS Microbiol Lett. 1990;66:203–208. doi: 10.1016/0378-1097(90)90283-v. [DOI] [PubMed] [Google Scholar]
- 3.Brückner R. Gene replacement in Staphylococcus carnosus and Staphylococcus xylosus. FEMS Microbiol Lett. 1997;151:1–8. doi: 10.1111/j.1574-6968.1997.tb10387.x. [DOI] [PubMed] [Google Scholar]
- 4.Brückner R. A series of shuttle vectors for Bacillus subtilis and Escherichia coli. Gene. 1992;122:187–192. doi: 10.1016/0378-1119(92)90048-t. [DOI] [PubMed] [Google Scholar]
- 5.Cole J. The rapid accumulation of large quantities of ammonia during nitrite reduction by Escherichia coli. FEMS Microbiol Lett. 1978;4:327–329. [Google Scholar]
- 6.Cole J A. Nitrate reduction to ammonia by enteric bacteria: redundancy, or a strategy for survival during oxygen starvation? FEMS Microbiol Lett. 1996;136:1–11. doi: 10.1111/j.1574-6968.1996.tb08017.x. [DOI] [PubMed] [Google Scholar]
- 7.Darwin A J, Stewart V. The NAR modulon systems: nitrate and nitrite regulation of anaerobic gene expression. In: Lin E C C, Lynch A S, editors. Regulation of anaerobic gene expression in Escherichia coli. R. G. Austin, Tex: Landes Company; 1996. pp. 343–359. [Google Scholar]
- 8.Dulley J R, Grieve P A. A simple technique for eliminating interference by detergents in the Lowry method of protein determination. Anal Biochem. 1975;64:136–140. doi: 10.1016/0003-2697(75)90415-7. [DOI] [PubMed] [Google Scholar]
- 9.Götz F, Schumacher B. Improvements of protoplast transformation in Staphylococcus carnosus. FEMS Microbiol Lett. 1987;40:285–288. [Google Scholar]
- 10.Guest J R, Green J, Irvine A S, Spiro S. The FNR modulon and FNR-regulated gene expression. In: Lin E C C, Lynch A S, editors. Regulation of anaerobic gene expression in Escherichia coli. R. G. Austin, Tex: Landes Company; 1996. pp. 317–342. [Google Scholar]
- 11.Harbourne N R, Griffiths L, Busby S J, Cole J A. Transcriptional control, translation and function of the products of the five open reading frames of the Escherichia coli nir operon. Mol Microbiol. 1992;6:2805–2813. doi: 10.1111/j.1365-2958.1992.tb01460.x. [DOI] [PubMed] [Google Scholar]
- 12.Jackson R H, Cornish-Bowden A, Cole J A. Prosthetic groups of the NADH-dependent nitrite reductase from Escherichia coli K12. Biochem J. 1981;193:861–867. doi: 10.1042/bj1930861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lowry O H, Rosebrough N J, Farr A J, Randall R J. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 14.Luque J, Flores E, Herrero A. Nitrate and nitrite transport in the cyanobacterium Synechococcus sp. PCC 7942 are mediated by the same permease. Biochim Biophys Acta. 1994;1184:296–298. [Google Scholar]
- 15.Marmur J. A procedure for isolation of deoxyribonucleic acid from microorganisms. J Mol Biol. 1961;3:208–218. [Google Scholar]
- 16.Morett E, Segovia L. The ς54 bacterial enhancer-binding protein family: mechanism of action and phylogenetic relationship of their functional domains. J Bacteriol. 1993;175:6067–6074. doi: 10.1128/jb.175.19.6067-6074.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Neubauer H, Götz F. Physiology and interaction of nitrate and nitrite reduction in Staphylococcus carnosus. J Bacteriol. 1996;178:2005–2009. doi: 10.1128/jb.178.7.2005-2009.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nicholas D J D, Nason A. Determination of nitrate and nitrite. Methods Enzymol. 1957;3:981–984. [Google Scholar]
- 19.Page L, Griffiths L, Cole J A. Different physiological roles of two independent pathways for nitrite reduction to ammonia by enteric bacteria. Arch Microbiol. 1990;154:349–354. doi: 10.1007/BF00276530. [DOI] [PubMed] [Google Scholar]
- 20.Pantel I, Lindgren P-E, Neubauer H, Götz F. Identification and characterization of the Staphylococcus carnosus narGHJI operon. Mol Gen Genet. 1998;259:105–114. doi: 10.1007/s004380050794. [DOI] [PubMed] [Google Scholar]
- 21.Peakman T, Crouzet J, Majaux J, Busby S, Mohan S, Harborne N, Wootton J, Nicolson R, Cole J. Nucleotide sequence, organization and structural analysis of the products of genes in the nirB-cysG region of the Escherichia coli K-12 chromosome. Eur J Biochem. 1990;191:315–323. doi: 10.1111/j.1432-1033.1990.tb19125.x. [DOI] [PubMed] [Google Scholar]
- 22.Rabin R S, Stewart V. Dual response regulators (NarL and NarP) interact with dual sensors (NarX and NarQ) to control nitrate- and nitrite-regulated gene expression in Escherichia coli K-12. J Bacteriol. 1993;175:3259–3268. doi: 10.1128/jb.175.11.3259-3268.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 24.Schleifer K H, Fischer U. Description of a new species of the genus Staphylococcus: Staphylococcus carnosus. Int J Syst Bacteriol. 1982;32:153–156. [Google Scholar]
- 25.Showe M K, DeMoss J A. Localization and regulation of synthesis of nitrate reductase in Escherichia coli. J Bacteriol. 1968;95:1305–1313. doi: 10.1128/jb.95.4.1305-1313.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sizemore C, Buchner E, Rygus T, Witke C, Götz F, Hillen W. Organization, promoter analysis and transcriptional regulation of the Staphylococcus xylosus xylose utilization operon. Mol Gen Genet. 1991;227:377–384. doi: 10.1007/BF00273926. [DOI] [PubMed] [Google Scholar]
- 27.Spencer J B, Stolowich N J, Roessner C A, Scott A I. The Escherichia coli cysG gene encodes the multifunctional protein, siroheme synthase. FEBS Lett. 1993;1:57–60. doi: 10.1016/0014-5793(93)80438-z. [DOI] [PubMed] [Google Scholar]
- 28.Tyson K L, Bell A I, Cole J A, Busby S J W. Definition of nitrite and nitrate responsive elements at the anaerobically inducible Escherichia coli nirB promoter: interactions between FNR and NarL. Mol Microbiol. 1993;7:151–157. doi: 10.1111/j.1365-2958.1993.tb01106.x. [DOI] [PubMed] [Google Scholar]
- 29.Vieira J, Messing J. The pUC plasmids, an M13mp7-derived system for insertion mutagenesis and sequencing with synthetic universal primers. Gene. 1982;19:259–268. doi: 10.1016/0378-1119(82)90015-4. [DOI] [PubMed] [Google Scholar]
- 30.Wagner E, Götz F, Brückner R. Cloning and characterization of the scrA gene encoding the sucrose-specific enzyme II of the phosphotransferase system from Staphylococcus xylosus. Mol Gen Genet. 1993;241:33–41. doi: 10.1007/BF00280198. [DOI] [PubMed] [Google Scholar]
- 31.Warren M J, Bolt E L, Roessner C A, Scott A I, Spencer J B, Woodcock S C. Gene dissection demonstrates that the Escherichia coli cysG gene encodes a multifunctional protein. Biochem J. 1994;302:837–844. doi: 10.1042/bj3020837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Williams S B, Stewart V. Nitrate- and nitrite-sensing protein NarX of Escherichia coli K-12: mutational analysis of the amino-terminal tail and first transmembrane segment. J Bacteriol. 1997;179:721–729. doi: 10.1128/jb.179.3.721-729.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Youngman P, Poth H, Green B, York K, Olmedo G, Smith K. Methods for genetic manipulation, cloning and functional analysis of sporulation genes in Bacillus subtilis. In: Smith I, Slepecky R A, Setlow P, editors. Regulation of procaryotic development. Washington, D.C: ASM Press; 1989. pp. 65–69. [Google Scholar]